
The Liver under the Spotlight: Bile Acids and Oxysterols as Pivotal Actors Controlling Metabolism



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
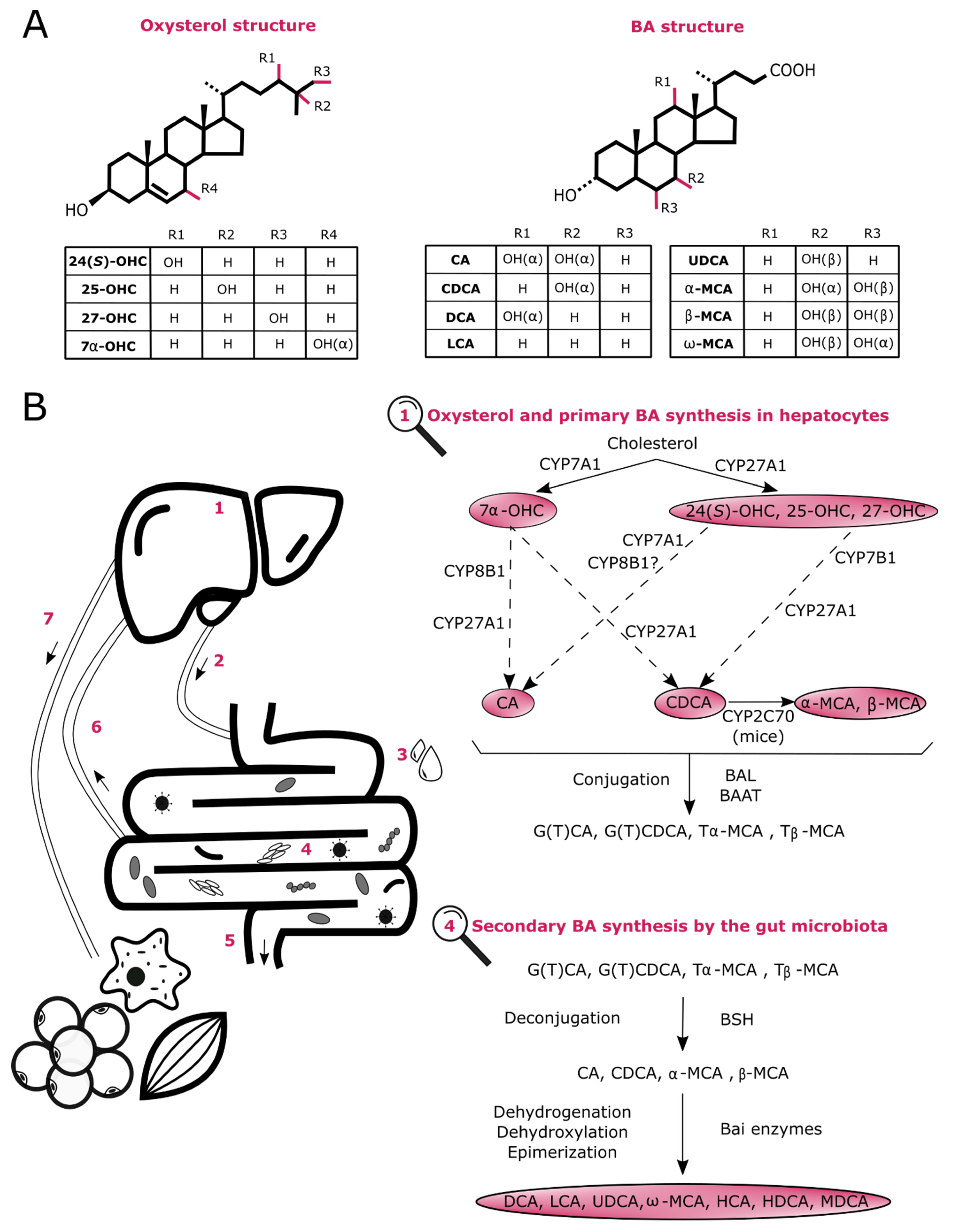
2. Biosynthesis of Oxysterols and BAs
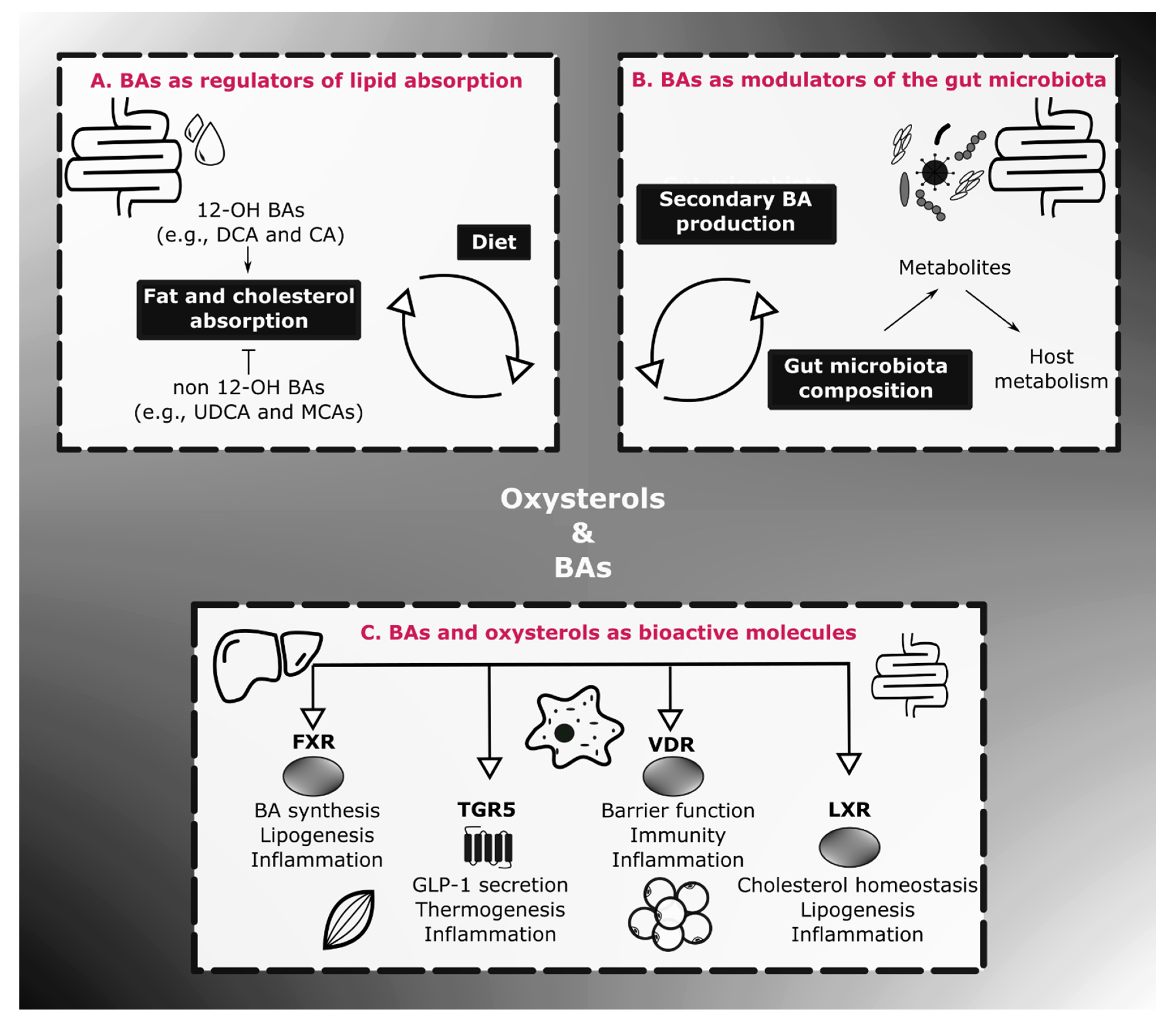
3. Physiological Roles of BAs on Energy Homeostasis and Inflammation
3.1. BA Profile in Inflammatory and Metabolic Diseases
3.2. BAs as Endocrine Molecules
3.2.1. Farnesoid X Receptor (FXR)
- FXR and BA Regulation
- FXR, Energy Homeostasis and Inflammation
3.2.2. Takeda G-Protein Coupled Receptor 5 (TGR5)
3.2.3. Vitamin D Receptor (VDR)
3.3. BAs, Gut Microbes and Energy Homeostasis
4. Oxysterols in Energy Homeostasis and Inflammation
4.1. Oxysterol Profile in Inflammatory and Metabolic Diseases
4.2. Oxysterols as Endocrine Molecules
Liver X Receptor (LXR)
- LXR and Cholesterol Homeostasis
- LXR, Glucose and Lipid Metabolism
- LXR and Inflammation
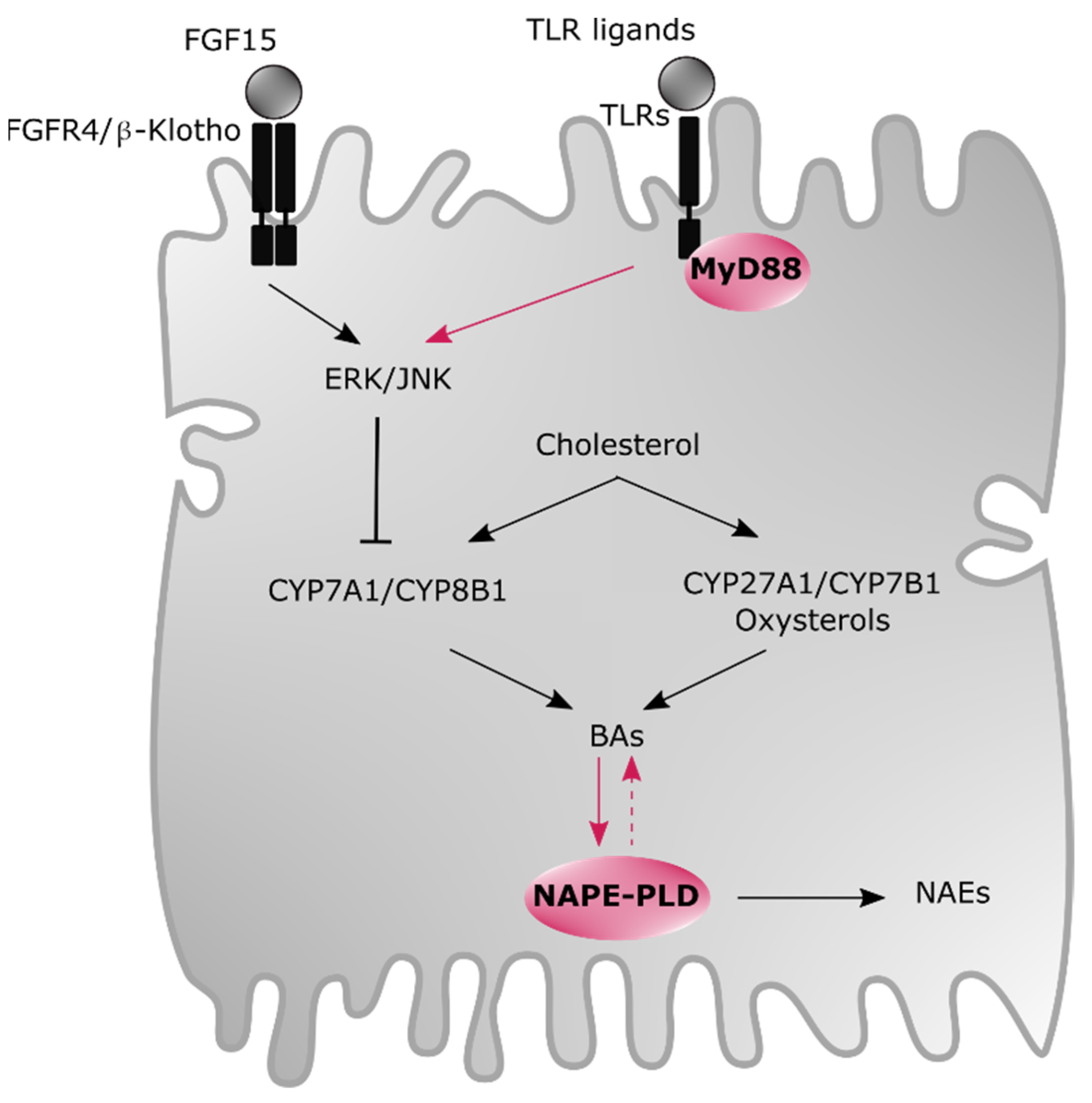
5. Newly Identified Modulators of BAs and Oxysterols
6. Therapeutic Strategies
7. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ntambi, J.M. Hepatic de Novo Lipogenesis and Regulation of Metabolism; Springer: Berlin/Heidelberg, Germany, 2015. [Google Scholar]
- Sanders, F.W.B.; Griffin, J.L. De novo lipogenesis in the liver in health and disease: More than just a shunting yard for glucose. Biol. Rev. 2016, 91, 452–468. [Google Scholar] [CrossRef] [PubMed]
- Malarkey, D.E.; Johnson, K.; Ryan, L.; Boorman, G.; Maronpot, R.R. New Insights into Functional Aspects of Liver Morphology. Toxicol. Pathol. 2005, 33, 27–34. [Google Scholar] [CrossRef]
- Kuntz, E.; Kuntz, H.-D. Morphology of the Liver. In Hepatology Textbook and Atlas; Springer International Publishing: Berlin/Heidelberg, Germany, 2008; pp. 15–33. [Google Scholar]
- Smedsrød, B.; Pertoft, H.; Gustafson, S.; Laurent, T.C. Scavenger functions of the liver endothelial cell. Biochem. J. 1990, 266, 313–327. [Google Scholar] [CrossRef] [PubMed]
- Horst, A.K.; Neumann, K.; Diehl, L.; Tiegs, G. Modulation of liver tolerance by conventional and nonconventional antigen-presenting cells and regulatory immune cells. Cell. Mol. Immunol. 2016, 13, 277–292. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, T.; Friedman, T.T.S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef]
- Vallim, T.Q.D.A.; Tarling, E.J.; Edwards, P.A. Pleiotropic Roles of Bile Acids in Metabolism. Cell Metab. 2013, 17, 657–669. [Google Scholar] [CrossRef]
- Guillemot-Legris, O.; Mutemberezi, V.; Muccioli, G.G. Oxysterols in Metabolic Syndrome: From Bystander Molecules to Bioactive Lipids. Trends Mol. Med. 2016, 22, 594–614. [Google Scholar] [CrossRef]
- Willinger, T. Oxysterols in intestinal immunity and inflammation. J. Intern. Med. 2018, 285, 367–380. [Google Scholar] [CrossRef]
- Griffiths, W.J.; Abdel-Khalik, J.; Hearn, T.; Yutuc, E.; Morgan, A.H.; Wang, Y. Current trends in oxysterol research. Biochem. Soc. Trans. 2016, 44, 652–658. [Google Scholar] [CrossRef]
- Luu, W.; Sharpe, L.J.; Capell-Hattam, I.; Gelissen, I.C.; Brown, A.J. Oxysterols: Old Tale, New Twists. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 447–467. [Google Scholar] [CrossRef]
- Russell, D.W. The Enzymes, Regulation, and Genetics of Bile Acid Synthesis. Annu. Rev. Biochem. 2003, 72, 137–174. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Chiang, J.Y.L. Bile Acid Signaling in Metabolic Disease and Drug Therapy. Pharmacol. Rev. 2014, 66, 948–983. [Google Scholar] [CrossRef]
- Wahlström, A.; Sayin, S.I.; Marschall, H.-U.; Bäckhed, F. Intestinal Crosstalk between Bile Acids and Microbiota and Its Impact on Host Metabolism. Cell Metab. 2016, 24, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Schaap, F.G.; Trauner, M.; Jansen, P.L.M. Bile acid receptors as targets for drug development. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Li-Hawkins, J.; Gåfvels, M.; Olin, M.; Lund, E.G.; Andersson, U.; Schuster, G.; Bjorkhem, I.; Russell, D.W.; Eggertsen, G. Cholic acid mediates negative feedback regulation of bile acid synthesis in mice. J. Clin. Investig. 2002, 110, 1191–1200. [Google Scholar] [CrossRef]
- Pandak, W.M.; Kakiyama, G. The acidic pathway of bile acid synthesis: Not just an alternative pathway. Liver Res. 2019, 3, 88–98. [Google Scholar] [CrossRef]
- Honda, A.; Miyazaki, T.; Iwamoto, J.; Hirayama, T.; Morishita, Y.; Monma, T.; Ueda, H.; Mizuno, S.; Sugiyama, F.; Takahashi, S.; et al. Regulation of bile acid metabolism in mouse models with hydrophobic bile acid composition. J. Lipid Res. 2020, 61, 54–69. [Google Scholar] [CrossRef]
- Chiang, J.Y.L.; Ferrell, J.M. Bile acid receptors FXR and TGR5 signaling in fatty liver diseases and therapy. Am. J. Physiol. Liver Physiol. 2020, 318, G554–G573. [Google Scholar] [CrossRef] [PubMed]
- Monte, M.J.; Marin, J.J.G.; Antelo, A.; Vazquez-Tato, J. Bile acids: Chemistry, physiology, and pathophysiology. World J. Gastroenterol. 2009, 15, 804–816. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.Q.-H.; Tazuma, S.; Cohen, D.E.; Carey, M.C. Feeding natural hydrophilic bile acids inhibits intestinal cholesterol absorption: Studies in the gallstone-susceptible mouse. Am. J. Physiol. Liver Physiol. 2003, 285, G494–G502. [Google Scholar] [CrossRef]
- Chávez-Talavera, O.; Tailleux, A.; Lefebvre, P.; Staels, B. Bile Acid Control of Metabolism and Inflammation in Obesity, Type 2 Diabetes, Dyslipidemia, and Nonalcoholic Fatty Liver Disease. Gastroenterology 2017, 152, 1679–1694. [Google Scholar] [CrossRef]
- Mouzaki, M.; Wang, A.Y.; Bandsma, R.; Comelli, E.M.; Arendt, B.M.; Zhang, L.; Fung, S.; Fischer, S.E.; McGilvray, I.G.; Allard, J.P. Bile Acids and Dysbiosis in Non-Alcoholic Fatty Liver Disease. PLoS ONE 2016, 11, e0151829. [Google Scholar] [CrossRef] [PubMed]
- Ferslew, B.C.; Xie, G.; Johnston, C.K.; Su, M.; Stewart, P.W.; Jia, W.; Brouwer, K.L.R.; Barritt, A.S. Altered Bile Acid Metabolome in Patients with Nonalcoholic Steatohepatitis. Dig. Dis. Sci. 2015, 60, 3318–3328. [Google Scholar] [CrossRef] [PubMed]
- Jiao, N.; Baker, S.S.; Chapa-Rodriguez, A.; Liu, W.; Nugent, C.A.; Tsompana, M.; Mastrandrea, L.; Buck, M.J.; Baker, R.D.; Genco, R.J.; et al. Suppressed hepatic bile acid signalling despite elevated production of primary and secondary bile acids in NAFLD. Gut 2018, 67, 1881–1891. [Google Scholar] [CrossRef] [PubMed]
- Aranha, M.M.; Cortez-Pinto, H.; Costa, A.; Da Silva, I.B.M.; Camilo, M.E.; De Moura, M.C.; Rodrigues, C.M. Bile acid levels are increased in the liver of patients with steatohepatitis. Eur. J. Gastroenterol. Hepatol. 2008, 20, 519–525. [Google Scholar] [CrossRef]
