
Role of Gonadotropin-Releasing Hormone (GnRH) in Ovarian Cancer



Department of Gynecology and Obstetrics, University Medicine Göttingen, 37075 Göttingen, Germany
*
Author to whom correspondence should be addressed.
Cells 2021, 10(2), 437; https://doi.org/10.3390/cells10020437
Submission received: 22 December 2020
/
Revised: 28 January 2021
/
Accepted: 17 February 2021
/
Published: 18 February 2021
(This article belongs to the Special Issue Hormone Dependent Cancers: Molecular Mechanisms and Therapeutical Implications)
Abstract
:The hypothalamus–pituitary–gonadal (HPG) axis is the endocrine regulation system that controls the woman’s cycle. The gonadotropin-releasing hormone (GnRH) plays the central role. In addition to the gonadotrophic cells of the pituitary, GnRH receptors are expressed in other reproductive organs, such as the ovary and in tumors originating from the ovary. In ovarian cancer, GnRH is involved in the regulation of proliferation and metastasis. The effects on ovarian tumors can be indirect or direct. GnRH acts indirectly via the HPG axis and directly via GnRH receptors on the surface of ovarian cancer cells. In this systematic review, we will give an overview of the role of GnRH in ovarian cancer development, progression and therapy.
1. Introduction
Ovarian carcinomas are the eighth most common malignant tumor disease in women diagnosed in the world [1]. Although the risk of developing ovarian cancer in course of their life is only 1.3% for every woman [2], ovarian cancer is the fifth most common cancer-associated cause of death in women [3]. With a 5-year survival rate of 43%, ovarian cancer is the deadliest gynecological disease. Due to the lack of methods for early detection, the disease is diagnosed at an advanced stage in 70% of cases. Only 20% are discovered in stage I and are associated with 5-year survival rates of 90% [4]. Another reason for the late diagnosis is the unspecific symptoms. About 50% of the patients suffer from decreased performance or weight loss [5]. Ovarian cancer is a complex disease that originates from multiple sites [6,7,8]. Besides ovarian surface epithelium, ovarian cancers originate from fallopian tubes [9,10,11,12].
Ovarian cancer is a hormone-dependent disease that is influenced by hormonal signaling pathways [13,14]. This effect asserts itself after the illness and could therefore also have therapeutic potential. Gonadotropin-releasing hormone (GnRH) is pulsatile released from neurons in the hypothalamus and induces expression and pulsatile release of follicle-stimulating hormone (FSH) and luteinizing hormone (LH) from anterior pituitary. Both, FSH and LH in turn promote maturation of follicles, ovulation, corpus luteum formation and synthesis of estrogen and progesterone. In the following, we give an overview on the role of GnRH, the key player in the regulation of ovarian function, in ovarian cancer development and progression. In addition, we shed light on the use of GnRH receptors expressed on the ovarian cancer cell surface as a target of therapy in ovarian cancer.
2. Methods
We performed a literature search of PubMed using the terms “GnRH” or “LHRH” or “gonadotropin” and “ovarian cancer”, identifying 381 articles. Abstracts and full papers were screened independently by both authors excluding publications if they were not related to the topic, were double publications or not in English. The remaining 83 publications are described and discussed in this review. Important studies on other tumor entities (42) were included when no such studies on ovarian cancer were available and in addition, its use was necessary for better understanding. Furthermore, publications were discussed which deal with GnRH receptor mechanisms and signaling pathways independent of specific tumor entities (31). Due to the heterogeneity and the limitations of the clinical data, a meta-analysis was not appropriate. Completed and published clinical trials were discussed. A search on clinicaltrials.gov for currently ongoing trials on GnRH and ovarian cancer did not yield any results [15]. Only ongoing studies with other tumor entities were found.
3. Results and Discussion
3.1. Expression of GnRH and Its Receptor in Ovarian Cancer
GnRH is a peptide consisting of 10 amino acids that plays a main function in the control of human reproduction. The GnRH receptor belongs to class A of the G-protein-coupled receptor family with 7 transmembrane domains [16]. In addition to expression in gonadotropic cells of the pituitary gland and reproductive organs such as myometrium [17] or ovaries [18], expression of GnRH and GnRH receptor could also be detected in some human carcinomas of the urogenital tract. This includes cancers of the ovary [19], endometrium, urinary bladder and prostate [20]. In initial studies, GnRH receptors were found in almost 80% of human epithelial ovarian carcinomas [20,21,22,23]. The serous-papillary ovarian tumor is the most common epithelial tumor disease of the ovary. Histologically, the malignant form is a serous-papillary cystadenocarcinoma. This is divided into the less aggressive low-grade and the more common and more aggressive high-grade subtype. A recent study showed that nearly 90% of patients with high-grade serous ovarian cancer were GnRH receptor-positive [24]. In another study, it was demonstrated that all low-grade (100%) and about 82% high-grade serous ovarian cancer showed GnRH receptor expression [25]. The receptor binding abilities differ between gonadotropic cells in the pituitary and cancer cells. Two types of GnRH coupling sites were found in ovarian cancer cells. The first site has a low affinity and a high capacity. The second binding site has a high affinity with low capacity and is comparable to the GnRH receptor expressed in the pituitary gonadrotrophs [20,26,27]. The low-affinity binding site is similar to the binding site found in the human placenta and corpus luteum. It cannot differentiate between GnRH antagonists and high active GnRH agonists [28]. In addition, the GnRH receptor with low affinity can only be activated with high concentrations of GnRH agonists, while the GnRH receptor with high affinity is already completely activated at low concentrations of GnRH agonists. After the GnRH receptor gene sequence in human pituitary gonadotrophs was shown in 1992 [29], intensive research work was carried out which led to the detection of high-affinity GnRH receptors in ovarian cancer cell lines and in about 80% of primary ovarian cancers [29,30,31,32]. High affinity/low capacity binding sites closely related to the GnRH receptor in the pituitary gonadotrophs have been found in samples of ovarian carcinomas and cell lines expressing mRNA for the GnRH receptor found in the anterior pituitary [30,31,33,34,35,36,37]. Kakar et al. [38] demonstrated that the gene sequence of the GnRH receptor isolated from human ovarian cancers does not differ from that in the pituitary gland. Not only the GnRH receptor was found in these tumors, but also its ligand, GnRH. First of all expression of GnRH mRNA was found in two human breast cancer cell lines [39]. Later, GnRH immunoreactivity, biological activity, and mRNA expression by cell lines and in most tissue samples of ovarian cancers were demonstrated [33,36,37]. As ovarian cancers show expression of GnRH and the GnRH receptor, it seems plausible to speculate about the existence of a paracrine system based on GnRH in many of these cancers. A first very important indication was, on one hand, that treatment with authentic GnRH inhibited the growth of cell lines derived from ovarian cancer. On the other hand, by removing GnRH secreted by the tumor cells by using an anti-GnRH antiserum, proliferation of these cancer cells was upregulated [19].
3.2. Direct Effects of GnRH on Ovarian Cancer
Time- and dose-dependent anti-proliferative actions of GnRH agonists have been observed in many cell lines from different cancer entities of the reproductive tract. This includes ovarian cancer [20,21,40,41,42]. GnRH antagonists also show significant growth-inhibiting actions on most GnRH receptor-positive cancer cell lines [20,21]. This indicates that GnRH agonists and antagonists may not be differentiated in the GnRH system in cancer cells. In addition, the signaling mechanisms of the GnRH receptor known to be activated in gonadotropic cells of the pituitary, are not connected in the transmission of the proliferation inhibiting actions of GnRH analogs in cancer cells. In pituitary gonadotrophs, specific binding of GnRH to its receptor leads to a conformational change and subsequent activation of the heterotrimeric G-protein subunit αq/11, which leads to calcium-dependent signal transduction [43,44]. Activation of phospholipase C (PLC) causes the hydrolysis of phosphatidylinositol-4,5-bisphosphate (PIP2). This creates diacylglycerol (DAG) and inositol-1,4,5-trisphosphate (IP3) [45,46,47]. IP3 diffuses from the plasma membrane and activates receptors on the endoplasmic reticulum (ER). This leads to an outflow of calcium from the ER into the cytosol [48,49]. DAG remains in the membrane and induces Ca2+-dependent protein kinase C (PKC), which in the course of the induction of mitogen-activated protein kinases (MAPK) [50,51] leads to gondotropin synthesis (LH and FSH) and secretion [50,52,53,54]. The signaling found to be activated by GnRH agonists in the pituitary is not turned on in cancers of the ovary, although PLC, PKC and AC can be activated in these cells by pharmacologic stimulation. [40,55].
In contrast to the pituitary gland, the GnRH receptor-mediated signal transduction in gynecological tumors takes place via coupling with G-protein αi instead of G-protein αq, which may result in variable receptor conformations and signaling complexes [55,56,57] (Figure 1). Since the gene sequence of GnRH receptors in human carcinomas of the ovary is the same as that in the pituitary gland, different receptor conformations may explain why GnRH receptors in ovarian cancer show different mechanisms compared with cells derived from the pituitary. After binding to G-protein αi, the activated GnRH receptor induces a phosphotyrosine phosphatase (PTP) and prevents the signaling of growth factor receptors, which leads to a reduction in proliferation of cancer cells [40,55,58,59,60,61]. Growth factor-driven mitogenic signal transduction is prevented which leads to downregulation of growth factor-mediated activation of mitogen-activated protein kinase (MAPK) [40], c-fos expression [62], and growth factor-induced proliferation [63]. These findings are in agreement with other research on GnRH analogs showing reduction of growth factor receptor expression [64,65,66] and/or growth factor-induced tyrosine kinase activity [40,58,59,61,65,67,68,69]. Induction of apoptosis does not appear to be involved in the down-regulation of cancer cell proliferation by GnRH agonists [20]. In contrast, Imai et al. have reported, that GnRH agonist leuprorelin stimulated intratumoral expression of apoptosis-inducing Fas ligand in GnRH receptor-positive tumor cells. At a concentration of 10 µM, leuprorelin induced up to a 90% reduction in cell number preceded by Fas ligand production [70,71,72,73]. However, GnRH agonist triptorelin did not induce apoptosis. On the contrary, by activating NFκB, triptorelin even seems to protect against apoptosis [74,75].
In human cells from ovarian cancer, GnRH agonists act not only on the mitogenic signaling of growth factor receptors. They also induce the activation of activator protein-1 (AP-1). In addition, GnRH agonists promote activation of JNK, which triggers AP-1 [76]. In ovarian cancer cells, GnRH agonists stimulate neither phospholipase C (PLC) nor protein kinase C (PKC) [40]. In addition, GnRH agonists inhibit the mitogen-activated protein kinase (MAPK, ERK) activity induced by growth factors [40]. Therefore, GnRH-induced JNK/AP-1 activation depends not on the AP-1 activators PKC or MAPK (ERK).
Since the JNK/c-jun signal transduction is activated by antiproliferative GnRH agonists and since JNK/c-jun is integrated into the reduction of cell growth in different systems, it seems plausible that the JNK/c-jun signal transduction participates in the inhibiting effects of GnRH agonists. GnRH agonists have also been shown to induce the binding of JunD to DNA, resulting in a reduction of cell growth, as evidenced by an increased G0/1 phase of the cell cycle and reduced synthesis of DNA [77].
Expression of the immediate-early response gene c-fos is induced by 17β-estradiol (E2) in estrogen receptor α (ERα)-positive ovarian cancer cell lines [78,79,80,81,82,83,84,85]. Via MAPK-dependent phosphorylation of Elk-1 the serum response element (SRE) is activated [86,87]. Since GnRH agonists inhibit EGF-induced cell proliferation and expression of c-fos through Ras/MAPK, it was interesting to know whether activation of SRE and expression of c-fos induced by E2 in ERα-positive cancer cells is also influenced by GnRH agonists and whether GnRH inhibits E2-driven cell proliferation [20]. Resting ERα-positive/ERβ-positive cancer cell lines were stimulated to proliferate by treatment with E2. Simultaneous treatment with GnRH agonists prevented this effect in a time- and dose-dependent manner [88]. ERα-negative/ERβ-positive cell lines were not affected by treatment with E2. Furthermore, E2 activates serum response element (SRE) and expression of c-fos in ERα-positive / ERβ-positive cell lines. This was inhibited by GnRH agonists [88]. GnRH agonists had no influence on the activation of estrogen response element (ERE) caused by E2. Transcriptional activation of SRE by ERα is due to activation of the mitogen-activated protein kinase (MAPK) pathway. GnRH inhibits this signal transduction, resulting in a decrease in E2-induced SRE activation followed by a decrease in E2-mediated c-fos expression. This is causative for a reduction in E2-induced cell growth [88].
