
Why Cells and Viruses Cannot Survive without an ESCRT




Abstract
:1. Introduction
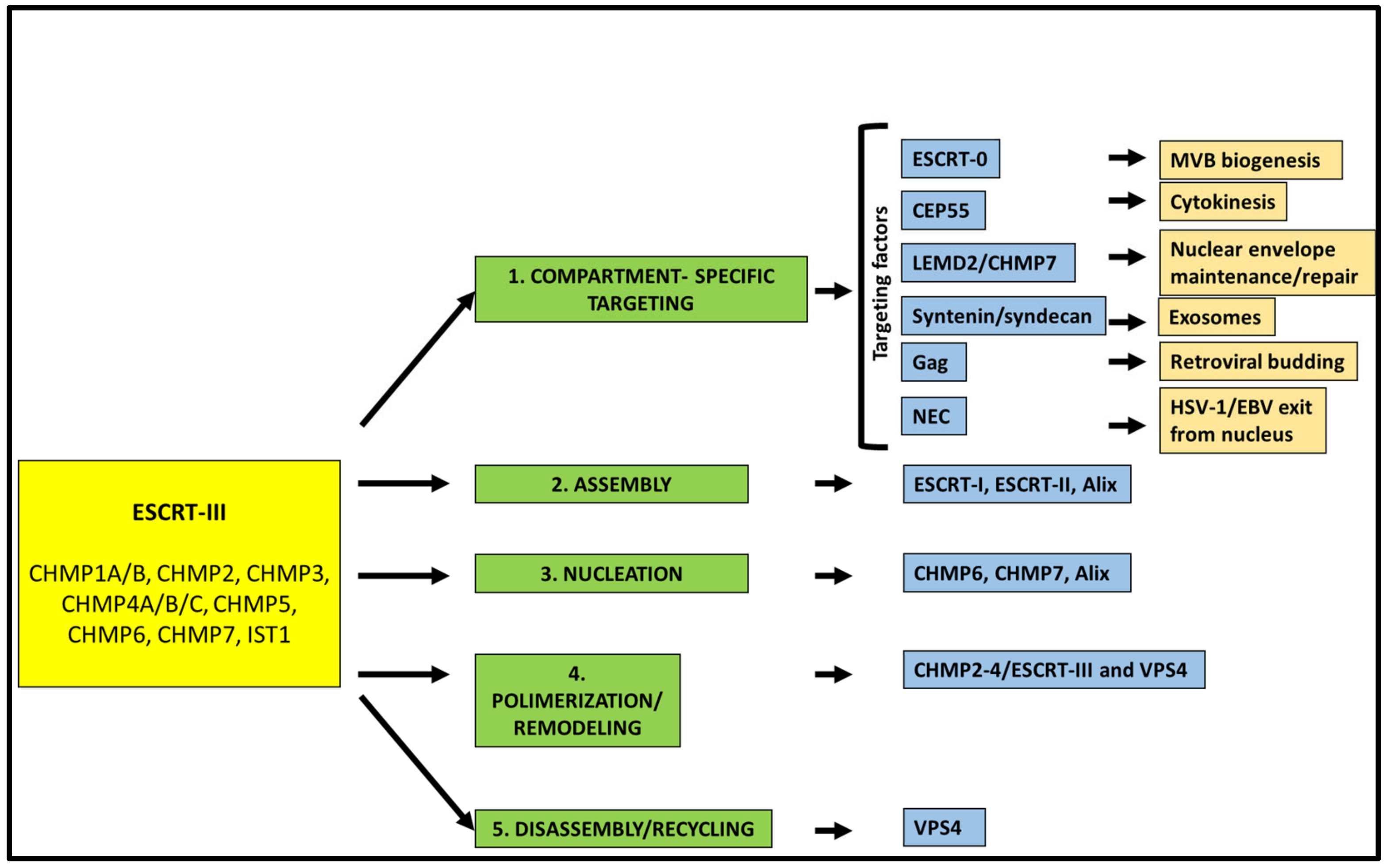
2. The ESCRT Machinery: An Overview
3. ESCRT Machinery Functions in Fundamental Cellular Pathways
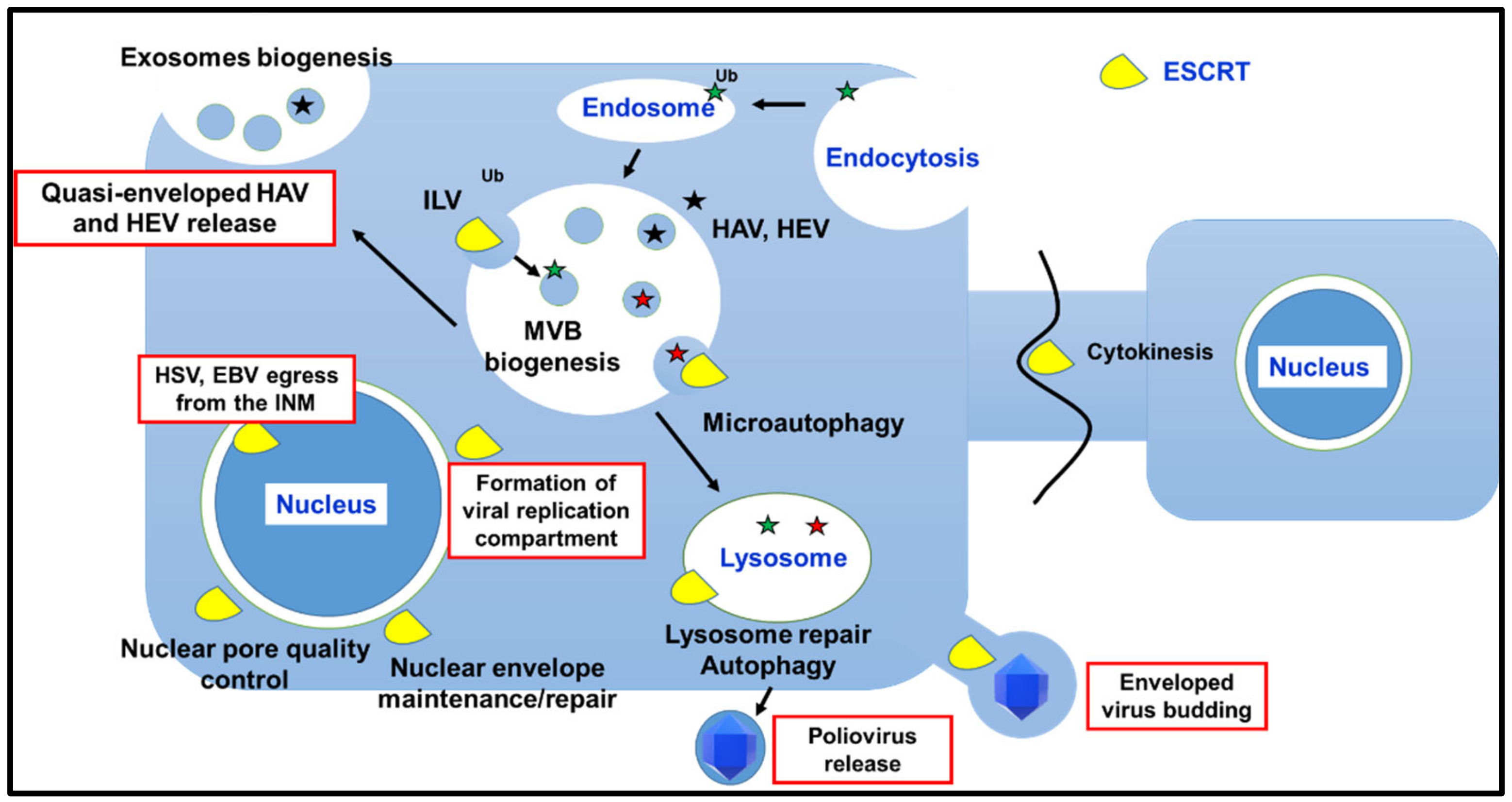
3.1. ESCRTs Involvement in MVB Biogenesis
3.2. Role of ESCRT Machinery in Autophagy
3.3. Role of the ESCRT Machinery in Cytokinesis
3.4. Involvement of the ESCRT Machinery in Damage Repair of Cellular Membranes
3.4.1. Plasma Membrane Repair
3.4.2. Nuclear Envelope Maintenance and Repair
3.4.3. Endolysosomal Membrane Repair
4. ESCRT Machinery and Viral Replication Cycle
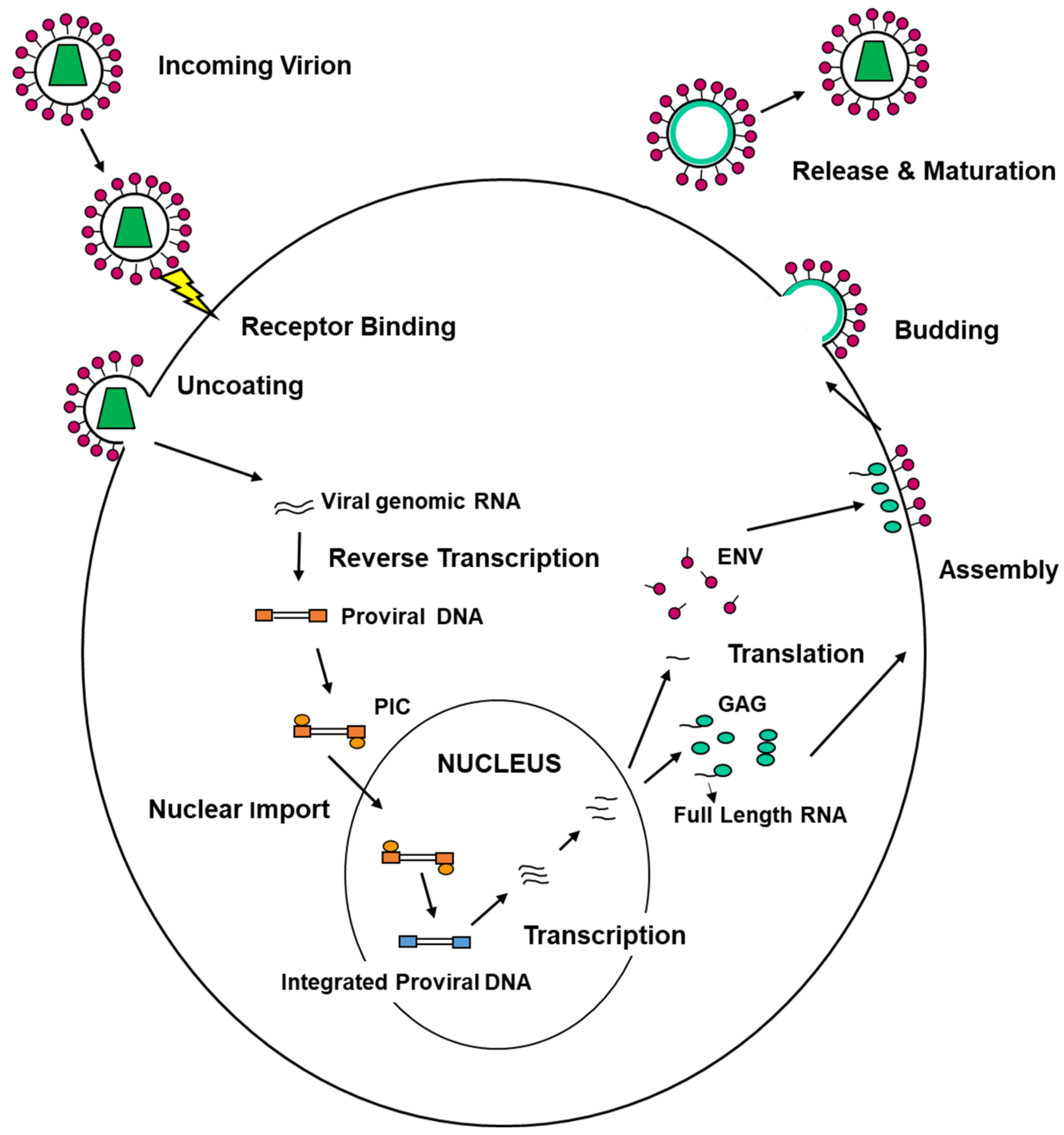
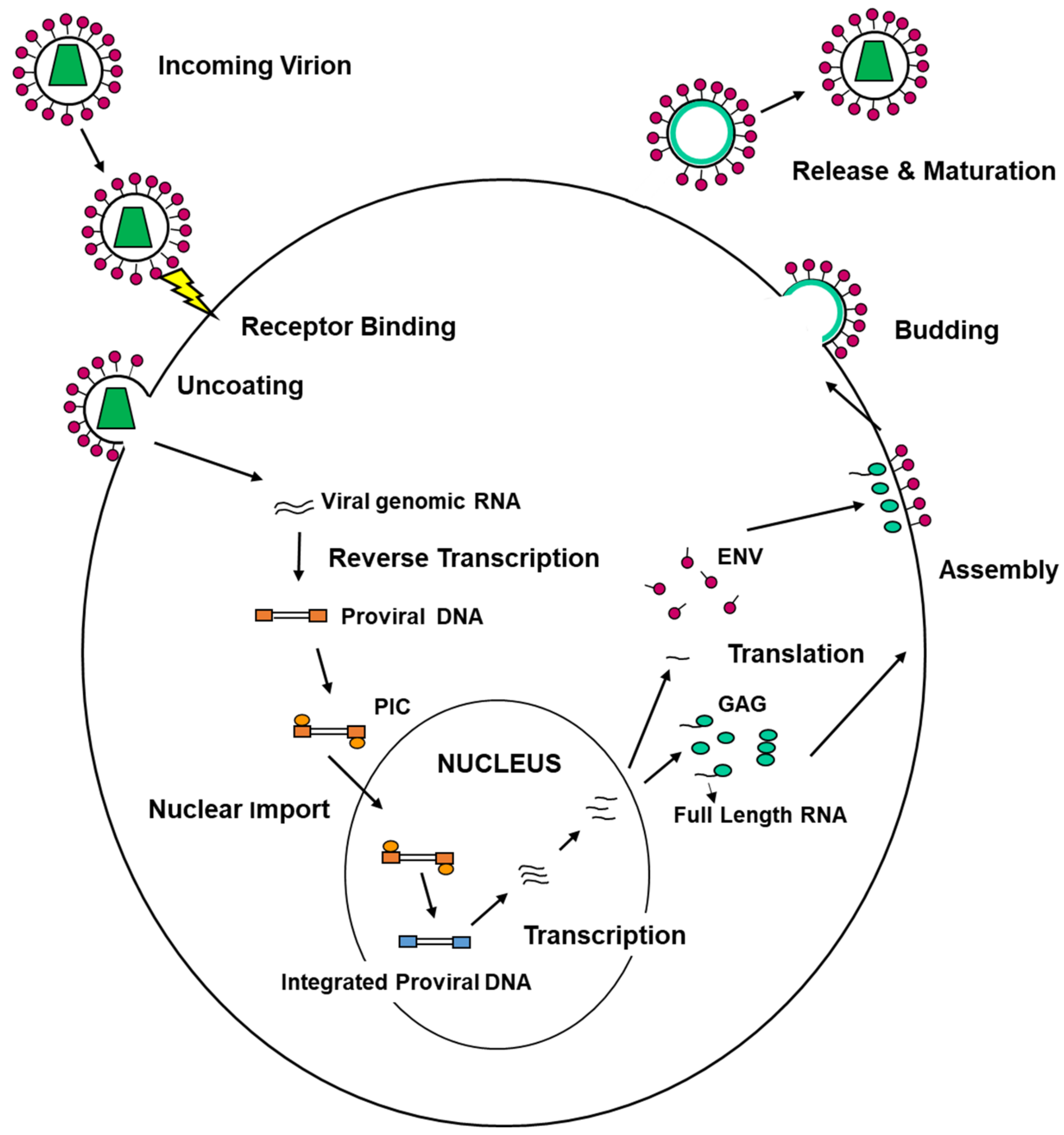
4.1. ESCRT Machinery and Viral Entry
4.2. ESCRT Machinery Involvement in the Formation of Viral Replication/Assembly Compartments
4.3. ESCRT Machinery and Viral Egress from Infected Cells
4.4. EVs and Autophagy in Viral Transmission
5. The Travel of Herpes Simplex Virus Type 1 from the Nucleus to the Extracellular Environment: Is There a Role for the ESCRT Machinery?
6. Conclusions and Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Rajwar, A.; Morya, V.; Kharbanda, S.; Bhatia, D. DNA Nanodevices to Probe and Program Membrane Organization, Dynamics, and Applications. J. Membr. Biol. 2020, 253, 577–587. [Google Scholar] [CrossRef] [PubMed]
- Vietri, M.; Radulovic, M.; Stenmark, H. The many functions of ESCRTs. Nat. Rev. Mol. Cell Biol. 2020, 21, 25–42. [Google Scholar] [CrossRef]
- Campsteijn, C.; Vietri, M.; Stenmark, H. Novel ESCRT functions in cell biology: Spiraling out of control? Curr. Opin. Cell Biol. 2016, 41, 1–8. [Google Scholar] [CrossRef]
- Robinson, M.; Schor, S.; Barouch-Bentov, R.; Einav, S. Viral journeys on the intracellular highways. Cell Mol. Life Sci. 2018, 75, 3693–3714. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.; Zhou, S.; Gao, S.; Deng, H. Remodeling of host membranes during herpesvirus assembly and egress. Protein Cell 2019, 10, 315–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Lan, Y.; Sanyal, S. Membrane heist: Coronavirus host membrane remodeling during replication. Biochimie 2020, 179, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.; Krijnse-Locker, J. Modification of intracellular membrane structures for virus replication. Nat. Rev. Microbiol. 2008, 6, 363–374. [Google Scholar] [CrossRef]
- Martins, S.T.; Alves, L.R. Extracellular Vesicles in Viral Infections: Two Sides of the Same Coin? Front. Cell Infect. Microbiol. 2020, 10, 593170. [Google Scholar] [CrossRef]
- Katzmann, D.J.; Babst, M.; Emr, S.D. Ubiquitin-dependent sorting into the multivesicular body pathway requires the function of a conserved endosomal protein sorting complex, ESCRT-I. Cell 2001, 106, 145–155. [Google Scholar] [CrossRef] [Green Version]
- Schoneberg, J.; Lee, I.H.; Iwasa, J.H.; Hurley, J.H. Reverse-topology membrane scission by the ESCRT proteins. Nat. Rev. Mol. Cell Biol. 2017, 18, 5–17. [Google Scholar] [CrossRef]
- Hanson, P.I.; Cashikar, A. Multivesicular body morphogenesis. Annu. Rev. Cell Dev. Biol. 2012, 28, 337–362. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, I.; Akram, Z.; Iqbal, H.M.N.; Munn, A.L. The regulation of Endosomal Sorting Complex Required for Transport and accessory proteins in multivesicular body sorting and enveloped viral budding—An overview. Int. J. Biol. Macromol. 2019, 127, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mast, F.D.; Herricks, T.; Strehler, K.M.; Miller, L.R.; Saleem, R.A.; Rachubinski, R.A.; Aitchison, J.D. ESCRT-III is required for scissioning new peroxisomes from the endoplasmic reticulum. J. Cell Biol. 2018, 217, 2087–2102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kostelansky, M.S.; Sun, J.; Lee, S.; Kim, J.; Ghirlando, R.; Hierro, A.; Emr, S.D.; Hurley, J.H. Structural and functional organization of the ESCRT-I trafficking complex. Cell 2006, 125, 113–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kostelansky, M.S.; Schluter, C.; Tam, Y.Y.; Lee, S.; Ghirlando, R.; Beach, B.; Conibear, E.; Hurley, J.H. Molecular architecture and functional model of the complete yeast ESCRT-I heterotetramer. Cell 2007, 129, 485–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weissenhorn, W.; Gottlinger, H. ESCRT-I part II: Forming the real ESCRT-I complex. Cell Host Microbe 2007, 2, 1–2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hierro, A.; Sun, J.; Rusnak, A.S.; Kim, J.; Prag, G.; Emr, S.D.; Hurley, J.H. Structure of the ESCRT-II endosomal trafficking complex. Nature 2004, 431, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Horvath, P.; Muller-Reichert, T. A Structural View on ESCRT-Mediated Abscission. Front. Cell Dev. Biol. 2020, 8, 586880. [Google Scholar] [CrossRef] [PubMed]
- Gill, D.J.; Teo, H.; Sun, J.; Perisic, O.; Veprintsev, D.B.; Emr, S.D.; Williams, R.L. Structural insight into the ESCRT-I/-II link and its role in MVB trafficking. EMBO J. 2007, 26, 600–612. [Google Scholar] [CrossRef] [Green Version]
- Strack, B.; Calistri, A.; Craig, S.; Popova, E.; Gottlinger, H.G. AIP1/ALIX is a binding partner for HIV-1 p6 and EIAV p9 functioning in virus budding. Cell 2003, 114, 689–699. [Google Scholar] [CrossRef]
- von Schwedler, U.K.; Stuchell, M.; Muller, B.; Ward, D.M.; Chung, H.Y.; Morita, E.; Wang, H.E.; Davis, T.; He, G.P.; Cimbora, D.M.; et al. The protein network of HIV budding. Cell 2003, 114, 701–713. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Sitaraman, S.; Hierro, A.; Beach, B.M.; Odorizzi, G.; Hurley, J.H. Structural basis for endosomal targeting by the Bro1 domain. Dev. Cell 2005, 8, 937–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, N.; Zhang, L.; Taylor, S.; Mironov, A.; Urbe, S.; Woodman, P. Recruitment of UBPY and ESCRT exchange drive HD-PTP-dependent sorting of EGFR to the MVB. Curr. Biol. 2013, 23, 453–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parkinson, M.D.; Piper, S.C.; Bright, N.A.; Evans, J.L.; Boname, J.M.; Bowers, K.; Lehner, P.J.; Luzio, J.P. A non-canonical ESCRT pathway, including histidine domain phosphotyrosine phosphatase (HD-PTP), is used for down-regulation of virally ubiquitinated MHC class I. Biochem. J. 2015, 471, 79–88. [Google Scholar] [CrossRef] [Green Version]
- Zhai, Q.; Landesman, M.B.; Robinson, H.; Sundquist, W.I.; Hill, C.P. Structure of the Bro1 domain protein BROX and functional analyses of the ALIX Bro1 domain in HIV-1 budding. PLoS ONE 2011, 6, e27466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monroe, N.; Hill, C.P. Meiotic Clade AAA ATPases: Protein Polymer Disassembly Machines. J. Mol. Biol. 2016, 428, 1897–1911. [Google Scholar] [CrossRef] [Green Version]
