
New Insights into the Link between Melanoma and Thyroid Cancer: Role of Nucleocytoplasmic Trafficking


 , , and
, , and
Abstract
:1. The Connection between Melanoma and Thyroid Cancer: Our Up-to-date Knowledge
2. Dysregulation of the Nucleocytoplasmic Trafficking
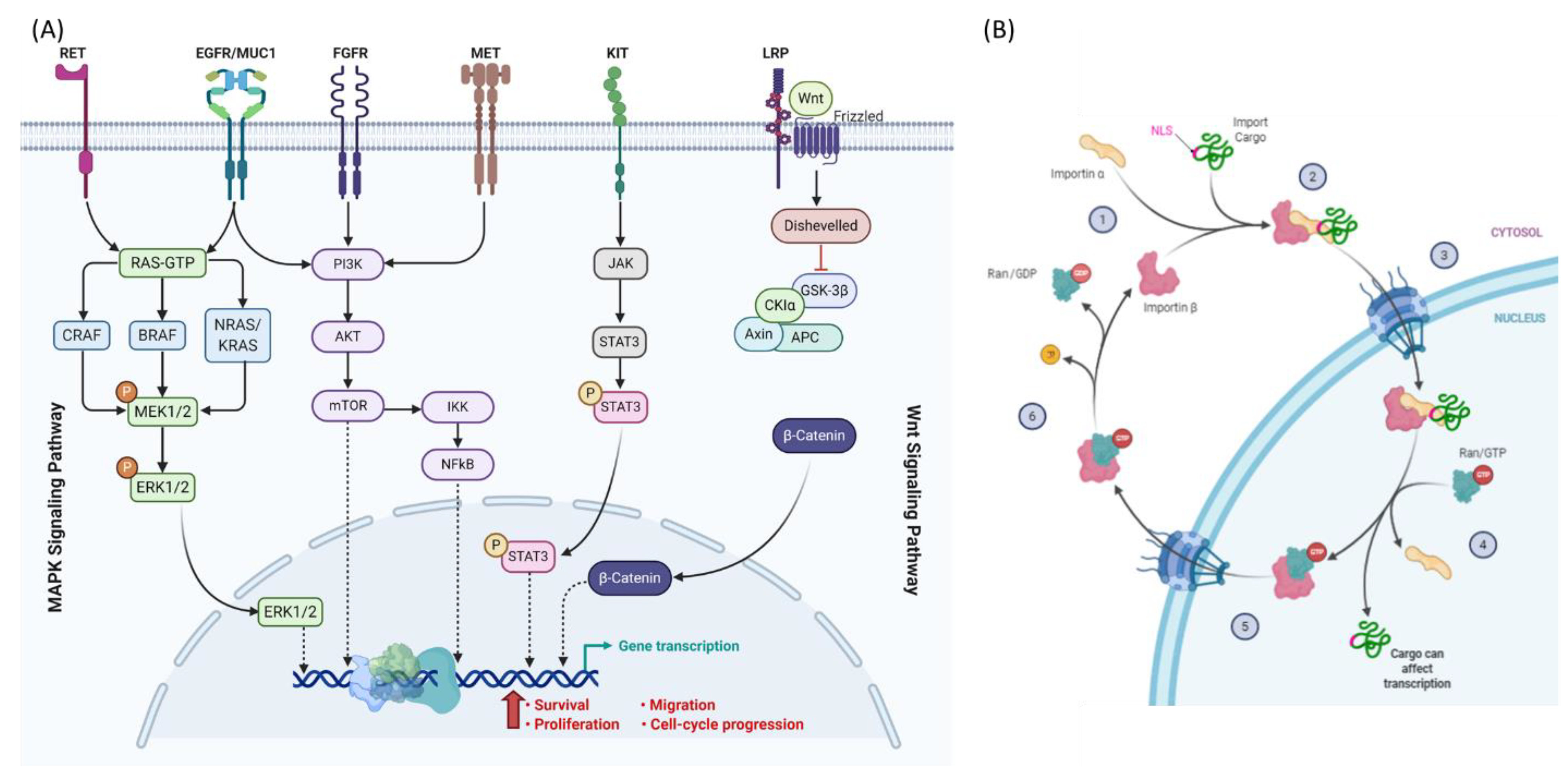
3. Dysregulation of Nucleocytoplasmic Transport in Melanoma and Thyroid Cancer
4. Nucleocytoplasmic Transport and Mechanisms of Resistance in Cancer
- Galectin-3: It interacts with a wide range of partners and has multiple activities in cancer cells. Subcellular localization of Galectin-3 is important for its function as a regulator of apoptosis [97]. Phosphorylated cytoplasmic Galectin-3 activates ERK and c-Jun N-terminal kinase (JNK), resulting in subsequent suppression of apoptosis in cancerous cells. Treatment with cisplatin, a pro-apoptotic agent, can lead to movement of Galectin-3 to the cytoplasm, resulting in drug resistance. CRM1 inhibition by leptomycin B prevents nuclear export of Galectin-3 and restores cisplatin-induced apoptosis in cancer cells [98].
- Topoisomerase IIα: Cancer cells can develop drug resistance to the cytotoxic effects of topoisomerase II inhibitors like doxorubicin by exporting topoisomerase IIα from the nucleus to the cytoplasm by a CRM1-mediated mechanism. Topoisomerase IIα participates in DNA replication and transcription. Doxorubicin targets topoisomerase IIα, producing DNA-cleavable complexes and cell death. For DNA damage to occur, topoisomerase IIα must be localized in the nucleus. CRM1 inhibition can block the nuclear export of topoisomerase IIα and sensitize cancer cells to treatment with doxorubicin [99].
- Bcr-Abl: The chromosomal translocation between chromosomes 9 and 22 leads to the formation of a new gene called Bcr-Abl. This gene produces the tyrosine kinase Bcr-Abl protein, which is localized in the cytoplasm where it activates proliferative and anti-apoptotic signaling pathways. However, the presence of Bcr-Abl kinase protein in the nucleus followed by its activation along with p73 will result in DNA damage-induced apoptosis. Targeting of Bcr-Abl kinase by imatinib in combination with leptomycin B leads to nuclear retention of Bcr-Abl kinase and promotes apoptosis in imatinib-resistant chronic myeloid leukemia (CML) cells [100].