Activation of PTP by GnRH also inhibits Src/MMP/HB-EGF signaling via the G-protein βγ subunit of G-protein coupled estrogen receptor 1 (GPER1), a membrane-bound estrogen receptor [89,90,91,92]. Due to the inhibition of GPER signaling, cancer cell proliferation by E2 in breast cancer cells without expression of ERα was inhibited [89,90,91]. In human breast cancer cells, GnRH analogs counteract EGF-dependent proliferation. GnRH analogs likely disrupt the change in growth regulation from estrogen dependency to EGF dependence that occurs after the acquisition of secondary resistance against 4OH-tamoxifen. This disruption of EGF receptor signal transduction re-sensitized the resistant cell lines for a therapeutical use of 4OH-Tamoxifen [93]. Especially in breast cancer, GnRH is not only involved in cancer cell proliferation. In metastatic breast cancer cells, GnRH inhibits cell invasiveness in vitro and metastasis in vivo [94,95,96,97,98]. In addition, it was shown that GnRH is involved in epithelial to mesenchymal transition (EMT) [98]. However, these effects of GnRH still have to be researched in ovarian cancer in the future.
3.3. Expression of GnRH-II and Its Receptor in Ovarian Cancer
In addition to the well-known GnRH, many mammals express a second structural version of GnRH. The so-called GnRH-II is completely identical in its amino acid sequence from fish to mammal and differs from GnRH in three amino acids. A specific, functionally active receptor for GnRH-II has been found in many species. This also includes non-human primates [99,100,101,102]. Whether there is a GnRH-II receptor in humans is controversial [100,101,103,104,105,106,107,108]. There are, however, several indications for a functional GnRH-II receptor in humans [109] (Figure 2A). GnRH-II has inhibitory actions on human ovarian cancer cell proliferation that are significantly larger than those of the high active GnRH-I agonist triptorelin [104]. As well as GnRH-I agonists, GnRH-II agonists inhibit mitogenic signaling from growth factor receptors by activating a PTP. This leads to a down-regulation of the proliferation of cancer cells [55,62,110]. In contrast to GnRH-I and GnRH-II agonists and in addition GnRH-I antagonist cetrorelix, GnRH-II antagonists promote apoptosis in human ovarian cancer cells [111]. GnRH-II antagonist-induced apotosis is permitted via the intrinsic apoptotic pathway. This happens via the MAPKs p38- and JNK-induced activity of the pro-apoptotic protein Bax, followed by the loss of the mitochondrial membrane potential and subsequent cytochrome c release followed by caspase-3 activation [111,112]. Confirming these anti-tumor effects in nude mice it was shown that antagonistic analogs of GnRH-II significantly reduced the growth of human ovarian cancer xenografts in mice with no noticeable side effects [111].
SK-OV-3 cells are a cell line derived from ovarian cancer without GnRH-I receptor expression [23]. Here, the GnRH-I agonist triptorelin showed no effects on cell proliferation [23]. The GnRH-I antagonist Cetrorelix and GnRH-II, on the other hand, had a strong antiproliferative effect on this ovarian cancer cell line [113]. In addition, it was shown that in cell lines in which both, the GnRH-I agonist triptorelin and the GnRH-I antagonist cetrorelix act, only the effects of the former were abolished when the GnRH-I receptor was knocked down. The effects of cetrorelix and GnRH-II, however, were retained [113]. These results suggest that the proliferation reducing effects of the cetrorelix and of GnRH-II are not permitted via the GnRH-I receptor. These findings are in accord with results published by other researchers. Enomoto et al. [114] have shown that the human GnRH-II receptor is working and its splicing variant indicates the way of the cell response that follows GnRH-II stimulation. Expression of GnRH-II was detected in normal neoplastic cells of the ovarian surface epithelium and in cancers developed from these cells. Furthermore, they could show that GnRH-II had antiproliferative effects in immortalized cells from the ovarian surface epithelial [115].
However, at first glance, these findings appear to be in contrast to the fact that the human GnRH-II receptor gene has a stop codon in exon 2 [104,108]. The complete human GnRH-II receptor is known to be a 7-transmembrane receptor. Therefore, it was speculated that a working GnRH-II receptor might be a truncated 5-transmembrane domain receptor lacking transmembrane regions 1 and 2 [101]. This is in accord with data showing the expression of a GnRH-II receptor-like antigen in human ovarian cancer cells [109]. Photochemical reaction of 125I-labelled (4-azidobenzoyl)-N-hydroxysuccinimide-[D-Lys6]-GnRH-II with isolated cell membranes of human ovarian cancer cells provided a signal at about 43 kDa. A Western blot of the same gel showed that this signal could be a GnRH-II receptor-like antigen, which would indicate the existence of a shortened GnRH-II receptor [109]. In competition experiments, treatment with the GnRH-I agonist triptorelin led to a slight decrease in the binding of 125I-labelled (4-azidobenzoyl)-N-hydroxysuccinimide-[D-Lys6]-GnRH-II to its binding site. Treatment with the GnRH-I antagonist cetrorelix led to a significantly greater decrease, while the GnRH-II agonist [D-Lys6]-GnRH-II was the strongest competitor [109]. Indeed, these data suggest that the GnRH-II receptor-like antigen shown may be the specific binding site for GnRH-II sought.
On the other hand, it could be shown that GnRH-II antagonists as well bind to the GnRH-I receptor with affinities for the GnRH-I receptor that are comparable to those of the cetrorelix [112,116]. It was also shown that [D-Lys6]-GnRH-II has agonistic characteristics at the GnRH-I receptor, while GnRH-II antagonists clearly act as antagonists on the GnRH-I receptor [112]. This would mean that GnRH-I and -II antagonists, as clear antagonists, should not have any effects and are only block the GnRH-I receptor. Therefore, it was speculated that, besides the known autocrine GnRH-I system, a further autocrine system based on a GnRH-II could be present in human ovarian cancers.
Although there are the above mentioned indications for the presence of a working GnRH-II receptor in humans [104,109,113], other groups assumed that the human GnRH-II receptor is not functional. Kim et al. were able to show that the actions of GnRH-I and GnRH-II can be undone by transfecting siRNA in order to abolish expression of GnRH-I receptor [117]. These results suggest that the effects of GnRH-I and GnRH-II are permitted through the GnRH-I receptor (Figure 2B). These contradictions are a little confusing. Ultimately, it has not yet been possible to clarify whether there is a functional GnRH-II receptor in humans.
3.4. GnRH Analogs as a Possible Therapy for Ovarian Cancer
3.4.1. Suppression of Pituitary Gonadotropin Secretion by GnRH Analogs
The most obvious strategy is the suppression of gonadotropin secretion by downregulation of the HPG axis using GnRH analogs. It was shown that in cancers of the ovary and ovarian sex-cord-stromal tumors, GnRH agonists might have antitumor effects permitted by inhibition of secretion of the gonadotrophins (reversible medical hypophysectomy) [26]. First, Parmar et al. reported a patient with advanced ovarian cancer who got a relapse after surgery, chemotherapy and radiation therapy and was then treated with triptorelin [118,119] (Table 1). Simultaneously with the suppression of gonadotropins, there was a significant shrinkage of the tumor mass over 12 months. Subsequently, several researchers showed that the reduction in LH and FSH levels achieved by administration of GnRH agonists in 10–50% of patients with advanced ovarian cancer who have relapsed after standard treatment, led to objective remissions or stable disease [118,119,120,121,122,123] (Table 1). Based on these promising data from in vitro experiments and clinical pilot studies, a prospective double-blind randomized clinical study was started in patients with advanced ovarian cancer who received surgical treatment and chemotherapy [124]. Unfortunately, this study showed no benefits of suppression of gonadotropins by treatment with GnRH agonist triptorelin in progression-free and overall survival [124] (Table 1). In a more recent phase II trial using tamoxifen and goserelin in a patient with recurrent epithelial ovarian cancer, no consistent correlation between suppression of LH and FSH and tumor response was found [125] (Table 1).
3.4.2. Direct Treatment of Ovarian Cancer
In addition to the indirect use of GnRH analogs for suppression of LH and FSH, the expression of tumor cell GnRH receptors can be used to treat cancer cells directly. The easiest way to use the GnRH receptor as a therapeutic target is to use GnRH agonists or antagonists. Different GnRH agonists were developed and tested including clinical trials i.e., Triptorelin, Buserelin, and Goserelin. However, combinations of GnRH agonists with cytotoxic chemotherapy were not more efficacious than cytotoxic chemotherapy alone. This may be due to the fact that the doses were too low for a direct effect on the tumor cells.
Since GnRH agonists could not meet the expectations in the clinical settings, further trials were focused on the use of high-dose GnRH antagonists. GnRH antagonist Cetrorelix is able to safely and effectively inhibit the secretion of gonadotropins. Due to its suppression of LH and sex steroid hormones, Cetrorelix has been used to treat hormone-dependent cancers. Cetrorelix is also used to inhibit the premature LH surge in controlled ovarian hyperstimulation [136]. Cetrorelix has also been shown to have a direct anti-tumor effect in GnRH receptor-positive cancer cells. In addition, Cetrorelix had better anti-tumor efficacy than GnRH agonists in in vivo models of human cancers. Therefore, a phase II clinical trial with a high dose of GnRH-I antagonist Cetrorelix was performed in patients with ovarian or müllerian carcinoma refractory to platinum chemotherapy [137]. Eligible patients received 10 mg of cetrorelix subcutaneously every day. Three of 17 evaluable patients treated with Cetrorelix, obtained partial remission (18%) which lasted for 2 to 6 months. Six patients did undergo disease stabilization (35%) for up to 1 year [137]. In this very refractory patient group, these results are quite impressive when compared with chemotherapy under palliative situations. Only minimal toxicity was found, except for potential anaphylactoid reactions [137]. One patient had a grade 4 anaphylactoid reaction controlled by cortisol and cimetidine, two patients developed a grade 2 histamine reaction, one patient had a grade 2 arthralgia, and two patients had a 20% cholesterol increase. In addition, minor hot flushes, headaches, or local skin reactions at the site of injection were observed [137].
3.4.3. GnRH Receptor Target Therapies
A third strategy was the use of GnRH receptors for a targeted therapy. Expression of GnRH receptors could only be detected in a few normal tissues such as organs from the reproductive tract and cells from the anterior pituitary. Other tissues and hematopoietic stem cells, on the other hand, do not express the GnRH receptor. In addition, ovaries, fallopian tubes and uterus are usually removed during surgery in ovarian cancer therapy [145]. The GnRH receptor is therefore very suitable as a target for therapies with improved anti-tumor effects and reduced side effects.
Cytotoxic GnRH agonists have been developed in which a cytotoxic substance is covalently linked to a GnRH agonist [146]. These so-called cytotoxic GnRH analogs bind specifically to the GnRH receptor with their peptide part and act as a chemotherapeutical drug after internalization of the receptor–ligand complex into the cell [146]. Therefore, these cytotoxic GnRH analogs only act in cells with GnRH receptors on the cell surface. As a result, they have significantly fewer side effects than non-conjugated cytotoxic substances [146]. The cytotoxic GnRH agonist zoptarelin doxorubicin (AEZS-108, AN-152) is such a fusion molecule. In zoptarelin doxorubicin, the cytotoxic part doxorubicin is covalently bound to the GnRH agonist [D-Lys6] GnRH. After GnRH receptor-mediated internalization of zoptarelin doxorubicin into the cell, the active part is split off and the now free doxorubicin accumulates selectively in the cell nucleus. The competitive inhibition of the uptake of zoptarelin doxorubicin by an excess of another GnRH agonist proves that zoptarelin doxorubicin enters the cell specifically via the GnRH receptor. In tumor cell lines that have no GnRH receptors, no intracellular zoptarelin doxorubicin could be found [147]. In addition, zoptarelin doxorubicin was significantly more effective than doxorubicin at the same dose at inhibiting cell growth in most ovarian cancer cell lines with GnRH receptor expression. Another advantage of this therapeutic strategy is the bypassing of the multidrug resistance protein-1 (MDR-1) resistance mechanism [148]. This mechanism of entry of zoptarelin doxorubicin may overcome the resistance conveyed by MDR-1, which is a major drawback of systemic therapies [148]. In addition, the targeting of zoptarelin doxorubicin to GnRH receptor-positive cancers makes this analog more effective and less toxic than doxorubicin [148].