- Hurley, J.H.; Yang, D. MIT domainia. Dev. Cell 2008, 14, 6–8. [Google Scholar] [CrossRef] [Green Version]
- Monroe, N.; Han, H.; Gonciarz, M.D.; Eckert, D.M.; Karren, M.A.; Whitby, F.G.; Sundquist, W.I.; Hill, C.P. The oligomeric state of the active Vps4 AAA ATPase. J. Mol. Biol. 2014, 426, 510–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obita, T.; Saksena, S.; Ghazi-Tabatabai, S.; Gill, D.J.; Perisic, O.; Emr, S.D.; Williams, R.L. Structural basis for selective recognition of ESCRT-III by the AAA ATPase Vps4. Nature 2007, 449, 735–739. [Google Scholar] [CrossRef]
- Stuchell-Brereton, M.D.; Skalicky, J.J.; Kieffer, C.; Karren, M.A.; Ghaffarian, S.; Sundquist, W.I. ESCRT-III recognition by VPS4 ATPases. Nature 2007, 449, 740–744. [Google Scholar] [CrossRef] [PubMed]
- Christ, L.; Raiborg, C.; Wenzel, E.M.; Campsteijn, C.; Stenmark, H. Cellular Functions and Molecular Mechanisms of the ESCRT Membrane-Scission Machinery. Trends Biochem. Sci. 2017, 42, 42–56. [Google Scholar] [CrossRef]
- Samson, R.Y.; Dobro, M.J.; Jensen, G.J.; Bell, S.D. The Structure, Function and Roles of the Archaeal ESCRT Apparatus. Subcell Biochem. 2017, 84, 357–377. [Google Scholar] [CrossRef]
- Ghazi-Tabatabai, S.; Obita, T.; Pobbati, A.V.; Perisic, O.; Samson, R.Y.; Bell, S.D.; Williams, R.L. Evolution and assembly of ESCRTs. Biochem Soc. Trans. 2009, 37, 151–155. [Google Scholar] [CrossRef]
- Samson, R.Y.; Bell, S.D. Ancient ESCRTs and the evolution of binary fission. Trends Microbiol. 2009, 17, 507–513. [Google Scholar] [CrossRef]
- Samson, R.Y.; Obita, T.; Hodgson, B.; Shaw, M.K.; Chong, P.L.; Williams, R.L.; Bell, S.D. Molecular and structural basis of ESCRT-III recruitment to membranes during archaeal cell division. Mol. Cell 2011, 41, 186–196. [Google Scholar] [CrossRef] [Green Version]
- Samson, R.Y.; Obita, T.; Freund, S.M.; Williams, R.L.; Bell, S.D. A role for the ESCRT system in cell division in archaea. Science 2008, 322, 1710–1713. [Google Scholar] [CrossRef] [Green Version]
- Bissig, C.; Gruenberg, J. ALIX and the multivesicular endosome: ALIX in Wonderland. Trends Cell Biol. 2014, 24, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Tabernero, L.; Woodman, P. Dissecting the role of His domain protein tyrosine phosphatase/PTPN23 and ESCRTs in sorting activated epidermal growth factor receptor to the multivesicular body. Biochem. Soc. Trans. 2018, 46, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Henne, W.M.; Borbat, P.P.; Buchkovich, N.J.; Freed, J.H.; Mao, Y.; Fromme, J.C.; Emr, S.D. Structural basis for activation, assembly and membrane binding of ESCRT-III Snf7 filaments. Elife 2015, 4. [Google Scholar] [CrossRef]
- Tang, S.; Buchkovich, N.J.; Henne, W.M.; Banjade, S.; Kim, Y.J.; Emr, S.D. ESCRT-III activation by parallel action of ESCRT-I/II and ESCRT-0/Bro1 during MVB biogenesis. Elife 2016, 5. [Google Scholar] [CrossRef] [Green Version]
- Banjade, S.; Tang, S.; Shah, Y.H.; Emr, S.D. Electrostatic lateral interactions drive ESCRT-III heteropolymer assembly. Elife 2019, 8. [Google Scholar] [CrossRef]
- Schoneberg, J.; Pavlin, M.R.; Yan, S.; Righini, M.; Lee, I.H.; Carlson, L.A.; Bahrami, A.H.; Goldman, D.H.; Ren, X.; Hummer, G.; et al. ATP-dependent force generation and membrane scission by ESCRT-III and Vps4. Science 2018, 362, 1423–1428. [Google Scholar] [CrossRef] [Green Version]
- Peng, X.; Yang, L.; Ma, Y.; Li, Y.; Li, H. Focus on the morphogenesis, fate and the role in tumor progression of multivesicular bodies. Cell Commun. Signal. 2020, 18, 122. [Google Scholar] [CrossRef]
- Eitan, E.; Suire, C.; Zhang, S.; Mattson, M.P. Impact of lysosome status on extracellular vesicle content and release. Ageing Res. Rev. 2016, 32, 65–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latifkar, A.; Ling, L.; Hingorani, A.; Johansen, E.; Clement, A.; Zhang, X.; Hartman, J.; Fischbach, C.; Lin, H.; Cerione, R.A.; et al. Loss of Sirtuin 1 Alters the Secretome of Breast Cancer Cells by Impairing Lysosomal Integrity. Dev. Cell 2019, 49, 393–408.e397. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Kovalenko, A.; Bogdanov, K.; Wallach, D. MLKL, the Protein that Mediates Necroptosis, Also Regulates Endosomal Trafficking and Extracellular Vesicle Generation. Immunity 2017, 47, 51–65.e57. [Google Scholar] [CrossRef] [Green Version]
- Jeppesen, D.K.; Fenix, A.M.; Franklin, J.L.; Higginbotham, J.N.; Zhang, Q.; Zimmerman, L.J.; Liebler, D.C.; Ping, J.; Liu, Q.; Evans, R.; et al. Reassessment of Exosome Composition. Cell 2019, 177, 428–445.e418. [Google Scholar] [CrossRef] [Green Version]
- Raiborg, C.; Rusten, T.E.; Stenmark, H. Protein sorting into multivesicular endosomes. Curr. Opin. Cell Biol. 2003, 15, 446–455. [Google Scholar] [CrossRef]
- Raiborg, C.; Stenmark, H. The ESCRT machinery in endosomal sorting of ubiquitylated membrane proteins. Nature 2009, 458, 445–452. [Google Scholar] [CrossRef]
- Calistri, A.; Munegato, D.; Carli, I.; Parolin, C.; Palu, G. The ubiquitin-conjugating system: Multiple roles in viral replication and infection. Cells 2014, 3, 386–417. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, D.; Riezman, H. Proteasome-independent functions of ubiquitin in endocytosis and signaling. Science 2007, 315, 201–205. [Google Scholar] [CrossRef] [Green Version]
- Piper, R.C.; Luzio, J.P. Ubiquitin-dependent sorting of integral membrane proteins for degradation in lysosomes. Curr. Opin. Cell Biol. 2007, 19, 459–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bache, K.G.; Brech, A.; Mehlum, A.; Stenmark, H. HRS regulates multivesicular body formation via ESCRT recruitment to endosomes. J. Cell Biol. 2003, 162, 435–442. [Google Scholar] [CrossRef] [Green Version]
- Raiborg, C.; Bache, K.G.; Gillooly, D.J.; Madshus, I.H.; Stang, E.; Stenmark, H. HRS sorts ubiquitinated proteins into clathrin-coated microdomains of early endosomes. Nat. Cell Biol. 2002, 4, 394–398. [Google Scholar] [CrossRef]
- Gillooly, D.J.; Morrow, I.C.; Lindsay, M.; Gould, R.; Bryant, N.J.; Gaullier, J.M.; Parton, R.G.; Stenmark, H. Localization of phosphatidylinositol 3-phosphate in yeast and mammalian cells. EMBO J. 2000, 19, 4577–4588. [Google Scholar] [CrossRef]
- Mayers, J.R.; Fyfe, I.; Schuh, A.L.; Chapman, E.R.; Edwardson, J.M.; Audhya, A. ESCRT-0 assembles as a heterotetrameric complex on membranes and binds multiple ubiquitinylated cargoes simultaneously. J. Biol. Chem. 2011, 286, 9636–9645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henne, W.M.; Buchkovich, N.J.; Zhao, Y.; Emr, S.D. The endosomal sorting complex ESCRT-II mediates the assembly and architecture of ESCRT-III helices. Cell 2012, 151, 356–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mierzwa, B.E.; Chiaruttini, N.; Redondo-Morata, L.; von Filseck, J.M.; Konig, J.; Larios, J.; Poser, I.; Muller-Reichert, T.; Scheuring, S.; Roux, A.; et al. Dynamic subunit turnover in ESCRT-III assemblies is regulated by Vps4 to mediate membrane remodelling during cytokinesis. Nat. Cell Biol. 2017, 19, 787–798. [Google Scholar] [CrossRef]
