
Impacts of Environmental Factors on Head and Neck Cancer Pathogenesis and Progression




{kind=link}
{kind=link}
Abstract
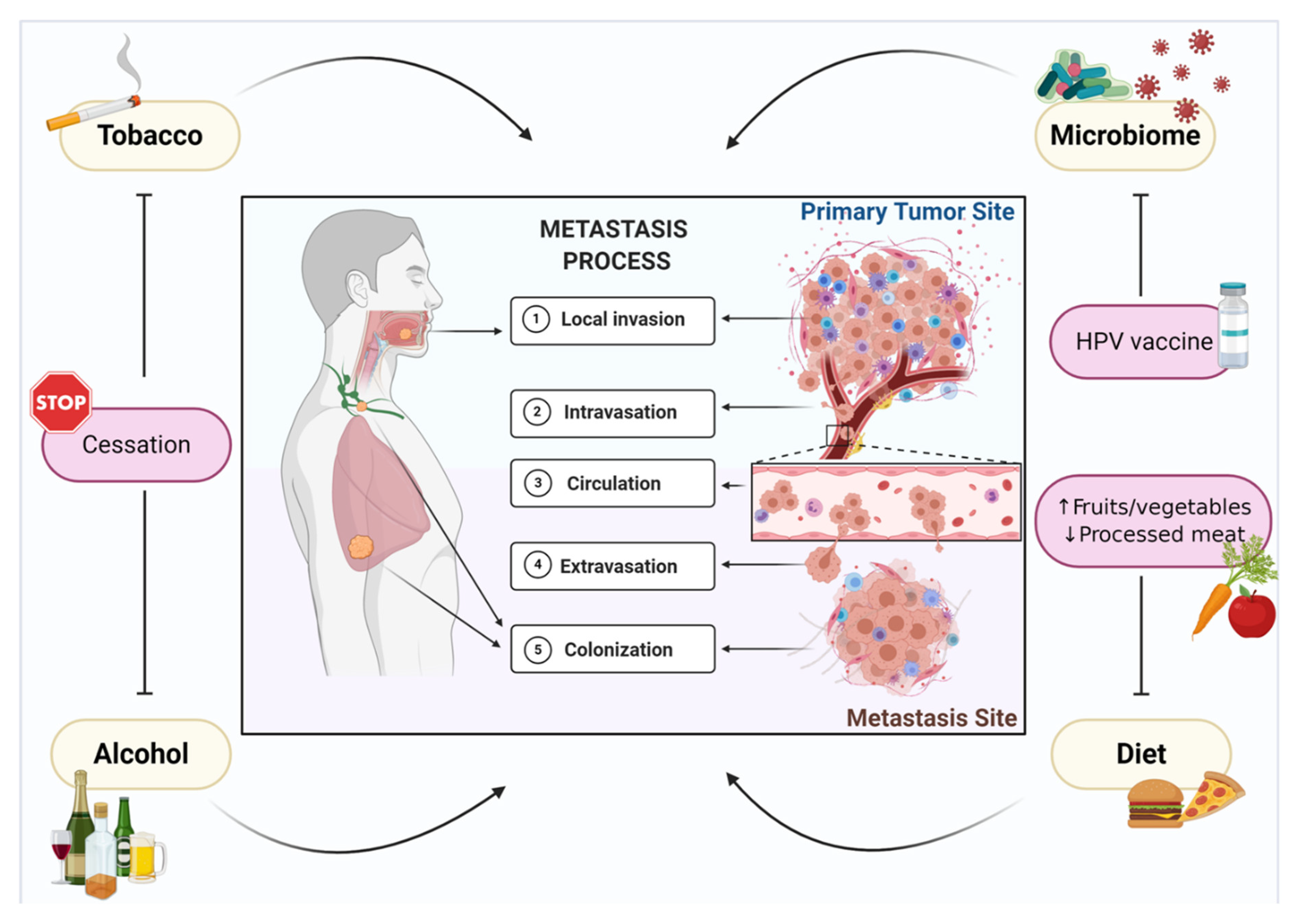
:1. Introduction
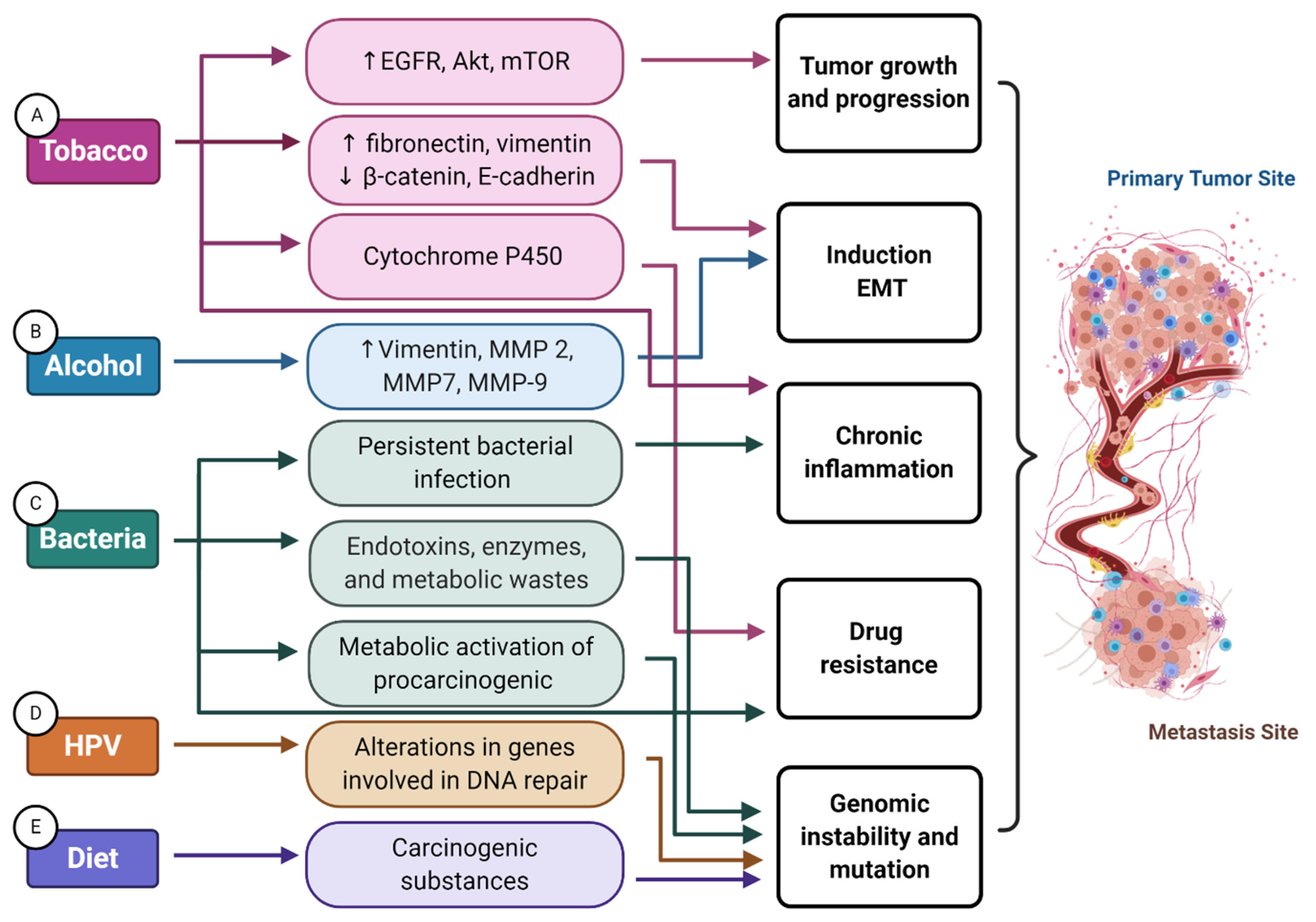
2. Tobacco Smoking
3. Alcohol
4. Microbiome
4.1. Viruses
4.1.1. EBV
4.1.2. HPV
4.2. Bacteria
5. Diet and Nutrition
5.1. Vegetables and Fruits
5.2. Red Meat and Processed Meat
6. The Influence of Environmental Factors during Cancer Treatment
7. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AICR | American Institute for Cancer Research |
| Akt BDNF | protein kinase B brain-derived neurotrophic factor |
| CDKN2 | cyclin-dependent kinase inhibitor 2A |
| COX-2 | cyclooxygenase 2 |
| CTLA-4 | cytotoxic T-lymphocyte-associated protein 4 |
| CYP | cytochrome P450 |
| DNA | deoxyribonucleic acid |
| EBV | Epstein–Barr virus |
| EGFR | epidermal growth factor receptor |
| EMT | epithelial–mesenchymal transition |
| FGF-2 | fibroblast growth factor 2 |
| GSTs | glutathione-S-transferases |
| HAA | heterocyclic aromatic amines |
| HBV | hepatitis B virus |
| HCV | hepatitis C virus |
| HHV | human herpes virus |
| HIF-1α | hypoxia-inducible factor 1-alpha |
| HIV | human immunodeficiency virus |
| HNSCC | head and neck squamous cell carcinoma |
| H. pylori | Helicobacter pylori |
| HPV | human papillomavirus |
| hTERT | human telomerase catalytic subunit |
| HTLV-1 | human T-lymphotropic virus |
| IARC | International Agency for Research on Cancer |
| MALT | mucosa-associated lymphoid tissue |
| MMP | matrix metalloproteinase |
| MUC1 | mucin 1 |
| mTOR | mammalian target of rapamycin |
| nAChRs | nicotinic acetylcholine receptors |
| NF-κB | nuclear factor kappa B |
| NGF | nerve growth factor |
| NNN | N′-nitrosonornicotine |
| NOC | N-nitroso-compounds |
| OSCC | oral squamous cell carcinoma |
| PAHs | polycyclic aromatic hydrocarbons |
| PD-L1 | programmed death-ligand 1 |
| PNI | perineural invasion |
| pRb | retinoblastoma protein |
| PRKDC | protein kinase, DNA-activated, catalytic subunit |
| RNA | ribonucleic acid |
| ROS | reactive oxygen species |
| Trk | tropomyosin-related kinase |
| TSNA | nitrosamines |
| UGTs | uridine-5′-diphosphate-glucuronosyltransferases |
| VEGF | vascular endothelial growth factor |
| WCRF | World Cancer Research Fund |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- El-Naggar, A.K.; Chan, C.J.; Grandis, J.R.; Takata, T.; Slootweg, P.J. WHO Classification of Head and Neck Tumours; IARC: Lyon, France, 2017. [Google Scholar]
- Network, C.G.A. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leemans, C.R.; Snijders, P.J.F.; Brakenhoff, R.H. The molecular landscape of head and neck cancer. Nat. Rev. Cancer 2018, 18, 269–282. [Google Scholar] [CrossRef] [PubMed]
- Braakhuis, B.J.; Brakenhoff, R.H.; Leemans, C.R. Treatment choice for locally advanced head and neck cancers on the basis of risk factors: Biological risk factors. Ann. Oncol. 2012, 23 (Suppl. 10), x173–x177. [Google Scholar] [CrossRef]
- Chow, L.Q.M. Head and Neck Cancer. N. Engl. J. Med. 2020, 382, 60–72. [Google Scholar] [CrossRef]
- Crile, G. Landmark article Dec 1, 1906: Excision of cancer of the head and neck. With special reference to the plan of dissection based on one hundred and thirty-two operations. By George Crile. JAMA 1987, 258, 3286–3293. [Google Scholar] [CrossRef]
- Fares, J.; Fares, M.Y.; Khachfe, H.H.; Salhab, H.A.; Fares, Y. Molecular principles of metastasis: A hallmark of cancer revisited. Signal. Transduct. Target. Ther. 2020, 5, 28. [Google Scholar] [CrossRef]
- Doll, R.; Hill, A.B. Smoking and carcinoma of the lung; preliminary report. Br. Med. J. 1950, 2, 739–748. [Google Scholar] [CrossRef] [Green Version]
- ARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Tobacco Smoke and Involuntary Smoking; World Health Organization: Geneva, Switzerland, 2004; Volume 83, pp. 1–1438. [Google Scholar]
- Hecht, S.S. Biochemistry, biology, and carcinogenicity of tobacco-specific N-nitrosamines. Chem. Res. Toxicol. 1998, 11, 559–603. [Google Scholar] [CrossRef]
- Jethwa, A.R.; Khariwala, S.S. Tobacco-related carcinogenesis in head and neck cancer. Cancer Metastasis Rev. 2017, 36, 411–423. [Google Scholar] [CrossRef] [PubMed]
- Hashibe, M.; Brennan, P.; Benhamou, S.; Castellsague, X.; Chen, C.; Curado, M.P.; Dal Maso, L.; Daudt, A.W.; Fabianova, E.; Fernandez, L.; et al. Alcohol drinking in never users of tobacco, cigarette smoking in never drinkers, and the risk of head and neck cancer: Pooled analysis in the International Head and Neck Cancer Epidemiology Consortium. J. Natl. Cancer Inst. 2007, 99, 777–789. [Google Scholar] [CrossRef]
- Sharp, L.; McDevitt, J.; Carsin, A.E.; Brown, C.; Comber, H. Smoking at diagnosis is an independent prognostic factor for cancer-specific survival in head and neck cancer: Findings from a large, population-based study. Cancer Epidemiol. Biomark. Prev. 2014, 23, 2579–2590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beynon, R.A.; Lang, S.; Schimansky, S.; Penfold, C.M.; Waylen, A.; Thomas, S.J.; Pawlita, M.; Waterboer, T.; Martin, R.M.; May, M.; et al. Tobacco smoking and alcohol drinking at diagnosis of head and neck cancer and all-cause mortality: Results from head and neck 5000, a prospective observational cohort of people with head and neck cancer. Int. J. Cancer 2018, 143, 1114–1127. [Google Scholar] [CrossRef]
- Marron, M.; Boffetta, P.; Zhang, Z.F.; Zaridze, D.; Wünsch-Filho, V.; Winn, D.M.; Wei, Q.; Talamini, R.; Szeszenia-Dabrowska, N.; Sturgis, E.M.; et al. Cessation of alcohol drinking, tobacco smoking and the reversal of head and neck cancer risk. Int. J. Epidemiol. 2010, 39, 182–196. [Google Scholar] [CrossRef] [Green Version]
- McBride, S.M.; Ali, N.N.; Margalit, D.N.; Chan, A.W. Active tobacco smoking and distant metastasis in patients with oropharyngeal cancer. Int. J. Radiat. Oncol. Biol. Phys. 2012, 84, 183–188. [Google Scholar] [CrossRef]
- Maxwell, J.H.; Kumar, B.; Feng, F.Y.; Worden, F.P.; Lee, J.S.; Eisbruch, A.; Wolf, G.T.; Prince, M.E.; Moyer, J.S.; Teknos, T.N.; et al. Tobacco use in human papillomavirus-positive advanced oropharynx cancer patients related to increased risk of distant metastases and tumor recurrence. Clin. Cancer Res. 2010, 16, 1226–1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, R.; Ibaragi, S.; Eguchi, T.; Kuwajima, D.; Kodama, S.; Nishioka, T.; Okui, T.; Obata, K.; Takabatake, K.; Kawai, H.; et al. Nicotine promotes lymph node metastasis and cetuximab resistance in head and neck squamous cell carcinoma. Int. J. Oncol. 2019, 54, 283–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carracedo, D.G.; Rodrigo, J.P.; Nieto, C.S.; Gonzalez, M.V. Epithelial cell nicotinic acetylcholine receptor expression in head and neck squamous cell carcinoma pathogenesis. Anticancer Res. 2007, 27, 835–839. [Google Scholar]
- Maneckjee, R.; Minna, J.D. Opioid and nicotine receptors affect growth regulation of human lung cancer cell lines. Proc. Natl. Acad. Sci. USA 1990, 87, 3294–3298. [Google Scholar] [CrossRef] [Green Version]
- Dasgupta, P.; Rizwani, W.; Pillai, S.; Kinkade, R.; Kovacs, M.; Rastogi, S.; Banerjee, S.; Carless, M.; Kim, E.; Coppola, D.; et al. Nicotine induces cell proliferation, invasion and epithelial-mesenchymal transition in a variety of human cancer cell lines. Int. J. Cancer 2009, 124, 36–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumeister, P.; Welz, C.; Jacobi, C.; Reiter, M. Is Perineural Invasion of Head and Neck Squamous Cell Carcinomas Linked to Tobacco Consumption? Otolaryngol. Head Neck Surg. 2018, 158, 878–881. [Google Scholar] [CrossRef] [PubMed]