In in vivo experiments with tumor-bearing mice, zoptarelin doxorubicin showed lower toxicity than free doxorubicin. In addition, zoptarelin doxorubicin showed better efficacy in inhibition of the growth of GnRH receptor-positive tumors [145,149]. This is probably due to the uptake of zoptarelin doxorubicin via the GnRH receptor and the resulting reduced resistance [148,150]. Zoptarelin doxorubicin has been used in women with GnRH receptor positive cancer in clinical dose-escalation and pharmacokinetic studies. The maximum tolerated dose without supportive drugs was 267 mg/m2. This dose was proposed for the therapeutic phase II studies [151] (Table 2). In 2014, the first data from a multicenter phase II study showed that zoptarelin doxorubicin is an effective and safe compound [152]. The activity and toxicity of zoptarelin doxorubicin were evaluated in 42 women with platinum-refractory or resistant GnRH receptor-positive ovarian cancer. The patients were treated with a zoptarelin doxorubicin dose of 267 mg/m2. This is equimolar to 76.8 mg/m2 of free doxorubicin. Six (14.3%) of these 42 patients had a partial response, 16 (38%) had stable disease, and 16 (38%) had progressive disease. Four patients could not be evaluated. The median time to progression was 12 weeks (95% confidence interval (CI): 8-20 weeks). Median overall survival was 53 weeks (95% CI: 39-73 weeks). The side effects were tolerable [152] (Table 2). In a phase III advanced endometrial cancer trial with unknown GnRH receptor status, median overall survival was not improved with zoptarelin doxorubicin [153]. Unfortunately, zoptarelin doxorubicin was discontinued and no phase III trial was conducted in ovarian cancer. Therefore, it remains unclear whether zoptarelin doxorubicin would have had an advantage in ovarian cancer. In addition, it is unclear, whether changes in study design would have shown better results. However, the success of most GnRH receptor-mediated therapy concepts led to a number of other promising approaches.
Based on tumor-specific signaling of GnRH receptor in gynecological cancers including ovarian cancer and specific distribution pattern of GnRH receptors, gene therapy by using a GnRH analogs as an inducer for transcription of a therapeutic gene was successfully developed and tested in vitro and in athymic mice bearing xenografts of ovarian cancer cells [154].
EP-100 a fusion peptide in which 18-amino-acid cationic α-helical lytic peptide (CLIP-71) is covalently linked to GnRH, was created to deliver lytic peptides to GnRH receptor-positive cancer cells [155] (Table 2). It was recently shown, that EP-100 in combination with PARP inhibitor olaparib is a promising strategy for the therapy of ovarian cancer [156].
Expression of the GnRH receptor can not only be used for target therapies. The selective expression of the GnRH receptor is also suitable to detect metastases that also express the GnRH receptor. Liu et al. have successfully developed a selective fluorescence probe to detect peritoneal metastases of ovarian cancer. For this purpose indocyanine green was conjugated to a GnRH antagonist [157].
4. Conclusions
GnRH is an important player in the control of the HPG axis. In addition to this classical hypophysiotropic function, GnRH is important as a regulator of cell proliferation and invasion in a number of human malignancies, including ovarian carcinoma. Furthermore, the selective expression of GnRH receptors makes it an ideal target for new effective therapeutic strategies with little side effects. First studies using GnRH analogs for direct tumor treatment were only moderately successful. Probably the used dosages were too low. Therefore, further studies focused on the use of high-dose GnRH analogs showing better efficacies. Following the discovery of GnRH-II, analogs were developed and successfully tested in vitro and in vivo. Since the existence of a functional GnRH-II receptor in humans is still controversial, this strategy has not been continued. The most promising strategy so far is the use of the GnRH receptor for target therapy. Cytotoxic GnRH agonist zoptarelin doxorubicin was shown to be an effective and safe compound. No phase III trial was conducted in ovarian cancer so far. Even if a single phase III study was unsuccessful, it still makes sense to pursue the concept further. In addition, the specific expression of GnRH receptors could be used diagnostically to detect metastases.
Author Contributions
Literature search, C.G. and G.E.; Writing—original draft, C.G. and G.E. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Reid, B.M.; Permuth, J.B.; Sellers, T.A. Epidemiology of ovarian cancer: A review. Cancer Biol. Med. 2017, 14, 9–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torre, L.A.; Trabert, B.; DeSantis, C.E.; Mph, K.D.M.; Samimi, G.; Runowicz, C.D.; Gaudet, M.M.; Jemal, A.; Siegel, R.L. Ovarian cancer statistics, 2018. CA A Cancer J. Clin. 2018, 68, 284–296. [Google Scholar] [CrossRef]
- Cho, K.R.; Shih, I.-M. Ovarian Cancer. Annu. Rev. Pathol. Mech. Dis. 2009, 4, 287–313. [Google Scholar] [CrossRef]
- Gajjar, K.; Ogden, G.; Mujahid, M.I.; Razvi, K. Symptoms and Risk Factors of Ovarian Cancer: A Survey in Primary Care. ISRN Obstet. Gynecol. 2012, 2012, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Kurman, R.J.; Shih, I.-M. The Origin and Pathogenesis of Epithelial Ovarian Cancer: A Proposed Unifying Theory. Am. J. Surg. Pathol. 2010, 34, 433–443. [Google Scholar] [CrossRef] [Green Version]
- Nezhat, F.R.; Apostol, R.; Nezhat, C.; Pejovic, T. New insights in the pathophysiology of ovarian cancer and implications for screening and prevention. Am. J. Obstet. Gynecol. 2015, 213, 262–267. [Google Scholar] [CrossRef]
- Nezhat, F.R.; Pejovic, T.; Reis, F.M.; Guo, S.-W. The Link Between Endometriosis and Ovarian Cancer: Clinical Implications. Int. J. Gynecol. Cancer 2014, 24, 623–628. [Google Scholar] [CrossRef] [PubMed]
- Cress, R.D.; Chen, Y.S.; Morris, C.R.; Petersen, M.; Leiserowitz, G.S. Characteristics of Long-Term Survivors of Epithelial Ovarian Cancer. Obstet. Gynecol. 2015, 126, 491–497. [Google Scholar] [CrossRef]
- Falconer, H.; Yin, L.; Grönberg, H.; Altman, D. Ovarian Cancer Risk After Salpingectomy: A Nationwide Population-Based Study. J. Natl. Cancer Inst. 2014, 107, dju410. [Google Scholar] [CrossRef] [Green Version]
- Labidi-Galy, S.I.; Papp, E.; Hallberg, D.; Niknafs, N.; Adleff, V.; Noe, M.; Bhattacharya, R.; Novak, M.; Jones, S.; Phallen, J.; et al. High grade serous ovarian carcinomas originate in the fallopian tube. Nat. Commun. 2017, 8, 1093. [Google Scholar] [CrossRef] [PubMed]
- Perets, R.; Drapkin, R. It’s Totally Tubular…Riding The New Wave of Ovarian Cancer Research. Cancer Res. 2016, 76, 10–17. [Google Scholar] [CrossRef] [Green Version]
- Langdon, S.; Crew, A.; Ritchie, A.; Muir, M.; Wakeling, A.; Smyth, J.; Miller, W. Growth inhibition of oestrogen receptor-positive human ovarian carcinoma by anti-oestrogens in vitro and in a xenograft model. Eur. J. Cancer 1994, 30, 682–686. [Google Scholar] [CrossRef]
- Langdon, S.; Hirst, G.; Miller, E.; Hawkins, R.; Tesdale, A.; Smyth, J.; Miller, W. The regulation of growth and protein expression by estrogen in vitro: A study of 8 human ovarian carcinoma cell lines. J. Steroid Biochem. Mol. Biol. 1994, 50, 131–135. [Google Scholar] [CrossRef]
- ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US). Available online: https://www.clinicaltrials.gov (accessed on 18 December 2020).