5. Targeting Nucleocytoplasmic Transport
5.1. Targeting Nuclear Import
5.2. Targeting Nuclear Export
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Oakley, G.M.; Curtin, K.; Layfield, L.; Jarboe, E.; Buchmann, L.O.; Hunt, J.P. Increased melanoma risk in individuals with papillary thyroid carcinoma. JAMA Otolaryngol. Head Neck Surg. 2014, 140, 423–427. [Google Scholar] [CrossRef] [Green Version]
- Cheng, L.; Lopez-Beltran, A.; Massari, F.; MacLennan, G.T.; Montironi, R. Molecular testing for BRAF mutations to inform melanoma treatment decisions: A move toward precision medicine. Mod. Pathol. 2018, 31, 24–38. [Google Scholar] [CrossRef]
- Xu, X.; Quiros, R.M.; Gattuso, P.; Ain, K.B.; Prinz, R.A. High prevalence of BRAF gene mutation in papillary thyroid carcinomas and thyroid tumor cell lines. Cancer Res. 2003, 63, 4561–4567. [Google Scholar]
- Tavares, C.; Melo, M.; Cameselle-Teijeiro, J.M.; Soares, P.; Sobrinho-Simoes, M. ENDOCRINE TUMOURS: Genetic predictors of thyroid cancer outcome. Eur. J. Endocrinol. 2016, 174, R117–R126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cicenas, J.; Tamosaitis, L.; Kvederaviciute, K.; Tarvydas, R.; Staniute, G.; Kalyan, K.; Meskinyte-Kausiliene, E.; Stankevicius, V.; Valius, M. KRAS, NRAS and BRAF mutations in colorectal cancer and melanoma. Med. Oncol. 2017, 34, 26. [Google Scholar] [CrossRef] [PubMed]
- Wiesner, T.; He, J.; Yelensky, R.; Esteve-Puig, R.; Botton, T.; Yeh, I.; Lipson, D.; Otto, G.; Brennan, K.; Murali, R.; et al. Kinase fusions are frequent in Spitz tumours and spitzoid melanomas. Nat. Commun. 2014, 5, 3116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macara, I.G. Transport into and out of the nucleus. Microbiol Mol. Biol. Rev. 2001, 65, 570–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hung, M.C.; Link, W. Protein localization in disease and therapy. J. Cell Sci. 2011, 124, 3381–3392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Tanani, M.; Dakirel, H.; Raynor, B.; Morgan, R. Mechanisms of Nuclear Export in Cancer and Resistance to Chemotherapy. Cancers 2016, 8, 35. [Google Scholar] [CrossRef] [Green Version]
- Stelma, T.; Chi, A.; van der Watt, P.J.; Verrico, A.; Lavia, P.; Leaner, V.D. Targeting nuclear transporters in cancer: Diagnostic, prognostic and therapeutic potential. IUBMB Life 2016, 68, 268–280. [Google Scholar] [CrossRef] [Green Version]
- Turner, J.G.; Dawson, J.; Sullivan, D.M. Nuclear export of proteins and drug resistance in cancer. Biochem. Pharm. 2012, 83, 1021–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vuorinen, E.M.; Rajala, N.K.; Ihalainen, T.O.; Kallioniemi, A. Depletion of nuclear import protein karyopherin alpha 7 (KPNA7) induces mitotic defects and deformation of nuclei in cancer cells. BMC Cancer 2018, 18, 325. [Google Scholar] [CrossRef] [Green Version]
- Gorlich, D.; Mattaj, I.W. Nucleocytoplasmic transport. Science 1996, 271, 1513–1518. [Google Scholar] [CrossRef] [Green Version]
- Kau, T.R.; Way, J.C.; Silver, P.A. Nuclear transport and cancer: From mechanism to intervention. Nat. Rev. Cancer 2004, 4, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Mor, A.; White, M.A.; Fontoura, B.M. Nuclear trafficking in health and disease. Curr. Opin. Cell Biol. 2014, 28, 28–35. [Google Scholar] [CrossRef] [Green Version]
- Tedesco, M.; La Sala, G.; Barbagallo, F.; De Felici, M.; Farini, D. STRA8 shuttles between nucleus and cytoplasm and displays transcriptional activity. J. Biol. Chem. 2009, 284, 35781–35793. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.M.; Aizawa, K.; Jiang, J.; Kung, S.K.P.; Jain, R. JLP-centrosome is essential for the microtubule-mediated nucleocytoplasmic transport induced by extracellular stimuli. Sci. Adv. 2019, 5, eaav0318. [Google Scholar] [CrossRef] [Green Version]
- Luo, P.; Xu, Z.; Li, G.; Yan, H.; Zhu, Y.; Zhu, H.; Ma, S.; Yang, B.; He, Q. HMGB1 represses the anti-cancer activity of sunitinib by governing TP53 autophagic degradation via its nucleus-to-cytoplasm transport. Autophagy 2018, 14, 2155–2170. [Google Scholar] [CrossRef] [Green Version]
- Khan, H.Y.; Ge, J.; Nagasaka, M.; Aboukameel, A.; Mpilla, G.; Muqbil, I.; Szlaczky, M.; Chaker, M.; Baloglu, E.; Landesman, Y.; et al. Targeting XPO1 and PAK4 in 8505C Anaplastic Thyroid Cancer Cells: Putative Implications for Overcoming Lenvatinib Therapy Resistance. Int. J. Mol. Sci. 2019, 21, 237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zerfaoui, M.; Errami, Y.; Naura, A.S.; Suzuki, Y.; Kim, H.; Ju, J.; Liu, T.; Hans, C.P.; Kim, J.G.; Abd Elmageed, Z.Y.; et al. Poly(ADP-ribose) polymerase-1 is a determining factor in Crm1-mediated nuclear export and retention of p65 NF-kappa B upon TLR4 stimulation. J. Immunol. 2010, 185, 1894–1902. [Google Scholar] [CrossRef]
- Cagatay, T.; Chook, Y.M. Karyopherins in cancer. Curr. Opin. Cell Biol. 2018, 52, 30–42. [Google Scholar] [CrossRef]