- Amit, M.; Na’ara, S.; Gil, Z. Mechanisms of cancer dissemination along nerves. Nat. Rev. Cancer 2016, 16, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Kolokythas, A.; Cox, D.P.; Dekker, N.; Schmidt, B.L. Nerve growth factor and tyrosine kinase A receptor in oral squamous cell carcinoma: Is there an association with perineural invasion? J. Oral Maxillofac. Surg. 2010, 68, 1290–1295. [Google Scholar] [CrossRef]
- de Moraes, J.K.; Wagner, V.P.; Fonseca, F.P.; Vargas, P.A.; de Farias, C.B.; Roesler, R.; Martins, M.D. Uncovering the role of brain-derived neurotrophic factor/tyrosine kinase receptor B signaling in head and neck malignancies. J. Oral Pathol. Med. 2018, 47, 221–227. [Google Scholar] [CrossRef]
- Yilmaz, T.; Jiffar, T.; de la Garza, G.; Lin, H.; Milas, Z.; Takahashi, Y.; Hanna, E.; MacIntyre, T.; Brown, J.L.; Myers, J.N.; et al. Theraputic targeting of Trk supresses tumor proliferation and enhances cisplatin activity in HNSCC. Cancer Biol. Ther. 2010, 10, 644–653. [Google Scholar] [CrossRef] [Green Version]
- Du, X.; Squier, C.A.; Kremer, M.J.; Wertz, P.W. Penetration of N-nitrosonornicotine (NNN) across oral mucosa in the presence of ethanol and nicotine. J. Oral Pathol. Med. 2000, 29, 80–85. [Google Scholar] [CrossRef]
- Hashibe, M.; Brennan, P.; Chuang, S.C.; Boccia, S.; Castellsague, X.; Chen, C.; Curado, M.P.; Dal Maso, L.; Daudt, A.W.; Fabianova, E.; et al. Interaction between tobacco and alcohol use and the risk of head and neck cancer: Pooled analysis in the International Head and Neck Cancer Epidemiology Consortium. Cancer Epidemiol. Biomark. Prev. 2009, 18, 541–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marziliano, A.; Teckie, S.; Diefenbach, M.A. Alcohol-related head and neck cancer: Summary of the literature. Head Neck 2020, 42, 732–738. [Google Scholar] [CrossRef]
- Lin, P.Y.; Yu, C.H.; Wang, J.T.; Chen, H.H.; Cheng, S.J.; Kuo, M.Y.; Chiang, C.P. Expression of hypoxia-inducible factor-1 alpha is significantly associated with the progression and prognosis of oral squamous cell carcinomas in Taiwan. J. Oral Pathol. Med. 2008, 37, 18–25. [Google Scholar] [CrossRef]
- Forsyth, C.B.; Tang, Y.; Shaikh, M.; Zhang, L.; Keshavarzian, A. Alcohol stimulates activation of Snail, epidermal growth factor receptor signaling, and biomarkers of epithelial-mesenchymal transition in colon and breast cancer cells. Alcohol. Clin. Exp. Res. 2010, 34, 19–31. [Google Scholar] [CrossRef]
- Xu, M.; Chen, G.; Fu, W.; Liao, M.; Frank, J.A.; Bower, K.A.; Fang, S.; Zhang, Z.; Shi, X.; Luo, J. Ethanol disrupts vascular endothelial barrier: Implication in cancer metastasis. Toxicol. Sci. 2012, 127, 42–53. [Google Scholar] [CrossRef] [Green Version]
- Thomas, A.M.; Gleber-Netto, F.O.; Fernandes, G.R.; Amorim, M.; Barbosa, L.F.; Francisco, A.L.; de Andrade, A.G.; Setubal, J.C.; Kowalski, L.P.; Nunes, D.N.; et al. Alcohol and tobacco consumption affects bacterial richness in oral cavity mucosa biofilms. BMC Microbiol. 2014, 14, 250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Epstein, M.A.; Achong, B.G.; Barr, Y.M. Virus particles in cultured lymphoblasts from burkitt’s lymphoma. Lancet 1964, 1, 702–703. [Google Scholar] [CrossRef]
- Krump, N.A.; You, J. Molecular mechanisms of viral oncogenesis in humans. Nat. Rev. Microbiol. 2018, 16, 684–698. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.K. Virus-associated neoplasms of the nasopharynx and sinonasal tract: Diagnostic problems. Mod. Pathol. 2017, 30, S68–S83. [Google Scholar] [CrossRef] [Green Version]