- Isberg, V.; De Graaf, C.; Bortolato, A.; Cherezov, V.; Katritch, V.; Marshall, F.H.; Mordalski, S.; Pin, J.-P.; Stevens, R.C.; Vriend, G.; et al. Generic GPCR residue numbers—aligning topology maps while minding the gaps. Trends Pharmacol. Sci. 2015, 36, 22–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chegini, N.; Rong, H.; Dou, Q.; Kipersztok, S.; Williams, R.S. Gonadotropin-releasing hormone (GnRH) and GnRH receptor gene expression in human myometrium and leiomyomata and the direct action of GnRH analogs on myometrial smooth muscle cells and interaction with ovarian steroids in vitro. J. Clin. Endocrinol. Metab. 1996, 81, 3215–3221. [Google Scholar] [CrossRef] [Green Version]
- Minaretzis, D.; Jakubowski, M.; Mortola, J.F.; Pavlou, S.N. Gonadotropin-releasing hormone receptor gene expression in human ovary and granulosa-lutein cells. J. Clin. Endocrinol. Metab. 1995, 80, 430–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emons, G.; Weiss, S.; Ortmann, O.; Gründker, C.; Schulz, K.D. LHRH might act as a negative autocrine regulator of proliferation of human ovarian cancer. Eur. J. Endocrinol. 2000, 142, 665–670. [Google Scholar] [CrossRef] [Green Version]
- Gründker, C.; Günthert, A.R.; Westphalen, S.; Emons, G. Biology of the gonadotropin-releasing hormone system in gynecological cancers. Eur. J. Endocrinol. 2002, 146, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Emons, G.; Grundker, C.; Gunthert, A.R.; Westphalen, S.; Kavanagh, J.; Verschraegen, C. GnRH antagonists in the treatment of gynecological and breast cancers. Endocr. Relat. Cancer 2003, 10, 291–299. [Google Scholar] [CrossRef] [Green Version]
- Emons, G.; Ortmann, O.; Schulz, K.-D.; Schally, A.V. Growth-inhibitory actions of analogues of Luteinizing Hormone Releasing Hormone on tumor cells. Trends Endocrinol. Metab. 1997, 8, 355–362. [Google Scholar] [CrossRef]
- Volker, P.; Grundker, C.; Schmidt, O.; Schulz, K.D.; Emons, G. Expression of receptors for luteinizing hor-mone-releasing hormone in human ovarian and endometrial cancers: Frequency, autoregulation, and corre-lation with direct antiproliferative activity of luteinizing hormone-releasing hormone analogues. Am. J. Obstet. Gynecol. 2002, 186, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Wentao, Y.; Bi, R.; Xiaojun, C.; Chen, X.; Yang, W.; Wu, X. A clinically applicable molecular classification for high-grade serous ovarian cancer based on hormone receptor expression. Sci. Rep. 2016, 6, 25408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Z.; Wen, H.; Ju, X.; Bi, R.; Chen, X.; Yang, W.; Wu, X. Expression of hypothalamic-pituitary-gonadal axis-related hormone receptors in low-grade serous ovarian cancer (LGSC). J. Ovarian Res. 2017, 10, 7. [Google Scholar] [CrossRef] [Green Version]
- Emons, G.; Schally, A.V. The use of luteinizing hormone releasing hormone agonists and antagonists in gynaecological cancers. Hum. Reprod. 1994, 9, 1364–1379. [Google Scholar] [CrossRef] [PubMed]
- Loop, S.M.; Gorder, C.A.; Lewis, S.M.; Saiers, J.H.; Drivdahl, R.H.; Ostenson, R.C. Growth inhibition of hu-man prostate tumor cells by an agonist of gonadotrophin-releasing hormone. Prostate 1995, 26, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Eidne, K.A.; Flanagan, C.A.; Harris, N.S.; Millar, R.P. Gonadotropin-releasing hormone (GnRH)-binding sites in human breast cancer cell lines and inhibitory effects of GnRH antagonists. J. Clin. Endocrinol. Metab. 1987, 64, 425–432. [Google Scholar] [CrossRef]
- Kakar, S.S.; Musgrove, L.C.; Devor, D.C.; Sellers, J.C.; Neill, J.D. Cloning, sequencing, and expression of hu-man gonadotropin releasing hormone (GnRH) receptor. Biochem. Biophys. Res. Commun. 1992, 189, 289–295. [Google Scholar] [CrossRef]
- Imai, A.; Ohno, T.; Iida, K.; Fuseya, T.; Furui, T.; Tamaya, T. Gonadotropin-releasing hormone receptor in gynecologic tumors. Frequent expression in adenocarcinoma histologic types. Cancer 1994, 74, 2555–2561. [Google Scholar] [CrossRef]
- Imai, A.; Ohno, T.; Iida, K.; Fuseya, T.; Furui, T.; Tamaya, T. Presence of Gonadotropin-Releasing Hormone Receptor and Its Messenger Ribonucleic Acid in Endometrial Carcinoma and Endometrium. Gynecol. Oncol. 1994, 55, 144–148. [Google Scholar] [CrossRef]
- Imai, A.; Ohno, T.; Ohsuye, K.; Tamaya, T. Expression of Gonadotropin-Releasing Hormone Receptor in Human Epithelial Ovarian Carcinoma. Ann. Clin. Biochem. Int. J. Lab. Med. 1994, 31, 550–555. [Google Scholar] [CrossRef] [Green Version]
- Irmer, G.; Bürger, C.; Müller, R.; Ortmann, O.; Peter, U.; Kakar, S.S.; Neill, J.D.; Schulz, K.D.; Emons, G. Ex-pression of the messenger RNAs for luteinizing hormone-releasing hormone (LHRH) and its receptor in human ovarian epithelial carcinoma. Cancer Res. 1995, 55, 817–822. [Google Scholar] [PubMed]
- Emons, G.; Ortmann, O.; Becker, M.; Irmer, G.; Springer, B.; Laun, R.; Hölzel, F.; Schulz, K.D.; Schally, A.V. High affinity binding and direct antiproliferative effects of LHRH analogues in human ovarian cancer cell lines. Cancer Res. 1993, 53, 5439–5446. [Google Scholar] [PubMed]
- Emons, G.; Schröder, B.; Ortmann, O.; Westphalen, S.; Schulz, K.D.; Schally, A.V. High affinity binding and direct antiproliferative effects of luteinizing hormone-releasing hormone analogs in human endometrial cancer cell lines. J. Clin. Endocrinol. Metab. 1993, 77, 1458–1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohno, T.; Imai, A.; Furui, T.; Takahashi, K.; Tamaya, T. Presence of gonadotropin-releasing hormone and its messenger ribonucleic acid in human ovarian epithelial carcinoma. Am. J. Obstet. Gynecol. 1993, 169, 605–610. [Google Scholar] [CrossRef]
- Irmer, G.; Bürger, C.; Ortmann, O.; Schulz, K.D.; Emons, G. Expression of luteinizing hormone releasing hormone and its mRNA in human endometrial cancer cell lines. J. Clin. Endocrinol. Metab. 1994, 79, 916–919. [Google Scholar] [CrossRef]
- Kakar, S.S.; Grizzle, W.E.; Neill, J.D. The nucleotide sequences of human GnRH receptors in breast and ovarian tumors are identical with that found in pituitary. Mol. Cell. Endocrinol. 1994, 106, 145–149. [Google Scholar] [CrossRef]
- Harris, N.; Dutlow, C.; Eidne, K.; Dong, K.W.; Roberts, J.; Millar, R. Gonadotropin-releasing hormone gene expression in MDA-MB-231 and ZR-75-1 breast carcinoma cell lines. Cancer Res. 1991, 51, 2577–2581. [Google Scholar]
- Dondi, D.; Limonta, P.; Moretti, R.M.; Marelli, M.M.; Garattini, E.; Motta, M. Antiproliferative effects of lu-teinizing hormone-releasing hormone (LHRH) agonists on human androgen-independent prostate cancer cell line DU 145: Evidence for an autocrine-inhibitory LHRH loop. Cancer Res. 1994, 54, 4091–4095. [Google Scholar]
- Limonta, P.; Dondi, D.; Moretti, R.M.; Maggi, R.; Motta, M. Antiproliferative effects of luteinizing hor-mone-releasing hormone agonists on the human prostatic cancer cell line LNCaP. J. Clin. Endocrinol. Metab. 1992, 75, 207–212. [Google Scholar]
- Limonta, P.; Moretti, R.M.; Dondi, D.; Marelli, M.M.; Motta, M. Androgen-dependent prostatic tumors: Bio-synthesis and possible actions of LHRH. J. Steroid Biochem. Mol. Biol. 1994, 49, 347–350. [Google Scholar] [CrossRef]
- Cheng, K.W.; Leung, P.C. The expression, regulation and signal transduction pathways of the mammalian gonadotropin-releasing hormone receptor. Can. J. Physiol. Pharm. 2000, 78, 1029–1052. [Google Scholar] [CrossRef]
- Naor, Z. Signal Transduction Mechanisms of Ca 2+ Mobilizing Hormones: The Case of Gonadotropin-Releasing Hormone. Endocr. Rev. 1990, 11, 326–353. [Google Scholar] [CrossRef]
- Kraus, S.; Naor, Z.; Seger, R. Intracellular Signaling Pathways Mediated by the Gonadotropin-Releasing Hormone (GnRH) Receptor. Arch. Med. Res. 2001, 32, 499–509. [Google Scholar] [CrossRef]
- McArdle, C.A.; Franklin, J.; Green, L.; Hislop, J.N. Signalling, cycling and desensitisation of gonadotrophin-releasing hormone receptors. J. Endocrinol. 2002, 173, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Ruf, F.; Fink, M.Y.; Sealfon, S.C. Structure of the GnRH receptor-stimulated signaling network: Insights from genomics. Front. Neuroendocr. 2003, 24, 181–199. [Google Scholar] [CrossRef]
- Berridge, M.J. Inositol trisphosphate and calcium signalling. Nat. Cell Biol. 1993, 361, 315–325. [Google Scholar] [CrossRef]
- Keizer, J.; Li, Y.X.; Stojilković, S.; Rinzel, J. InsP3-induced Ca2+ excitability of the endoplasmic reticulum. Mol. Biol. Cell 1995, 6, 945–951. [Google Scholar] [CrossRef]
- Harris, D.; Bonfil, D.; Chuderland, D.; Kraus, S.; Seger, R.; Naor, Z. Activation of MAPK Cascades by GnRH: ERK and Jun N-Terminal Kinase Are Involved in Basal and GnRH-Stimulated Activity of the Glycoprotein Hormone LHβ-Subunit Promoter. Endocrinology 2002, 143, 1018–1025. [Google Scholar] [CrossRef]
- Zhang, T.; Roberson, M.S. Role of MAP kinase phosphatases in GnRH-dependent activation of MAP kinases. J. Mol. Endocrinol. 2006, 36, 41–50. [Google Scholar] [CrossRef] [Green Version]
- Bonfil, D.; Chuderland, D.; Kraus, S.; Shahbazian, D.; Friedberg, I.; Seger, R.; Naor, Z. Extracellular Signal-Regulated Kinase, Jun N-Terminal Kinase, p38, and c-Src Are Involved in Gonadotropin-Releasing Hormone-Stimulated Activity of the Glycoprotein Hormone Follicle-Stimulating Hormone β-Subunit Promoter. Endocrinology 2004, 145, 2228–2244. [Google Scholar] [CrossRef] [Green Version]
- Levi, N.L.; Hanoch, T.; Benard, O.; Rozenblat, M.; Harris, D.; Reiss, N.; Naor, Z.; Seger, R. Stimulation of Jun N-Terminal Kinase (JNK) by Gonadotropin-Releasing Hormone in Pituitary αT3–1 Cell Line Is Mediated by Protein Kinase C, c-Src, and CDC42. Mol. Endocrinol. 1998, 12, 815–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberson, M.S.; Zhang, T.; Li, H.L.; Mulvaney, J.M. Activation of the p38 Mitogen-Activated Protein Kinase Pathway by Gonadotropin-Releasing Hormone. Endocrinology 1999, 140, 1310–1318. [Google Scholar] [CrossRef] [PubMed]
- Gründker, C.; Völker, P.; Emons, G. Antiproliferative signaling of luteinizing hormone-releasing hormone in human endometrial and ovarian cancer cells through G protein alpha(I)-mediated activation of phosphotyrosine phosphatase. Endocrinology 2001, 142, 2369–2380. [Google Scholar] [CrossRef]
- Limonta, P.; Moretti, R.M.; Marelli, M.M.; Dondi, D.; Parenti, M.; Motta, M. The luteinizing hormone-releasing hormone receptor in human prostate cancer cells: Messenger ribonucleic acid expression, molecular size, and signal transduction pathway. Endocrinology 1999, 140, 5250–5256. [Google Scholar] [CrossRef] [PubMed]