- Adell, M.A.; Vogel, G.F.; Pakdel, M.; Muller, M.; Lindner, H.; Hess, M.W.; Teis, D. Coordinated binding of Vps4 to ESCRT-III drives membrane neck constriction during MVB vesicle formation. J. Cell Biol. 2014, 205, 33–49. [Google Scholar] [CrossRef] [PubMed]
- Clague, M.J.; Liu, H.; Urbe, S. Governance of endocytic trafficking and signaling by reversible ubiquitylation. Dev. Cell 2012, 23, 457–467. [Google Scholar] [CrossRef] [Green Version]
- Dores, M.R.; Paing, M.M.; Lin, H.; Montagne, W.A.; Marchese, A.; Trejo, J. AP-3 regulates PAR1 ubiquitin-independent MVB/lysosomal sorting via an ALIX-mediated pathway. Mol. Biol. Cell 2012, 23, 3612–3623. [Google Scholar] [CrossRef]
- Dores, M.R.; Lin, H.; Neil, J.G.; Mendez, F.; Trejo, J. The alpha-arrestin ARRDC3 mediates ALIX ubiquitination and G protein-coupled receptor lysosomal sorting. Mol. Biol. Cell 2015, 26, 4660–4673. [Google Scholar] [CrossRef] [PubMed]
- Dores, M.R.; Grimsey, N.J.; Mendez, F.; Trejo, J. ALIX Regulates the Ubiquitin-Independent Lysosomal Sorting of the P2Y1 Purinergic Receptor via a YPX3L Motif. PLoS ONE 2016, 11, e0157587. [Google Scholar] [CrossRef]
- Baietti, M.F.; Zhang, Z.; Mortier, E.; Melchior, A.; Degeest, G.; Geeraerts, A.; Ivarsson, Y.; Depoortere, F.; Coomans, C.; Vermeiren, E.; et al. Syndecan-syntenin-ALIX regulates the biogenesis of exosomes. Nat. Cell Biol. 2012, 14, 677–685. [Google Scholar] [CrossRef]
- Altan-Bonnet, N. Extracellular vesicles are the Trojan horses of viral infection. Curr. Opin. Microbiol. 2016, 32, 77–81. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, Y.; He, H.; Tang, Z.; Hattori, T.; Liu, Y.; Young, M.M.; Serfass, J.M.; Chen, L.; Gebru, M.; Chen, C.; et al. An autophagy assay reveals the ESCRT-III component CHMP2A as a regulator of phagophore closure. Nat. Commun. 2018, 9, 2855. [Google Scholar] [CrossRef] [Green Version]
- Zhen, Y.; Spangenberg, H.; Munson, M.J.; Brech, A.; Schink, K.O.; Tan, K.W.; Sorensen, V.; Wenzel, E.M.; Radulovic, M.; Engedal, N.; et al. ESCRT-mediated phagophore sealing during mitophagy. Autophagy 2020, 16, 826–841. [Google Scholar] [CrossRef] [Green Version]
- Sahu, R.; Kaushik, S.; Clement, C.C.; Cannizzo, E.S.; Scharf, B.; Follenzi, A.; Potolicchio, I.; Nieves, E.; Cuervo, A.M.; Santambrogio, L. Microautophagy of cytosolic proteins by late endosomes. Dev. Cell 2011, 20, 131–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filimonenko, M.; Stuffers, S.; Raiborg, C.; Yamamoto, A.; Malerod, L.; Fisher, E.M.; Isaacs, A.; Brech, A.; Stenmark, H.; Simonsen, A. Functional multivesicular bodies are required for autophagic clearance of protein aggregates associated with neurodegenerative disease. J. Cell Biol. 2007, 179, 485–500. [Google Scholar] [CrossRef]
- Rusten, T.E.; Simonsen, A. ESCRT functions in autophagy and associated disease. Cell Cycle 2008, 7, 1166–1172. [Google Scholar] [CrossRef] [Green Version]
- Carlton, J.G.; Martin-Serrano, J. Parallels between cytokinesis and retroviral budding: A role for the ESCRT machinery. Science 2007, 316, 1908–1912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlton, J.G.; Agromayor, M.; Martin-Serrano, J. Differential requirements for Alix and ESCRT-III in cytokinesis and HIV-1 release. Proc. Natl. Acad. Sci. USA 2008, 105, 10541–10546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlton, J.G.; Caballe, A.; Agromayor, M.; Kloc, M.; Martin-Serrano, J. ESCRT-III governs the Aurora B-mediated abscission checkpoint through CHMP4C. Science 2012, 336, 220–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morita, E.; Sandrin, V.; Chung, H.Y.; Morham, S.G.; Gygi, S.P.; Rodesch, C.K.; Sundquist, W.I. Human ESCRT and ALIX proteins interact with proteins of the midbody and function in cytokinesis. EMBO J. 2007, 26, 4215–4227. [Google Scholar] [CrossRef] [Green Version]
- Guizetti, J.; Schermelleh, L.; Mantler, J.; Maar, S.; Poser, I.; Leonhardt, H.; Muller-Reichert, T.; Gerlich, D.W. Cortical constriction during abscission involves helices of ESCRT-III-dependent filaments. Science 2011, 331, 1616–1620. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.H.; Elia, N.; Ghirlando, R.; Lippincott-Schwartz, J.; Hurley, J.H. Midbody targeting of the ESCRT machinery by a noncanonical coiled coil in CEP55. Science 2008, 322, 576–580. [Google Scholar] [CrossRef] [Green Version]
- Mierzwa, B.; Gerlich, D.W. Cytokinetic abscission: Molecular mechanisms and temporal control. Dev. Cell 2014, 31, 525–538. [Google Scholar] [CrossRef] [Green Version]
- Karasmanis, E.P.; Hwang, D.; Nakos, K.; Bowen, J.R.; Angelis, D.; Spiliotis, E.T. A Septin Double Ring Controls the Spatiotemporal Organization of the ESCRT Machinery in Cytokinetic Abscission. Curr. Biol. 2019, 29, 2174–2182.e2177. [Google Scholar] [CrossRef] [PubMed]
- Goliand, I.; Nachmias, D.; Gershony, O.; Elia, N. Inhibition of ESCRT-II-CHMP6 interactions impedes cytokinetic abscission and leads to cell death. Mol. Biol. Cell 2014, 25, 3740–3748. [Google Scholar] [CrossRef] [Green Version]
- Christ, L.; Wenzel, E.M.; Liestol, K.; Raiborg, C.; Campsteijn, C.; Stenmark, H. ALIX and ESCRT-I/II function as parallel ESCRT-III recruiters in cytokinetic abscission. J. Cell Biol. 2016, 212, 499–513. [Google Scholar] [CrossRef] [PubMed]
- Hadders, M.A.; Agromayor, M.; Obita, T.; Perisic, O.; Caballe, A.; Kloc, M.; Lamers, M.H.; Williams, R.L.; Martin-Serrano, J. ESCRT-III binding protein MITD1 is involved in cytokinesis and has an unanticipated PLD fold that binds membranes. Proc. Natl. Acad. Sci. USA 2012, 109, 17424–17429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Chang, J.; Renvoise, B.; Tipirneni, A.; Yang, S.; Blackstone, C. MITD1 is recruited to midbodies by ESCRT-III and participates in cytokinesis. Mol. Biol. Cell. 2012, 23, 4347–4361. [Google Scholar] [CrossRef] [PubMed]
- Goliand, I.; Adar-Levor, S.; Segal, I.; Nachmias, D.; Dadosh, T.; Kozlov, M.M.; Elia, N. Resolving ESCRT-III Spirals at the Intercellular Bridge of Dividing Cells Using 3D STORM. Cell Rep. 2018, 24, 1756–1764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elia, N.; Sougrat, R.; Spurlin, T.A.; Hurley, J.H.; Lippincott-Schwartz, J. Dynamics of endosomal sorting complex required for transport (ESCRT) machinery during cytokinesis and its role in abscission. Proc. Natl. Acad. Sci. USA 2011, 108, 4846–4851. [Google Scholar] [CrossRef] [Green Version]