- Tsao, S.W.; Tsang, C.M.; Lo, K.W. Epstein-Barr virus infection and nasopharyngeal carcinoma. Philos. Trans. R. Soc. Lond B Biol. Sci. 2017, 372. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, Y.; Wakisaka, N.; Kondo, S.; Endo, K.; Sugimoto, H.; Hatano, M.; Ueno, T.; Ishikawa, K.; Yoshizaki, T. Progression of understanding for the role of Epstein-Barr virus and management of nasopharyngeal carcinoma. Cancer Metastasis Rev. 2017, 36, 435–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, C.; Li, L.; Xiang, Y.-Q.; Lung, M.L.; Zeng, T.; Lu, J.; Tsao, S.W.; Zeng, M.-S.; Yun, J.-P.; Kwong, D.L.W.; et al. Epstein–Barr Virus miRNA BART2-5p Promotes Metastasis of Nasopharyngeal Carcinoma by Suppressing RND3. Cancer Res. 2020, 80, 1957–1969. [Google Scholar] [CrossRef] [Green Version]
- Rampias, T.; Sasaki, C.; Weinberger, P.; Psyrri, A. E6 and e7 gene silencing and transformed phenotype of human papillomavirus 16-positive oropharyngeal cancer cells. J. Natl. Cancer Inst. 2009, 101, 412–423. [Google Scholar] [CrossRef] [Green Version]
- Münger, K.; Baldwin, A.; Edwards, K.M.; Hayakawa, H.; Nguyen, C.L.; Owens, M.; Grace, M.; Huh, K. Mechanisms of Human Papillomavirus-Induced Oncogenesis. J. Virol. 2004, 78, 11451–11460. [Google Scholar] [CrossRef] [Green Version]
- Gillison, M.L.; Chaturvedi, A.K.; Anderson, W.F.; Fakhry, C. Epidemiology of Human Papillomavirus-Positive Head and Neck Squamous Cell Carcinoma. J. Clin. Oncol. 2015, 33, 3235–3242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaturvedi, A.K.; Graubard, B.I.; Broutian, T.; Pickard, R.K.L.; Tong, Z.Y.; Xiao, W.; Kahle, L.; Gillison, M.L. Effect of Prophylactic Human Papillomavirus (HPV) Vaccination on Oral HPV Infections Among Young Adults in the United States. J. Clin. Oncol. 2018, 36, 262–267. [Google Scholar] [CrossRef]
- Gillison, M.L.; Koch, W.M.; Capone, R.B.; Spafford, M.; Westra, W.H.; Wu, L.; Zahurak, M.L.; Daniel, R.W.; Viglione, M.; Symer, D.E.; et al. Evidence for a causal association between human papillomavirus and a subset of head and neck cancers. J. Natl. Cancer Inst. 2000, 92, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Michaud, D.S.; Langevin, S.M.; Eliot, M.; Nelson, H.H.; Pawlita, M.; McClean, M.D.; Kelsey, K.T. High-risk HPV types and head and neck cancer. Int. J. Cancer 2014, 135, 1653–1661. [Google Scholar] [CrossRef] [PubMed]
- Ang, K.K.; Harris, J.; Wheeler, R.; Weber, R.; Rosenthal, D.I.; Nguyen-Tân, P.F.; Westra, W.H.; Chung, C.H.; Jordan, R.C.; Lu, C.; et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N. Engl. J. Med. 2010, 363, 24–35. [Google Scholar] [CrossRef] [Green Version]
- Nichols, A.C.; Lang, P.; Prisman, E.; Berthelet, E.; Tran, E.; Hamilton, S.; Wu, J.; Fung, K.; de Almeida, J.R.; Bayley, A.; et al. Treatment de-escalation for HPV-associated oropharyngeal squamous cell carcinoma with radiotherapy vs. trans-oral surgery (ORATOR2): Study protocol for a randomized phase II trial. BMC Cancer 2020, 20, 125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindel, K.; Beer, K.T.; Laissue, J.; Greiner, R.H.; Aebersold, D.M. Human papillomavirus positive squamous cell carcinoma of the oropharynx: A radiosensitive subgroup of head and neck carcinoma. Cancer 2001, 92, 805–813. [Google Scholar] [CrossRef]
- Braakhuis, B.J.; Tabor, M.P.; Kummer, J.A.; Leemans, C.R.; Brakenhoff, R.H. A genetic explanation of Slaughter’s concept of field cancerization: Evidence and clinical implications. Cancer Res. 2003, 63, 1727–1730. [Google Scholar]
- Welters, M.J.P.; Ma, W.; Santegoets, S.; Goedemans, R.; Ehsan, I.; Jordanova, E.S.; van Ham, V.J.; van Unen, V.; Koning, F.; van Egmond, S.I.; et al. Intratumoral HPV16-Specific T Cells Constitute a Type I-Oriented Tumor Microenvironment to Improve Survival in HPV16-Driven Oropharyngeal Cancer. Clin. Cancer Res. 2018, 24, 634–647. [Google Scholar] [CrossRef] [Green Version]