- Cavazza, T.; Vernos, I. The RanGTP Pathway: From Nucleo-Cytoplasmic Transport to Spindle Assembly and Beyond. Front. Cell Dev. Biol. 2015, 3, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gravina, G.L.; Senapedis, W.; McCauley, D.; Baloglu, E.; Shacham, S.; Festuccia, C. Nucleo-cytoplasmic transport as a therapeutic target of cancer. J. Hematol. Oncol. 2014, 7, 85. [Google Scholar] [CrossRef] [Green Version]
- Kosyna, F.K.; Depping, R. Controlling the Gatekeeper: Therapeutic Targeting of Nuclear Transport. Cells 2018, 7, 221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senapedis, W.T.; Baloglu, E.; Landesman, Y. Clinical translation of nuclear export inhibitors in cancer. Semin. Cancer Biol. 2014, 27, 74–86. [Google Scholar] [CrossRef]
- Tan, D.S.; Bedard, P.L.; Kuruvilla, J.; Siu, L.L.; Razak, A.R. Promising SINEs for embargoing nuclear-cytoplasmic export as an anticancer strategy. Cancer Discov. 2014, 4, 527–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muqbil, I.; Azmi, A.S.; Mohammad, R.M. Nuclear Export Inhibition for Pancreatic Cancer Therapy. Cancers 2018, 10, 138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahipal, A.; Malafa, M. Importins and exportins as therapeutic targets in cancer. Pharmacology 2016, 164, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Subhash, V.V.; Yeo, M.S.; Wang, L.; Tan, S.H.; Wong, F.Y.; Thuya, W.L.; Tan, W.L.; Peethala, P.C.; Soe, M.Y.; Tan, D.S.P.; et al. Anti-tumor efficacy of Selinexor (KPT-330) in gastric cancer is dependent on nuclear accumulation of p53 tumor suppressor. Sci. Rep. 2018, 8, 12248. [Google Scholar] [CrossRef]
- Beck, M.; Schirmacher, P.; Singer, S. Alterations of the nuclear transport system in hepatocellular carcinoma-New basis for therapeutic strategies. J. Hepatol. 2017, 67, 1051–1061. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Yang, H.L.; Xu, J.W.; Wang, J.Z.; Nie, R.H.; Li, C.F. Artemisinin-naphthoquine combination versus chloroquine-primaquine to treat vivax malaria: An open-label randomized and non-inferiority trial in Yunnan Province, China. Malar. J. 2013, 12, 409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, X.; Liang, C.; Li, F.; Wang, L.; Wu, X.; Lu, A.; Xiao, G.; Zhang, G. The Rules and Functions of Nucleocytoplasmic Shuttling Proteins. Int. J. Mol. Sci. 2018, 19, 1445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhim, J.S. Neoplastic transformation of human cells in vitro. Crit. Rev. Oncog. 1993, 4, 313–335. [Google Scholar]
- Berger, C.M.; Gaume, X.; Bouvet, P. The roles of nucleolin subcellular localization in cancer. Biochimie 2015, 113, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Hill, R.; Cautain, B.; de Pedro, N.; Link, W. Targeting nucleocytoplasmic transport in cancer therapy. Oncotarget 2014, 5, 11–28. [Google Scholar] [CrossRef] [Green Version]
- Wainstein, E.; Seger, R. The dynamic subcellular localization of ERK: Mechanisms of translocation and role in various organelles. Curr. Opin. Cell Biol. 2016, 39, 15–20. [Google Scholar] [CrossRef]
- Conforti, F.; Wang, Y.; Rodriguez, J.A.; Alberobello, A.T.; Zhang, Y.W.; Giaccone, G. Molecular Pathways: Anticancer Activity by Inhibition of Nucleocytoplasmic Shuttling. Clin. Cancer Res. 2015, 21, 4508–4513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.; Chang, F.; Lehmann, B.; Terrian, D.M.; Milella, M.; Tafuri, A.; et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim. Biophys. Acta 2007, 1773, 1263–1284. [Google Scholar] [CrossRef] [Green Version]
- Roelli, M.A.; Ruffieux-Daidie, D.; Stooss, A.; ElMokh, O.; Phillips, W.A.; Dettmer, M.S.; Charles, R.P. PIK3CA(H1047R)-induced paradoxical ERK activation results in resistance to BRAF(V600E) specific inhibitors in BRAF(V600E) PIK3CA(H1047R) double mutant thyroid tumors. Oncotarget 2017, 8, 103207–103222. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.Y.; Lee, S.H.; Oh, C.W. Cutaneous malignant melanoma associated with papillary thyroid cancer. Ann. Derm. 2010, 22, 370–372. [Google Scholar] [CrossRef]
- Lazzara, D.R.; Zarkhin, S.G.; Rubenstein, S.N.; Glick, B.P. Melanoma and Thyroid Carcinoma: Our Current Understanding. J. Clin. Aesthet Derm. 2019, 12, 39–41. [Google Scholar]
- Ellerhorst, J.A.; Sendi-Naderi, A.; Johnson, M.K.; Cooke, C.P.; Dang, S.M.; Diwan, A.H. Human melanoma cells express functional receptors for thyroid-stimulating hormone. Endocr. Relat. Cancer 2006, 13, 1269–1277. [Google Scholar] [CrossRef]
- Janz, T.A.; Neskey, D.M.; Nguyen, S.A.; Lentsch, E.J. Is the incidence of anaplastic thyroid cancer increasing: A population based epidemiology study. World J. Otorhinolaryngol. Head Neck Surg. 2019, 5, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.D. The Rel/NF-kappa B family: Friend and foe. Trends Biochem. Sci. 2000, 25, 434–440. [Google Scholar] [CrossRef]