- Reid, E.; Connell, J.; Edwards, T.L.; Duley, S.; Brown, S.E.; Sanderson, C.M. The hereditary spastic paraplegia protein spastin interacts with the ESCRT-III complex-associated endosomal protein CHMP1B. Hum. Mol. Genet. 2005, 14, 19–38. [Google Scholar] [CrossRef]
- Yang, D.; Rismanchi, N.; Renvoise, B.; Lippincott-Schwartz, J.; Blackstone, C.; Hurley, J.H. Structural basis for midbody targeting of spastin by the ESCRT-III protein CHMP1B. Nat. Struct. Mol. Biol. 2008, 15, 1278–1286. [Google Scholar] [CrossRef] [PubMed]
- Lindas, A.C.; Karlsson, E.A.; Lindgren, M.T.; Ettema, T.J.; Bernander, R. A unique cell division machinery in the Archaea. Proc. Natl. Acad. Sci. USA 2008, 105, 18942–18946. [Google Scholar] [CrossRef] [Green Version]
- Thoresen, S.B.; Campsteijn, C.; Vietri, M.; Schink, K.O.; Liestol, K.; Andersen, J.S.; Raiborg, C.; Stenmark, H. ANCHR mediates Aurora-B-dependent abscission checkpoint control through retention of VPS4. Nat. Cell Biol. 2014, 16, 550–560. [Google Scholar] [CrossRef] [PubMed]
- Caballe, A.; Wenzel, D.M.; Agromayor, M.; Alam, S.L.; Skalicky, J.J.; Kloc, M.; Carlton, J.G.; Labrador, L.; Sundquist, W.I.; Martin-Serrano, J. ULK3 regulates cytokinetic abscission by phosphorylating ESCRT-III proteins. Elife 2015, 4, e06547. [Google Scholar] [CrossRef] [Green Version]
- Sadler, J.B.A.; Wenzel, D.M.; Williams, L.K.; Guindo-Martinez, M.; Alam, S.L.; Mercader, J.M.; Torrents, D.; Ullman, K.S.; Sundquist, W.I.; Martin-Serrano, J. A cancer-associated polymorphism in ESCRT-III disrupts the abscission checkpoint and promotes genome instability. Proc. Natl. Acad. Sci. USA 2018, 115, E8900–E8908. [Google Scholar] [CrossRef] [Green Version]
- Lens, S.M.A.; Medema, R.H. Cytokinesis defects and cancer. Nat. Rev. Cancer 2019, 19, 32–45. [Google Scholar] [CrossRef] [PubMed]
- Tandon, D.; Banerjee, M. Centrosomal protein 55: A new paradigm in tumorigenesis. Eur. J. Cell Biol. 2020, 99, 151086. [Google Scholar] [CrossRef] [PubMed]
- Hatch, E.M.; Fischer, A.H.; Deerinck, T.J.; Hetzer, M.W. Catastrophic nuclear envelope collapse in cancer cell micronuclei. Cell 2013, 154, 47–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vietri, M.; Schultz, S.W.; Bellanger, A.; Jones, C.M.; Petersen, L.I.; Raiborg, C.; Skarpen, E.; Pedurupillay, C.R.J.; Kjos, I.; Kip, E.; et al. Unrestrained ESCRT-III drives micronuclear catastrophe and chromosome fragmentation. Nat. Cell Biol. 2020, 22, 856–867. [Google Scholar] [CrossRef]
- Tanaka, N.; Kyuuma, M.; Sugamura, K. Endosomal sorting complex required for transport proteins in cancer pathogenesis, vesicular transport, and non-endosomal functions. Cancer Sci. 2008, 99, 1293–1303. [Google Scholar] [CrossRef]
- Mattissek, C.; Teis, D. The role of the endosomal sorting complexes required for transport (ESCRT) in tumorigenesis. Mol. Membr. Biol. 2014, 31, 111–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alfred, V.; Vaccari, T. When membranes need an ESCRT: Endosomal sorting and membrane remodelling in health and disease. Swiss Med. Wkly. 2016, 146, w14347. [Google Scholar] [CrossRef]
- Andrews, N.W.; Almeida, P.E.; Corrotte, M. Damage control: Cellular mechanisms of plasma membrane repair. Trends Cell Biol. 2014, 24, 734–742. [Google Scholar] [CrossRef] [Green Version]
- Jimenez, A.J.; Maiuri, P.; Lafaurie-Janvore, J.; Divoux, S.; Piel, M.; Perez, F. ESCRT machinery is required for plasma membrane repair. Science 2014, 343, 1247136. [Google Scholar] [CrossRef] [PubMed]
- Grassel, L.; Fast, L.A.; Scheffer, K.D.; Boukhallouk, F.; Spoden, G.A.; Tenzer, S.; Boller, K.; Bago, R.; Rajesh, S.; Overduin, M.; et al. The CD63-Syntenin-1 Complex Controls Post-Endocytic Trafficking of Oncogenic Human Papillomaviruses. Sci. Rep. 2016, 6, 32337. [Google Scholar] [CrossRef] [Green Version]
- Scheffer, L.L.; Sreetama, S.C.; Sharma, N.; Medikayala, S.; Brown, K.J.; Defour, A.; Jaiswal, J.K. Mechanism of Ca(2)(+)-triggered ESCRT assembly and regulation of cell membrane repair. Nat. Commun. 2014, 5, 5646. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.; Zhou, X.; Corvera, J.; Gallick, G.E.; Lin, S.H.; Kuang, J. ALG-2 activates the MVB sorting function of ALIX through relieving its intramolecular interaction. Cell Discov. 2015, 1, 15018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruhl, S.; Shkarina, K.; Demarco, B.; Heilig, R.; Santos, J.C.; Broz, P. ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science 2018, 362, 956–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, Y.N.; Guy, C.; Olauson, H.; Becker, J.U.; Yang, M.; Fitzgerald, P.; Linkermann, A.; Green, D.R. ESCRT-III Acts Downstream of MLKL to Regulate Necroptotic Cell Death and Its Consequences. Cell 2017, 169, 286–300.e216. [Google Scholar] [CrossRef] [Green Version]
- Robijns, J.; Houthaeve, G.; Braeckmans, K.; De Vos, W.H. Loss of Nuclear Envelope Integrity in Aging and Disease. Int Rev. Cell Mol. Biol. 2018, 336, 205–222. [Google Scholar] [CrossRef]
- Guttinger, S.; Laurell, E.; Kutay, U. Orchestrating nuclear envelope disassembly and reassembly during mitosis. Nat. Rev. Mol. Cell Biol. 2009, 10, 178–191. [Google Scholar] [CrossRef]
- Vietri, M.; Schink, K.O.; Campsteijn, C.; Wegner, C.S.; Schultz, S.W.; Christ, L.; Thoresen, S.B.; Brech, A.; Raiborg, C.; Stenmark, H. Spastin and ESCRT-III coordinate mitotic spindle disassembly and nuclear envelope sealing. Nature 2015, 522, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Olmos, Y.; Hodgson, L.; Mantell, J.; Verkade, P.; Carlton, J.G. ESCRT-III controls nuclear envelope reformation. Nature 2015, 522, 236–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ventimiglia, L.N.; Cuesta-Geijo, M.A.; Martinelli, N.; Caballe, A.; Macheboeuf, P.; Miguet, N.; Parnham, I.M.; Olmos, Y.; Carlton, J.G.; Weissenhorn, W.; et al. CC2D1B Coordinates ESCRT-III Activity during the Mitotic Reformation of the Nuclear Envelope. Dev. Cell 2018, 47, 547–563.e546. [Google Scholar] [CrossRef] [Green Version]
- Martinelli, N.; Hartlieb, B.; Usami, Y.; Sabin, C.; Dordor, A.; Miguet, N.; Avilov, S.V.; Ribeiro, E.A., Jr.; Gottlinger, H.; Weissenhorn, W. CC2D1A is a regulator of ESCRT-III CHMP4B. J. Mol. Biol. 2012, 419, 75–88. [Google Scholar] [CrossRef] [Green Version]
- Pieper, G.H.; Sprenger, S.; Teis, D.; Oliferenko, S. ESCRT-III/Vps4 Controls Heterochromatin-Nuclear Envelope Attachments. Dev. Cell 2020, 53, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Maciejowski, J.; Hatch, E.M. Nuclear Membrane Rupture and Its Consequences. Annu. Rev. Cell Dev. Biol. 2020, 36, 85–114. [Google Scholar] [CrossRef] [PubMed]