- Galvis, M.M.; Borges, G.A.; Oliveira, T.B.; Toledo, I.P.; Castilho, R.M.; Guerra, E.N.S.; Kowalski, L.P.; Squarize, C.H. Immunotherapy improves efficacy and safety of patients with HPV positive and negative head and neck cancer: A systematic review and meta-analysis. Crit Rev. Oncol. Hematol. 2020, 150, 102966. [Google Scholar] [CrossRef]
- O’Sullivan, B.; Huang, S.H.; Siu, L.L.; Waldron, J.; Zhao, H.; Perez-Ordonez, B.; Weinreb, I.; Kim, J.; Ringash, J.; Bayley, A.; et al. Deintensification candidate subgroups in human papillomavirus-related oropharyngeal cancer according to minimal risk of distant metastasis. J. Clin. Oncol. 2013, 31, 543–550. [Google Scholar] [CrossRef]
- Sinha, P.; Thorstad, W.T.; Nussenbaum, B.; Haughey, B.H.; Adkins, D.R.; Kallogjeri, D.; Lewis, J.S., Jr. Distant metastasis in p16-positive oropharyngeal squamous cell carcinoma: A critical analysis of patterns and outcomes. Oral Oncol. 2014, 50, 45–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trosman, S.J.; Koyfman, S.A.; Ward, M.C.; Al-Khudari, S.; Nwizu, T.; Greskovich, J.F.; Lamarre, E.D.; Scharpf, J.; Khan, M.J.; Lorenz, R.R.; et al. Effect of human papillomavirus on patterns of distant metastatic failure in oropharyngeal squamous cell carcinoma treated with chemoradiotherapy. JAMA Otolaryngol. Head Neck Surg. 2015, 141, 457–462. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.H.; Perez-Ordonez, B.; Weinreb, I.; Hope, A.; Massey, C.; Waldron, J.N.; Kim, J.; Bayley, A.J.; Cummings, B.; Cho, B.C.; et al. Natural course of distant metastases following radiotherapy or chemoradiotherapy in HPV-related oropharyngeal cancer. Oral Oncol. 2013, 49, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.H.; Perez-Ordonez, B.; Liu, F.F.; Waldron, J.; Ringash, J.; Irish, J.; Cummings, B.; Siu, L.L.; Kim, J.; Weinreb, I.; et al. Atypical clinical behavior of p16-confirmed HPV-related oropharyngeal squamous cell carcinoma treated with radical radiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 2012, 82, 276–283. [Google Scholar] [CrossRef]
- Hanna, G.J.; Kacew, A.; Chau, N.G.; Shivdasani, P.; Lorch, J.H.; Uppaluri, R.; Haddad, R.I.; MacConaill, L.E. Improved outcomes in PI3K-pathway-altered metastatic HPV oropharyngeal cancer. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Dok, R.; Glorieux, M.; Holacka, K.; Bamps, M.; Nuyts, S. Dual role for p16 in the metastasis process of HPV positive head and neck cancers. Mol. Cancer 2017, 16, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogtmann, E.; Goedert, J.J. Epidemiologic studies of the human microbiome and cancer. Br. J. Cancer 2016, 114, 237–242. [Google Scholar] [CrossRef] [Green Version]
- Parsonnet, J.; Friedman, G.D.; Vandersteen, D.P.; Chang, Y.; Vogelman, J.H.; Orentreich, N.; Sibley, R.K. Helicobacter pylori infection and the risk of gastric carcinoma. N. Engl. J. Med. 1991, 325, 1127–1131. [Google Scholar] [CrossRef]
- Troppan, K.; Wenzl, K.; Neumeister, P.; Deutsch, A. Molecular Pathogenesis of MALT Lymphoma. Gastroenterol. Res. Pract. 2015, 2015, 102656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Domenico, E.G.; Cavallo, I.; Pontone, M.; Toma, L.; Ensoli, F. Biofilm Producing Salmonella Typhi: Chronic Colonization and Development of Gallbladder Cancer. Int. J. Mol. Sci. 2017, 18, 1887. [Google Scholar] [CrossRef]
- Lamont, R.J.; Koo, H.; Hajishengallis, G. The oral microbiota: Dynamic communities and host interactions. Nat. Rev. Microbiol. 2018, 16, 745–759. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Preston, R.; Godoy-Vitorino, F.; Jedlicka, A.; Rodríguez-Hilario, A.; González, H.; Bondy, J.; Lawson, F.; Folawiyo, O.; Michailidi, C.; Dziedzic, A.; et al. 16S rRNA amplicon sequencing identifies microbiota associated with oral cancer, human papilloma virus infection and surgical treatment. Oncotarget 2016, 7, 51320–51334. [Google Scholar] [CrossRef] [Green Version]