- Dobkin-Bekman, M.; Naidich, M.; Pawson, A.J.; Millar, R.P.; Seger, R.; Naor, Z. Activation of mitogen-activated protein kinase (MAPK) by GnRH is cell-context dependent. Mol. Cell Endocrinol. 2006, 252, 184–190. [Google Scholar] [CrossRef]
- Lee, M.T.; Liebow, C.; Kamer, A.R.; Schally, A.V. Effects of epidermal growth factor and analogues of lute-inizing hormone-releasing hormone and somatostatin on phosphorylation and dephosphorylation of tyro-sine residues of specific protein substrates in various tumors. Proc. Natl. Acad. Sci. USA 1991, 88, 1656–1660. [Google Scholar] [CrossRef] [Green Version]
- Furui, T.; Imai, A.; Takagi, H.; Horibe, S.; Fuseya, T.; Tamaya, T. Phosphotyrosine Phosphatase-Activity in Membranes from Endometrial Carcinoma. Oncol. Rep. 1995, 2, 1055–1057. [Google Scholar] [CrossRef]
- Imai, A.; Takagi, H.; Horibe, S.; Fuseya, T.; Tamaya, T. Coupling of gonadotropin-releasing hormone recep-tor to Gi protein in human reproductive tract tumors. J. Clin. Endocrinol. Metab. 1996, 81, 3249–3253. [Google Scholar] [PubMed] [Green Version]
- Imai, A.; Takagi, H.; Furui, T.; Horibe, S.; Fuseya, T.; Tamaya, T. Evidence for coupling of phosphotyrosine phosphatase to gonadotropin-releasing hormone receptor in ovarian carcinoma membrane. Cancer 1996, 77, 132–137. [Google Scholar] [CrossRef]
- Gründker, C.; Völker, P.; Schulz, K.-D.; Emons, G. Luteinizing Hormone—Releasing Hormone Agonist Triptorelin and Antagonist Cetrorelix Inhibit EGF-Induced c-fos Expression in Human Gynecological Cancers. Gynecol. Oncol. 2000, 78, 194–202. [Google Scholar] [CrossRef]
- Miller, W.R.; Scott, W.N.; Morris, R.; Fraser, H.M.; Sharpe, R.M. Growth of human breast cancer cells inhib-ited by a luteinizing hormone-releasing hormone agonist. Nature 1985, 313, 231–233. [Google Scholar] [CrossRef] [PubMed]
- Yano, T.; Pinski, J.; Halmos, G.; Szepesházi, K.; Groot, K.; Schally, A.V. Inhibition of growth of OV-1063 human epithelial ovarian cancer xenografts in nude mice by treatment with luteinizing hormone-releasing hormone antagonist SB-75. Proc. Natl. Acad. Sci. 1994, 91, 7090–7094. [Google Scholar] [CrossRef] [Green Version]
- Moretti, R.M.; Marelli, M.M.; Dondi, D.; Poletti, A.; Martini, L.; Motta, M.; Limonta, P. Luteinizing hormone-releasing hormone agonists interfere with the stimulatory actions of epidermal growth factor in human prostatic cancer cell lines, LNCaP and DU 145. J. Clin. Endocrinol. Metab. 1996, 81, 3930–3937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirahige, Y.; Cook, C.; Pinski, J.; Halmos, G.; Nair, R.; Schally, A. Treatment with Luteinizing-Hormone-Releasing Hormone Antagonist sb-75 Decreases Levels of Epidermal Growth-Factor Receptor and its Messenger-RNA in ov-1063 Human Epithelial Ovarian-Cancer Xenografts in Nude-Mice. Int. J. Oncol. 1994, 5, 1031–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kéri, G.; Balogh, Á.; Szöke, B.; Teplán, I.; Csuka, O. Gonadotropin-Releasing Hormone Analogues Inhibit Cell Proliferation and Activate Signal Transduction Pathways in MDA-MB-231 Human Breast Cancer Cell Line. Tumor Biol. 1991, 12, 61–67. [Google Scholar] [CrossRef]
- Liebow, C.; Lee, M.T.; Kamer, A.R.; Schally, A.V. Regulation of luteinizing hormone-releasing hormone re-ceptor binding by heterologous and autologous receptor-stimulated tyrosine phosphorylation. Proc. Natl. Acad. Sci. USA 1991, 88, 2244–2248. [Google Scholar] [CrossRef] [Green Version]
- Hershkovitz, E.; Marbach, M.; Bosin, E.; Levy, J.; Roberts, C.T., Jr.; LeRoith, D.; Schally, A.V.; Sharoni, Y. Lu-teinizing hormone-releasing hormone antagonists interfere with autocrine and paracrine growth stimula-tion of MCF-7 mammary cancer cells by insulin-like growth factors. J. Clin. Endocrinol. Metab. 1993, 77, 963–968. [Google Scholar]
- Fuseya, T.; Imai, A.; Horibe, S.; Takagi, A.; Tamaya, T. Evidence for common signalling pathways of GnRH receptor and Fas in tumors. Oncol. Rep. 1996, 3, 1111–1113. [Google Scholar] [CrossRef] [PubMed]
- Imai, A.; Horibe, S.; Takagi, A.; Ohno, T.; Tamaya, T. Frequent expression of Fas in gonadotropin-releasing hormone receptor-bearing tumors. Eur. J. Obstet. Gynecol. Reprod. Biol. 1997, 74, 73–78. [Google Scholar] [CrossRef]
- Imai, A.; Takagi, A.; Horibe, S.; Takagi, H.; Tamaya, T. Fas and Fas ligand system may mediate antiproliferative activity of gonadotropin-releasing hormone receptor in endometrial cancer cells. Int. J. Oncol. 1998, 13, 97–197. [Google Scholar] [CrossRef]
- Imai, A.; Takagi, A.; Horibe, S.; Takagi, H.; Tamaya, T. Evidence for Tight Coupling of Gonadotropin-Releasing Hormone Receptor to Stimulated Fas Ligand Expression in Reproductive Tract Tumors: Possible Mechanism for Hormonal Control of Apoptotic Cell Death 1. J. Clin. Endocrinol. Metab. 1998, 83, 427–431. [Google Scholar] [CrossRef] [Green Version]
- Gunthert, A.R.; Grundker, C.; Bottcher, B.; Emons, G. Luteinizing hormone-releasing hormone (LHRH) in-hibits apoptosis induced by cytotoxic agent and UV-light but not apoptosis mediated through CD95 in hu-man ovarian and endometrial cancer cells. Anticancer Res. 2004, 24, 1727–1732. [Google Scholar]
- Gründker, C.; Schulz, K.; Günthert, A.R.; Emons, G. Luteinizing Hormone-Releasing Hormone Induces Nuclear Factorκ B-Activation and Inhibits Apoptosis in Ovarian Cancer Cells. J. Clin. Endocrinol. Metab. 2000, 85, 3815–3820. [Google Scholar] [CrossRef] [Green Version]
- Grundker, C.; Schlotawa, L.; Viereck, V.; Emons, G. Protein kinase C-independent stimulation of activator protein-1 and c-Jun N-terminal kinase activity in human endometrial cancer cells by the LHRH agonist triptorelin. Eur. J. Endocrinol. 2001, 145, 651–658. [Google Scholar] [CrossRef]
- Günthert, A.R.; Gründker, C.; Hollmann, K.; Emons, G. Luteinizing hormone-releasing hormone induces JunD—DNA binding and extends cell cycle in human ovarian cancer cells. Biochem. Biophys. Res. Commun. 2002, 294, 11–15. [Google Scholar] [CrossRef]
- Bonapace, I.M.; Addeo, R.; Altucci, L.; Cicatiello, L.; Bifulco, M.; Laezza, C.; Salzano, S.; Sica, V.; Bresciani, F.; Weisz, A. 17 beta-Estradiol overcomes a G1 block induced by HMG-CoA reductase inhibitors and fosters cell cycle progression without inducing ERK-1 and -2 MAP kinases activation. Oncogene 1996, 12, 753–763. [Google Scholar] [PubMed]
- Doucas, V.; Spyrou, G.; Yaniv, M. Unregulated expression of c-Jun or c-Fos proteins but not Jun D inhibits oestrogen receptor activity in human breast cancer derived cells. EMBO J. 1991, 10, 2237–2245. [Google Scholar] [CrossRef]
- Duan, R.; Porter, W.; Safe, S. Estrogen-Induced c-fos Protooncogene Expression in MCF-7 Human Breast Cancer Cells: Role of Estrogen Receptor Sp1 Complex Formation. Endocrinology 1998, 139, 1981–1990. [Google Scholar] [CrossRef] [PubMed]
- Van Der Burg, B.; De Groot, R.P.; Isbrücker, L.; Kruijer, W.; De Laat, S.W. Stimulation of TPA-Responsive Element Activity by a Cooperative Action of Insulin and Estrogen in Human Breast Cancer Cells. Mol. Endocrinol. 1990, 4, 1720–1726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Burg, B.; de Groot, R.P.; Isbrucker, L.; Kruijer, W.; de Laat, S.W. Oestrogen directly stimulates growth factor signal transduction pathways in human breast cancer cells. J. Steroid Biochem. Mol. Biol. 1991, 40, 215–221. [Google Scholar] [CrossRef]
- Van Der Burg, B.; Van Selm-Miltenburg, A.J.; De Laat, S.W.; Van Zoelen, E.J. Direct effects of estrogen on c-fos and c-myc protooncogene expression and cellular proliferation in human breast cancer cells. Mol. Cell. Endocrinol. 1989, 64, 223–228. [Google Scholar] [CrossRef]
- Weisz, A.; Bresciani, F. Estrogen regulation of proto-oncogenes coding for nuclear proteins. Crit. Rev. Oncog. 1993, 4, 361–388. [Google Scholar]
- Wilding, G.; Lippman, M.E.; Gelmann, E.P. Effects of steroid hormones and peptide growth factors on pro-tooncogene c-fos expression in human breast cancer cells. Cancer Res. 1988, 48, 802–805. [Google Scholar]
- Duan, R.; Xie, W.; Burghardt, R.C.; Safe, S. Estrogen Receptor-mediated Activation of the Serum Response Element in MCF-7 Cells through MAPK-dependent Phosphorylation of Elk-1. J. Biol. Chem. 2001, 276, 11590–11598. [Google Scholar] [CrossRef] [Green Version]
- Duan, R.; Xie, W.; Li, X.; McDougal, A.; Safe, S. Estrogen regulation of c-fos gene expression through phosphatidylinositol-3-kinase-dependent activation of serum response factor in MCF-7 breast cancer cells. Biochem. Biophys. Res. Commun. 2002, 294, 384–394. [Google Scholar] [CrossRef]
- Grundker, C.; Gunthert, A.R.; Hellriegel, M.; Emons, G. Gonadotropin-releasing hormone (GnRH) agonist triptorelin inhibits estradiol-induced serum response element (SRE) activation and c-fos expression in hu-man endometrial, ovarian and breast cancer cells. Eur. J. Endocrinol. 2004, 151, 619–628. [Google Scholar] [CrossRef]
- Girgert, R.; Emons, G.; Gründker, C. Inactivation of GPR30 reduces growth of triple-negative breast cancer cells: Possible application in targeted therapy. Breast Cancer Res. Treat. 2012, 134, 199–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girgert, R.; Emons, G.; Gründker, C. Inhibition of GPR30 by estriol prevents growth stimulation of triple-negative breast cancer cells by 17β-estradiol. BMC Cancer 2014, 14, 935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girgert, R.; Emons, G.; Gründker, C. 17β-estradiol-induced growth of triple-negative breast cancer cells is prevented by the reduction of GPER expression after treatment with gefitinib. Oncol. Rep. 2016, 37, 1212–1218. [Google Scholar] [CrossRef] [PubMed]
- Luttrell, L.M.; Daaka, Y.; Lefkowitz, R.J. Regulation of tyrosine kinase cascades by G-protein-coupled recep-tors. Curr. Opin. Cell Biol. 1999, 11, 177–183. [Google Scholar] [CrossRef]
- Günthert, A.R.; Gründker, C.; Olota, A.; Läsche, J.; Eicke, N.; Emons, G. Analogs of GnRH-I and GnRH-II inhibit epidermal growth factor-induced signal transduction and resensitize resistant human breast cancer cells to 4OH-tamoxifen. Eur. J. Endocrinol. 2005, 153, 613–625. [Google Scholar] [CrossRef]
- Von Alten, J.; Fister, S.; Schulz, H.; Viereck, V.; Frosch, K.-H.; Emons, G.; Gründker, C. GnRH analogs reduce invasiveness of human breast cancer cells. Breast Cancer Res. Treat. 2006, 100, 13–21. [Google Scholar] [CrossRef]
- Ziegler, E.; Hansen, M.-T.; Haase, M.; Emons, G.; Gründker, C. Generation of MCF-7 cells with aggressive metastatic potential in vitro and in vivo. Breast Cancer Res. Treat. 2014, 148, 269–277. [Google Scholar] [CrossRef]
