
The Metabolic Stability of the Nicotinic Acetylcholine Receptor at the Neuromuscular Junction


{kind=link}
Abstract
1. Introduction
2. Turnover Rates of Achrs at Aneural, Developing and Mature NMJS
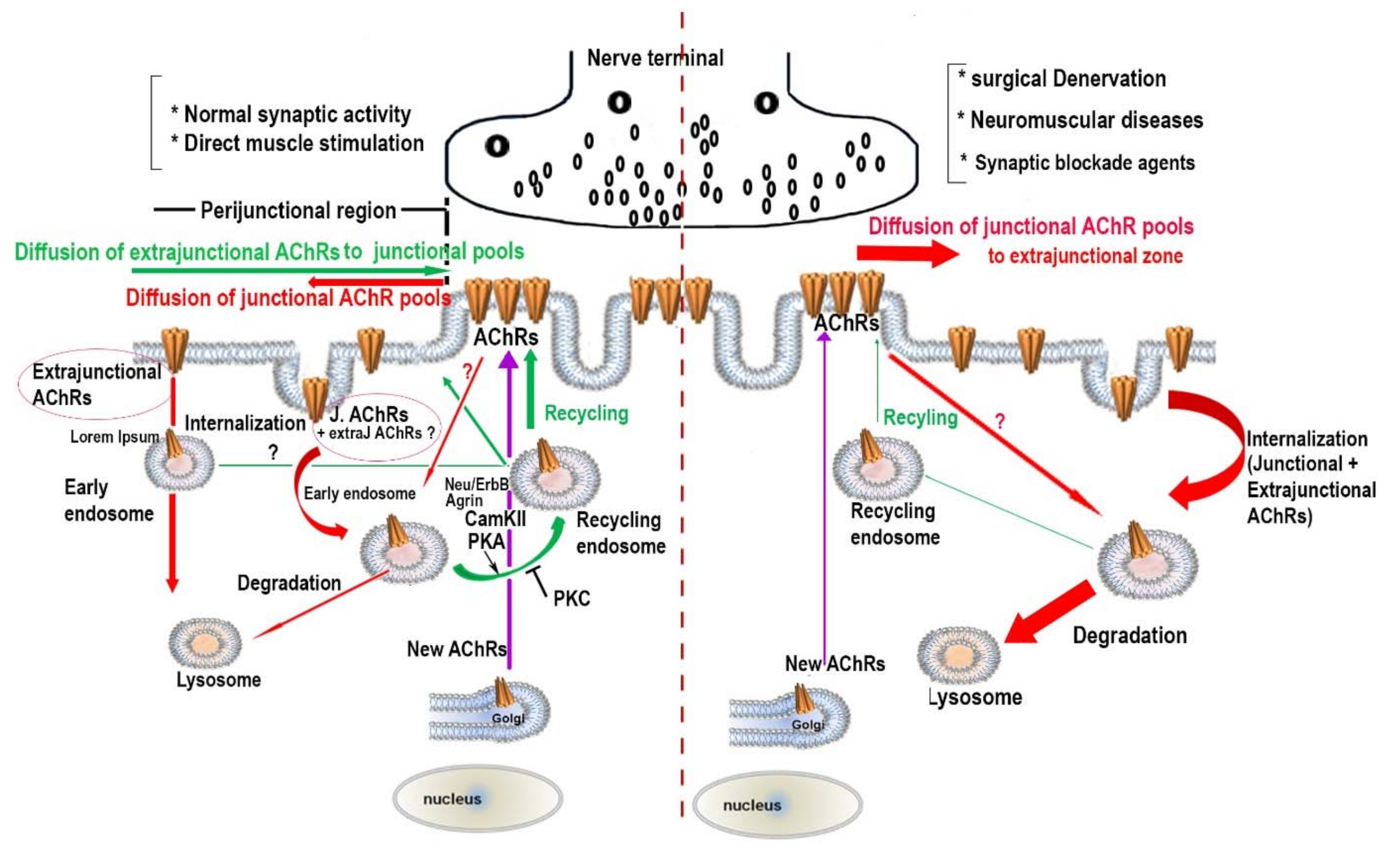
3. The Effect of Synaptic Activity on the Metabolic Stability of AChR Pools
4. The Regulatory Signaling Molecules Involved in the Metabolic Stability of AChRs at the NMJ
5. Concluding Remarks
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wu, H.; Xiong, W.C.; Mei, L. To build a synapse: Signaling pathways in neuromuscular junction assembly. Development 2010, 137, 1017–1033. [Google Scholar] [CrossRef] [PubMed]
- Sanes, J.R.; Lichtman, J.W. Development of the Vertebrate Neuromuscular Junction. Annu. Rev. Neurosci. 1999, 22, 389–442. [Google Scholar] [CrossRef] [PubMed]
- Tintignac, L.A.; Brenner, H.-R.; Rüegg, M.A. Mechanisms Regulating Neuromuscular Junction Development and Function and Causes of Muscle Wasting. Physiol. Rev. 2015, 95, 809–852. [Google Scholar] [CrossRef] [PubMed]
- Changeux, J.-P.; Devillers-Thiéry, A.; Galzi, J.-L.; Revah, F. The Acetylcholine Receptor: A Model of an Allosteric Membrane Protein Mediating Intercellular Communication. Novartis Found. Symp. 2007, 164, 66–97. [Google Scholar] [CrossRef]
- Mishina, M.; Takai, T.; Imoto, K.; Noda, M.; Takahashi, T.; Numa, S.; Methfessel, C.; Sakmann, B. Molecular distinction between fetal and adult forms of muscle acetylcholine receptor. Nat. Cell Biol. 1986, 321, 406–411. [Google Scholar] [CrossRef]
- Missias, A.C.; Mudd, J.; Cunningham, J.M.; Steinbach, J.H.; Merlie, J.P.; Sanes, J.R. Deficient development and maintenance of postsynaptic specializations in mutant mice lacking an ’adult’ acetylcholine receptor subunit. Development 1997, 124, 5075–5086. [Google Scholar]
- Sakmann, B.; Brenner, H.R. Change in synaptic channel gating during neuromuscular development. Nat. Cell Biol. 1978, 276, 401–402. [Google Scholar] [CrossRef]
- Yampolsky, P.; Gensler, S.; McArdle, J.; Witzemann, V. AChR channel conversion and AChR-adjusted neuronal survival during embryonic development. Mol. Cell. Neurosci. 2008, 37, 634–645. [Google Scholar] [CrossRef]
- Witzemann, V.; Chevessier, F.; Pacifici, P.G.; Yampolsky, P. The neuromuscular junction: Selective remodeling of synaptic regulators at the nerve/muscle interface. Mech. Dev. 2013, 130, 402–411. [Google Scholar] [CrossRef]
- Fischbach, G.D.; Schuetze, S.M. A post-natal decrease in acetylcholine channel open time at rat end-plates. J. Physiol. 1980, 303, 125–137. [Google Scholar] [CrossRef]
- Yampolsky, P.; Pacifici, P.G.; Lomb, L.; Giese, G.; Rudolf, R.; Röder, I.V.; Witzemann, V. Time Lapse in Vivo Visualization of Developmental Stabilization of Synaptic Receptors at Neuromuscular Junctions. J. Biol. Chem. 2010, 285, 34589–34596. [Google Scholar] [CrossRef]
- Geng, L.; Zhang, H.; Peng, H.B. The formation of acetylcholine receptor clusters visualized with quantum dots. BMC Neurosci. 2009, 10, 80. [Google Scholar] [CrossRef] [PubMed]