- Ambrosini, G.; Do, C.; Tycko, B.; Realubit, R.B.; Karan, C.; Musi, E.; Carvajal, R.D.; Chua, V.; Aplin, A.E.; Schwartz, G.K. Inhibition of NF-kappaB-Dependent Signaling Enhances Sensitivity and Overcomes Resistance to BET Inhibition in Uveal Melanoma. Cancer Res. 2019, 79, 2415–2425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abd Elmageed, Z.Y.; Moore, R.F.; Tsumagari, K.; Lee, M.M.; Sholl, A.B.; Friedlander, P.; Al-Qurayshi, Z.; Hassan, M.; Wang, A.R.; Boulares, H.A.; et al. Prognostic Role of BRAF(V600E) Cellular Localization in Melanoma. J. Am. Coll. Surg. 2018, 226, 526–537. [Google Scholar] [CrossRef]
- Greger, J.G.; Eastman, S.D.; Zhang, V.; Bleam, M.R.; Hughes, A.M.; Smitheman, K.N.; Dickerson, S.H.; Laquerre, S.G.; Liu, L.; Gilmer, T.M. Combinations of BRAF, MEK, and PI3K/mTOR inhibitors overcome acquired resistance to the BRAF inhibitor GSK2118436 dabrafenib, mediated by NRAS or MEK mutations. Mol. Cancer 2012, 11, 909–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savoia, P.; Fava, P.; Casoni, F.; Cremona, O. Targeting the ERK Signaling Pathway in Melanoma. Int J. Mol. Sci 2019, 20, 1483. [Google Scholar] [CrossRef] [Green Version]
- Song, H.; Zhang, J.; Ning, L.; Zhang, H.; Chen, D.; Jiao, X.; Zhang, K. The MEK1/2 Inhibitor AZD6244 Sensitizes BRAF-Mutant Thyroid Cancer to Vemurafenib. Med. Sci. Monit. 2018, 24, 3002–3010. [Google Scholar] [CrossRef] [Green Version]
- Wei, W.J.; Sun, Z.K.; Shen, C.T.; Song, H.J.; Zhang, X.Y.; Qiu, Z.L.; Luo, Q.Y. Obatoclax and LY3009120 Efficiently Overcome Vemurafenib Resistance in Differentiated Thyroid Cancer. Theranostics 2017, 7, 987–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaeger, R.; Corcoran, R.B. Targeting Alterations in the RAF-MEK Pathway. Cancer Discov. 2019, 9, 329–341. [Google Scholar] [CrossRef] [Green Version]
- Mao, Y.; Xing, M. Recent incidences and differential trends of thyroid cancer in the USA. Endocr. Relat. Cancer 2016, 23, 313–322. [Google Scholar] [CrossRef]
- Ringel, M.D. Molecular markers of aggressiveness of thyroid cancer. Curr. Opin. Endocrinol. Diabetes Obes. 2009, 16, 361–366. [Google Scholar] [CrossRef] [Green Version]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Garnett, M.J.; Marais, R. Guilty as charged: B-RAF is a human oncogene. Cancer Cell 2004, 6, 313–319. [Google Scholar] [CrossRef] [Green Version]
- Kakarmath, S.; Heller, H.T.; Alexander, C.A.; Cibas, E.S.; Krane, J.F.; Barletta, J.A.; Lindeman, N.I.; Frates, M.C.; Benson, C.B.; Gawande, A.A.; et al. Clinical, Sonographic, and Pathological Characteristics of RAS-Positive Versus BRAF-Positive Thyroid Carcinoma. J. Clin. Endocrinol. Metab. 2016, 101, 4938–4944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adachi, M.; Fukuda, M.; Nishida, E. Nuclear export of MAP kinase (ERK) involves a MAP kinase kinase (MEK)-dependent active transport mechanism. J. Cell Biol. 2000, 148, 849–856. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, N.; Ramaswamy, S.; Vazquez, F.; Signoretti, S.; Loda, M.; Sellers, W.R. Forkhead transcription factors are critical effectors of cell death and cell cycle arrest downstream of PTEN. Mol. Cell Biol. 2000, 20, 8969–8982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takaishi, H.; Konishi, H.; Matsuzaki, H.; Ono, Y.; Shirai, Y.; Saito, N.; Kitamura, T.; Ogawa, W.; Kasuga, M.; Kikkawa, U.; et al. Regulation of nuclear translocation of forkhead transcription factor AFX by protein kinase B. Proc. Natl. Acad. Sci. USA 1999, 96, 11836–11841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, E.D.; Nunez, G.; Barr, F.G.; Guan, K.L. Negative regulation of the forkhead transcription factor FKHR by Akt. J. Biol. Chem. 1999, 274, 16741–16746. [Google Scholar] [CrossRef] [Green Version]
- Medema, R.H.; Kops, G.J.; Bos, J.L.; Burgering, B.M. AFX-like Forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27kip1. Nature 2000, 404, 782–787. [Google Scholar] [CrossRef]
- Benvenga, S.; Koch, C.A. Molecular pathways associated with aggressiveness of papillary thyroid cancer. Curr. Genom. 2014, 15, 162–170. [Google Scholar] [CrossRef] [Green Version]
- Ciavardelli, D.; Bellomo, M.; Consalvo, A.; Crescimanno, C.; Vella, V. Metabolic Alterations of Thyroid Cancer as Potential Therapeutic Targets. Biomed. Res. Int. 2017, 2017, 2545031. [Google Scholar] [CrossRef] [Green Version]
- Hanly, E.K.; Tuli, N.Y.; Bednarczyk, R.B.; Suriano, R.; Geliebter, J.; Moscatello, A.L.; Darzynkiewicz, Z.; Tiwari, R.K. Hyperactive ERK and persistent mTOR signaling characterize vemurafenib resistance in papillary thyroid cancer cells. Oncotarget 2016, 7, 8676–8687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.; Xing, M. TERT promoter mutations in thyroid cancer. Endocr. Relat. Cancer 2016, 23, R143–R155. [Google Scholar] [CrossRef] [Green Version]
- Shinohara, M.; Chung, Y.J.; Saji, M.; Ringel, M.D. AKT in thyroid tumorigenesis and progression. Endocrinology 2007, 148, 942–947. [Google Scholar] [CrossRef] [Green Version]