- Bolz, J.; Dosá, E.; Schubert, J.; Eckert, A.W. Bacterial colonization of microbial biofilms in oral squamous cell carcinoma. Clin. Oral Investig. 2014, 18, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, Y.; Zheng, H.J.; Zhang, C.P. The Oral Microbiota May Have Influence on Oral Cancer. Front. Cell. Infect. Microbiol. 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Galvis, M.; Teng, Y. Targeting Hypoxia-Driven Metabolic Reprogramming to Constrain Tumor Progression and Metastasis. Int. J. Mol. Sci. 2020, 21, 5487. [Google Scholar] [CrossRef]
- Hooper, S.J.; Wilson, M.J.; Crean, S.J. Exploring the link between microorganisms and oral cancer: A systematic review of the literature. Head Neck 2009, 31, 1228–1239. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, A.; Moissl-Eichinger, C.; Perras, A.; Koskinen, K.; Tomazic, P.V.; Thurnher, D. The salivary microbiome as an indicator of carcinogenesis in patients with oropharyngeal squamous cell carcinoma: A pilot study. Sci. Rep. 2017, 7, 5867. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Funchain, P.; Bebek, G.; Altemus, J.; Zhang, H.; Niazi, F.; Peterson, C.; Lee, W.T.; Burkey, B.B.; Eng, C. Microbiomic differences in tumor and paired-normal tissue in head and neck squamous cell carcinomas. Genome Med. 2017, 9, 14. [Google Scholar] [CrossRef] [Green Version]
- Vétizou, M.; Pitt, J.M.; Daillère, R.; Lepage, P.; Waldschmitt, N.; Flament, C.; Rusakiewicz, S.; Routy, B.; Roberti, M.P.; Duong, C.P.; et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 2015, 350, 1079–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Lei, Y.M.; Jabri, B.; Alegre, M.L.; et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 2015, 350, 1084–1089. [Google Scholar] [CrossRef] [Green Version]
- Key, T.J.; Bradbury, K.E.; Perez-Cornago, A.; Sinha, R.; Tsilidis, K.K.; Tsugane, S. Diet, nutrition, and cancer risk: What do we know and what is the way forward? BMJ 2020, 368, m511. [Google Scholar] [CrossRef] [Green Version]
- Collaborators, G.D. Health effects of dietary risks in 195 countries, 1990-2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2019, 393, 1958–1972. [Google Scholar] [CrossRef] [Green Version]
- World Cancer Research Fund/American Institute for Cancer Research. Diet, Nutrition, Physical Activity and Cancer: A Global Perspective; Continuous Update Project Expert Report; American Institute for Cancer Research: Washington, DC, USA, 1997. [Google Scholar]
- World Cancer Research Fund/American Institute for Cancer Research. Diet, Nutrition, Physical Activity and Cancer: A Global Perspective; Continuous Update Project Expert Report; American Institute for Cancer Research: Washington, DC, USA, 2018. [Google Scholar]
- Galvão De Podestá, O.P.; Peres, S.V.; Salaroli, L.B.; Cattafesta, M.; De Podestá, J.R.V.; von Zeidler, S.L.V.; de Oliveira, J.C.; Kowalski, L.P.; Ikeda, M.K.; Brennan, P.; et al. Consumption of minimally processed foods as protective factors in the genesis of squamous cell carcinoma of the head and neck in Brazil. PLoS ONE 2019, 14, e0220067. [Google Scholar] [CrossRef] [PubMed]
- Butler, C.; Lee, Y.A.; Li, S.; Li, Q.; Chen, C.J.; Hsu, W.L.; Lou, P.J.; Zhu, C.; Pan, J.; Shen, H.; et al. Diet and the risk of head-and-neck cancer among never-smokers and smokers in a Chinese population. Cancer Epidemiol. 2017, 46, 20–26. [Google Scholar] [CrossRef]
- Macfarlane, G.J.; Zheng, T.; Marshall, J.R.; Boffetta, P.; Niu, S.; Brasure, J.; Merletti, F.; Boyle, P. Alcohol, tobacco, diet and the risk of oral cancer: A pooled analysis of three case-control studies. Eur. J. Cancer B Oral Oncol. 1995, 31, 181–187. [Google Scholar] [CrossRef]