- Barik, A.; Zhang, B.; Sohal, G.S.; Xiong, W.-C.; Mei, L. Crosstalk between Agrin and Wnt signaling pathways in development of vertebrate neuromuscular junction. Dev. Neurobiol. 2014, 74, 828–838. [Google Scholar] [CrossRef] [PubMed]
- Singhal, N.; Martin, P.T. Role of extracellular matrix proteins and their receptors in the development of the vertebrate neuromuscular junction. Dev. Neurobiol. 2011, 71, 982–1005. [Google Scholar] [CrossRef] [PubMed]
- Grady, R.M.; Zhou, H.; Cunningham, J.M.; Henry, M.D.; Campbell, K.P.; Sanes, J.R. Maturation and maintenance of the neuromuscular synapse: Genetic evidence for roles of the dystrophin--glycoprotein complex. Neuron 2000, 25, 279–293. [Google Scholar] [CrossRef]
- Adams, M.E.; Kramarcy, N.R.; Krall, S.P.; Rossi, S.G.; Rotundo, R.L.; Sealock, R.; Froehner, S.C. Absence of α-Syntrophin Leads to Structurally Aberrant Neuromuscular Synapses Deficient in Utrophin. J. Cell Biol. 2000, 150, 1385–1398. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela, I.M.-P.Y.; Mouslim, C.; Pires-Oliveira, M.; Adams, M.E.; Froehner, S.C.; Akaaboune, M. Nicotinic Acetylcholine Receptor Stability at the NMJ Deficient in -Syntrophin In Vivo. J. Neurosci. 2011, 31, 15586–15596. [Google Scholar] [CrossRef]
- Rafael, J.A.; Townsend, E.R.; Squire, S.E.; Potter, A.C.; Chamberlain, J.S.; Davies, K.E. Dystrophin and utrophin influence fiber type composition and post-synaptic membrane structure. Hum. Mol. Genet. 2000, 9, 1357–1367. [Google Scholar] [CrossRef]
- Lyons, P.R.; Slater, C.R. Structure and Function of the Neuromuscular-Junction in Young-Adult Mdx Mice. J. Neurocytol. 1991, 20, 969–981. [Google Scholar] [CrossRef]
- Bruneau, E.G.; Brenner, D.S.; Kuwada, J.Y.; Akaaboune, M. Acetylcholine Receptor Clustering Is Required for the Accumulation and Maintenance of Scaffolding Proteins. Curr. Biol. 2008, 18, 109–115. [Google Scholar] [CrossRef]
- Chen, P.-J.; Valenzuela, I.M.-P.Y.; Aittaleb, M.; Akaaboune, M. AChRs Are Essential for the Targeting of Rapsyn to the Postsynaptic Membrane of NMJs in Living Mice. J. Neurosci. 2016, 36, 5680–5685. [Google Scholar] [CrossRef]
- Park, J.-Y.; Ikeda, H.; Ikenaga, T.; Ono, F. Acetylcholine Receptors Enable the Transport of Rapsyn from the Golgi Complex to the Plasma Membrane. J. Neurosci. 2012, 32, 7356–7363. [Google Scholar] [CrossRef]
- Ono, F.; Mandel, G.; Brehm, P. Acetylcholine Receptors Direct Rapsyn Clusters to the Neuromuscular Synapse in Zebrafish. J. Neurosci. 2004, 24, 5475–5481. [Google Scholar] [CrossRef]
- Witzemann, V.; Schwarz, H.; Koenen, M.; Berberich, C.; Villarroel, A.; Wernig, A.; Brenner, H.R.; Sakmann, B. Acetylcholine receptor epsilon-subunit deletion causes muscle weakness and atrophy in juvenile and adult mice. Proc. Natl. Acad. Sci. USA 1996, 93, 13286–13291. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, H.; Giese, G.; Müller, H.; Koenen, M.; Witzemann, V. Different functions of fetal and adult AChR subtypes for the formation and maintenance of neuromuscular synapses revealed in epsilon-subunit-deficient mice. Eur. J. Neurosci. 2000, 12, 3107–3116. [Google Scholar] [CrossRef]