- Tang, K.T.; Lee, C.H. BRAF mutation in papillary thyroid carcinoma: Pathogenic role and clinical implications. J. Chin. Med. Assoc. 2010, 73, 113–128. [Google Scholar] [CrossRef] [Green Version]
- Omholt, K.; Platz, A.; Ringborg, U.; Hansson, J. Cytoplasmic and nuclear accumulation of beta-catenin is rarely caused by CTNNB1 exon 3 mutations in cutaneous malignant melanoma. Int J. Cancer 2001, 92, 839–842. [Google Scholar] [CrossRef] [PubMed]
- Kourtidis, A.; Lu, R.; Pence, L.J.; Anastasiadis, P.Z. A central role for cadherin signaling in cancer. Exp. Cell Res. 2017, 358, 78–85. [Google Scholar] [CrossRef]
- Ebert, M.P.; Yu, J.; Hoffmann, J.; Rocco, A.; Rocken, C.; Kahmann, S.; Muller, O.; Korc, M.; Sung, J.J.; Malfertheiner, P. Loss of beta-catenin expression in metastatic gastric cancer. J. Clin. Oncol 2003, 21, 1708–1714. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Massague, J. Nucleocytoplasmic shuttling of signal transducers. Nat. Rev. Mol. Cell Biol. 2004, 5, 209–219. [Google Scholar] [CrossRef]
- Chien, A.J.; Moore, E.C.; Lonsdorf, A.S.; Kulikauskas, R.M.; Rothberg, B.G.; Berger, A.J.; Major, M.B.; Hwang, S.T.; Rimm, D.L.; Moon, R.T. Activated Wnt/beta-catenin signaling in melanoma is associated with decreased proliferation in patient tumors and a murine melanoma model. Proc. Natl. Acad. Sci. USA 2009, 106, 1193–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kageshita, T.; Hamby, C.V.; Ishihara, T.; Matsumoto, K.; Saida, T.; Ono, T. Loss of beta-catenin expression associated with disease progression in malignant melanoma. Br. J. Derm. 2001, 145, 210–216. [Google Scholar] [CrossRef]
- Kuphal, S.; Bosserhoff, A.K. Phosphorylation of beta-catenin results in lack of beta-catenin signaling in melanoma. Int J. Oncol. 2011, 39, 235–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Forno, P.D.; Pringle, J.H.; Hutchinson, P.; Osborn, J.; Huang, Q.; Potter, L.; Hancox, R.A.; Fletcher, A.; Saldanha, G.S. WNT5A expression increases during melanoma progression and correlates with outcome. Clin. Cancer Res. 2008, 14, 5825–5832. [Google Scholar] [CrossRef] [Green Version]
- Sharpless, N.E.; DePinho, R.A. p53: Good cop/bad cop. Cell 2002, 110, 9–12. [Google Scholar] [CrossRef] [Green Version]
- Brooks, C.L.; Gu, W. Ubiquitination, phosphorylation and acetylation: The molecular basis for p53 regulation. Curr. Opin. Cell Biol. 2003, 15, 164–171. [Google Scholar] [CrossRef]
- Liang, S.H.; Clarke, M.F. Regulation of p53 localization. Eur. J. Biochem. 2001, 268, 2779–2783. [Google Scholar] [CrossRef]
- Moll, U.M.; Ostermeyer, A.G.; Haladay, R.; Winkfield, B.; Frazier, M.; Zambetti, G. Cytoplasmic sequestration of wild-type p53 protein impairs the G1 checkpoint after DNA damage. Mol. Cell Biol. 1996, 16, 1126–1137. [Google Scholar] [CrossRef] [Green Version]
- Gerasimov, G.; Bronstein, M.; Troshina, K.; Alexandrova, G.; Dedov, I.; Jennings, T.; Kallakury, B.V.; Izquierdo, R.; Boguniewicz, A.; Figge, H.; et al. Nuclear p53 immunoreactivity in papillary thyroid cancers is associated with two established indicators of poor prognosis. Exp. Mol. Pathol. 1995, 62, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xiong, Y. Control of p53 ubiquitination and nuclear export by MDM2 and ARF. Cell Growth. Differ. 2001, 12, 175–186. [Google Scholar]
- French, A.D.; Fiori, J.L.; Camilli, T.C.; Leotlela, P.D.; O’Connell, M.P.; Frank, B.P.; Subaran, S.; Indig, F.E.; Taub, D.D.; Weeraratna, A.T. PKC and PKA phosphorylation affect the subcellular localization of claudin-1 in melanoma cells. Int J. Med. Sci. 2009, 6, 93–101. [Google Scholar] [CrossRef] [Green Version]
- Leotlela, P.D.; Wade, M.S.; Duray, P.H.; Rhode, M.J.; Brown, H.F.; Rosenthal, D.T.; Dissanayake, S.K.; Earley, R.; Indig, F.E.; Nickoloff, B.J.; et al. Claudin-1 overexpression in melanoma is regulated by PKC and contributes to melanoma cell motility. Oncogene 2007, 26, 3846–3856. [Google Scholar] [CrossRef] [Green Version]
- Zwanziger, D.; Badziong, J.; Ting, S.; Moeller, L.C.; Schmid, K.W.; Siebolts, U.; Wickenhauser, C.; Dralle, H.; Fuehrer, D. The impact of CLAUDIN-1 on follicular thyroid carcinoma aggressiveness. Endocr. Relat. Cancer 2015, 22, 819–830. [Google Scholar] [CrossRef] [Green Version]
- Anyetei-Anum, C.S.; Roggero, V.R.; Allison, L.A. Thyroid hormone receptor localization in target tissues. J. Endocrinol. 2018, 237, R19–R34. [Google Scholar] [CrossRef]