- Maasland, D.H.; van den Brandt, P.A.; Kremer, B.; Goldbohm, R.A.; Schouten, L.J. Consumption of vegetables and fruits and risk of subtypes of head-neck cancer in the Netherlands Cohort Study. Int. J. Cancer 2015, 136, E396–E409. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Lee, W.T.; Lee, Y.C.; Huang, C.C.; Ou, C.Y.; Lin, Y.H.; Huang, J.S.; Wong, T.Y.; Chen, K.C.; Hsiao, J.R.; et al. Investigating the association between diet and risk of head and neck cancer in Taiwan. Oncotarget 2017, 8, 98865–98875. [Google Scholar] [CrossRef] [Green Version]
- Llewellyn, C.D.; Linklater, K.; Bell, J.; Johnson, N.W.; Warnakulasuriya, S. An analysis of risk factors for oral cancer in young people: A case-control study. Oral Oncol. 2004, 40, 304–313. [Google Scholar] [CrossRef]
- Bradshaw, P.T.; Siega-Riz, A.M.; Campbell, M.; Weissler, M.C.; Funkhouser, W.K.; Olshan, A.F. Associations between dietary patterns and head and neck cancer: The Carolina head and neck cancer epidemiology study. Am. J. Epidemiol. 2012, 175, 1225–1233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chuang, S.C.; Jenab, M.; Heck, J.E.; Bosetti, C.; Talamini, R.; Matsuo, K.; Castellsague, X.; Franceschi, S.; Herrero, R.; Winn, D.M.; et al. Diet and the risk of head and neck cancer: A pooled analysis in the INHANCE consortium. Cancer Causes Control. 2012, 23, 69–88. [Google Scholar] [CrossRef] [PubMed]
- Nosrati, N.; Bakovic, M.; Paliyath, G. Molecular Mechanisms and Pathways as Targets for Cancer Prevention and Progression with Dietary Compounds. Int. J. Mol. Sci. 2017, 18, 2050. [Google Scholar] [CrossRef] [Green Version]
- Julia, C.; Meunier, N.; Touvier, M.; Ahluwalia, N.; Sapin, V.; Papet, I.; Cano, N.; Hercberg, S.; Galan, P.; Kesse-Guyot, E. Dietary patterns and risk of elevated C-reactive protein concentrations 12 years later. Br. J. Nutr. 2013, 110, 747–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hakimuddin, F.; Paliyath, G.; Meckling, K. Treatment of mcf-7 breast cancer cells with a red grape wine polyphenol fraction results in disruption of calcium homeostasis and cell cycle arrest causing selective cytotoxicity. J. Agric. Food Chem. 2006, 54, 7912–7923. [Google Scholar] [CrossRef]
- Leoncini, E.; Edefonti, V.; Hashibe, M.; Parpinel, M.; Cadoni, G.; Ferraroni, M.; Serraino, D.; Matsuo, K.; Olshan, A.F.; Zevallos, J.P.; et al. Carotenoid intake and head and neck cancer: A pooled analysis in the International Head and Neck Cancer Epidemiology Consortium. Eur. J. Epidemiol. 2016, 31, 369–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouvard, V.; Loomis, D.; Guyton, K.Z.; Grosse, Y.; Ghissassi, F.E.; Benbrahim-Tallaa, L.; Guha, N.; Mattock, H.; Straif, K. Carcinogenicity of consumption of red and processed meat. Lancet Oncol. 2015, 16, 1599–1600. [Google Scholar] [CrossRef] [Green Version]
- Perloy, A.; Maasland, D.H.E.; van den Brandt, P.A.; Kremer, B.; Schouten, L.J. Intake of meat and fish and risk of head-neck cancer subtypes in the Netherlands Cohort Study. Cancer Causes Control. 2017, 28, 647–656. [Google Scholar] [CrossRef] [Green Version]
- Steffen, A.; Bergmann, M.M.; Sánchez, M.J.; Chirlaque, M.D.; Jakszyn, P.; Amiano, P.; Quirós, J.R.; Barricarte Gurrea, A.; Ferrari, P.; Romieu, I.; et al. Meat and heme iron intake and risk of squamous cell carcinoma of the upper aero-digestive tract in the European Prospective Investigation into Cancer and Nutrition (EPIC). Cancer Epidemiol. Biomark. Prev. 2012, 21, 2138–2148. [Google Scholar] [CrossRef] [Green Version]
- Peppone, L.J.; Mustian, K.M.; Morrow, G.R.; Dozier, A.M.; Ossip, D.J.; Janelsins, M.C.; Sprod, L.K.; McIntosh, S. The effect of cigarette smoking on cancer treatment-related side effects. Oncologist 2011, 16, 1784–1792. [Google Scholar] [CrossRef] [Green Version]