- Lukas, R.J.; Morimoto, H.; Hanley, M.R.; Bennett, E.L. Radiolabeled. alpha.-bungarotoxin derivatives: Kinetic interaction with nicotinic acetylcholine receptors. Biochemistry 1981, 20, 7373–7378. [Google Scholar] [CrossRef] [PubMed]
- Brockes, J.P.; Hall, Z.W. Acetylcholine receptors in normal and denervated rat diaphragm muscle. II. Comparison of junctional and extrajunctional receptors. Biochemistry 1975, 14, 2100–2106. [Google Scholar] [CrossRef] [PubMed]
- Bevan, S.; Steinbach, J.H. The distribution of alpha-bungarotoxin binding sites of mammalian skeletal muscle developing in vivo. J. Physiol. 1977, 267, 195–213. [Google Scholar] [CrossRef] [PubMed]
- Berg, D.K.; Hall, Z.W. Loss of alpha-bungarotoxin from junctional and extrajunctional acetylcholine receptors in rat diaphragm muscle in vivo and in organ culture. J. Physiol. 1975, 252, 771–789. [Google Scholar] [CrossRef] [PubMed]
- Brockes, J.P.; Hall, Z.W. Acetylcholine receptors in normal and denervated rat diaphragm muscle. I. Purification and interaction with [125I]-alpha-bungarotoxin. Biochemistry 1975, 14, 2092–2099. [Google Scholar] [CrossRef] [PubMed]
- Patrick, J.; Heinemann, S.F.; Lindström, J.; Schubert, D.; Steinbach, J.H. Appearance of Acetylcholine Receptors During Differentiation of a Myogenic Cell Line. Proc. Natl. Acad. Sci. USA 1972, 69, 2762–2766. [Google Scholar] [CrossRef]
- Akaaboune, M.; Culican, S.M.; Turney, S.G.; Lichtman, J.W.; Faure, S.; Meyer, L.; Costagliola, D.; Vaneensberghe, C.; Genin, E.; Autran, B.; et al. Rapid and Reversible Effects of Activity on Acetylcholine Receptor Density at the Neuromuscular Junction in Vivo. Science 1999, 286, 503–507. [Google Scholar] [CrossRef] [PubMed]
- Akaaboune, M.; Grady, R.; Turney, S.; Sanes, J.R.; Lichtman, J.W. Neurotransmitter Receptor Dynamics Studied In Vivo by Reversible Photo-Unbinding of Fluorescent Ligands. Neuron 2002, 34, 865–876. [Google Scholar] [CrossRef]
- Fumagalli, G.; Engel, A.G.; Lindstrom, J. Ultrastructural Aspects of Acetylcholine Receptor Turnover at the Normal End-plate and in Autoimmune Myasthenia Gravis. J. Neuropathol. Exp. Neurol. 1982, 41, 567–579. [Google Scholar] [CrossRef]
- Bruneau, E.G.; Akaaboune, M. Running to stand still: Ionotropic receptor dynamics at central and peripheral synapses. Mol. Neurobiol. 2006, 34, 137–151. [Google Scholar] [CrossRef]
- Levitt, T.A.; Loring, R.H.; Salpeter, M.M. Neuronal control of acetylcholine receptor turnover rate at a vertebrate neuromuscular junction. Science 1980, 210, 550–551. [Google Scholar] [CrossRef] [PubMed]
- Bruneau, E.; Sutter, D.; Hume, R.I.; Akaaboune, M.; David, C. Identification of Nicotinic Acetylcholine Receptor Recycling and Its Role in Maintaining Receptor Density at the Neuromuscular Junction In Vivo. J. Neurosci. 2005, 25, 9949–9959. [Google Scholar] [CrossRef]