- Olbrich, T.; Ziegler, E.; Türk, G.; Schubert, A.; Emons, G.; Gründker, C. Kisspeptin-10 inhibits bone-directed migration of GPR54-positive breast cancer cells: Evidence for a dose-window effect. Gynecol. Oncol. 2010, 119, 571–578. [Google Scholar] [CrossRef]
- Magliocco, A.; Egan, C. Breast Cancer Metastasis: Advances Trough the Use of In Vitro Co-Culture Model Systems. In Breast Cancer-Focusing Tumor Micriinvironment, Stem Cells and Metastasis; Gunduz, M., Gunduz, E., Eds.; InTech: Rijeka, Croatia, 2011; Volume 1. [Google Scholar]
- Hellinger, J.W.; Hüchel, S.; Goetz, L.; Bauerschmitz, G.; Emons, G.; Gründker, C. Inhibition of CYR61-S100A4 Axis Limits Breast Cancer Invasion. Front. Oncol. 2019, 9, 1074. [Google Scholar] [CrossRef]
- Millar, R.; Lowe, S.; Conklin, D.; Pawson, A.; Maudsley, S.; Troskie, B.; Ott, T.; Millar, M.; Lincoln, G.; Sellar, R.; et al. A novel mammalian receptor for the evolutionarily conserved type II GnRH. Proc. Natl. Acad. Sci. 2001, 98, 9636–9641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neill, J.D.; Duck, L.; Sellers, J.C.; Musgrove, L.C. A Gonadotropin-Releasing Hormone (GnRH) Receptor Specific for GnRH II in Primates. Biochem. Biophys. Res. Commun. 2001, 282, 1012–1018. [Google Scholar] [CrossRef] [PubMed]
- Neill, J.D.; Musgrove, L.C.; Duck, L.W. Newly recognized GnRH receptors: Function and relative role. Trends Endocrinol. Metab. TEM 2004, 15, 383–392. [Google Scholar] [CrossRef]
- Stewart, A.J.; Katz, A.A.; Millar, R.P.; Morgan, K. Retention and Silencing of Prepro-GnRH-II and Type II GnRH Receptor Genes in Mammals. Neuroendocrinology 2009, 90, 416–432. [Google Scholar] [CrossRef] [PubMed]
- Gründker, C.; Föst, C.; Fister, S.; Nolte, N.; Günthert, A.R.; Emons, G. Gonadotropin-releasing hormone type II antagonist induces apoptosis in MCF-7 and triple-negative MDA-MB-231 human breast cancer cells in vitro and in vivo. Breast Cancer Res. 2010, 12, R49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gründker, C.; Günthert, A.R.; Millar, R.P.; Emons, G. Expression of Gonadotropin-Releasing Hormone II (GnRH-II) Receptor in Human Endometrial and Ovarian Cancer Cells and Effects of GnRH-II on Tumor Cell Proliferation. J. Clin. Endocrinol. Metab. 2002, 87, 1427–1430. [Google Scholar] [CrossRef] [Green Version]
- Millar, R.P. GnRH II and type II GnRH receptors. Trends Endocrinol. Metab. 2003, 14, 35–43. [Google Scholar] [CrossRef]
- Millar, R.; Conklin, D.; Lofton-Day, C.; Hutchinson, E.; Troskie, B.; Illing, N.; Sealfon, S.C.; Hapgood, J. A novel human GnRH receptor homolog gene: Abundant and wide tissue distribution of the antisense tran-script. J. Endocrinol. 1999, 162, 117–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, K.; Wang, P.; Zhao, J.; Wu, Y.L.; Cheng, Z.J.; Wu, G.X.; Hu, W.; Ma, L.; Pei, G. Five-transmembrane do-mains appear sufficient for a G protein-coupled receptor: Functional five-transmembrane domain chemo-kine receptors. Proc. Natl. Acad. Sci. USA 1999, 96, 7922–7927. [Google Scholar] [CrossRef] [Green Version]
- Morgan, K.; Conklin, D.; Pawson, A.J.; Sellar, R.; Ott, T.R.; Millar, R.P. A Transcriptionally Active Human Type II Gonadotropin-Releasing Hormone Receptor Gene Homolog Overlaps Two Genes in the Antisense Orientation on Chromosome 1q.12. Endocrinology 2003, 144, 423–436. [Google Scholar] [CrossRef]
- Eicke, N.; Günthert, A.R.; Viereck, V.; Siebold, D.; Béhé, M.; Becker, T.; Emons, G.; Gründker, C. GnRH-II receptor-like antigenicity in human placenta and in cancers of the human reproductive organs. Eur. J. Endocrinol. 2005, 153, 605–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emons, G.; Müller, V.; Ortmann, O.; Schulz, K.-D. Effects of LHRH-analogues on mitogenic signal transduction in cancer cells. J. Steroid Biochem. Mol. Biol. 1998, 65, 199–206. [Google Scholar] [CrossRef]
- Fister, S.; Günthert, A.R.; Emons, G.; Gründker, C. Gonadotropin-Releasing Hormone Type II Antagonists Induce Apoptotic Cell Death in Human Endometrial and Ovarian Cancer Cells In vitro and In vivo. Cancer Res. 2007, 67, 1750–1756. [Google Scholar] [CrossRef] [Green Version]
- Fister, S.; Günthert, A.R.; Aicher, B.; Paulini, K.W.; Emons, G.; Gründker, C. GnRH-II Antagonists Induce Apoptosis in Human Endometrial, Ovarian, and Breast Cancer Cells via Activation of Stress-Induced MAPKs p38 and JNK and Proapoptotic Protein Bax. Cancer Res. 2009, 69, 6473–6481. [Google Scholar] [CrossRef] [Green Version]
- Gründker, C.; Schlotawa, L.; Viereck, V.; Eicke, N.; Horst, A.; Kairies, B.; Emons, G. Antiproliferative effects of the GnRH antagonist cetrorelix and of GnRH-II on human endometrial and ovarian cancer cells are not mediated through the GnRH type I receptor. Eur. J. Endocrinol. 2004, 151, 141–149. [Google Scholar] [CrossRef] [Green Version]
- Enomoto, M.; Endo, D.; Kawashima, S.; Park, M.K. Human Type II GnRH Receptor Mediates Effects of GnRH on Cell Proliferation. Zoöl. Sci. 2004, 21, 763–770. [Google Scholar] [CrossRef] [Green Version]
- Choi, K.-C.; Auersperg, N.; Leung, P.C.K. Expression and Antiproliferative Effect of a Second Form of Gonadotropin-Releasing Hormone in Normal and Neoplastic Ovarian Surface Epithelial Cells. J. Clin. Endocrinol. Metab. 2001, 86, 5075. [Google Scholar] [CrossRef] [PubMed]
- Mangia, A.; Tommasi, S.; Reshkin, S.J.; Simone, G.; Stea, B.; Schittulli, F.; Paradiso, A. Gonadotropin releas-ing hormone receptor expression in primary breast cancer: Comparison of immunohistochemical, radiolig-and and Western blot analyses. Oncol. Rep. 2002, 9, 1127–1132. [Google Scholar] [PubMed] [Green Version]
- Kim, K.Y.; Choi, K.C.; Auersperg, N.; Leung, P.C. Mechanism of gonadotropin-releasing hormone (GnRH)-I and -II-induced cell growth inhibition in ovarian cancer cells: Role of the GnRH-I receptor and protein ki-nase C pathway. Endocr. Relat. Cancer 2006, 13, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Parmar, H.; Phillips, R.H.; Rustin, G.; Lightman, S.L.; Schally, A.V. Therapy of advanced ovarian cancer with D-Trp-6-LH-RH (decapeptyl) microcapsules. Biomed. Pharmacother. 1988, 42, 531–538. [Google Scholar]
- Parmar, H.; Rustin, G.; Lightman, S.L.; Phillips, R.H.; Hanham, I.W.; Schally, A.V. Response to D-Trp-6-luteinising hormone releasing hormone (Decapeptyl) microcapsules in advanced ovarian cancer. Br. Med J. Clin. Res. Ed. 1988, 296, 1229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruckner, H.W.; Motwani, B.T. Treatment of advanced refractory ovarian carcinoma with a gonadotropin-releasing hormone analogue. Am. J. Obstet. Gynecol. 1989, 161, 1216–1218. [Google Scholar] [CrossRef]
- Jäger, W.; Wildt, L.; Lang, N. Some observations on the effect of a GnRH analog in ovarian cancer. Eur. J. Obstet. Gynecol. Reprod. Biol. 1989, 32, 137–148. [Google Scholar] [CrossRef]
- Kavanagh, J.J.; Roberts, W.; Townsend, P.; Hewitt, S. Leuprolide acetate in the treatment of refractory or persistent epithelial ovarian cancer. J. Clin. Oncol. 1989, 7, 115–118. [Google Scholar] [CrossRef]
- Vavra, N.; Barrada, M.; Fitz, R.; Sevelda, P.; Baur, M.; Dittrich, C. Goserelin—A new form of hormone therapy in ovarian cancer. Gynakol. Rundsch. 1990, 30, 61–63. [Google Scholar] [CrossRef] [PubMed]
- Emons, G.; Ortmann, O.; Teichert, H.M.; Fassl, H.; Lohrs, U.; Kullander, S.; Kauppila, A.; Ayalon, D.; Schally, A.; Oberheuser, F. Luteinizing hormone-releasing hormone agonist triptorelin in combination with cytotox-ic chemotherapy in patients with advanced ovarian carcinoma. A prospective double blind randomized tri-al. Decapeptyl Ovarian Cancer Study Group. Cancer 1996, 78, 1452–1460. [Google Scholar] [CrossRef]
- Hasan, J.; Ton, N.; Mullamitha, S.; Clamp, A.; McNeilly, A.; Marshall, E.; Jayson, G.C. Phase II trial of tamoxifen and goserelin in recurrent epithelial ovarian cancer. Br. J. Cancer 2005, 93, 647–651. [Google Scholar] [CrossRef] [Green Version]
- Lind, M.; Cantwell, B.; Millward, M.; Robinson, A.; Proctor, M.; Simmons, D.; Carmichael, J.; Harris, A. A phase II trial of goserelin (Zoladex) in relapsed epithelial ovarian cancer. Br. J. Cancer 1992, 65, 621–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, D.S.; Brady, M.F.; Barrett, R.J. A Phase II Trial of Leuprolide Acetate in Patients with Advanced Epithelial Ovarian Carcinoma. Am. J. Clin. Oncol. 1992, 15, 125–128. [Google Scholar] [CrossRef]
- Carnino, F.; Iskra, L.; Fuda, G.; Foglia, G.; Odicino, F.; Bruzzone, M.; Chiara, S.; Gadducci, A.; Ragni, N. The treatment of progressive ovarian carcinoma with D-Trp-LHRH (decapeptyl). Eur. J. Cancer 1994, 30, 1903–1904. [Google Scholar] [CrossRef]
- Marinaccio, M.; D’Addario, V.; Serratì, A.; Pinto, V.; Cagnazzo, G. Leuprolide acetate as a salvage-therapy in relapsed epithelial ovarian cancer. Eur. J. Gynaecol. Oncol. 1996, 17, 286–288. [Google Scholar]
- Ron, I.G.; Wigler, N.; Merimsky, O.; Inbar, M.J.; Chaitchik, S. A Phase II Trial of D-Trp-6-LHRH (Decapeptyl) in Pretreated Patients with Advanced Epithelial Ovarian Cancer. Cancer Investig. 1995, 13, 272–275. [Google Scholar] [CrossRef]
- Duffaud, F.; El Van Der Burg, M.; Namer, M.; Vergote, I.; Willemse, P.B.; Huinink, W.T.B.; Guastalla, J.P.; Nooij, M.A.; Kerbrat, P.; Piccart, M.; et al. D-TRP-6-LHRH (Triptorelin) is not effective in ovarian carcinoma: An EORTC Gynaecological Cancer Co-operative Group Study. Anticancer Drugs 2001, 12, 159–162. [Google Scholar] [CrossRef]
- Paskeviciute, L.; Roed, H.; Engelholm, S.A. No rules without exception: Long-term complete remission observed in a study using a LH-RH agonist in platinum-refractory ovarian cancer. Gynecol. Oncol. 2002, 86, 297–301. [Google Scholar] [CrossRef]
- Du Bois, A.; Meier, W.; Lück, H.J.; Emons, G.; Moebus, V.; Schroeder, W.; Costa, S.; Bauknecht, T.; Olbricht, S.; Jackisch, C.; et al. Chemotherapy versus hormonal treatment in platinum- and paclitaxel-refractory ovarian cancer: A randomised trial of the German Arbeitsgemeinschaft Gynaekologische Onkologie (AGO) Study Group Ovarian Cancer. Ann. Oncol. 2002, 13, 251–257. [Google Scholar] [CrossRef]
- Balbi, G.; Piano, L.D.; Cardone, A.; Cirelli, G. Second-line therapy of advanced ovarian cancer with GnRH analogs. Int. J. Gynecol. Cancer 2004, 14, 799–803. [Google Scholar] [CrossRef]
- Sevelda, P.; Vavra, N.; Fitz, R.; Barrada, M.; Salzer, H.; Baur, M.; Dittrich, C. Goserelin a GnRH-analogue as third-line therapy of refractory epithelial ovarian cancer. Int. J. Gynecol. Cancer 1992, 2, 160–162. [Google Scholar] [CrossRef] [PubMed]