- Petros, W.P.; Younis, I.R.; Ford, J.N.; Weed, S.A. Effects of tobacco smoking and nicotine on cancer treatment. Pharmacotherapy 2012, 32, 920–931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Malley, M.; King, A.N.; Conte, M.; Ellingrod, V.L.; Ramnath, N. Effects of cigarette smoking on metabolism and effectiveness of systemic therapy for lung cancer. J. Thorac. Oncol. 2014, 9, 917–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Browman, G.P.; Wong, G.; Hodson, I.; Sathya, J.; Russell, R.; McAlpine, L.; Skingley, P.; Levine, M.N. Influence of cigarette smoking on the efficacy of radiation therapy in head and neck cancer. N. Engl. J. Med. 1993, 328, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Tilak, T.; Bakhshi, S.; Raina, V.; Kumar, L.; Chaudhary, S.P.; Sahoo, R.K.; Gupta, R.; Thulkar, S. Lactobacillus brevis CD2 lozenges prevent oral mucositis in patients undergoing high dose chemotherapy followed by haematopoietic stem cell transplantation. ESMO Open 2017, 1, e000138. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Rath, G.K.; Chaudhary, S.P.; Thakar, A.; Mohanti, B.K.; Bahadur, S. Lactobacillus brevis CD2 lozenges reduce radiation- and chemotherapy-induced mucositis in patients with head and neck cancer: A randomized double-blind placebo-controlled study. Eur. J. Cancer 2012, 48, 875–881. [Google Scholar] [CrossRef]
- Fessler, J.; Matson, V.; Gajewski, T.F. Exploring the emerging role of the microbiome in cancer immunotherapy. J. Immunother. Cancer 2019, 7, 108. [Google Scholar] [CrossRef]
- Guan, W.; Zhang, X.; Wang, X.; Lu, S.; Yin, J.; Zhang, J. Employing Parasite Against Cancer: A Lesson From the Canine Tapeworm Echinococcus Granulocus. Front. Pharmacol. 2019, 10, 1137. [Google Scholar] [CrossRef]
- Callejas, B.E.; Martínez-Saucedo, D.; Terrazas, L.I. Parasites as negative regulators of cancer. Biosci. Rep. 2018, 38, BSR20180935. [Google Scholar] [CrossRef]
- Mittelman, S.D. The Role of Diet in Cancer Prevention and Chemotherapy Efficacy. Annu. Rev. Nutr. 2020, 40, 273–297. [Google Scholar] [CrossRef]
- Tucci, J.; Alhushki, W.; Chen, T.; Sheng, X.; Kim, Y.M.; Mittelman, S.D. Switch to low-fat diet improves outcome of acute lymphoblastic leukemia in obese mice. Cancer Metab. 2018, 6, 15. [Google Scholar] [CrossRef] [Green Version]
- Allen, B.G.; Bhatia, S.K.; Buatti, J.M.; Brandt, K.E.; Lindholm, K.E.; Button, A.M.; Szweda, L.I.; Smith, B.J.; Spitz, D.R.; Fath, M.A. Ketogenic diets enhance oxidative stress and radio-chemo-therapy responses in lung cancer xenografts. Clin. Cancer Res. 2013, 19, 3905–3913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klement, R.J. Restricting carbohydrates to fight head and neck cancer-is this realistic? Cancer Biol. Med. 2014, 11, 145–161. [Google Scholar] [CrossRef] [PubMed]
- Spring, B.; King, A.C.; Pagoto, S.L.; Van Horn, L.; Fisher, J.D. Fostering multiple healthy lifestyle behaviors for primary prevention of cancer. Am. Psychol. 2015, 70, 75–90. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miranda-Galvis, M.; Loveless, R.; Kowalski, L.P.; Teng, Y. Impacts of Environmental Factors on Head and Neck Cancer Pathogenesis and Progression. Cells 2021, 10, 389. https://doi.org/10.3390/cells10020389
Miranda-Galvis M, Loveless R, Kowalski LP, Teng Y. Impacts of Environmental Factors on Head and Neck Cancer Pathogenesis and Progression. Cells. 2021; 10(2):389. https://doi.org/10.3390/cells10020389
Chicago/Turabian StyleMiranda-Galvis, Marisol, Reid Loveless, Luiz Paulo Kowalski, and Yong Teng. 2021. "Impacts of Environmental Factors on Head and Neck Cancer Pathogenesis and Progression" Cells 10, no. 2: 389. https://doi.org/10.3390/cells10020389
APA StyleMiranda-Galvis, M., Loveless, R., Kowalski, L. P., & Teng, Y. (2021). Impacts of Environmental Factors on Head and Neck Cancer Pathogenesis and Progression. Cells, 10(2), 389. https://doi.org/10.3390/cells10020389