- Bruneau, E.G.; Akaaboune, M. The dynamics of recycled acetylcholine receptors at the neuromuscular junction in vivo. Dev. 2006, 133, 4485–4493. [Google Scholar] [CrossRef]
- Dreyer, F.; Walther, C.; Peper, K. Junctional and Extrajunctional Acetylcholine Receptors in Normal and Denervated Frog Muscle-Fibers—Noise-Analysis Experiments with Different Agonists. Pflug. Arch.-Eur. J. Physiol. 1976, 366, 1–9. [Google Scholar] [CrossRef]
- Chang, C.C.; Huang, M.C. Turnover of junctional and extrajunctional acetylcholine receptors of the rat diaphragm. Nat. Cell Biol. 1975, 253, 643–644. [Google Scholar] [CrossRef] [PubMed]
- Reiness, C.G.; Hall, Z.W.; Weinberg, C.B. Antibody to acetylcholine receptor increases degradation of junctional and extrajunctional receptors in adult muscle. Nat. Cell Biol. 1978, 274, 68–70. [Google Scholar] [CrossRef]
- Linden, D.C.; Fambrough, D. Biosynthesis and degradation of acetylcholine receptors in rat skeletal muscles. Effects of electrical stimulation. Neuroscience 1979, 4, 527–538. [Google Scholar] [CrossRef]
- Miller, J.B. Regulation of acetylcholine receptors in the mouse muscle cell line, C2*1. Exp. Cell Res. 1984, 154, 256–269. [Google Scholar] [CrossRef]
- Trinidad, J.C.; Cohen, J.B. Neuregulin Inhibits Acetylcholine Receptor Aggregation in Myotubes. J. Biol. Chem. 2004, 279, 31622–31628. [Google Scholar] [CrossRef]
- O’Malley, J.P.; Rubin, L.L.; Salpeter, M.M. Two Populations of AChR in Rat Myotubes Have Different Degradation Rates and Responses to cAMP. Exp. Cell Res. 1993, 208, 44–47. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Nelson, P.G. Involvement of calpains in the destabilization of the acetylcholine receptor clusters in rat myotubes. J. Neurobiol. 2000, 42, 22–32. [Google Scholar] [CrossRef]
- Bruneau, E.; MacPherson, P.C.; Goldman, D.; Hume, R.I.; Akaaboune, M. The effect of agrin and laminin on acetylcholine receptor dynamics in vitro. Dev. Biol. 2005, 288, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Devreotes, P.N.; Fambrough, D.M. Acetylcholine receptor turnover in membranes of developing muscle fibers. J. Cell Biol. 1975, 65, 335–358. [Google Scholar] [CrossRef]
- Burden, S. Development of the neuromuscular junction in the chick embryo: The number, distribution, and stability of acetylcholine receptors. Dev. Biol. 1977, 57, 317–329. [Google Scholar] [CrossRef]
- Steinbach, J.H.; Merlie, J.; Heinemann, S.; Bloch, R. Degradation of junctional and extrajunctional acetylcholine receptors by developing rat skeletal muscle. Proc. Natl. Acad. Sci. USA 1979, 76, 3547–3551. [Google Scholar] [CrossRef]
- Burden, S. Acetylcholine receptors at the neuromuscular junction: Developmental change in receptor turnover. Dev. Biol. 1977, 61, 79–85. [Google Scholar] [CrossRef]
- Stanley, E.F.; Drachman, D.B. Denervation accelerates the degradation of junctional acetylcholine receptors. Exp. Neurol. 1981, 73, 390–396. [Google Scholar] [CrossRef]