- Caussy, C.; Hsu, C.; Singh, S.; Bassirian, S.; Kolar, J.; Faulkner, C.; Sinha, N.; Bettencourt, R.; Gara, N.; Valasek, M.A.; et al. Serum bile acid patterns are associated with the presence of NAFLD in twins, and dose-dependent changes with increase in fibrosis stage in patients with biopsy-proven NAFLD. Aliment. Pharmacol. Ther. 2019, 49, 183–193. [Google Scholar] [CrossRef]
- Puri, P.; Daita, K.; Joyce, A.; Mirshahi, F.; Santhekadur, P.K.; Cazanave, S.; Luketic, V.A.; Siddiqui, M.S.; Boyett, S.; Min, H.; et al. The presence and severity of nonalcoholic steatohepatitis is associated with specific changes in circulating bile acids. Hepatology 2018, 67, 534–548. [Google Scholar] [CrossRef]
- Molinaro, A.; Wahlström, A.; Marschall, H.-U. Role of Bile Acids in Metabolic Control. Trends Endocrinol. Metab. 2018, 29, 31–41. [Google Scholar] [CrossRef]
- Lavelle, A.; Sokol, H. Gut microbiota-derived metabolites as key actors in inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 223–237. [Google Scholar] [CrossRef]
- Duboc, H.; Rajca, S.; Rainteau, D.; Benarous, D.; Maubert, M.-A.; Quervain, E.; Thomas, G.; Barbu, V.; Humbert, L.; Despras, G.; et al. Connecting dysbiosis, bile-acid dysmetabolism and gut inflammation in inflammatory bowel diseases. Gut 2013, 62, 531–539. [Google Scholar] [CrossRef]
- Franzosa, E.A.; Sirota-Madi, A.; Avila-Pacheco, J.; Fornelos, N.; Haiser, H.J.; Reinker, S.; Vatanen, T.; Hall, A.B.; Mallick, H.; McIver, L.J.; et al. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat. Microbiol. 2019, 4, 293–305. [Google Scholar] [CrossRef]
- Matsuoka, K.; Suzuki, M.; Honda, C.; Endo, K.; Moroi, Y. Micellization of conjugated chenodeoxy- and ursodeoxycholates and solubilization of cholesterol into their micelles: Comparison with other four conjugated bile salts species. Chem. Phys. Lipids 2006, 139, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Murphy, C.; Parini, P.; Wang, J.; Björkhem, I.; Eggertsen, G.; Gåfvels, M. Cholic acid as key regulator of cholesterol synthesis, intestinal absorption and hepatic storage in mice. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2005, 1735, 167–175. [Google Scholar] [CrossRef]
- Zhang, Y.; Jiang, R.; Zheng, X.; Lei, S.; Huang, F.; Xie, G.; Kwee, S.; Yu, H.; Farrar, C.; Sun, B.; et al. Ursodeoxycholic acid accelerates bile acid enterohepatic circulation. Br. J. Pharmacol. 2019, 176, 2848–2863. [Google Scholar] [CrossRef] [PubMed]
- Haeusler, R.A.; Astiarraga, B.; Camastra, S.; Accili, D.; Ferrannini, E. Human Insulin Resistance Is Associated With Increased Plasma Levels of 12 -Hydroxylated Bile Acids. Diabetes 2013, 62, 4184–4191. [Google Scholar] [CrossRef]
- Wei, M.; Huang, F.; Zhao, L.; Zhang, Y.; Yang, W.; Wang, S.; Li, M.; Han, X.; Ge, K.; Qu, C.; et al. A dysregulated bile acid-gut microbiota axis contributes to obesity susceptibility. EBioMedicine 2020, 55, 102766. [Google Scholar] [CrossRef]
- Biemann, R.; Penner, M.; Borucki, K.; Westphal, S.; Luley, C.; Rönicke, R.; Biemann, K.; Weikert, C.; Lux, A.; Goncharenko, N.; et al. Serum bile acids and GLP-1 decrease following telemetric induced weight loss: Results of a randomized controlled trial. Sci. Rep. 2016, 6, 30173. [Google Scholar] [CrossRef]
- Jia, W.; Wei, M.; Rajani, C.; Zheng, X. Targeting the alternative bile acid synthetic pathway for metabolic diseases. Protein Cell 2020, 1–15. [Google Scholar] [CrossRef]
- Parks, D.J.; Blanchard, S.G.; Bledsoe, R.K.; Chandra, G.; Consler, T.G.; Kliewer, S.A.; Stimmel, J.B.; Willson, T.M.; Zavacki, A.M.; Moore, D.D.; et al. Bile Acids: Natural Ligands for an Orphan Nuclear Receptor. Science 1999, 284, 1365–1368. [Google Scholar] [CrossRef] [PubMed]
- Makishima, M.; Okamoto, A.Y.; Repa, J.J.; Tu, H.; Learned, R.M.; Luk, A.; Hull, M.V.; Lustig, K.D.; Mangelsdorf, D.J.; Shan, B. Identification of a Nuclear Receptor for Bile Acids. Science 1999, 284, 1362–1365. [Google Scholar] [CrossRef]
- Vaquero, J.; Monte, M.J.; Dominguez, M.; Muntané, J.; Marin, J.J. Differential activation of the human farnesoid X receptor depends on the pattern of expressed isoforms and the bile acid pool composition. Biochem. Pharmacol. 2013, 86, 926–939. [Google Scholar] [CrossRef]
- Kanda, T.; Foucand, L.; Nakamura, Y.; Niot, I.; Besnard, P.; Fujita, M.; Sakai, Y.; Hatakeyama, K.; Ono, T.; Fujii, H. Regulation of expression of human intestinal bile acid-binding protein in Caco-2 cells. Biochem. J. 1998, 330 Pt 1, 261–265. [Google Scholar] [CrossRef]
- Song, P.; Rockwell, C.E.; Cui, J.Y.; Klaassen, C.D. Individual bile acids have differential effects on bile acid signaling in mice. Toxicol. Appl. Pharmacol. 2015, 283, 57–64. [Google Scholar] [CrossRef]
- Yang, Z.-X.; Shen, W.; Sun, H. Effects of nuclear receptor FXR on the regulation of liver lipid metabolism in patients with non-alcoholic fatty liver disease. Hepatol. Int. 2010, 4, 741–748. [Google Scholar] [CrossRef] [PubMed]
- Sayin, S.I.; Wahlström, A.; Felin, J.; Jäntti, S.; Marschall, H.-U.; Bamberg, K.; Angelin, B.; Hyötyläinen, T.; Orešič, M.; Bäckhed, F. Gut Microbiota Regulates Bile Acid Metabolism by Reducing the Levels of Tauro-beta-muricholic Acid, a Naturally Occurring FXR Antagonist. Cell Metab. 2013, 17, 225–235. [Google Scholar] [CrossRef]
- Chiang, J.Y.L. Bile acids: Regulation of synthesis. J. Lipid Res. 2009, 50, 1955–1966. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.C.; Wang, M.; Blackmore, C.; Desnoyers, L.R. Liver-specific Activities of FGF19 Require Klotho beta. J. Biol. Chem. 2007, 282, 27277–27284. [Google Scholar] [CrossRef]
- Kong, B.; Wang, L.; Chiang, J.Y.L.; Zhang, Y.; Klaassen, C.D.; Guo, G.L. Mechanism of tissue-specific farnesoid X receptor in suppressing the expression of genes in bile-acid synthesis in mice. Hepatology 2012, 56, 1034–1043. [Google Scholar] [CrossRef] [PubMed]
- Sinal, C.J.; Tohkin, M.; Miyata, M.; Ward, J.M.; Lambert, G.; Gonzalez, F.J. Targeted Disruption of the Nuclear Receptor FXR/BAR Impairs Bile Acid and Lipid Homeostasis. Cell 2000, 102, 731–744. [Google Scholar] [CrossRef]
- Kok, T.; Hulzebos, C.V.; Wolters, H.; Havinga, R.; Agellon, L.B.; Stellaard, F.; Shan, B.; Schwarz, M.; Kuipers, F. Enterohepatic Circulation of Bile Salts in Farnesoid X Receptor-deficient Mice. J. Biol. Chem. 2003, 278, 41930–41937. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lee, F.Y.; Barrera, G.; Lee, H.; Vales, C.; Gonzalez, F.J.; Willson, T.M.; Edwards, P.A. Activation of the nuclear receptor FXR improves hyperglycemia and hyperlipidemia in diabetic mice. Proc. Natl. Acad. Sci. USA 2006, 103, 1006–1011. [Google Scholar] [CrossRef]