- Crispo, F.; Notarangelo, T.; Pietrafesa, M.; Lettini, G.; Storto, G.; Sgambato, A.; Maddalena, F.; Landriscina, M. BRAF Inhibitors in Thyroid Cancer: Clinical Impact, Mechanisms of Resistance and Future Perspectives. Cancers 2019, 11, 1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Castro, T.P.; Penha, R.C.C.; Buexm, L.A.; de Carvalho, F.N.; Oliveira, R.V.C.; Agarez, F.V.; Pinto, L.W.; Carvalho, D.P. Molecular Predictors for Advanced Papillary Thyroid Carcinoma Recurrence. Front. Endocrinol. 2019, 10, 839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fakhruddin, N.; Jabbour, M.; Novy, M.; Tamim, H.; Bahmad, H.; Farhat, F.; Zaatari, G.; Aridi, T.; Kriegshauser, G.; Oberkanins, C.; et al. BRAF and NRAS Mutations in Papillary Thyroid Carcinoma and Concordance in BRAF Mutations Between Primary and Corresponding Lymph Node Metastases. Sci. Rep. 2017, 7, 4666. [Google Scholar] [CrossRef] [Green Version]
- Luebker, S.A.; Koepsell, S.A. Diverse Mechanisms of BRAF Inhibitor Resistance in Melanoma Identified in Clinical and Preclinical Studies. Front. Oncol. 2019, 9, 268. [Google Scholar] [CrossRef] [Green Version]
- Dessars, B.; De Raeve, L.E.; El Housni, H.; Debouck, C.J.; Sidon, P.J.; Morandini, R.; Roseeuw, D.; Ghanem, G.E.; Vassart, G.; Heimann, P. Chromosomal translocations as a mechanism of BRAF activation in two cases of large congenital melanocytic nevi. J. Invest. Derm. 2007, 127, 1468–1470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlino, M.S.; Gowrishankar, K.; Saunders, C.A.; Pupo, G.M.; Snoyman, S.; Zhang, X.D.; Saw, R.; Becker, T.M.; Kefford, R.F.; Long, G.V.; et al. Antiproliferative effects of continued mitogen-activated protein kinase pathway inhibition following acquired resistance to BRAF and/or MEK inhibition in melanoma. Mol. Cancer 2013, 12, 1332–1342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webster, M.R.; Fane, M.E.; Alicea, G.M.; Basu, S.; Kossenkov, A.V.; Marino, G.E.; Douglass, S.M.; Kaur, A.; Ecker, B.L.; Gnanapradeepan, K.; et al. Paradoxical Role for Wild-Type p53 in Driving Therapy Resistance in Melanoma. Mol. Cell 2020, 77, 681. [Google Scholar] [CrossRef] [Green Version]
- Rosebeck, S.; Alonge, M.M.; Kandarpa, M.; Mayampurath, A.; Volchenboum, S.L.; Jasielec, J.; Dytfeld, D.; Maxwell, S.P.; Kraftson, S.J.; McCauley, D.; et al. Synergistic Myeloma Cell Death via Novel Intracellular Activation of Caspase-10-Dependent Apoptosis by Carfilzomib and Selinexor. Mol. Cancer 2016, 15, 60–71. [Google Scholar] [CrossRef] [Green Version]
- Pathria, G.; Wagner, C.; Wagner, S.N. Inhibition of CRM1-mediated nucleocytoplasmic transport: Triggering human melanoma cell apoptosis by perturbing multiple cellular pathways. J. Invest. Derm. 2012, 132, 2780–2790. [Google Scholar] [CrossRef] [Green Version]
- Salas Fragomeni, R.A.; Chung, H.W.; Landesman, Y.; Senapedis, W.; Saint-Martin, J.R.; Tsao, H.; Flaherty, K.T.; Shacham, S.; Kauffman, M.; Cusack, J.C. CRM1 and BRAF inhibition synergize and induce tumor regression in BRAF-mutant melanoma. Mol. Cancer 2013, 12, 1171–1179. [Google Scholar] [CrossRef] [Green Version]
- Turner, J.G.; Dawson, J.; Cubitt, C.L.; Baz, R.; Sullivan, D.M. Inhibition of CRM1-dependent nuclear export sensitizes malignant cells to cytotoxic and targeted agents. Semin. Cancer Biol. 2014, 27, 62–73. [Google Scholar] [CrossRef] [Green Version]
- Takenaka, Y.; Fukumori, T.; Yoshii, T.; Oka, N.; Inohara, H.; Kim, H.R.; Bresalier, R.S.; Raz, A. Nuclear export of phosphorylated galectin-3 regulates its antiapoptotic activity in response to chemotherapeutic drugs. Mol. Cell Biol. 2004, 24, 4395–4406. [Google Scholar] [CrossRef] [Green Version]
- Openo, K.P.; Kadrofske, M.M.; Patterson, R.J.; Wang, J.L. Galectin-3 expression and subcellular localization in senescent human fibroblasts. Exp. Cell Res. 2000, 255, 278–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, J.G.; Marchion, D.C.; Dawson, J.L.; Emmons, M.F.; Hazlehurst, L.A.; Washausen, P.; Sullivan, D.M. Human multiple myeloma cells are sensitized to topoisomerase II inhibitors by CRM1 inhibition. Cancer Res. 2009, 69, 6899–6905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aloisi, A.; Di Gregorio, S.; Stagno, F.; Guglielmo, P.; Mannino, F.; Sormani, M.P.; Bruzzi, P.; Gambacorti-Passerini, C.; Saglio, G.; Venuta, S.; et al. BCR-ABL nuclear entrapment kills human CML cells: Ex vivo study on 35 patients with the combination of imatinib mesylate and leptomycin B. Blood 2006, 107, 1591–1598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Campistrous, A.; Thiesen, A.; Gill, A.J.; Ghosh, S.; McMullen, T.P. Loss of nuclear localization of thyroid transcription factor 1 and adverse outcomes in papillary thyroid cancer. Hum. Pathol. 2019, 91, 36–42. [Google Scholar] [CrossRef]
- Lin, Y.Z.; Yao, S.Y.; Veach, R.A.; Torgerson, T.R.; Hawiger, J. Inhibition of nuclear translocation of transcription factor NF-kappa B by a synthetic peptide containing a cell membrane-permeable motif and nuclear localization sequence. J. Biol. Chem. 1995, 270, 14255–14258. [Google Scholar] [CrossRef] [Green Version]
- Ambrus, G.; Whitby, L.R.; Singer, E.L.; Trott, O.; Choi, E.; Olson, A.J.; Boger, D.L.; Gerace, L. Small molecule peptidomimetic inhibitors of importin alpha/beta mediated nuclear transport. Bioorg. Med. Chem. 2010, 18, 7611–7620. [Google Scholar] [CrossRef] [Green Version]