- Pumplin, D.W.; Fambrough, D.M. Turnover of Acetylcholine Receptors in Skeletal Muscle. Annu. Rev. Physiol. 1982, 44, 319–335. [Google Scholar] [CrossRef]
- Devreotes, P.N.; Fambrough, D.M. Turnover of Acetylcholine Receptors in Skeletal Muscle. Cold Spring Harb. Symp. Quant. Biol. 1976, 40, 237–251. [Google Scholar] [CrossRef]
- Fumagalli, G.; Balbi, S.; Cangiano, A.; Lømo, T. Regulation of turnover and number of acetylcholine receptors at neuromuscular junctions. Neuron 1990, 4, 563–569. [Google Scholar] [CrossRef]
- Engel, A.G.; Lindstrom, J.M.; Lambert, E.H.; Lennon, V.A. Ultrastructural localization of the acetylcholine receptor in myasthenia gravis and in its experimental autoimmune model. Neurology 1977, 27, 307. [Google Scholar] [CrossRef]
- Fambrough, D.M.; Drachman, D.B.; Satyamurti, S. Neuromuscular Junction in Myasthenia Gravis: Decreased Acetylcholine Receptors. Science 1973, 182, 293–295. [Google Scholar] [CrossRef]
- Heinemann, S.; Merlie, J.; Lindström, J. Modulation of acetylcholine receptor in rat diaphragm by anti-receptor sera. Nat. Cell Biol. 1978, 274, 65–68. [Google Scholar] [CrossRef]
- Hudgson, P.; McAdams, M.W.; Pericak-Vance, M.A.; Edwards, A.M.; Roses, A.D. Effect of sera from myasthenia gravis patients on acetylcholine receptors in myotube cultures. J. Neurol. Sci. 1983, 59, 37–45. [Google Scholar] [CrossRef]
- Martinez-Pena, Y.V.I.; Akaaboune, M. The disassembly of the neuromuscular synapse in high-fat diet-induced obese male mice. Mol. Metab. 2020, 36, 100979. [Google Scholar] [CrossRef] [PubMed]
- Brenner, H.R.; Rudin, W. On the effect of muscle activity on the end-plate membrane in denervated mouse muscle. J. Physiol. 1989, 410, 501–512. [Google Scholar] [CrossRef]
- Rotzler, S.; Brenner, H.R. Metabolic stabilization of acetylcholine receptors in vertebrate neuromuscular junction by muscle activity. J. Cell Biol. 1990, 111, 655–661. [Google Scholar] [CrossRef]
- Shyng, S.L.; Salpeter, M.M. Degradation rate of acetylcholine receptors inserted into denervated vertebrate neuromuscular junctions. J. Cell Biol. 1989, 108, 647–651. [Google Scholar] [CrossRef]
- Rotzler, S.; Schramek, H.; Brenner, H.R. Metabolic stabilization of endplate acetylcholine receptors regulated by Ca2+ influx associated with muscle activity. Nat. Cell Biol. 1991, 349, 337–339. [Google Scholar] [CrossRef] [PubMed]
- Andreose, J.; Xu, R.; Lomo, T.; Salpeter, M.; Fumagalli, G. Degradation of two AChR populations at rat neuromuscular junctions: Regulation in vivo by electrical stimulation. J. Neurosci. 1993, 13, 3433–3438. [Google Scholar] [CrossRef]
- Valenzuela, I.M.-P.Y.; Mouslim, C.; Akaaboune, M. Calcium/Calmodulin Kinase II-Dependent Acetylcholine Receptor Cycling at the Mammalian Neuromuscular Junction In Vivo. J. Neurosci. 2010, 30, 12455–12465. [Google Scholar] [CrossRef]