- Ma, K.; Saha, P.K.; Chan, L.; Moore, D.D. Farnesoid X receptor is essential for normal glucose homeostasis. J. Clin. Investig. 2006, 116, 1102–1109. [Google Scholar] [CrossRef] [PubMed]
- Pircher, P.C.; Kitto, J.L.; Petrowski, M.L.; Tangirala, R.K.; Bischoff, E.D.; Schulman, I.G.; Westin, S.K. Farnesoid X Receptor Regulates Bile Acid-Amino Acid Conjugation. J. Biol. Chem. 2003, 278, 27703–27711. [Google Scholar] [CrossRef] [PubMed]
- Ananthanarayanan, M.; Balasubramanian, N.; Makishima, M.; Mangelsdorf, D.J.; Suchy, F.J. Human Bile Salt Export Pump Promoter Is Transactivated by the Farnesoid X Receptor/Bile Acid Receptor. J. Biol. Chem. 2001, 276, 28857–28865. [Google Scholar] [CrossRef]
- Denson, L.A.; Sturm, E.; Echevarria, W.; Zimmerman, T.L.; Makishima, M.; Mangelsdorf, D.J.; Karpen, S.J. The Orphan Nuclear Receptor, shp, Mediates Bile Acid-Induced Inhibition of the Rat Bile Acid Transporter, ntcp. Gastroenterology 2001, 121, 140–147. [Google Scholar] [CrossRef]
- Watanabe, M.; Houten, S.M.; Wang, L.; Moschetta, A.; Mangelsdorf, D.J.; Heyman, R.A.; Moore, D.D.; Auwerx, J. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J. Clin. Investig. 2004, 113, 1408–1418. [Google Scholar] [CrossRef]
- Caron, S.; Samanez, C.H.; Dehondt, H.; Ploton, M.; Briand, O.; Lien, F.; Dorchies, E.; Dumont, J.; Postic, C.; Cariou, B.; et al. Farnesoid X Receptor Inhibits the Transcriptional Activity of Carbohydrate Response Element Binding Protein in Human Hepatocytes. Mol. Cell. Biol. 2013, 33, 2202–2211. [Google Scholar] [CrossRef] [PubMed]
- Torra, I.P.; Claudel, T.; Duval, C.; Kosykh, V.; Fruchart, J.-C.; Staels, B. Bile Acids Induce the Expression of the Human Peroxisome Proliferator-Activated Receptor α Gene via Activation of the Farnesoid X Receptor. Mol. Endocrinol. 2003, 17, 259–272. [Google Scholar] [CrossRef]
- Massafra, V.; Van Mil, S.W. Farnesoid X receptor: A “homeostat” for hepatic nutrient metabolism. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 45–59. [Google Scholar] [CrossRef]
- Xi, Y.; Li, H. Role of farnesoid X receptor in hepatic steatosis in nonalcoholic fatty liver disease. Biomed. Pharmacother. 2020, 121, 109609. [Google Scholar] [CrossRef]
- Yamagata, K.; Daitoku, H.; Shimamoto, Y.; Matsuzaki, H.; Hirota, K.; Ishida, J.; Fukamizu, A. Bile Acids Regulate Gluconeogenic Gene Expression via Small Heterodimer Partner-mediated Repression of Hepatocyte Nuclear Factor 4 and Foxo1. J. Biol. Chem. 2004, 279, 23158–23165. [Google Scholar] [CrossRef]
- Stayrook, K.R.; Bramlett, K.S.; Savkur, R.S.; Ficorilli, J.; Cook, T.; Christe, M.E.; Michael, L.F.; Burris, T.P. Regulation of Carbohydrate Metabolism by the Farnesoid X Receptor. Endocrinology 2005, 146, 984–991. [Google Scholar] [CrossRef] [PubMed]
- Cariou, B.; Van Harmelen, K.; Duran-Sandoval, D.; Van Dijk, T.H.; Grefhorst, A.; Abdelkarim, M.; Caron, S.; Torpier, G.; Fruchart, J.-C.; Gonzalez, F.J.; et al. The Farnesoid X Receptor Modulates Adiposity and Peripheral Insulin Sensitivity in Mice. J. Biol. Chem. 2006, 281, 11039–11049. [Google Scholar] [CrossRef]
- Wang, Y.-D.; Chen, W.-D.; Wang, M.; Yu, D.; Forman, B.M.; Huang, W. Farnesoid X receptor antagonizes nuclear factor κB in hepatic inflammatory response. Hepatology 2008, 48, 1632–1643. [Google Scholar] [CrossRef]
- Mencarelli, A.; Renga, B.; Distrutti, E.; Fiorucci, S. Antiatherosclerotic effect of farnesoid X receptor. Am. J. Physiol. Circ. Physiol. 2009, 296, H272–H281. [Google Scholar] [CrossRef] [PubMed]
- Massafra, V.; Pellicciari, R.; Gioiello, A.; Van Mil, S.W. Progress and challenges of selective Farnesoid X Receptor modulation. Pharmacol. Ther. 2018, 191, 162–177. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Investig. 2004, 114, 1752–1761. [Google Scholar] [CrossRef]
- Cai, D.; Yuan, M.; Frantz, D.F.; Melendez, P.A.; Hansen, L.; Lee, J.; Shoelson, S.E. Local and systemic insulin resistance resulting from hepatic activation of IKK-β and NF-κB. Nat. Med. 2005, 11, 183–190. [Google Scholar] [CrossRef]
- Prawitt, J.; Abdelkarim, M.; Stroeve, J.H.M.; Popescu, I.; Duez, H.; Velagapudi, V.R.; Dumont, J.; Bouchaert, E.; Van Dijk, T.H.; Lucas, A.; et al. Farnesoid X Receptor Deficiency Improves Glucose Homeostasis in Mouse Models of Obesity. Diabetes 2011, 60, 1861–1871. [Google Scholar] [CrossRef]
- Parséus, A.; Sommer, N.; Sommer, F.; Caesar, R.; Molinaro, A.; Ståhlman, M.; Greiner, T.U.; Perkins, R.; Bäckhed, F. Microbiota-induced obesity requires farnesoid X receptor. Gut 2017, 66, 429–437. [Google Scholar] [CrossRef]
- Watanabe, M.; Horai, Y.; Houten, S.M.; Morimoto, K.; Sugizaki, T.; Arita, E.; Mataki, C.; Sato, H.; Tanigawara, Y.; Schoonjans, K.; et al. Lowering Bile Acid Pool Size with a Synthetic Farnesoid X Receptor (FXR) Agonist Induces Obesity and Diabetes through Reduced Energy Expenditure. J. Biol. Chem. 2011, 286, 26913–26920. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Xie, C.; Li, F.; Zhang, L.; Nichols, R.G.; Krausz, K.W.; Cai, J.; Qi, Y.; Fang, Z.-Z.; Takahashi, S.; et al. Intestinal farnesoid X receptor signaling promotes nonalcoholic fatty liver disease. J. Clin. Investig. 2015, 125, 386–402. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Xie, C.; Lv, Y.; Li, J.; Krausz, K.W.; Shi, J.; Brocker, C.N.; Desai, D.; Amin, S.G.; Bisson, W.H.; et al. Intestine-selective farnesoid X receptor inhibition improves obesity-related metabolic dysfunction. Nat. Commun. 2015, 6, 10166. [Google Scholar] [CrossRef] [PubMed]
- Fang, S.; Suh, J.M.; Reilly, S.M.; Yu, E.; Osborn, O.; Lackey, D.; Yoshihara, E.; Perino, A.; Jacinto, S.; Lukasheva, Y.; et al. Intestinal FXR agonism promotes adipose tissue browning and reduces obesity and insulin resistance. Nat. Med. 2015, 21, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Pathak, P.; Xie, C.; Nichols, R.G.; Ferrell, J.M.; Boehme, S.; Krausz, K.W.; Patterson, A.D.; Gonzalez, F.J.; Chiang, J.Y.L. Intestine farnesoid X receptor agonist and the gut microbiota activate G-protein bile acid receptor-1 signaling to improve metabolism. Hepatology 2018, 68, 1574–1588. [Google Scholar] [CrossRef]
- Schmitt, J.; Kong, B.; Stieger, B.; Tschopp, O.; Schultze, S.M.; Rau, M.; Weber, A.; Muellhaupt, B.; Guo, G.L.; Geier, A. Protective effects of farnesoid X receptor (FXR) on hepatic lipid accumulation are mediated by hepatic FXR and independent of intestinal FGF15 signal. Liver Int. 2015, 35, 1133–1144. [Google Scholar] [CrossRef]