- Tan, O.; Bukulmez, O. Biochemistry, molecular biology and cell biology of gonadotropin-releasing hormone antagonists. Curr. Opin. Obstet. Gynecol. 2011, 23, 238–244. [Google Scholar] [CrossRef]
- Verschraegen, C.F.; Westphalen, S.; Hu, W.; Loyer, E.; Kudelka, A.; Völker, P.; Kavanagh, J.; Steger, M.; Schulz, K.-D.; Emons, G. Phase II study of cetrorelix, a luteinizing hormone-releasing hormone antagonist in patients with platinum-resistant ovarian cancer. Gynecol. Oncol. 2003, 90, 552–559. [Google Scholar] [CrossRef]
- Cho, N.; Harada, M.; Imaeda, T.; Imada, T.; Matsumoto, H.; Hayase, Y.; Sasaki, S.; Furuya, S.; Suzuki, N.; Okubo, S.; et al. Discovery of a Novel, Potent, and Orally Active Nonpeptide Antagonist of the Human Luteinizing Hormone-Releasing Hormone (LHRH) Receptor. J. Med. Chem. 1998, 41, 4190–4195. [Google Scholar] [CrossRef]
- Hara, T.; Araki, H.; Kusaka, M.; Harada, M.; Cho, N.; Suzuki, N.; Furuya, S.; Fujino, M. Suppression of a Pituitary-Ovarian Axis by Chronic Oral Administration of a Novel Nonpeptide Gonadotropin-Releasing Hormone Antagonist, TAK-013, in Cynomolgus Monkeys. J. Clin. Endocrinol. Metab. 2003, 88, 1697–1704. [Google Scholar] [CrossRef] [Green Version]
- Millar, R.P.; Zhu, Y.-F.; Struthers, R.S. Progress towards the development of non-peptide orally-active gonadotropin-releasing hormone (GnRH) antagonists: Therapeutic implications. Br. Med. Bull. 2000, 56, 761–772. [Google Scholar] [CrossRef] [Green Version]
- Miwa, K.; Hitaka, T.; Imada, T.; Sasaki, S.; Yoshimatsu, M.; Kusaka, M.; Tanaka, A.; Nakata, D.; Furuya, S.; Endo, S.; et al. Discovery of 1-{4-[1-(2,6-Difluorobenzyl)-5-[(dimethylamino)methyl]-3-(6-methoxypyridazin-3-yl)-2,4-dioxo-1,2,3,4-tetrahydrothieno[2,3-d]pyrimidin-6-yl]phenyl}-3-methoxyurea (TAK-385) as a Potent, Orally Active, Non-Peptide Antagonist of the Human Gonadotropin-Releasing Hormone Receptor. J. Med. Chem. 2011, 54, 4998–5012. [Google Scholar] [CrossRef] [PubMed]
- Nakata, D.; Masaki, T.; Tanaka, A.; Yoshimatsu, M.; Akinaga, Y.; Asada, M.; Sasada, R.; Takeyama, M.; Miwa, K.; Watanabe, T.; et al. Suppression of the hypothalamic-pituitary-gonadal axis by TAK-385 (relugolix), a novel, investigational, orally active, small molecule gonadotropin-releasing hormone (GnRH) antagonist: Studies in human GnRH receptor knock-in mice. Eur. J. Pharmacol. 2014, 723, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Millar, R.P.; Newton, C.L. Current and future applications of GnRH, kisspeptin and neurokinin B analogues. Nat. Rev. Endocrinol. 2013, 9, 451–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tukun, F.-L.; Olberg, D.E.; Riss, P.J.; Haraldsen, I.; Kaass, A.; Klaveness, J. Recent Development of Non-Peptide GnRH Antagonists. Molecules 2017, 22, 2188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grundker, C.; Volker, P.; Griesinger, F.; Ramaswamy, A.; Nagy, A.; Schally, A.V.; Emons, G. Antitumor ef-fects of the cytotoxic luteinizing hormone-releasing hormone analog AN-152 on human endometrial and ovarian cancers xenografted into nude mice. Am. J. Obstet. Gynecol. 2002, 187, 528–537. [Google Scholar] [CrossRef] [PubMed]
- Schally, A.V.; Nagy, A. Cancer chemotherapy based on targeting of cytotoxic peptide conjugates to their re-ceptors on tumors. Eur. J. Endocrinol. 1999, 141, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Westphalen, S.; Kotulla, G.; Kaiser, F.; Krauss, W.; Werning, G.; Elsasser, H.P.; Nagy, A.; Schulz, K.D.; Gründker, C.; Schally, A.V.; et al. Receptor mediated antiproliferative effects of the cytotoxic LHRH agonist AN-152 in human ovarian and endometrial cancer cell lines. Int. J. Oncol. 2000, 17, 1063–1069. [Google Scholar] [CrossRef]
- Günthert, A.R.; Gründker, C.; Bongertz, T.; Schlott, T.; Nagy, A.; Schally, A.V.; Emons, G. Internalization of cytotoxic analog AN-152 of luteinizing hormone-releasing hormone induces apoptosis in human endometrial and ovarian cancer cell lines independent of multidrug resistance-1 (MDR-1) system. Am. J. Obstet. Gynecol. 2004, 191, 1164–1172. [Google Scholar] [CrossRef]
- Engel, J.B.; Schally, A.V.; Dietl, J.; Rieger, L.; Hönig, A. Targeted Therapy of Breast and Gynecological Cancers with Cytotoxic Analogues of Peptide Hormones. Mol. Pharm. 2007, 4, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Gumlnthert, A.R.; Grumlndker, C.; Bongertz, T.; Nagy, A.; Schally, A.V.; Emons, G. Induction of apoptosis by AN-152, a cytotoxic analog of luteinizing hormone-releasing hormone (LHRH), in LHRH-R positive human breast cancer cells is independent of multidrug resistance-1 (MDR-1) system. Breast Cancer Res. Treat. 2004, 87, 255–264. [Google Scholar] [CrossRef]
- Emons, G.; Kaufmann, M.; Gorchev, G.; Tsekova, V.; Gründker, C.; Günthert, A.R.; Hanker, L.C.; Velikova, M.; Sindermann, H.; Engel, J.; et al. Dose escalation and pharmacokinetic study of AEZS-108 (AN-152), an LHRH agonist linked to doxorubicin, in women with LHRH receptor-positive tumors. Gynecol. Oncol. 2010, 119, 457–461. [Google Scholar] [CrossRef]
- Emons, G.; Gorchev, G.; Sehouli, J.; Wimberger, P.; Stähle, A.; Hanker, L.; Hilpert, F.; Sindermann, H.; Gründker, C.; Harter, P. Efficacy and safety of AEZS-108 (INN: Zoptarelin Doxorubicin Acetate) an LHRH agonist linked to doxorubicin in women with platinum refractory or resistant ovarian cancer expressing LHRH receptors: A multicenter Phase II trial of the ago-study group (AGO GYN 5). Gynecol. Oncol. 2014, 133, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.S.; Scambia, G.; Bondarenko, I.; Westermann, A.M.; Oaknin, A.; Oza, A.M.; Lisyanskaya, A.S.; Vergote, I.; Wenham, R.M.; Temkin, S.M.; et al. ZoptEC: Phase III randomized controlled study comparing zoptarelin with doxorubicin as second line therapy for locally advanced, recurrent, or metastatic endometrial cancer (NCT01767155). J. Clin. Oncol. 2018, 36, 5503. [Google Scholar] [CrossRef]
- Gründker, C.; Nia, A.H.; Emons, G. Gonadotropin-releasing hormone receptor-targeted gene therapy of gynecologic cancers. Mol. Cancer Ther. 2005, 4, 225–231. [Google Scholar]
- Curtis, K.K.; Sarantopoulos, J.; Northfelt, D.W.; Weiss, G.J.; Barnhart, K.M.; Whisnant, J.K.; Leuschner, C.; Alila, H.; Borad, M.J.; Ramanathan, R.K. Novel LHRH-receptor-targeted cytolytic peptide, EP-100: First-in-human phase I study in patients with advanced LHRH-receptor-expressing solid tumors. Cancer Chemother. Pharmacol. 2014, 73, 931–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, S.; Pradeep, S.; Villar-Prados, A.; Wen, Y.; Bayraktar, E.; Mangala, L.S.; Kim, M.S.; Wu, S.Y.; Hu, W.; Rodriguez-Aguayo, C.; et al. GnRH-R—Targeted Lytic Peptide Sensitizes BRCA Wild-type Ovarian Cancer to PARP Inhibition. Mol. Cancer Ther. 2019, 18, 969–979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Zhou, X.; Feng, W.; Pu, T.; Li, X.; Li, F.; Kang, Y.; Zhang, X.; Xu, C. Gonadotropin-Releasing Hormone Receptor-Targeted Near-Infrared Fluorescence Probe for Specific Recognition and Localization of Peritoneal Metastases of Ovarian Cancer. Front. Oncol. 2020, 10, 266. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Schematic representation of GnRH-I receptor signaling in ovarian cancer cells. Binding of GnRH-I and GnRH-I analogs causes G-protein αi-mediated activation of a PTP, resulting in dephosphorylated EGF-R and inhibition of EGF-R signaling through ERK1/2 or PI3K/AKT pathway leading to reduction of proliferation. GnRH-I-induced activation of PTP also inhibits the signal transduction of GPER and membrane-associated ERα through transactivation of EGF-R. In addition, GnRH induces activation of NFκB to reduce and FAS-ligand to increase apoptosis. Finally, GnRH agonists activate the JNK/AP-1 pathway resulting in increased G0/1 phase of the cell cycle and decreased synthesis of DNA. Further details are described in the text. Abbreviations: Akt, protein kinase B (PKB); AP-1, activator protein-1; E2, estradiol; EGF, epidermal growth factor; EGF-R, EGF receptor; ERα, estrogen receptor α; ERK1/2, p44/42 mitogen-activated protein (MAP) kinase; FAS, Fas receptor; FasL, Fas ligand; GDP, guanosine diphosphate; GnRH, gonadotropin-releasing hormone; GPER, G-protein coupled estrogen receptor; GRB2, growth factor receptor-bound protein 2; GTP, guanosine triphosphate; HB-EGF, heparin-binding EGF-like growth factor; JNK, c-Jun N-terminal kinase; JunD, transcription factor JunD; MEK, mitogen-activated protein kinase kinase (MAP2K); MMP, matrix metalloproteinase; NFkB, nucleus factor kB; PI3K, phosphoinositide 3-kinase; PTP, protein tyrosine phosphatase; RAS, G-protein rat sarcoma; SOS, guanine nucleotide exchange factor “son of sevenless”; SRE, serum response element; Src, tyrosine kinase cellular sarcoma; SRF, serum response factor; TAM, tamoxifen.
Figure 1.
Schematic representation of GnRH-I receptor signaling in ovarian cancer cells. Binding of GnRH-I and GnRH-I analogs causes G-protein αi-mediated activation of a PTP, resulting in dephosphorylated EGF-R and inhibition of EGF-R signaling through ERK1/2 or PI3K/AKT pathway leading to reduction of proliferation. GnRH-I-induced activation of PTP also inhibits the signal transduction of GPER and membrane-associated ERα through transactivation of EGF-R. In addition, GnRH induces activation of NFκB to reduce and FAS-ligand to increase apoptosis. Finally, GnRH agonists activate the JNK/AP-1 pathway resulting in increased G0/1 phase of the cell cycle and decreased synthesis of DNA. Further details are described in the text. Abbreviations: Akt, protein kinase B (PKB); AP-1, activator protein-1; E2, estradiol; EGF, epidermal growth factor; EGF-R, EGF receptor; ERα, estrogen receptor α; ERK1/2, p44/42 mitogen-activated protein (MAP) kinase; FAS, Fas receptor; FasL, Fas ligand; GDP, guanosine diphosphate; GnRH, gonadotropin-releasing hormone; GPER, G-protein coupled estrogen receptor; GRB2, growth factor receptor-bound protein 2; GTP, guanosine triphosphate; HB-EGF, heparin-binding EGF-like growth factor; JNK, c-Jun N-terminal kinase; JunD, transcription factor JunD; MEK, mitogen-activated protein kinase kinase (MAP2K); MMP, matrix metalloproteinase; NFkB, nucleus factor kB; PI3K, phosphoinositide 3-kinase; PTP, protein tyrosine phosphatase; RAS, G-protein rat sarcoma; SOS, guanine nucleotide exchange factor “son of sevenless”; SRE, serum response element; Src, tyrosine kinase cellular sarcoma; SRF, serum response factor; TAM, tamoxifen.