- Lockshin, R.A.; Zakeri, Z. Apoptosis, autophagy, and more. Int. J. Biochem. Cell Biol. 2004, 36, 2405–2419. [Google Scholar] [CrossRef]
- Kumari, S.; Borroni, V.; Chaudhry, A.; Chanda, B.; Massol, R.; Mayor, S.; Barrantes, F.J. Nicotinic acetylcholine receptor is internalized via a Rac-dependent, dynamin-independent endocytic pathway. J. Cell Biol. 2008, 181, 1179–1193. [Google Scholar] [CrossRef]
- Schmidt, N.; Akaaboune, M.; Gajendran, N.; Valenzuela, I.M.-P.Y.; Wakefield, S.; Thurnheer, R.; Brenner, H.-R. Neuregulin/ErbB regulate neuromuscular junction development by phosphorylation of α-dystrobrevin. J. Cell Biol. 2011, 195, 1171–1184. [Google Scholar] [CrossRef] [PubMed]
- Rudolf, R.; Straka, T. Nicotinic acetylcholine receptor at vertebrate motor endplates: Endocytosis, recycling, and degradation. Neurosci. Lett. 2019, 711, 134434. [Google Scholar] [CrossRef]
- Khan, M.M.; Strack, S.; Wild, F.; Hanashima, A.; Gasch, A.; Brohm, K.; Reischl, M.; Carnio, S.; Labeit, D.; Sandri, M.; et al. Role of autophagy, SQSTM1, SH3GLB1, and TRIM63 in the turnover of nicotinic acetylcholine receptors. Autophagy 2014, 10, 123–136. [Google Scholar] [CrossRef]
- Rudolf, R.; Bogomolovas, J.; Strack, S.; Choi, K.-R.; Khan, M.M.; Wagner, A.; Brohm, K.; Hanashima, A.; Gasch, A.; Labeit, D.; et al. Regulation of nicotinic acetylcholine receptor turnover by MuRF1 connects muscle activity to endo/lysosomal and atrophy pathways. AGE 2012, 35, 1663–1674. [Google Scholar] [CrossRef]
- Wild, F.; Khan, M.M.; Straka, T.; Rudolf, R. Progress of endocytic CHRN to autophagic degradation is regulated by RAB5-GTPase and T145 phosphorylation of SH3GLB1 at mouse neuromuscular junctions in vivo. Autophagy 2016, 12, 2300–2310. [Google Scholar] [CrossRef]
- Valenzuela, I.M.-P.Y.; Pires-Oliveira, M.; Akaaboune, M. PKC and PKA Regulate AChR Dynamics at the Neuromuscular Junction of Living Mice. PLoS ONE 2013, 8, e81311. [Google Scholar] [CrossRef]
- Röder, I.V.; Choi, K.R.; Reischl, M.; Petersen, Y.; Diefenbacher, M.E.; Zaccolo, M.; Pozzan, T.; Rudolf, R. Myosin Va cooperates with PKA RIalpha to mediate maintenance of the endplate in vivo. Proc. Natl. Acad. Sci. USA 2010, 107, 2031–2036. [Google Scholar] [CrossRef]
- Choi, K.-R.; Berrera, M.; Reischl, M.; Strack, S.; Albrizio, M.; Roder, I.V.; Wägner, A.; Petersen, Y.; Häfner, M.; Zaccolo, M.; et al. Rapsyn mediates subsynaptic anchoring of PKA type I and stabilisation of acetylcholine receptor in vivo. J. Cell Sci. 2012, 125, 714–723. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.M.; Lustrino, D.; Silveira, W.A.; Wild, F.; Straka, T.; Issop, Y.; O’Connor, E.; Cox, D.J.; Reischl, M.; Marquardt, T.; et al. Sympathetic innervation controls homeostasis of neuromuscular junctions in health and disease. Proc. Natl. Acad. Sci. USA 2016, 113, 746–750. [Google Scholar] [CrossRef]
- Laufer, R.; Changeux, J.P. Calcitonin gene-related peptide elevates cyclic AMP levels in chick skeletal muscle: PossibleS neurotrophic role for a coexisting neuronal messenger. EMBO J. 1987, 6, 901–906. [Google Scholar] [CrossRef]