- Van Nierop, F.S.; Scheltema, M.J.; Eggink, H.M.; Pols, T.W.; Sonne, D.P.; Knop, F.K.; Soeters, M.R. Clinical relevance of the bile acid receptor TGR5 in metabolism. Lancet Diabetes Endocrinol. 2017, 5, 224–233. [Google Scholar] [CrossRef]
- Cipriani, S.; Mencarelli, A.; Chini, M.G.; Distrutti, E.; Renga, B.; Bifulco, G.; Baldelli, F.; Donini, A.; Fiorucci, S. The Bile Acid Receptor GPBAR-1 (TGR5) Modulates Integrity of Intestinal Barrier and Immune Response to Experimental Colitis. PLoS ONE 2011, 6, e25637. [Google Scholar] [CrossRef]
- Maruyama, T.; Miyamoto, Y.; Nakamura, T.; Tamai, Y.; Okada, H.; Sugiyama, E.; Nakamura, T.; Itadani, H.; Tanaka, K. Identification of membrane-type receptor for bile acids (M-BAR). Biochem. Biophys. Res. Commun. 2002, 298, 714–719. [Google Scholar] [CrossRef]
- Kawamata, Y.; Fujii, R.; Hosoya, M.; Harada, M.; Yoshida, H.; Miwa, M.; Fukusumi, S.; Habata, Y.; Itoh, T.; Shintani, Y.; et al. A G Protein-coupled Receptor Responsive to Bile Acids. J. Biol. Chem. 2003, 278, 9435–9440. [Google Scholar] [CrossRef]
- Cani, P.D.; Van Hul, M.; Lefort, C.; Depommier, C.; Rastelli, M.; Everard, A. Microbial regulation of organismal energy homeostasis. Nat. Metab. 2019, 1, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Houten, S.M.; Mataki, C.; Christoffolete, M.A.; Kim, B.W.; Sato, H.; Messaddeq, N.; Harney, J.W.; Ezaki, O.; Kodama, T.; et al. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nat. Cell Biol. 2006, 439, 484–489. [Google Scholar] [CrossRef]
- Katsuma, S.; Hirasawa, A.; Tsujimoto, G. Bile acids promote glucagon-like peptide-1 secretion through TGR5 in a murine enteroendocrine cell line STC-1. Biochem. Biophys. Res. Commun. 2005, 329, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.; Gioiello, A.; Noriega, L.; Strehle, A.; Oury, J.; Rizzo, G.; Macchiarulo, A.; Yamamoto, H.; Mataki, C.; Pruzanski, M.; et al. TGR5-Mediated Bile Acid Sensing Controls Glucose Homeostasis. Cell Metab. 2009, 10, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Pols, T.W.; Nomura, M.; Harach, T.; Sasso, G.L.; Oosterveer, M.H.; Thomas, C.; Rizzo, G.; Gioiello, A.; Adorini, L.; Pellicciari, R.; et al. TGR5 Activation Inhibits Atherosclerosis by Reducing Macrophage Inflammation and Lipid Loading. Cell Metab. 2011, 14, 747–757. [Google Scholar] [CrossRef]
- Wang, Y.-D.; Chen, W.-D.; Yu, D.; Forman, B.M.; Huang, W. The G-Protein-coupled bile acid receptor, Gpbar1 (TGR5), negatively regulates hepatic inflammatory response through antagonizing nuclear factor kappa light-chain enhancer of activated B cells (NF-κB) in mice. Hepatol. 2011, 54, 1421–1432. [Google Scholar] [CrossRef]
- Sinha, S.R.; Haileselassie, Y.; Nguyen, L.P.; Tropini, C.; Wang, M.; Becker, L.S.; Sim, D.; Jarr, K.; Spear, E.T.; Singh, G.; et al. Dysbiosis-Induced Secondary Bile Acid Deficiency Promotes Intestinal Inflammation. Cell Host Microbe 2020, 27, 659–670. [Google Scholar] [CrossRef]
- Bakke, D.; Chatterjee, I.; Agrawal, A.; Dai, Y.; Sun, A.J. Regulation of Microbiota by Vitamin D Receptor: A Nuclear Weapon in Metabolic Diseases. Nucl. Recept. Res. 2018, 5. [Google Scholar] [CrossRef]
- White, J.H. Vitamin D metabolism and signaling in the immune system. Rev. Endocr. Metab. Disord. 2011, 13, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Makishima, M.; Lu, T.T.; Xie, W.; Whitfield, G.K.; Domoto, H.; Evans, R.M.; Haussler, M.R.; Mangelsdorf, D.J. Vitamin D Receptor as an Intestinal Bile Acid Sensor. Science 2002, 296, 1313–1316. [Google Scholar] [CrossRef]
- White, J.H. Vitamin D deficiency and the pathogenesis of Crohn’s disease. J. Steroid Biochem. Mol. Biol. 2018, 175, 23–28. [Google Scholar] [CrossRef]
- Sun, J.; Mustafi, R.; Cerda, S.; Chumsangsri, A.; Xia, Y.R.; Li, Y.C.; Bissonnette, M. Lithocholic acid down-regulation of NF-κB activity through vitamin D receptor in colonic cancer cells. J. Steroid Biochem. Mol. Biol. 2008, 111, 37–40. [Google Scholar] [CrossRef] [PubMed]
- Gurlek, A.; Pittelkow, M.R.; Kumar, R. Modulation of Growth Factor/Cytokine Synthesis and Signaling by 1α,25-Dihydroxyvitamin D3: Implications in Cell Growth and Differentiation. Endocr. Rev. 2002, 23, 763–786. [Google Scholar] [CrossRef]
- Yu, X.P.; Bellido, T.; Manolagas, S.C. Down-regulation of NF-kappa B protein levels in activated human lymphocytes by 1,25-dihydroxyvitamin D3. Proc. Natl. Acad. Sci. USA 1995, 92, 10990–10994. [Google Scholar] [CrossRef]
- Song, X.; Sun, X.; Oh, S.F.; Wu, M.; Zhang, Y.; Zheng, W.; Geva-Zatorsky, N.; Jupp, R.; Mathis, D.; Benoist, C.; et al. Microbial bile acid metabolites modulate gut RORγ+ regulatory T cell homeostasis. Nat. Cell Biol. 2020, 577, 410–415. [Google Scholar] [CrossRef]
- Hang, S.; Paik, D.; Yao, L.; Kim, E.; Trinath, J.; Lu, J.; Ha, S.; Nelson, B.N.; Kelly, S.P.; Wu, L.; et al. Bile acid metabolites control TH17 and Treg cell differentiation. Nat. Cell Biol. 2019, 576, 143–148. [Google Scholar] [CrossRef]
- Chatterjee, I.; Lu, R.; Zhang, Y.; Zhang, J.; Dai, Y.; Xia, Y.; Sun, J. Vitamin D receptor promotes healthy microbial metabolites and microbiome. Sci. Rep. 2020, 10, 7340. [Google Scholar] [CrossRef] [PubMed]
- Jia, W.; Xie, G.; Jia, W. Bile acid–microbiota crosstalk in gastrointestinal inflammation and carcinogenesis. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 111–128. [Google Scholar] [CrossRef]
- Xie, W.; Radominska-Pandya, A.; Shi, Y.; Simon, C.M.; Nelson, M.C.; Ong, E.S.; Waxman, D.J.; Evans, R.M. An essential role for nuclear receptors SXR/PXR in detoxification of cholestatic bile acids. Proc. Natl. Acad. Sci. USA 2001, 98, 3375–3380. [Google Scholar] [CrossRef] [PubMed]
- Staudinger, J.L.; Goodwin, B.; Jones, S.A.; Hawkins-Brown, D.; MacKenzie, K.I.; Latour, A.; Liui, Y.; Klaasseni, C.D.; Brown, K.K.; Reinhard, J.; et al. The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc. Natl. Acad. Sci. USA 2001, 98, 3369–3374. [Google Scholar] [CrossRef]
- Sato, H.; Macchiarulo, A.; Thomas, C.; Gioiello, A.; Une, M.; Hofmann, A.F.; Saladin, R.; Schoonjans, K.; Pellicciari, R.; Auwerx, J. Novel Potent and Selective Bile Acid Derivatives as TGR5 Agonists: Biological Screening, Structure−Activity Relationships, and Molecular Modeling Studies. J. Med. Chem. 2008, 51, 1831–1841. [Google Scholar] [CrossRef]
- Paone, P.; Cani, P.D. Mucus barrier, mucins and gut microbiota: The expected slimy partners? Gut 2020, 69, 2232–2243. [Google Scholar] [CrossRef]