- Denais, C.M.; Gilbert, R.M.; Isermann, P.; McGregor, A.L.; te Lindert, M.; Weigelin, B.; Davidson, P.M.; Friedl, P.; Wolf, K.; Lammerding, J. Nuclear envelope rupture and repair during cancer cell migration. Science 2016, 352, 353–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raab, M.; Gentili, M.; de Belly, H.; Thiam, H.R.; Vargas, P.; Jimenez, A.J.; Lautenschlaeger, F.; Voituriez, R.; Lennon-Dumenil, A.M.; Manel, N.; et al. ESCRT III repairs nuclear envelope ruptures during cell migration to limit DNA damage and cell death. Science 2016, 352, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Halfmann, C.T.; Sears, R.M.; Katiyar, A.; Busselman, B.W.; Aman, L.K.; Zhang, Q.; O’Bryan, C.S.; Angelini, T.E.; Lele, T.P.; Roux, K.J. Repair of nuclear ruptures requires barrier-to-autointegration factor. J. Cell Biol. 2019, 218, 2136–2149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webster, B.M.; Colombi, P.; Jager, J.; Lusk, C.P. Surveillance of nuclear pore complex assembly by ESCRT-III/Vps4. Cell 2014, 159, 388–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skowyra, M.L.; Schlesinger, P.H.; Naismith, T.V.; Hanson, P.I. Triggered recruitment of ESCRT machinery promotes endolysosomal repair. Science 2018, 360. [Google Scholar] [CrossRef] [Green Version]
- Radulovic, M.; Schink, K.O.; Wenzel, E.M.; Nahse, V.; Bongiovanni, A.; Lafont, F.; Stenmark, H. ESCRT-mediated lysosome repair precedes lysophagy and promotes cell survival. EMBO J. 2018, 37. [Google Scholar] [CrossRef] [PubMed]
- Radulovic, M.; Stenmark, H. ESCRTs in membrane sealing. Biochem. Soc. Trans. 2018, 46, 773–778. [Google Scholar] [CrossRef] [Green Version]
- Mansilla Pareja, M.E.; Bongiovanni, A.; Lafont, F.; Colombo, M.I. Alterations of the Coxiella burnetii Replicative Vacuole Membrane Integrity and Interplay with the Autophagy Pathway. Front. Cell Infect. Microbiol. 2017, 7, 112. [Google Scholar] [CrossRef] [Green Version]
- Mittal, E.; Skowyra, M.L.; Uwase, G.; Tinaztepe, E.; Mehra, A.; Koster, S.; Hanson, P.I.; Philips, J.A. Mycobacterium tuberculosis Type VII Secretion System Effectors Differentially Impact the ESCRT Endomembrane Damage Response. mBio 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, K.U.; Krempler, A.; Qi, Y.; Park, K.; Henry, M.D.; Triplett, A.A.; Riedlinger, G.; Rucker, I.E.; Hennighausen, L. Tsg101 is essential for cell growth, proliferation, and cell survival of embryonic and adult tissues. Mol. Cell Biol. 2003, 23, 150–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Handschuh, K.; Feenstra, J.; Koss, M.; Ferretti, E.; Risolino, M.; Zewdu, R.; Sahai, M.A.; Benazet, J.D.; Peng, X.P.; Depew, M.J.; et al. ESCRT-II/Vps25 constrains digit number by endosome-mediated selective modulation of FGF-SHH signaling. Cell Rep. 2014, 9, 674–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shim, J.H.; Xiao, C.; Hayden, M.S.; Lee, K.Y.; Trombetta, E.S.; Pypaert, M.; Nara, A.; Yoshimori, T.; Wilm, B.; Erdjument-Bromage, H.; et al. CHMP5 is essential for late endosome function and down-regulation of receptor signaling during mouse embryogenesis. J. Cell Biol. 2006, 172, 1045–1056. [Google Scholar] [CrossRef]
- Thompson, B.J.; Mathieu, J.; Sung, H.H.; Loeser, E.; Rorth, P.; Cohen, S.M. Tumor suppressor properties of the ESCRT-II complex component Vps25 in Drosophila. Dev. Cell 2005, 9, 711–720. [Google Scholar] [CrossRef] [PubMed]
- Vaccari, T.; Rusten, T.E.; Menut, L.; Nezis, I.P.; Brech, A.; Stenmark, H.; Bilder, D. Comparative analysis of ESCRT-I, ESCRT-II and ESCRT-III function in Drosophila by efficient isolation of ESCRT mutants. J. Cell Sci. 2009, 122, 2413–2423. [Google Scholar] [CrossRef] [Green Version]
- Spitzer, C.; Schellmann, S.; Sabovljevic, A.; Shahriari, M.; Keshavaiah, C.; Bechtold, N.; Herzog, M.; Muller, S.; Hanisch, F.G.; Hulskamp, M. The Arabidopsis elch mutant reveals functions of an ESCRT component in cytokinesis. Development 2006, 133, 4679–4689. [Google Scholar] [CrossRef] [Green Version]
- Razi, M.; Futter, C.E. Distinct roles for Tsg101 and HRS in multivesicular body formation and inward vesiculation. Mol. Biol. Cell 2006, 17, 3469–3483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bache, K.G.; Stuffers, S.; Malerod, L.; Slagsvold, T.; Raiborg, C.; Lechardeur, D.; Walchli, S.; Lukacs, G.L.; Brech, A.; Stenmark, H. The ESCRT-III subunit hVps24 is required for degradation but not silencing of the epidermal growth factor receptor. Mol. Biol. Cell 2006, 17, 2513–2523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stuffers, S.; Sem Wegner, C.; Stenmark, H.; Brech, A. Multivesicular endosome biogenesis in the absence of ESCRTs. Traffic 2009, 10, 925–937. [Google Scholar] [CrossRef] [PubMed]
- Capalbo, L.; Montembault, E.; Takeda, T.; Bassi, Z.I.; Glover, D.M.; D’Avino, P.P. The chromosomal passenger complex controls the function of endosomal sorting complex required for transport-III Snf7 proteins during cytokinesis. Open Biol. 2012, 2, 120070. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.; Cai, W. Autophagy and Viral Infection. Adv. Exp. Med. Biol. 2020, 1207, 425–432. [Google Scholar] [CrossRef]
- Purvinsh, L.; Gorshkov, A.; Brodskaia, A.; Vasin, A. Extracellular Vesicles in Viral Pathogenesis: A Case of Dr. Jekyll and Mr. Hyde. Life 2021, 11, 45. [Google Scholar] [CrossRef]
- Raymond, A.D.; Diaz, P.; Chevelon, S.; Agudelo, M.; Yndart-Arias, A.; Ding, H.; Kaushik, A.; Jayant, R.D.; Nikkhah-Moshaie, R.; Roy, U.; et al. Microglia-derived HIV Nef+ exosome impairment of the blood-brain barrier is treatable by nanomedicine-based delivery of Nef peptides. J. Neurovirol. 2016, 22, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Arenaccio, C.; Anticoli, S.; Manfredi, F.; Chiozzini, C.; Olivetta, E.; Federico, M. Latent HIV-1 is activated by exosomes from cells infected with either replication-competent or defective HIV-1. Retrovirology 2015, 12, 87. [Google Scholar] [CrossRef] [Green Version]
- Arenaccio, C.; Chiozzini, C.; Columba-Cabezas, S.; Manfredi, F.; Affabris, E.; Baur, A.; Federico, M. Exosomes from human immunodeficiency virus type 1 (HIV-1)-infected cells license quiescent CD4+ T lymphocytes to replicate HIV-1 through a Nef- and ADAM17-dependent mechanism. J. Virol. 2014, 88, 11529–11539. [Google Scholar] [CrossRef] [Green Version]
- Lenassi, M.; Cagney, G.; Liao, M.; Vaupotic, T.; Bartholomeeusen, K.; Cheng, Y.; Krogan, N.J.; Plemenitas, A.; Peterlin, B.M. HIV Nef is secreted in exosomes and triggers apoptosis in bystander CD4+ T cells. Traffic 2010, 11, 110–122. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Santini, P.A.; Sullivan, J.S.; He, B.; Shan, M.; Ball, S.C.; Dyer, W.B.; Ketas, T.J.; Chadburn, A.; Cohen-Gould, L.; et al. HIV-1 evades virus-specific IgG2 and IgA responses by targeting systemic and intestinal B cells via long-range intercellular conduits. Nat. Immunol. 2009, 10, 1008–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mack, M.; Kleinschmidt, A.; Bruhl, H.; Klier, C.; Nelson, P.J.; Cihak, J.; Plachy, J.; Stangassinger, M.; Erfle, V.; Schlondorff, D. Transfer of the chemokine receptor CCR5 between cells by membrane-derived microparticles: A mechanism for cellular human immunodeficiency virus 1 infection. Nat. Med. 2000, 6, 769–775. [Google Scholar] [CrossRef] [PubMed]