- Cansizoglu, A.E.; Lee, B.J.; Zhang, Z.C.; Fontoura, B.M.; Chook, Y.M. Structure-based design of a pathway-specific nuclear import inhibitor. Nat. Struct. Mol. Biol. 2007, 14, 452–454. [Google Scholar] [CrossRef]
- Hintersteiner, M.; Ambrus, G.; Bednenko, J.; Schmied, M.; Knox, A.J.; Meisner, N.C.; Gstach, H.; Seifert, J.M.; Singer, E.L.; Gerace, L.; et al. Identification of a small molecule inhibitor of importin beta mediated nuclear import by confocal on-bead screening of tagged one-bead one-compound libraries. ACS Chem. Biol. 2010, 5, 967–979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soderholm, J.F.; Bird, S.L.; Kalab, P.; Sampathkumar, Y.; Hasegawa, K.; Uehara-Bingen, M.; Weis, K.; Heald, R. Importazole, a small molecule inhibitor of the transport receptor importin-beta. ACS Chem. Biol. 2011, 6, 700–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Watt, P.J.; Chi, A.; Stelma, T.; Stowell, C.; Strydom, E.; Carden, S.; Angus, L.; Hadley, K.; Lang, D.; Wei, W.; et al. Targeting the Nuclear Import Receptor Kpnbeta1 as an Anticancer Therapeutic. Mol. Cancer 2016, 15, 560–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishi, K.; Yoshida, M.; Fujiwara, D.; Nishikawa, M.; Horinouchi, S.; Beppu, T. Leptomycin B targets a regulatory cascade of crm1, a fission yeast nuclear protein, involved in control of higher order chromosome structure and gene expression. J. Biol. Chem. 1994, 269, 6320–6324. [Google Scholar] [CrossRef]
- Neville, M.; Stutz, F.; Lee, L.; Davis, L.I.; Rosbash, M. The importin-beta family member Crm1p bridges the interaction between Rev and the nuclear pore complex during nuclear export. Curr. Biol. 1997, 7, 767–775. [Google Scholar] [CrossRef] [Green Version]
- Stade, K.; Ford, C.S.; Guthrie, C.; Weis, K. Exportin 1 (Crm1p) is an essential nuclear export factor. Cell 1997, 90, 1041–1050. [Google Scholar] [CrossRef] [Green Version]
- Ossareh-Nazari, B.; Bachelerie, F.; Dargemont, C. Evidence for a role of CRM1 in signal-mediated nuclear protein export. Science 1997, 278, 141–144. [Google Scholar] [CrossRef]
- Newlands, E.S.; Rustin, G.J.; Brampton, M.H. Phase I trial of elactocin. Br. J. Cancer 1996, 74, 648–649. [Google Scholar] [CrossRef] [Green Version]
- Burzlaff, A.; Kalesse, M.; Kasper, C.; Scheper, T. Multi parameter in vitro testing of ratjadone using flow cytometry. Appl. Microbiol. Biotechnol. 2003, 62, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Fleta-Soriano, E.; Martinez, J.P.; Hinkelmann, B.; Gerth, K.; Washausen, P.; Diez, J.; Frank, R.; Sasse, F.; Meyerhans, A. The myxobacterial metabolite ratjadone A inhibits HIV infection by blocking the Rev/CRM1-mediated nuclear export pathway. Microb. Cell Fact. 2014, 13, 17. [Google Scholar] [CrossRef] [Green Version]
- Mutka, S.C.; Yang, W.Q.; Dong, S.D.; Ward, S.L.; Craig, D.A.; Timmermans, P.B.; Murli, S. Identification of nuclear export inhibitors with potent anticancer activity in vivo. Cancer Res. 2009, 69, 510–517. [Google Scholar] [CrossRef] [Green Version]
- Saito, N.; Sakakibara, K.; Sato, T.; Friedman, J.M.; Kufe, D.W.; VonHoff, D.D.; Kawabe, T. CBS9106-induced CRM1 degradation is mediated by cullin ring ligase activity and the neddylation pathway. Mol. Cancer 2014, 13, 3013–3023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parikh, K.; Cang, S.; Sekhri, A.; Liu, D. Selective inhibitors of nuclear export (SINE)-A novel class of anti-cancer agents. J. Hematol. Oncol. 2014, 7, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vercruysse, T.; De Bie, J.; Neggers, J.E.; Jacquemyn, M.; Vanstreels, E.; Schmid-Burgk, J.L.; Hornung, V.; Baloglu, E.; Landesman, Y.; Senapedis, W.; et al. The Second-Generation Exportin-1 Inhibitor KPT-8602 Demonstrates Potent Activity against Acute Lymphoblastic Leukemia. Clin. Cancer Res. 2017, 23, 2528–2541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brose, M.S.; Cabanillas, M.E.; Cohen, E.E.; Wirth, L.J.; Riehl, T.; Yue, H.; Sherman, S.I.; Sherman, E.J. Vemurafenib in patients with BRAF(V600E)-positive metastatic or unresectable papillary thyroid cancer refractory to radioactive iodine: A non-randomised, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 1272–1282. [Google Scholar] [CrossRef] [Green Version]
- Brose, M.S.; Nutting, C.M.; Jarzab, B.; Elisei, R.; Siena, S.; Bastholt, L.; de la Fouchardiere, C.; Pacini, F.; Paschke, R.; Shong, Y.K.; et al. Sorafenib in radioactive iodine-refractory, locally advanced or metastatic differentiated thyroid cancer: A randomised, double-blind, phase 3 trial. Lancet 2014, 384, 319–328. [Google Scholar] [CrossRef] [Green Version]
- Bononi, A.; Pinton, P. Study of PTEN subcellular localization. Methods 2015, 77–78, 92–103. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Transport Factor | Cargoes |