- Allegretti, J.R.; Kassam, Z.; Mullish, B.H.; Chiang, A.; Carrellas, M.; Hurtado, J.; Marchesi, J.R.; McDonald, J.A.; Pechlivanis, A.; Barker, G.F.; et al. Effects of Fecal Microbiota Transplantation with Oral Capsules in Obese Patients. Clin. Gastroenterol. Hepatol. 2020, 18, 855–863. [Google Scholar] [CrossRef]
- Gérard, P. Metabolism of Cholesterol and Bile Acids by the Gut Microbiota. Pathogens 2013, 3, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Kenny, D.J.; Plichta, D.R.; Shungin, D.; Koppel, N.; Hall, A.B.; Fu, B.; Vasan, R.S.; Shaw, S.Y.; Vlamakis, H.; Balskus, E.P.; et al. Cholesterol Metabolism by Uncultured Human Gut Bacteria Influences Host Cholesterol Level. Cell Host Microbe 2020, 28, 245–257. [Google Scholar] [CrossRef]
- Kurdi, P.; Kawanishi, K.; Mizutani, K.; Yokota, A. Mechanism of Growth Inhibition by Free Bile Acids in Lactobacilli and Bifidobacteria. J. Bacteriol. 2006, 188, 1979–1986. [Google Scholar] [CrossRef] [PubMed]
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B.; Bajaj, J.S. Bile acids and the gut microbiome. Curr. Opin. Gastroenterol. 2014, 30, 332–338. [Google Scholar] [CrossRef]
- Van Best, N.; Rolle-Kampczyk, U.; Schaap, F.G.; Basic, M.; Damink, S.W.M.O.; Bleich, A.; Savelkoul, P.H.M.; Von Bergen, M.; Penders, J.; Hornef, M.W. Bile acids drive the newborn’s gut microbiota maturation. Nat. Commun. 2020, 11, 3692. [Google Scholar] [CrossRef]
- Tremblay-Franco, M.; Zerbinati, C.; Pacelli, A.; Palmaccio, G.; Lubrano, C.; Ducheix, S.; Guillou, H.; Iuliano, L. Effect of obesity and metabolic syndrome on plasma oxysterols and fatty acids in human. Steroids 2015, 99, 287–292. [Google Scholar] [CrossRef]
- Guillemot-Legris, O.; Mutemberezi, V.; Cani, P.; Muccioli, G.G. Obesity is associated with changes in oxysterol metabolism and levels in mice liver, hypothalamus, adipose tissue and plasma. Sci. Rep. 2016, 6, 19694. [Google Scholar] [CrossRef] [PubMed]
- Mutemberezi, V.; Masquelier, J.; Guillemot-Legris, O.; Muccioli, G.G. Development and validation of an HPLC-MS method for the simultaneous quantification of key oxysterols, endocannabinoids, and ceramides: Variations in metabolic syndrome. Anal. Bioanal. Chem. 2015, 408, 733–745. [Google Scholar] [CrossRef]
- Murakami, H.; Tamasawa, N.; Matsui, J.; Yasujima, M.; Suda, T. Plasma oxysterols and tocopherol in patients with diabetes mellitus and hyperlipidemia. Lipids 2000, 35, 333–338. [Google Scholar] [CrossRef]
- Bertolotti, M.; Del Puppo, M.; Corna, F.; Anzivino, C.; Gabbi, C.; Baldelli, E.; Carulli, L.; Loria, P.; Kienle, M.G.; Carulli, N. Increased appearance rate of 27-hydroxycholesterol in vivo in hypercholesterolemia: A possible compensatory mechanism. Nutr. Metab. Cardiovasc. Dis. 2012, 22, 823–830. [Google Scholar] [CrossRef]
- Ikegami, T.; Hyogo, H.; Honda, A.; Miyazaki, T.; Tokushige, K.; Hashimoto, E.; Inui, K.; Matsuzaki, Y.; Tazuma, S. Increased serum liver X receptor ligand oxysterols in patients with non-alcoholic fatty liver disease. J. Gastroenterol. 2012, 47, 1257–1266. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, W.J.; Wang, Y. Oxysterol research: A brief review. Biochem. Soc. Trans. 2019, 47, 517–526. [Google Scholar] [CrossRef]
- Guillemot-Legris, O.; Mutemberezi, V.; Buisseret, B.; Paquot, A.; Palmieri, V.; Bottemanne, P.; Lemaire, J.; Rahier, J.-F.; Alhouayek, M.; Muccioli, G.G. Colitis Alters Oxysterol Metabolism and is Affected by 4β-Hydroxycholesterol Administration. J. Crohn’s Colitis 2018, 13, 218–229. [Google Scholar] [CrossRef]
- Setchell, K.D.R.; Schwarz, M.; O’Connell, N.C.; Lund, E.G.; Davis, D.L.; Lathe, R.; Thompson, H.R.; Tyson, R.W.; Sokol, R.J.; Russell, D.W. Identification of a new inborn error in bile acid synthesis: Mutation of the oxysterol 7alpha-hydroxylase gene causes severe neonatal liver disease. J. Clin. Investig. 1998, 102, 1690–1703. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.; Mills, P.B.; Footitt, E.; Gissen, P.; McClean, P.; Stahlschmidt, J.; Coupry, I.; Lavie, J.; Mochel, F.; Goizet, C.; et al. Liver disease in infancy caused by oxysterol 7α-hydroxylase deficiency: Successful treatment with chenodeoxycholic acid. J. Inherit. Metab. Dis. 2014, 37, 851–861. [Google Scholar] [CrossRef]
- Ueki, I.; Kimura, A.; Nishiyori, A.; Chen, H.-L.; Takei, H.; Nittono, H.; Kurosawa, T. Neonatal Cholestatic Liver Disease in an Asian Patient with a Homozygous Mutation in the Oxysterol 7α-hydroxylase Gene. J. Pediatr. Gastroenterol. Nutr. 2008, 46, 465–469. [Google Scholar] [CrossRef] [PubMed]
- Raselli, T.; Hearn, T.; Wyss, A.; Atrott, K.; Peter, A.; Frey-Wagner, I.; Spalinger, M.R.; Maggio, E.M.; Sailer, A.W.; Schmitt, J.; et al. Elevated oxysterol levels in human and mouse livers reflect nonalcoholic steatohepatitis. J. Lipid Res. 2019, 60, 1270–1283. [Google Scholar] [CrossRef]
- Javitt, N. 26-Hydroxycholesterol: Synthesis, metabolism, and biologic activities. J. Lipid Res. 1990, 31, 1527–1533. [Google Scholar] [CrossRef]
- Salen, G.; Grundy, S.M. The Metabolism of Cholestanol, Cholesterol, and Bile Acids in Cerebrotendinous Xanthomatosis. J. Clin. Investig. 1973, 52, 2822–2835. [Google Scholar] [CrossRef]
- Spann, N.J.; Glass, C.K. Sterols and oxysterols in immune cell function. Nat. Immunol. 2013, 14, 893–900. [Google Scholar] [CrossRef] [PubMed]
- Mutemberezi, V.; Guillemot-Legris, O.; Muccioli, G.G. Oxysterols: From cholesterol metabolites to key mediators. Prog. Lipid Res. 2016, 64, 152–169. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Tontonoz, P. Liver X receptors in lipid signalling and membrane homeostasis. Nat. Rev. Endocrinol. 2018, 14, 452–463. [Google Scholar] [CrossRef]
- Hong, C.; Tontonoz, P. Liver X receptors in lipid metabolism: Opportunities for drug discovery. Nat. Rev. Drug Discov. 2014, 13, 433–444. [Google Scholar] [CrossRef]
- Goldstein, J.L.; Brown, M.S. The LDL Receptor. Arter. Thromb. Vasc. Biol. 2009, 29, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Mitsche, M.A.; Lütjohann, D.; Cohen, J.C.; Xie, X.-S.; Hobbs, H.H. Relative roles of ABCG5/ABCG8 in liver and intestine. J. Lipid Res. 2015, 56, 319–330. [Google Scholar] [CrossRef]
- Peet, D.J.; Turley, S.D.; Ma, W.; Janowski, B.A.; Lobaccaro, J.-M.A.; Hammer, R.E.; Mangelsdorf, D.J. Cholesterol and Bile Acid Metabolism Are Impaired in Mice Lacking the Nuclear Oxysterol Receptor LXRα. Cell 1998, 93, 693–704. [Google Scholar] [CrossRef]
- Chu, K.; Miyazaki, M.; Man, W.C.; Ntambi, J.M. Stearoyl-Coenzyme A Desaturase 1 Deficiency Protects against Hypertriglyceridemia and Increases Plasma High-Density Lipoprotein Cholesterol Induced by Liver X Receptor Activation. Mol. Cell. Biol. 2006, 26, 6786–6798. [Google Scholar] [CrossRef]