Figure 2.
Schematic representation of GnRH-II signaling in ovarian cancer cells assuming that a truncated GnRH-II receptor (A) or no GnRH-II receptor (B) exists. (A) Binding of GnRH-II agonists, GnRH-I antagonists and GnRH-II antagonists to the truncated GnRH-II receptor cause PTP activation leading to dephopsphorylation of activated EGF-R. In addition, GnRH-II antagonists induce apoptosis through activation of p38, JNK and the intrinsic apoptotic pathway. GnRH-I agonists only couple to the GnRH-I receptor. Signal transduction mechanisms and further details are described in Figure 1 and the text. (B) Signal transduction of GnRH-I and GnRH-II and their agonistic as well as antagonistic analogs through the GnRH-I-R if no GnRH-II-R exists. Signaling and details are described in the text. Abbreviations: AP-1, activator protein-1; Bax, B-cell lymphoma 2 (Bcl-2)-associated X protein; EGF, epidermal growth factor; EGF-R, EGF receptor; ERK1/2, p44/42 mitogen-activated protein (MAP) kinase; GDP, guanosine diphosphate; GnRH, gonadotropin-releasing hormone; JNK, c-Jun N-terminal kinase; JunD, transcription factor JunD; P38, mitogen-activated protein kinase P38; PI3K, phosphoinositide 3-kinase; PTP, protein tyrosine phosphatase. Signal transduction pathway affected by GnRH-I agonists, GnRH-I antagonists, and GnRH-II agonists are marked brown. GnRH-II antagonist-induced signaling is marked red.
Figure 2.
Schematic representation of GnRH-II signaling in ovarian cancer cells assuming that a truncated GnRH-II receptor (A) or no GnRH-II receptor (B) exists. (A) Binding of GnRH-II agonists, GnRH-I antagonists and GnRH-II antagonists to the truncated GnRH-II receptor cause PTP activation leading to dephopsphorylation of activated EGF-R. In addition, GnRH-II antagonists induce apoptosis through activation of p38, JNK and the intrinsic apoptotic pathway. GnRH-I agonists only couple to the GnRH-I receptor. Signal transduction mechanisms and further details are described in Figure 1 and the text. (B) Signal transduction of GnRH-I and GnRH-II and their agonistic as well as antagonistic analogs through the GnRH-I-R if no GnRH-II-R exists. Signaling and details are described in the text. Abbreviations: AP-1, activator protein-1; Bax, B-cell lymphoma 2 (Bcl-2)-associated X protein; EGF, epidermal growth factor; EGF-R, EGF receptor; ERK1/2, p44/42 mitogen-activated protein (MAP) kinase; GDP, guanosine diphosphate; GnRH, gonadotropin-releasing hormone; JNK, c-Jun N-terminal kinase; JunD, transcription factor JunD; P38, mitogen-activated protein kinase P38; PI3K, phosphoinositide 3-kinase; PTP, protein tyrosine phosphatase. Signal transduction pathway affected by GnRH-I agonists, GnRH-I antagonists, and GnRH-II agonists are marked brown. GnRH-II antagonist-induced signaling is marked red.
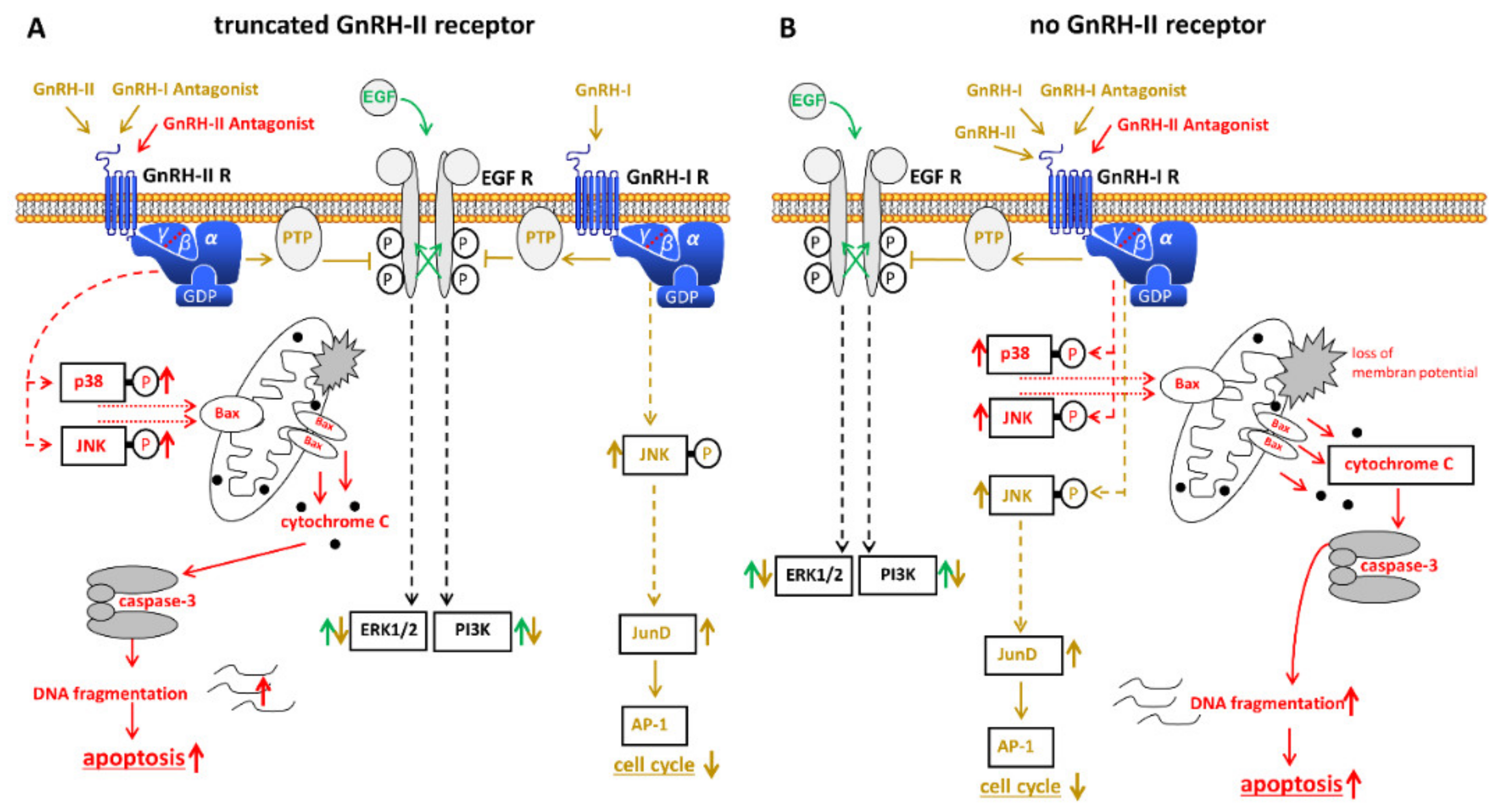

{kind=link}
{kind=link}
Table 1.
Clinical trials—Suppression of pituitary gonadotropin secretion through GnRH-analogs as treatment for epithelial ovarian cancer.
Table 1.
Clinical trials—Suppression of pituitary gonadotropin secretion through GnRH-analogs as treatment for epithelial ovarian cancer.
| Reference | Pat. | Age | Diagnosis | Drug | Results |
|---|---|---|---|---|---|
| [118] | 41 | 42–79 | advanced ovarian cancer | Triptorelin depot | PR: 6 SD: 5 |
| [120] | 5 | 50–64 | refractory ovarian cancer | Leuprolide acetate 1 mg/day | CR: 1 PR: 4 |
| [121] | 19 11 untreated | 38–73 | progressive ovarian cancer | Triptorelin 0.1 mg/day or 3.2 mg/month depot | SD: 12 |
| [122] | 18 | 21–68 | progressive ovarian cancer | Leuprolide acetate 1.0 mg/day | PR: 4 SD: 2 NE: 5 |
| [124] | 69 66 placebo | <44–>75 | stage III or IV epithelial ovarian cancer | Triptorelin 3.75 mg/month depot | No significant differences in progression free and overall survival |
| [125] | 26 | 49–79 | advanced ovarian cancer | Tamoxifen 20 mg/month Goserelin 3.6 mg/month | CR: 1 PR: 2 SD: 10 |
| [126] | 30 | 38–90 | relapsed epithelial ovarian cancer | Goserelin 3.6 mg/month | PR: 2 SD: 5 |
| [127] | 25 | median 65.5 | advanced epithelial ovarian cancer | Leuprolide acetate | PR: 4 SD:15 |
| [128] | 20 | average 60 | progressive ovarian cancer | Triptorelin 3.75 mg/4 weeks | SD: 14 |
| [129] | 32 | relapsed epithelial ovarian cancer | Leuprolide acetate 3.75 mg depot | PR: 4 SD: 5 | |
| [130] | 14 | 47–77 | advanced epithelial ovarian cancer | Triptorelin 3.2 mg/28 days | SD: 8 |
| [131] | 68 | 42–87 | relapsed ovarian cancer, mostly refractory to platinum | Triptorelin 3.75 mg/on days 1, 8 and 28 followed by 4-weekly | SD: 11 |
| [132] | 32 | 32–77 | platinum-refractory ovarian cancer | Leuprolide 3.75 mg/month | CR: 1 PR: 2 SD: 4 |
| [133] | 37 | 27–75 | platinum- and paclitaxel-refractory ovarian cancer | Leuprolide acetate 3.75 mg/4 weeks | SD: 4 |
| [134] | 12 | 45–68 | advanced ovarian cancer | Leuprolide acetate 3.75 mg on days 1, 8, 28 followed by 28-day intervals | PR: 1 SD: 3 |
| [135] | 23 | 44–72 | refractory ovarian cancer | Goserelin 3.6 mg/month | PR: 4 SD: 7 |
CR, complete response; PR, partial response; SD, stable disease; NE, not evaluable.
Table 2.
Clinical trials—GnRH receptor target therapy for treatment of epithelial ovarian cancer.
| Reference | Pat. | Age | Diagnosis | Drug | Treatment | Results |
|---|---|---|---|---|---|---|
| [151] | 4 | 55 +/− 11 | epithelial cancers of ovary, endometrium, or breast | Zoptarelin-Doxorubicin | 10, 20, 40, or 80 mg/m2 | maximum tolerated dose: 267 mg/m2 |
| 6 | 59 +/− 5 | 160 mg/m2 | ||||
| 7 | 48 +/− 11 | 267 mg/m2 | ||||
| [152] | 42 | 49 +/− 10 | epithelial ovarian, fallopian tube or primary peritoneal cancer | Zoptarelin-Doxorubicin | 267 mg/m2 | PR: 6 (14.3%) SD: 16 (38.1%) PD: 16 (38.1%) NE: 4 (9.5%) |
| [155] | 28 (F) 9 (M) | 59 (39–80) 61 (39–80) | ovarian, endometrial, breast and other cancers | EP-100 | Initial cohorts: 1–7, 0.6–7.8 mg/m2, n = 21 Later cohorts:: 7–11, 7.8–40 mg/m2, n = 16 | recommended phase II dose: 40 mg/m2 |
F, female; M, male; PR, partial response; SD, stable disease; PD, progressive disease; NE, not evaluable.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Gründker, C.; Emons, G. Role of Gonadotropin-Releasing Hormone (GnRH) in Ovarian Cancer. Cells 2021, 10, 437. https://doi.org/10.3390/cells10020437
AMA Style
Gründker C, Emons G. Role of Gonadotropin-Releasing Hormone (GnRH) in Ovarian Cancer. Cells. 2021; 10(2):437. https://doi.org/10.3390/cells10020437
Chicago/Turabian StyleGründker, Carsten, and Günter Emons. 2021. "Role of Gonadotropin-Releasing Hormone (GnRH) in Ovarian Cancer" Cells 10, no. 2: 437. https://doi.org/10.3390/cells10020437
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.