- Rozmyslowicz, T.; Majka, M.; Kijowski, J.; Murphy, S.L.; Conover, D.O.; Poncz, M.; Ratajczak, J.; Gaulton, G.N.; Ratajczak, M.Z. Platelet- and megakaryocyte-derived microparticles transfer CXCR4 receptor to CXCR4-null cells and make them susceptible to infection by X4-HIV. AIDS 2003, 17, 33–42. [Google Scholar] [CrossRef]
- Narayanan, A.; Iordanskiy, S.; Das, R.; Van Duyne, R.; Santos, S.; Jaworski, E.; Guendel, I.; Sampey, G.; Dalby, E.; Iglesias-Ussel, M.; et al. Exosomes derived from HIV-1-infected cells contain trans-activation response element RNA. J. Biol. Chem. 2013, 288, 20014–20033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khatua, A.K.; Taylor, H.E.; Hildreth, J.E.; Popik, W. Exosomes packaging APOBEC3G confer human immunodeficiency virus resistance to recipient cells. J. Virol. 2009, 83, 512–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bridgeman, A.; Maelfait, J.; Davenne, T.; Partridge, T.; Peng, Y.; Mayer, A.; Dong, T.; Kaever, V.; Borrow, P.; Rehwinkel, J. Viruses transfer the antiviral second messenger cGAMP between cells. Science 2015, 349, 1228–1232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dias, M.V.S.; Costa, C.S.; da Silva, L.L.P. The Ambiguous Roles of Extracellular Vesicles in HIV Replication and Pathogenesis. Front. Microbiol. 2018, 9, 2411. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Li, S.; Wu, S.; Chen, L. Exosomes Modulate the Viral Replication and Host Immune Responses in HBV Infection. Biomed. Res. Int. 2019, 2019, 2103943. [Google Scholar] [CrossRef] [Green Version]
- Jaworski, E.; Narayanan, A.; Van Duyne, R.; Shabbeer-Meyering, S.; Iordanskiy, S.; Saifuddin, M.; Das, R.; Afonso, P.V.; Sampey, G.C.; Chung, M.; et al. Human T-lymphotropic virus type 1-infected cells secrete exosomes that contain Tax protein. J. Biol. Chem. 2014, 289, 22284–22305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miura, T.A. Respiratory epithelial cells as master communicators during viral infections. Curr. Clin. Microbiol. Rep. 2019, 6, 10–17. [Google Scholar] [CrossRef]
- Hassanpour, M.; Rezaie, J.; Nouri, M.; Panahi, Y. The role of extracellular vesicles in COVID-19 virus infection. Infect. Genet. Evol 2020, 85, 104422. [Google Scholar] [CrossRef]
- Wang, J.; Li, J.; Cheng, C.; Liu, S. Angiotensin-converting enzyme 2 augments the effects of endothelial progenitor cells-exosomes on vascular smooth muscle cell phenotype transition. Cell Tissue Res. 2020, 382, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Cocozza, F.; Nevo, N.; Piovesana, E.; Lahaye, X.; Buchrieser, J.; Schwartz, O.; Manel, N.; Tkach, M.; Thery, C.; Martin-Jaular, L. Extracellular vesicles containing ACE2 efficiently prevent infection by SARS-CoV-2 Spike protein-containing virus. J. Extracell Vesicles 2020, 10, e12050. [Google Scholar] [CrossRef]
- Ipinmoroti, A.O.; Matthews, Q.L. Extracellular Vesicles: Roles in Human Viral Infections, Immune-Diagnostic, and Therapeutic Applications. Pathogens 2020, 9, 1056. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.C.; Li, C.Y.; Chang, W.T.; Cheng, W.C.; Yen, C.H.; Tu, W.Y.; Lin, Z.Y.; Lin, C.C.; Yeh, M.L.; Huang, C.F.; et al. Exosome-derived differentiation antagonizing non-protein coding RNA with risk of hepatitis C virus-related hepatocellular carcinoma recurrence. Liver Int. 2020. [Google Scholar] [CrossRef]
- Zhang, J.; Li, X.; Hu, J.; Cao, P.; Yan, Q.; Zhang, S.; Dang, W.; Lu, J. Long noncoding RNAs involvement in Epstein-Barr virus infection and tumorigenesis. Virol. J. 2020, 17, 51. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Li, H.; Sun, H.; Fan, H.; Hu, Y.; Liu, M.; Li, X.; Tang, H. Hepatitis B Virus-Encoded MicroRNA Controls Viral Replication. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pincetic, A.; Kuang, Z.; Seo, E.J.; Leis, J. The interferon-induced gene ISG15 blocks retrovirus release from cells late in the budding process. J. Virol. 2010, 84, 4725–4736. [Google Scholar] [CrossRef] [Green Version]
- Kuang, Z.; Seo, E.J.; Leis, J. Mechanism of inhibition of retrovirus release from cells by interferon-induced gene ISG15. J. Virol. 2011, 85, 7153–7161. [Google Scholar] [CrossRef] [Green Version]
- Bashirova, A.A.; Bleiber, G.; Qi, Y.; Hutcheson, H.; Yamashita, T.; Johnson, R.C.; Cheng, J.; Alter, G.; Goedert, J.J.; Buchbinder, S.; et al. Consistent effects of TSG101 genetic variability on multiple outcomes of exposure to human immunodeficiency virus type 1. J. Virol. 2006, 80, 6757–6763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strickland, M.; Ehrlich, L.S.; Watanabe, S.; Khan, M.; Strub, M.P.; Luan, C.H.; Powell, M.D.; Leis, J.; Tjandra, N.; Carter, C.A. Tsg101 chaperone function revealed by HIV-1 assembly inhibitors. Nat. Commun. 2017, 8, 1391. [Google Scholar] [CrossRef]
- Watanabe, S.M.; Ehrlich, L.S.; Strickland, M.; Li, X.; Soloveva, V.; Goff, A.J.; Stauft, C.B.; Bhaduri-McIntosh, S.; Tjandra, N.; Carter, C. Selective Targeting of Virus Replication by Proton Pump Inhibitors. Sci. Rep. 2020, 10, 4003. [Google Scholar] [CrossRef] [Green Version]
- Ketter, E.; Randall, G. Virus Impact on Lipids and Membranes. Annu. Rev. Virol. 2019, 6, 319–340. [Google Scholar] [CrossRef] [PubMed]
- Tuthill, T.J.; Bubeck, D.; Rowlands, D.J.; Hogle, J.M. Characterization of early steps in the poliovirus infection process: Receptor-decorated liposomes induce conversion of the virus to membrane-anchored entry-intermediate particles. J. Virol. 2006, 80, 172–180. [Google Scholar] [CrossRef] [Green Version]
- Lozach, P.Y.; Huotari, J.; Helenius, A. Late-penetrating viruses. Curr. Opin. Virol 2011, 1, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Votteler, J.; Sundquist, W.I. Virus budding and the ESCRT pathway. Cell Host Microbe 2013, 14, 232–241. [Google Scholar] [CrossRef] [Green Version]
- Wodrich, H.; Henaff, D.; Jammart, B.; Segura-Morales, C.; Seelmeir, S.; Coux, O.; Ruzsics, Z.; Wiethoff, C.M.; Kremer, E.J. A capsid-encoded PPxY-motif facilitates adenovirus entry. PLoS Pathog. 2010, 6, e1000808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiethoff, C.M.; Wodrich, H.; Gerace, L.; Nemerow, G.R. Adenovirus protein VI mediates membrane disruption following capsid disassembly. J. Virol. 2005, 79, 1992–2000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moyer, C.L.; Wiethoff, C.M.; Maier, O.; Smith, J.G.; Nemerow, G.R. Functional genetic and biophysical analyses of membrane disruption by human adenovirus. J. Virol. 2011, 85, 2631–2641. [Google Scholar] [CrossRef] [Green Version]