- Joseph, S.B.; Laffitte, B.A.; Patel, P.H.; Watson, M.A.; Matsukuma, K.E.; Walczak, R.; Collins, J.L.; Osborne, T.F.; Tontonoz, P. Direct and Indirect Mechanisms for Regulation of Fatty Acid Synthase Gene Expression by Liver X Receptors. J. Biol. Chem. 2002, 277, 11019–11025. [Google Scholar] [CrossRef]
- Yoshikawa, T.; Shimano, H.; Amemiya-Kudo, M.; Yahagi, N.; Hasty, A.H.; Matsuzaka, T.; Okazaki, H.; Tamura, Y.; Iizuka, Y.; Ohashi, K.; et al. Identification of Liver X Receptor-Retinoid X Receptor as an Activator of the Sterol Regulatory Element-Binding Protein 1c Gene Promoter. Mol. Cell. Biol. 2001, 21, 2991–3000. [Google Scholar] [CrossRef] [PubMed]
- Jakobsson, T.; Treuter, E.; Gustafsson, J.A.; Steffensen, K.R. Liver X receptor biology and pharmacology: New pathways, challenges and opportunities. Trends Pharmacol. Sci. 2012, 33, 394–404. [Google Scholar] [CrossRef]
- Patel, R.; Patel, M.; Tsai, R.; Lin, V.; Bookout, A.L.; Zhang, Y.; Magomedova, L.; Li, T.; Chan, J.F.; Budd, C.; et al. LXRβ is required for glucocorticoid-induced hyperglycemia and hepatosteatosis in mice. J. Clin. Investig. 2011, 121, 431–441. [Google Scholar] [CrossRef]
- Wang, Y.; Kumar, N.; Solt, L.A.; Richardson, T.I.; Helvering, L.M.; Crumbley, C.; Garcia-Ordonez, R.D.; Stayrook, K.R.; Zhang, X.; Novick, S.; et al. Modulation of Retinoic Acid Receptor-related Orphan Receptor α and γ Activity by 7-Oxygenated Sterol Ligands. J. Biol. Chem. 2010, 285, 5013–5025. [Google Scholar] [CrossRef] [PubMed]
- Kase, E.T.; Thoresen, G.H.; Westerlund, S.; Højlund, K.; Rustan, A.C.; Gaster, M. Liver X receptor antagonist reduces lipid formation and increases glucose metabolism in myotubes from lean, obese and type 2 diabetic individuals. Diabetologia 2007, 50, 2171–2180. [Google Scholar] [CrossRef]
- Shirouchi, B.; Kashima, K.; Horiuchi, Y.; Nakamura, Y.; Fujimoto, Y.; Tong, L.-T.; Sato, M. 27-Hydroxycholesterol suppresses lipid accumulation by down-regulating lipogenic and adipogenic gene expression in 3T3-L1 cells. Cytotechnology 2016, 69, 485–492. [Google Scholar] [CrossRef]
- Schulman, I.G. Liver X receptors link lipid metabolism and inflammation. FEBS Lett. 2017, 591, 2978–2991. [Google Scholar] [CrossRef]
- Medzhitov, R.; Hong, C.; Rong, X.; Zhu, X.; Tarling, E.J.; Hedde, P.N.; Gratton, E.; Parks, J.S.; Tontonoz, P. LXRs link metabolism to inflammation through Abca1-dependent regulation of membrane composition and TLR signaling. Elife 2015, 4, e08009. [Google Scholar] [CrossRef]
- Thomas, D.G.; Doran, A.C.; Fotakis, P.; Westerterp, M.; Antonson, P.; Jiang, H.; Jiang, X.-C.; Gustafsson, J.-Å.; Tabas, I.; Tall, A.R. LXR Suppresses Inflammatory Gene Expression and Neutrophil Migration through cis-Repression and Cholesterol Efflux. Cell Rep. 2018, 25, 3774–3785. [Google Scholar] [CrossRef]
- Jakobsson, T.; Vedin, L.-L.; Hassan, T.; Venteclef, N.; Greco, D.; D’Amato, M.; Treuter, E.; Gustafsson, J.A.; Steffensen, K.R. The oxysterol receptor LXRβ protects against DSS- and TNBS-induced colitis in mice. Mucosal Immunol. 2014, 7, 1416–1428. [Google Scholar] [CrossRef] [PubMed]
- Bai, Q.; Zhang, X.; Xu, L.; Kakiyama, G.; Heuman, D.M.; Sanyal, A.J.; Pandak, W.M.; Yin, L.; Xie, W.; Ren, S. Oxysterol sulfation by cytosolic sulfotransferase suppresses liver X receptor/sterol regulatory element binding protein–1c signaling pathway and reduces serum and hepatic lipids in mouse models of nonalcoholic fatty liver disease. Metabolism 2012, 61, 836–845. [Google Scholar] [CrossRef]
- Xu, L.; Kim, J.K.; Bai, Q.; Zhang, X.; Kakiyama, G.; Min, H.-K.; Sanyal, A.J.; Pandak, W.M.; Ren, S. 5-Cholesten-3β,25-Diol 3-Sulfate Decreases Lipid Accumulation in Diet-Induced Nonalcoholic Fatty Liver Disease Mouse Model. Mol. Pharmacol. 2012, 83, 648–658. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.; Li, X.; Rodriguez-Agudo, D.; Gil, G.; Hylemon, P.; Pandak, W.M. Sulfated oxysterol, 25HC3S, is a potent regulator of lipid metabolism in human hepatocytes. Biochem. Biophys. Res. Commun. 2007, 360, 802–808. [Google Scholar] [CrossRef]
- Janowski, B.A.; Grogan, M.J.; Jones, S.A.; Wisely, G.B.; Kliewer, S.A.; Corey, E.J.; Mangelsdorf, D.J. Structural requirements of ligands for the oxysterol liver X receptors LXR and LXR. Proc. Natl. Acad. Sci. USA 1999, 96, 266–271. [Google Scholar] [CrossRef] [PubMed]
- Hannedouche, S.; Zhang, J.; Yi, T.; Shen, W.; Nguyen, D.; Pereira, J.P.; Guerini, D.; Baumgarten, B.U.; Roggo, S.; Wen, B.; et al. Oxysterols direct immune cell migration via EBI2. Nat. Cell Biol. 2011, 475, 524–527. [Google Scholar] [CrossRef]
- Soroosh, P.; Wu, J.; Xue, X.; Song, J.; Sutton, S.W.; Sablad, M.; Yu, J.; Nelen, M.I.; Liu, X.; Castro, G.; et al. Oxysterols are agonist ligands of ROR t and drive Th17 cell differentiation. Proc. Natl. Acad. Sci. USA 2014, 111, 12163–12168. [Google Scholar] [CrossRef]
- Cyster, J.G.; Dang, E.V.; Reboldi, A.; Yi, T. 25-Hydroxycholesterols in innate and adaptive immunity. Nat. Rev. Immunol. 2014, 14, 731–743. [Google Scholar] [CrossRef]
- Jang, J.; Park, S.; Hur, H.J.; Cho, H.-J.; Hwang, I.; Kang, Y.P.; Im, I.; Lee, H.; Lee, E.; Yang, W.; et al. 25-hydroxycholesterol contributes to cerebral inflammation of X-linked adrenoleukodystrophy through activation of the NLRP3 inflammasome. Nat. Commun. 2016, 7, 13129. [Google Scholar] [CrossRef]
- Lefort, C.; Roumain, M.; Van Hul, M.; Rastelli, M.; Manco, R.; Leclercq, I.; Delzenne, N.M.; Di Marzo, V.; Flamand, N.; Luquet, S.; et al. Hepatic NAPE-PLD Is a Key Regulator of Liver Lipid Metabolism. Cells 2020, 9, 1247. [Google Scholar] [CrossRef] [PubMed]
- Lefort, C.; Van Hul, M.; Delzenne, N.M.; Everard, A.; Cani, P.D. Hepatic MyD88 regulates liver inflammation by altering synthesis of oxysterols. Am. J. Physiol. Metab. 2019, 317, E99–E108. [Google Scholar] [CrossRef] [PubMed]
- Everard, A.; Geurts, L.; Caesar, R.; Van Hul, M.; Matamoros, S.; Duparc, T.; Denis, R.G.P.; Cochez, P.; Pierard, F.; Castel, J.; et al. Intestinal epithelial MyD88 is a sensor switching host metabolism towards obesity according to nutritional status. Nat. Commun. 2014, 5, 5648. [Google Scholar] [CrossRef] [PubMed]
- Duparc, T.; Plovier, H.; Marrachelli, V.G.; Van Hul, M.; Essaghir, A.; Ståhlman, M.; Matamoros, S.; Geurts, L.; Pardo-Tendero, M.M.; Druart, C.; et al. Hepatocyte MyD88 affects bile acids, gut microbiota and metabolome contributing to regulate glucose and lipid metabolism. Gut 2016, 66, 620–632. [Google Scholar] [CrossRef] [PubMed]