|---|---|
| Exportins | |
| Exportin-1 (Crm1) | Leucin-rich NES cargoes, NF-kB, Cyclin D1, NFAT, p53, p21, IkB, BCR-ABL, FOXO3a, TOPO IIa, eIF4E, HIV genomic RNA |
| Cellular apoptosis susceptibility (CAS/XPO2) | Importin alpha |
| Exportin-t | tRNA |
| Exportin 5 | Pre-microRNA, tRNA, eEF-1A, ILF3, Staufen2, dsRNA-binding proteins, 60S pre-ribosomal subunits |
| Exportin 6 | Profilin, Actin |
| Exportin 7 | P50Rho-GAP, Histone 2A, Histone H3, 14-3-3 |
| Importins | |
| Importin β1 | Cargos with basic NLs via importin alpha, NFAT, PRPF31, CREB, p65, β-catenin, JAK1, STAT5, cyclin B1, SRY/SOX-9, PTHrP |
| Importin β2 | Histone, ribosomal proteins, FOXO4, FUS, hnRNAPA1 |
| Importin β3 | c-Jun, Histones, ribosomal proteins, IRF3, RASAL2, HPV E5 (16E2) |
| Importin 3 | HuR |
| Importin 4 | HIF1-alpha, Histones, ribosomal proteins, Vitamin D receptor |
| Importin 7 | c-Jun, CREB, Ribosomal proteins, SMAD3, HIV RTC, GR, Histone H1 |
| Importin 8 | SMADs, eIF4E, Signal Recognition Particle Protein 19 |
| Importin 9 | c-Jun, PP2A (PR65), NUAK1, nuclear actin, Histone, ribosomal proteins, |
| Importin 11 | UBE2E3, UBE2E1, PTEN, β-catenin, UBcM2, rpL12 |
| Importin 12 | SRSF1, CIRBP |
| Import/Export | |
| Importin 13 | Import: c-Jun, Mago-Y14, RBM8, Ubc9, Glucocorticoid Receptor, Pax6 Export: eIF1A |
| Exportin 4 | Import: Sox2, SRY Export: SMAD3, eIF5A |
| Non-characterized | |
| Ran BP6 | Undefined |
| Ran BP17 | Undefined |
| Signal Transducer | Translocation Effects | Oncogenic Role | Specific Cancer | References |
|---|---|---|---|---|
| FOXO1, FOXO3a, FOXO4, FOXO6 | Cytoplasmic mislocalization promoted by Akt. Nuclear localization of Akt in thyroid cells increases oncogenic expression, high metastatic invasion in lymph nodes and tumor aggression | Activate transcription of genes that triggers cellular proliferative, cell cycle, differentiation, and cell death. | Melanoma, thyroid cancer | Kau et al., 2004; Tang et al., 1999, Takaishi et al., 1999; Nakamura et al., 2000 |
| Claudin-1 | Translocation from nucleus to cytoplasm in melanoma cells and increased cytoplasmic expression in a PKC-dependent manner but altered migration by PKA Phosphorylation. | Increased expression, invasiveness in melanoma hence a marker of progression | Melanoma | French et al., 2009; Leotlela et al., 2007 |
| B-catenin | Nuclear expression | Tumor suppressor role in primary and secondary tumors | Melanoma, thyroid cancer | Chien et al., 2009 |
| Cyclin D1 | Cytoplasmic claudin-1 is highly expressed with more aggression and increased invasiveness in melanoma unlike benign nuclear claudin-1 | Accumulation of cells in the G1 phase of cell cycle. | Melanoma | French et al., 2009; Leotlela et al., 2007 |
| CDKN1B (p27) | Phosphorylated by Akt and exported from nucleus to cytoplasm. Cytoplasmic expression is associated with poor 5-year survival in metastatic melanoma | A cell-cycle inhibitor, blocks cell cycle in the G0/G1 differentiation signals or cellular stress —cell cycle, activation of PI3K and MEK-dependent kinases | Thyroid, melanoma | Kau et al., 2004 |
| p53 | Mutation, post-translational modification, or cytoplasmic mislocalization | Acts as a tumor suppressor and trigger cell cycle arrest, apoptosis, senescence, DNA repair, DNA damage and change the metabolism depending on physiological conditions. Also, known as Guardian of the genome. | Melanoma | Fabbro & Henderson, 2003; Webster et al., 2019 |
| NF-kB | Nuclear import of NF-κB leads to increased target gene expression leading to promotion of tumorigenesis and resistance to anticancer therapies | Activate NF-kB signaling and induce apoptosis of cancer cells. | Thyroid cancer | Kau et al., 2004 |
| Muc 1/EGFR | MUC1 confers survival advantage in melanoma, overexpression of EGFR and nuclear mislocalization is associated with aggressiveness | Induce oncogene expression through interaction with β-catenin and EGFR. | Melanoma and thyroid cancer | Zhao et al., 2014; Patel et al., 2005; Ward et al., 2007 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zerfaoui, M.; Dokunmu, T.M.; Toraih, E.A.; Rezk, B.M.; Abd Elmageed, Z.Y.; Kandil, E. New Insights into the Link between Melanoma and Thyroid Cancer: Role of Nucleocytoplasmic Trafficking. Cells 2021, 10, 367. https://doi.org/10.3390/cells10020367
Zerfaoui M, Dokunmu TM, Toraih EA, Rezk BM, Abd Elmageed ZY, Kandil E. New Insights into the Link between Melanoma and Thyroid Cancer: Role of Nucleocytoplasmic Trafficking. Cells. 2021; 10(2):367. https://doi.org/10.3390/cells10020367
Chicago/Turabian StyleZerfaoui, Mourad, Titilope Modupe Dokunmu, Eman Ali Toraih, Bashir M. Rezk, Zakaria Y. Abd Elmageed, and Emad Kandil. 2021. "New Insights into the Link between Melanoma and Thyroid Cancer: Role of Nucleocytoplasmic Trafficking" Cells 10, no. 2: 367. https://doi.org/10.3390/cells10020367