- Prchla, E.; Plank, C.; Wagner, E.; Blaas, D.; Fuchs, R. Virus-mediated release of endosomal content in vitro: Different behavior of adenovirus and rhinovirus serotype 2. J. Cell Biol. 1995, 131, 111–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farr, G.A.; Zhang, L.G.; Tattersall, P. Parvoviral virions deploy a capsid-tethered lipolytic enzyme to breach the endosomal membrane during cell entry. Proc. Natl. Acad. Sci. USA 2005, 102, 17148–17153. [Google Scholar] [CrossRef] [Green Version]
- Montespan, C.; Marvin, S.A.; Austin, S.; Burrage, A.M.; Roger, B.; Rayne, F.; Faure, M.; Campell, E.M.; Schneider, C.; Reimer, R.; et al. Multi-layered control of Galectin-8 mediated autophagy during adenovirus cell entry through a conserved PPxY motif in the viral capsid. PLoS Pathog. 2017, 13, e1006217. [Google Scholar] [CrossRef]
- Kumar, B.; Roy, A.; Veettil, M.V.; Chandran, B. Insight into the Roles of E3 Ubiquitin Ligase c-Cbl, ESCRT Machinery, and Host Cell Signaling in Kaposi’s Sarcoma-Associated Herpesvirus Entry and Trafficking. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Shtanko, O.; Nikitina, R.A.; Altuntas, C.Z.; Chepurnov, A.A.; Davey, R.A. Crimean-Congo hemorrhagic fever virus entry into host cells occurs through the multivesicular body and requires ESCRT regulators. PLoS Pathog. 2014, 10, e1004390. [Google Scholar] [CrossRef] [Green Version]
- Broniarczyk, J.; Bergant, M.; Gozdzicka-Jozefiak, A.; Banks, L. Human papillomavirus infection requires the TSG101 component of the ESCRT machinery. Virology 2014, 460–461, 83–90. [Google Scholar] [CrossRef]
- Karjalainen, M.; Rintanen, N.; Lehkonen, M.; Kallio, K.; Maki, A.; Hellstrom, K.; Siljamaki, V.; Upla, P.; Marjomaki, V. Echovirus 1 infection depends on biogenesis of novel multivesicular bodies. Cell Microbiol. 2011, 13, 1975–1995. [Google Scholar] [CrossRef] [PubMed]
- Pasqual, G.; Rojek, J.M.; Masin, M.; Chatton, J.Y.; Kunz, S. Old world arenaviruses enter the host cell via the multivesicular body and depend on the endosomal sorting complex required for transport. PLoS Pathog. 2011, 7, e1002232. [Google Scholar] [CrossRef]
- Silva-Ayala, D.; Lopez, T.; Gutierrez, M.; Perrimon, N.; Lopez, S.; Arias, C.F. Genome-wide RNAi screen reveals a role for the ESCRT complex in rotavirus cell entry. Proc. Natl. Acad. Sci. USA 2013, 110, 10270–10275. [Google Scholar] [CrossRef] [Green Version]
- Barajas, D.; Jiang, Y.; Nagy, P.D. A unique role for the host ESCRT proteins in replication of Tomato bushy stunt virus. PLoS Pathog. 2009, 5, e1000705. [Google Scholar] [CrossRef] [Green Version]
- Kovalev, N.; de Castro Martin, I.F.; Pogany, J.; Barajas, D.; Pathak, K.; Risco, C.; Nagy, P.D. Role of Viral RNA and Co-opted Cellular ESCRT-I and ESCRT-III Factors in Formation of Tombusvirus Spherules Harboring the Tombusvirus Replicase. J. Virol. 2016, 90, 3611–3626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, L.G.; Clendening, E.A.; Sheen, H.; Gidda, S.K.; White, K.A.; Mullen, R.T. A unique N-terminal sequence in the Carnation Italian ringspot virus p36 replicase-associated protein interacts with the host cell ESCRT-I component Vps23. J. Virol. 2014, 88, 6329–6344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz, A.; Zhang, J.; Ollwerther, A.; Wang, X.; Ahlquist, P. Host ESCRT proteins are required for bromovirus RNA replication compartment assembly and function. PLoS Pathog. 2015, 11, e1004742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabata, K.; Arimoto, M.; Arakawa, M.; Nara, A.; Saito, K.; Omori, H.; Arai, A.; Ishikawa, T.; Konishi, E.; Suzuki, R.; et al. Unique Requirement for ESCRT Factors in Flavivirus Particle Formation on the Endoplasmic Reticulum. Cell Rep. 2016, 16, 2339–2347. [Google Scholar] [CrossRef] [Green Version]
- Tandon, R.; AuCoin, D.P.; Mocarski, E.S. Human cytomegalovirus exploits ESCRT machinery in the process of virion maturation. J. Virol. 2009, 83, 10797–10807. [Google Scholar] [CrossRef] [Green Version]
- Lindenbach, B.D. Virion assembly and release. Curr. Top. Microbiol. Immunol. 2013, 369, 199–218. [Google Scholar] [CrossRef] [Green Version]
- Rossman, J.S.; Lamb, R.A. Viral membrane scission. Ann. Rev. Cell Dev. Biol. 2013, 29, 551–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottlinger, H.G.; Dorfman, T.; Sodroski, J.G.; Haseltine, W.A. Effect of mutations affecting the p6 gag protein on human immunodeficiency virus particle release. Proc. Natl. Acad. Sci. USA 1991, 88, 3195–3199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wills, J.W.; Cameron, C.E.; Wilson, C.B.; Xiang, Y.; Bennett, R.P.; Leis, J. An assembly domain of the Rous sarcoma virus Gag protein required late in budding. J. Virol. 1994, 68, 6605–6618. [Google Scholar] [CrossRef] [Green Version]
- Calistri, A.; Salata, C.; Parolin, C.; Palu, G. Role of multivesicular bodies and their components in the egress of enveloped RNA viruses. Rev. Med. Virol. 2009, 19, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Crump, C. Virus Assembly and Egress of HSV. Adv. Exp. Med. Biol. 2018, 1045, 23–44. [Google Scholar] [CrossRef] [PubMed]
- Calistri, A.; Sette, P.; Salata, C.; Cancellotti, E.; Forghieri, C.; Comin, A.; Gottlinger, H.; Campadelli-Fiume, G.; Palu, G.; Parolin, C. Intracellular trafficking and maturation of herpes simplex virus type 1 gB and virus egress require functional biogenesis of multivesicular bodies. J. Virol. 2007, 81, 11468–11478. [Google Scholar] [CrossRef] [Green Version]
- Calistri, A.; Munegato, D.; Toffoletto, M.; Celestino, M.; Franchin, E.; Comin, A.; Sartori, E.; Salata, C.; Parolin, C.; Palu, G. Functional Interaction Between the ESCRT-I Component TSG101 and the HSV-1 Tegument Ubiquitin Specific Protease. J. Cell Physiol. 2015, 230, 1794–1806. [Google Scholar] [CrossRef]
- Garcia, M.L.; Reynolds, T.D.; Mothes, W.; Robek, M.D. Functional characterization of the putative hepatitis B virus core protein late domain using retrovirus chimeras. PLoS ONE 2013, 8, e72845. [Google Scholar] [CrossRef] [Green Version]
- Hurley, J.H.; Hanson, P.I. Membrane budding and scission by the ESCRT machinery: it’s all in the neck. Nat. Rev. Mol. Cell Biol. 2010, 11, 556–566. [Google Scholar] [CrossRef] [PubMed]
- Ariumi, Y.; Kuroki, M.; Maki, M.; Ikeda, M.; Dansako, H.; Wakita, T.; Kato, N. The ESCRT system is required for hepatitis C virus production. PLoS ONE 2011, 6, e14517. [Google Scholar] [CrossRef] [Green Version]
- Rossman, J.S.; Jing, X.; Leser, G.P.; Lamb, R.A. Influenza virus M2 protein mediates ESCRT-independent membrane scission. Cell 2010, 142, 902–913. [Google Scholar] [CrossRef] [Green Version]