- Di Marzo, V. New approaches and challenges to targeting the endocannabinoid system. Nat. Rev. Drug Discov. 2018, 17, 623–639. [Google Scholar] [CrossRef]
- Cani, P.; Plovier, H.; Van Hul, M.; Geurts, L.; Delzenne, N.M.; Druart, C.; Everard, A. Endocannabinoids — at the crossroads between the gut microbiota and host metabolism. Nat. Rev. Endocrinol. 2016, 12, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Everard, A.; Plovier, H.; Rastelli, M.; Van Hul, M.; D’Oplinter, A.D.W.; Geurts, L.; Druart, C.; Robine, S.; Delzenne, N.M.; Muccioli, G.G.; et al. Intestinal epithelial N-acylphosphatidylethanolamine phospholipase D links dietary fat to metabolic adaptations in obesity and steatosis. Nat. Commun. 2019, 10, 457. [Google Scholar] [CrossRef] [PubMed]
- Geurts, L.; Everard, A.; Van Hul, M.; Essaghir, A.; Duparc, T.; Matamoros, S.; Plovier, H.; Castel, J.; Denis, R.G.P.; Bergiers, M.; et al. Adipose tissue NAPE-PLD controls fat mass development by altering the browning process and gut microbiota. Nat. Commun. 2015, 6, 6495. [Google Scholar] [CrossRef]
- Margheritis, E.; Castellani, B.; Magotti, P.; Peruzzi, S.; Romeo, E.; Natali, F.; Mostarda, S.; Gioiello, A.; Piomelli, D.; Garau, G. Bile Acid Recognition by NAPE-PLD. ACS Chem. Biol. 2016, 11, 2908–2914. [Google Scholar] [CrossRef]
- Setchell, K.D.; Rodrigues, C.M.; Clerici, C.; Solinas, A.; Morelli, A.; Gartung, C.; Boyer, J. Bile acid concentrations in human and rat liver tissue and in hepatocyte nuclei. Gastroenterology 1997, 112, 226–235. [Google Scholar] [CrossRef]
- Northfield, T.C.; McColl, I. Postprandial concentrations of free and conjugated bile acids down the length of the normal human small intestine. Gut 1973, 14, 513–518. [Google Scholar] [CrossRef]
- Aggarwal, G.; Zarrow, J.E.; Mashhadi, Z.; Flynn, C.R.; Vinson, P.; Weaver, C.D.; Davies, S.S. Symmetrically substituted dichlorophenes inhibit N-acyl-phosphatidylethanolamine phospholipase D. J. Biol. Chem. 2020, 295, 7289–7300. [Google Scholar] [CrossRef]
- Castellani, B.; Diamanti, E.; Pizzirani, D.; Tardia, P.; Maccesi, M.; Realini, N.; Magotti, P.; Garau, G.; Bakkum, T.; Rivara, S.; et al. Synthesis and characterization of the first inhibitor ofN-acylphosphatidylethanolamine phospholipase D (NAPE-PLD). Chem. Commun. 2017, 53, 12814–12817. [Google Scholar] [CrossRef]
- González-Muniesa, P.; Mártinez-González, M.-A.; Hu, F.B.; Després, J.-P.; Matsuzawa, Y.; Loos, R.J.F.; Moreno, L.A.; Bray, G.A.; Martinez, J.A. Obesity. Nat. Rev. Dis. Prim. 2017, 3, 17034. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, G.G.; Ng, S.C. Understanding and Preventing the Global Increase of Inflammatory Bowel Disease. Gastroenterology 2017, 152, 313–321. [Google Scholar] [CrossRef]
- Jackson, V.M.; Breen, D.M.; Fortin, J.-P.; Liou, A.; Kuzmiski, J.B.; Loomis, A.K.; Rives, M.-L.; Shah, B.; Carpino, P.A. Latest approaches for the treatment of obesity. Expert Opin. Drug Discov. 2015, 10, 825–839. [Google Scholar] [CrossRef]
- Han, C.Y. Update on FXR Biology: Promising Therapeutic Target? Int. J. Mol. Sci. 2018, 19, 2069. [Google Scholar] [CrossRef] [PubMed]
- Adorini, L.; Pruzanski, M.; Shapiro, D. Farnesoid X receptor targeting to treat nonalcoholic steatohepatitis. Drug Discov. Today 2012, 17, 988–997. [Google Scholar] [CrossRef] [PubMed]
- Cipriani, S.; Mencarelli, A.; Palladino, G.; Fiorucci, S. FXR activation reverses insulin resistance and lipid abnormalities and protects against liver steatosis in Zucker (fa/fa) obese rats. J. Lipid Res. 2010, 51, 771–784. [Google Scholar] [CrossRef]
- Roberts, S.B.; Ismail, M.; Kanagalingam, G.; Mason, A.L.; Swain, M.G.; Vincent, C.; Yoshida, E.M.; Tsien, C.; Flemming, J.A.; Janssen, H.L.; et al. Real-World Effectiveness of Obeticholic Acid in Patients with Primary Biliary Cholangitis. Hepatol. Commun. 2020, 4, 1332–1345. [Google Scholar] [CrossRef]
- Mudaliar, S.; Henry, R.R.; Sanyal, A.J.; Morrow, L.; Marschall, H.; Kipnes, M.; Adorini, L.; Sciacca, C.I.; Clopton, P.; Castelloe, E.; et al. Efficacy and Safety of the Farnesoid X Receptor Agonist Obeticholic Acid in Patients With Type 2 Diabetes and Nonalcoholic Fatty Liver Disease. Gastroenterology 2013, 145, 574–582. [Google Scholar] [CrossRef]
- Hindson, J. Obeticholic acid for the treatment of NASH. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 66. [Google Scholar] [CrossRef]
- Pencek, R.; Marmon, T.; Roth, J.D.; Liberman, A.; Hooshmand-Rad, R.; Young, M.A. Effects of obeticholic acid on lipoprotein metabolism in healthy volunteers. Diabetes Obes. Metab. 2016, 18, 936–940. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, G.; Passeri, D.; De Franco, F.; Ciaccioli, G.; Donadio, L.; Rizzo, G.; Orlandi, S.; Sadeghpour, B.; Wang, X.X.; Jiang, T.; et al. Functional Characterization of the Semisynthetic Bile Acid Derivative INT-767, a Dual Farnesoid X Receptor and TGR5 Agonist. Mol. Pharmacol. 2010, 78, 617–630. [Google Scholar] [CrossRef]
- D’Amore, C.; Di Leva, F.S.; Sepe, V.; Renga, B.; Del Gaudio, C.; D’Auria, M.V.; Zampella, A.; Fiorucci, S.; Limongelli, V. Design, Synthesis, and Biological Evaluation of Potent Dual Agonists of Nuclear and Membrane Bile Acid Receptors. J. Med. Chem. 2014, 57, 937–954. [Google Scholar] [CrossRef]
- Carino, A.; Cipriani, S.; Marchianò, S.; Biagioli, M.; Santorelli, C.; Donini, A.; Zampella, A.; Monti, M.C.; Fiorucci, S. BAR502, a dual FXR and GPBAR1 agonist, promotes browning of white adipose tissue and reverses liver steatosis and fibrosis. Sci. Rep. 2017, 7, 42801. [Google Scholar] [CrossRef] [PubMed]
- Pellicciari, R.; Gioiello, A.; Macchiarulo, A.; Thomas, C.; Rosatelli, E.; Natalini, B.; Sardella, R.; Pruzanski, M.; Roda, A.; Pastorini, E.; et al. Discovery of 6α-Ethyl-23(S)-methylcholic Acid (S-EMCA, INT-777) as a Potent and Selective Agonist for the TGR5 Receptor, a Novel Target for Diabesity. J. Med. Chem. 2009, 52, 7958–7961. [Google Scholar] [CrossRef] [PubMed]
- Sepe, V.; Renga, B.; Festa, C.; D’Amore, C.; Masullo, D.; Cipriani, S.; Di Leva, F.S.; Monti, M.C.; Novellino, E.; Limongelli, V.; et al. Modification on Ursodeoxycholic Acid (UDCA) Scaffold. Discovery of Bile Acid Derivatives As Selective Agonists of Cell-Surface G-Protein Coupled Bile Acid Receptor 1 (GP-BAR1). J. Med. Chem. 2014, 57, 7687–7701. [Google Scholar] [CrossRef]
- Di Leva, F.S.; Festa, C.; Renga, B.; Sepe, V.; Novellino, E.; Fiorucci, S.; Zampella, A.; Limongelli, V. Structure-based drug design targeting the cell membrane receptor GPBAR1: Exploiting the bile acid scaffold towards selective agonism. Sci. Rep. 2015, 5, 16605. [Google Scholar] [CrossRef] [PubMed]