- Shyng, S.; Xu, R.; Salpeter, M. Cyclic AMP stabilizes the degradation of original junctional acetylcholine receptors in denervated muscle. Neuron 1991, 6, 469–475. [Google Scholar] [CrossRef]
- Perkins, G.A.; Wang, L.; Huang, L.J.-S.; Humphries, K.M.; Yao, V.J.; Martone, M.; Deerinck, T.; Barraclough, D.M.; Violin, J.D.; Smith, D.; et al. PKA, PKC, and AKAP localization in and around the neuromuscular junction. BMC Neurosci. 2001, 2, 17. [Google Scholar] [CrossRef]
- Dai, Z.; Peng, H.B. A Role of Tyrosine Phosphatase in Acetylcholine Receptor Cluster Dispersal and Formation. J. Cell Biol. 1998, 141, 1613–1624. [Google Scholar] [CrossRef]
- Lee, C.W.; Han, J.; Bamburg, J.R.; Han, L.; Lynn, R.; Zheng, J.Q. Regulation of acetylcholine receptor clustering by ADF/cofilin-directed vesicular trafficking. Nat. Neurosci. 2009, 12, 848–856. [Google Scholar] [CrossRef] [PubMed]
- Brenner, H.-R.; Akaaboune, M. Recycling of acetylcholine receptors at ectopic postsynaptic clusters induced by exogenous agrin in living rats. Dev. Biol. 2014, 394, 122–128. [Google Scholar] [CrossRef][Green Version]
- Mouslim, C.; Aittaleb, M.; Hume, R.I.; Akaaboune, M. A Role for the Calmodulin Kinase II-Related Anchoring Protein (kap) in Maintaining the Stability of Nicotinic Acetylcholine Receptors. J. Neurosci. 2012, 32, 5177–5185. [Google Scholar] [CrossRef] [PubMed]
- y Valenzuela, I.M.; Aittaleb, M.; Chen, P.J.; Akaaboune, M. The knockdown of alphakap alters the postsynaptic apparatus of neuromuscular junctions in living mice. J. Neurosci. 2015, 35, 5118–5127. [Google Scholar] [CrossRef]
- Escher, P.; Lacazette, E.; Courtet, M.; Blindenbacher, A.; Landmann, L.; Bezakova, G.; Lloyd, K.C.; Mueller, U.; Brenner, H.R. Synapses Form in Skeletal Muscles Lacking Neuregulin Receptors. Science 2005, 308, 1920–1923. [Google Scholar] [CrossRef]
- Belhasan, D.C.; Akaaboune, M. The role of the dystrophin glycoprotein complex on the neuromuscular system. Neurosci. Lett. 2020, 722, 134833. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martinez-Pena y Valenzuela, I.; Akaaboune, M. The Metabolic Stability of the Nicotinic Acetylcholine Receptor at the Neuromuscular Junction. Cells 2021, 10, 358. https://doi.org/10.3390/cells10020358
Martinez-Pena y Valenzuela I, Akaaboune M. The Metabolic Stability of the Nicotinic Acetylcholine Receptor at the Neuromuscular Junction. Cells. 2021; 10(2):358. https://doi.org/10.3390/cells10020358
Chicago/Turabian StyleMartinez-Pena y Valenzuela, Isabel, and Mohammed Akaaboune. 2021. "The Metabolic Stability of the Nicotinic Acetylcholine Receptor at the Neuromuscular Junction" Cells 10, no. 2: 358. https://doi.org/10.3390/cells10020358
APA StyleMartinez-Pena y Valenzuela, I., & Akaaboune, M. (2021). The Metabolic Stability of the Nicotinic Acetylcholine Receptor at the Neuromuscular Junction. Cells, 10(2), 358. https://doi.org/10.3390/cells10020358