- Alemi, F.; Kwon, E.; Poole, D.P.; Lieu, T.; Lyo, V.; Cattaruzza, F.; Cevikbas, F.; Steinhoff, M.; Nassini, R.; Materazzi, S.; et al. The TGR5 receptor mediates bile acid–induced itch and analgesia. J. Clin. Investig. 2013, 123, 1513–1530. [Google Scholar] [CrossRef] [PubMed]
- Briere, D.A.; Ruan, X.; Cheng, C.C.; Siesky, A.M.; Fitch, T.E.; Dominguez, C.; Sanfeliciano, S.G.; Montero, C.; Suen, C.S.; Xu, Y.; et al. Novel Small Molecule Agonist of TGR5 Possesses Anti-Diabetic Effects but Causes Gallbladder Filling in Mice. PLoS ONE 2015, 10, e0136873. [Google Scholar] [CrossRef]
- Li, T.; Holmstrom, S.R.; Kir, S.; Umetani, M.; Schmidt, D.R.; Kliewer, S.A.; Mangelsdorf, D.J. The G Protein-Coupled Bile Acid Receptor, TGR5, Stimulates Gallbladder Filling. Mol. Endocrinol. 2011, 25, 1066–1071. [Google Scholar] [CrossRef]
- Lehmann, J.M.; McKee, D.D.; Watson, M.A.; Willson, T.M.; Moore, J.T.; Kliewer, S.A. The human orphan nuclear receptor PXR is activated by compounds that regulate CYP3A4 gene expression and cause drug interactions. J. Clin. Investig. 1998, 102, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Janowski, B.A.; Willy, P.J.; Devi, T.R.; Falck, J.R.; Mangelsdorf, D.J. An oxysterol signalling pathway mediated by the nuclear receptor LXRα. Nat. Cell Biol. 1996, 383, 728–731. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y. Recent Progress on Bile Acid Receptor Modulators for Treatment of Metabolic Diseases. J. Med. Chem. 2016, 59, 6553–6579. [Google Scholar] [CrossRef]
- Di Leva, F.S.; Festa, C.; Carino, A.; De Marino, S.; Marchianò, S.; Di Marino, D.; Finamore, C.; Monti, M.C.; Zampella, A.; Fiorucci, S.; et al. Discovery of ((1,2,4-oxadiazol-5-yl)pyrrolidin-3-yl)ureidyl derivatives as selective non-steroidal agonists of the G-protein coupled bile acid receptor-1. Sci. Rep. 2019, 9, 2504. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Chen, Z.-X.; Wang, K.; Ning, M.-M.; Zou, Q.-A.; Feng, Y.; Ye, Y.-L.; Leng, Y.; Shen, J.-H. Intestinally-targeted TGR5 agonists equipped with quaternary ammonium have an improved hypoglycemic effect and reduced gallbladder filling effect. Sci. Rep. 2016, 6, 28676. [Google Scholar] [CrossRef]
- LaSalle, M.; Hoguet, V.; Hennuyer, N.; Leroux, F.; Piveteau, C.; Belloy, L.; Lestavel, S.; Vallez, E.; Dorchies, E.; Duplan, I.; et al. Topical Intestinal Aminoimidazole Agonists of G-Protein-Coupled Bile Acid Receptor 1 Promote Glucagon Like Peptide-1 Secretion and Improve Glucose Tolerance. J. Med. Chem. 2017, 60, 4185–4211. [Google Scholar] [CrossRef]
- Alvarez-Sola, G.; Uriarte, I.; Latasa, M.U.; Fernandez-Barrena, M.G.; Urtasun, R.; Elizalde, M.; Barcena-Varela, M.; Jiménez, M.; Chang, H.C.; Barbero, R.; et al. Fibroblast growth factor 15/19 (FGF15/19) protects from diet-induced hepatic steatosis: Development of an FGF19-based chimeric molecule to promote fatty liver regeneration. Gut 2017, 66, 1818–1828. [Google Scholar] [CrossRef] [PubMed]
- Benoit, B.; Meugnier, E.; Castelli, M.; Chanon, S.; Vieille-Marchiset, A.; Durand, C.; Bendridi, N.; Pesenti, S.; Monternier, P.-A.; Durieux, A.-C.; et al. Fibroblast growth factor 19 regulates skeletal muscle mass and ameliorates muscle wasting in mice. Nat. Med. 2017, 23, 990–996. [Google Scholar] [CrossRef]
- Degirolamo, C.; Sabbà, C.; Moschetta, A. Therapeutic potential of the endocrine fibroblast growth factors FGF19, FGF21 and FGF23. Nat. Rev. Drug Discov. 2016, 15, 51–69. [Google Scholar] [CrossRef]
- Zucman-Rossi, J.; Villanueva, A.; Nault, J.-C.; Llovet, J.M. Genetic Landscape and Biomarkers of Hepatocellular Carcinoma. Gastroenterology 2015, 149, 1226–1239. [Google Scholar] [CrossRef]
- Miura, S.; Mitsuhashi, N.; Shimizu, H.; Kimura, F.; Yoshidome, H.; Otsuka, M.; Kato, A.; Shida, T.; Okamura, D.; Miyazaki, M. Fibroblast growth factor 19 expression correlates with tumor progression and poorer prognosis of hepatocellular carcinoma. BMC Cancer 2012, 12, 56. [Google Scholar] [CrossRef] [PubMed]
- Hansen, M.; Sonne, D.P.; Mikkelsen, K.H.; Gluud, L.L.; Vilsbøll, T.; Knop, F.K. Bile acid sequestrants for glycemic control in patients with type 2 diabetes: A systematic review with meta-analysis of randomized controlled trials. J. Diabetes Complicat. 2017, 31, 918–927. [Google Scholar] [CrossRef]
- Fonseca, V.A.; Handelsman, Y.; Staels, B. Colesevelam lowers glucose and lipid levels in type 2 diabetes: The clinical evidence. Diabetes Obes. Metab. 2010, 12, 384–392. [Google Scholar] [CrossRef]
- Rao, A.; Kosters, A.; Mells, J.E.; Zhang, W.; Setchell, K.D.R.; Amanso, A.M.; Wynn, G.M.; Xu, T.; Keller, B.T.; Yin, H.; et al. Inhibition of ileal bile acid uptake protects against nonalcoholic fatty liver disease in high-fat diet–fed mice. Sci. Transl. Med. 2016, 8, 357ra122. [Google Scholar] [CrossRef]
- Ge, M.-X.; Niu, W.-X.; Ren, J.-F.; Cai, S.-Y.; Yu, D.-K.; Liu, H.-T.; Zhang, N.; Zhang, Y.-X.; Wang, Y.-C.; Shao, R.-G.; et al. A novel ASBT inhibitor, IMB17-15, repressed nonalcoholic fatty liver disease development in high-fat diet-fed Syrian golden hamsters. Acta Pharmacol. Sin. 2019, 40, 895–907. [Google Scholar] [CrossRef] [PubMed]
- Saveleva, E.; Tyutrina, E.; Nakanishi, T.; Tamai, I.; Salmina, A. The inhibitors of the apical sodium-dependent bile acid transporter (ASBT) as promising drugs. Biomeditsinskaya Khimiya 2020, 66, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Al-Dury, S.; Marschall, H.-U. Ileal Bile Acid Transporter Inhibition for the Treatment of Chronic Constipation, Cholestatic Pruritus, and NASH. Front. Pharmacol. 2018, 9, 931. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lefort, C.; Cani, P.D. The Liver under the Spotlight: Bile Acids and Oxysterols as Pivotal Actors Controlling Metabolism. Cells 2021, 10, 400. https://doi.org/10.3390/cells10020400
Lefort C, Cani PD. The Liver under the Spotlight: Bile Acids and Oxysterols as Pivotal Actors Controlling Metabolism. Cells. 2021; 10(2):400. https://doi.org/10.3390/cells10020400
Chicago/Turabian StyleLefort, Charlotte, and Patrice D. Cani. 2021. "The Liver under the Spotlight: Bile Acids and Oxysterols as Pivotal Actors Controlling Metabolism" Cells 10, no. 2: 400. https://doi.org/10.3390/cells10020400
APA StyleLefort, C., & Cani, P. D. (2021). The Liver under the Spotlight: Bile Acids and Oxysterols as Pivotal Actors Controlling Metabolism. Cells, 10(2), 400. https://doi.org/10.3390/cells10020400