- Wirblich, C.; Bhattacharya, B.; Roy, P. Nonstructural protein 3 of bluetongue virus assists virus release by recruiting ESCRT-I protein Tsg101. J. Virol. 2006, 80, 460–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parent, L.J.; Bennett, R.P.; Craven, R.C.; Nelle, T.D.; Krishna, N.K.; Bowzard, J.B.; Wilson, C.B.; Puffer, B.A.; Montelaro, R.C.; Wills, J.W. Positionally independent and exchangeable late budding functions of the Rous sarcoma virus and human immunodeficiency virus Gag proteins. J. Virol. 1995, 69, 5455–5460. [Google Scholar] [CrossRef] [Green Version]
- Yuan, B.; Campbell, S.; Bacharach, E.; Rein, A.; Goff, S.P. Infectivity of Moloney murine leukemia virus defective in late assembly events is restored by late assembly domains of other retroviruses. J. Virol. 2000, 74, 7250–7260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin-Serrano, J.; Yarovoy, A.; Perez-Caballero, D.; Bieniasz, P.D. Divergent retroviral late-budding domains recruit vacuolar protein sorting factors by using alternative adaptor proteins. Proc. Natl. Acad. Sci. USA 2003, 100, 12414–12419. [Google Scholar] [CrossRef] [Green Version]
- Martin-Serrano, J.; Bieniasz, P.D. A bipartite late-budding domain in human immunodeficiency virus type 1. J. Virol. 2003, 77, 12373–12377. [Google Scholar] [CrossRef] [Green Version]
- Strack, B.; Calistri, A.; Accola, M.A.; Palu, G.; Gottlinger, H.G. A role for ubiquitin ligase recruitment in retrovirus release. Proc. Natl. Acad. Sci. USA 2000, 97, 13063–13068. [Google Scholar] [CrossRef] [Green Version]
- Staub, O.; Dho, S.; Henry, P.; Correa, J.; Ishikawa, T.; McGlade, J.; Rotin, D. WW domains of Nedd4 bind to the proline-rich PY motifs in the epithelial Na+ channel deleted in Liddle’s syndrome. EMBO J. 1996, 15, 2371–2380. [Google Scholar] [CrossRef]
- Staub, O.; Gautschi, I.; Ishikawa, T.; Breitschopf, K.; Ciechanover, A.; Schild, L.; Rotin, D. Regulation of stability and function of the epithelial Na+ channel (ENaC) by ubiquitination. EMBO J. 1997, 16, 6325–6336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strack, B.; Calistri, A.; Gottlinger, H.G. Late assembly domain function can exhibit context dependence and involves ubiquitin residues implicated in endocytosis. J. Virol. 2002, 76, 5472–5479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrus, J.E.; von Schwedler, U.K.; Pornillos, O.W.; Morham, S.G.; Zavitz, K.H.; Wang, H.E.; Wettstein, D.A.; Stray, K.M.; Cote, M.; Rich, R.L.; et al. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell 2001, 107, 55–65. [Google Scholar] [CrossRef] [Green Version]
- VerPlank, L.; Bouamr, F.; LaGrassa, T.J.; Agresta, B.; Kikonyogo, A.; Leis, J.; Carter, C.A. Tsg101, a homologue of ubiquitin-conjugating (E2) enzymes, binds the L domain in HIV type 1 Pr55(Gag). Proc. Natl. Acad. Sci. USA 2001, 98, 7724–7729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kikonyogo, A.; Bouamr, F.; Vana, M.L.; Xiang, Y.; Aiyar, A.; Carter, C.; Leis, J. Proteins related to the Nedd4 family of ubiquitin protein ligases interact with the L domain of Rous sarcoma virus and are required for gag budding from cells. Proc. Natl. Acad. Sci. USA 2001, 98, 11199–11204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, K.M.; Hirsch, V.M.; Bouamr, F. Budding of a Retrovirus: Some Assemblies Required. Viruses 2020, 12, 1188. [Google Scholar] [CrossRef]
- Pincetic, A.; Medina, G.; Carter, C.; Leis, J. Avian sarcoma virus and human immunodeficiency virus, type 1 use different subsets of ESCRT proteins to facilitate the budding process. J. Biol. Chem. 2008, 283, 29822–29830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langelier, C.; von Schwedler, U.K.; Fisher, R.D.; De Domenico, I.; White, P.L.; Hill, C.P.; Kaplan, J.; Ward, D.; Sundquist, W.I. Human ESCRT-II complex and its role in human immunodeficiency virus type 1 release. J. Virol. 2006, 80, 9465–9480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, B.; Ip, N.C.Y.; Abbink, T.E.M.; Kenyon, J.C.; Lever, A.M.L. ESCRT-II functions by linking to ESCRT-I in human immunodeficiency virus-1 budding. Cell Microbiol. 2020, 22, e13161. [Google Scholar] [CrossRef] [Green Version]
- Hurley, J.H.; Cada, A.K. Inside job: How the ESCRTs release HIV-1 from infected cells. Biochem. Soc. Trans. 2018, 46, 1029–1036. [Google Scholar] [CrossRef]
- Del Vecchio, C.; Celestino, M.; Celegato, M.; Palu, G.; Parolin, C.; Bouamr, F.; Calistri, A. Alix-Mediated Rescue of Feline Immunodeficiency Virus Budding Differs from That Observed with Human Immunodeficiency Virus. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [PubMed]
- Weiss, E.R.; Popova, E.; Yamanaka, H.; Kim, H.C.; Huibregtse, J.M.; Gottlinger, H. Rescue of HIV-1 release by targeting widely divergent NEDD4-type ubiquitin ligases and isolated catalytic HECT domains to Gag. PLoS Pathog. 2010, 6, e1001107. [Google Scholar] [CrossRef]
- Usami, Y.; Popov, S.; Popova, E.; Gottlinger, H.G. Efficient and specific rescue of human immunodeficiency virus type 1 budding defects by a Nedd4-like ubiquitin ligase. J. Virol. 2008, 82, 4898–4907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, H.Y.; Morita, E.; von Schwedler, U.; Muller, B.; Krausslich, H.G.; Sundquist, W.I. NEDD4L overexpression rescues the release and infectivity of human immunodeficiency virus type 1 constructs lacking PTAP and YPXL late domains. J. Virol. 2008, 82, 4884–4897. [Google Scholar] [CrossRef] [Green Version]
- Calistri, A.; Del Vecchio, C.; Salata, C.; Celestino, M.; Celegato, M.; Gottlinger, H.; Palu, G.; Parolin, C. Role of the feline immunodeficiency virus L-domain in the presence or absence of Gag processing: Involvement of ubiquitin and Nedd4-2s ligase in viral egress. J. Cell Physiol. 2009, 218, 175–182. [Google Scholar] [CrossRef] [Green Version]
- Sette, P.; Nagashima, K.; Piper, R.C.; Bouamr, F. Ubiquitin conjugation to Gag is essential for ESCRT-mediated HIV-1 budding. Retrovirology 2013, 10, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popov, S.; Popova, E.; Inoue, M.; Gottlinger, H.G. Divergent Bro1 domains share the capacity to bind human immunodeficiency virus type 1 nucleocapsid and to enhance virus-like particle production. J. Virol. 2009, 83, 7185–7193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dussupt, V.; Javid, M.P.; Abou-Jaoude, G.; Jadwin, J.A.; de La Cruz, J.; Nagashima, K.; Bouamr, F. The nucleocapsid region of HIV-1 Gag cooperates with the PTAP and LYPXnL late domains to recruit the cellular machinery necessary for viral budding. PLoS Pathog. 2009, 5, e1000339. [Google Scholar] [CrossRef] [Green Version]
- Bello, N.F.; Dussupt, V.; Sette, P.; Rudd, V.; Nagashima, K.; Bibollet-Ruche, F.; Chen, C.; Montelaro, R.C.; Hahn, B.H.; Bouamr, F. Budding of retroviruses utilizing divergent L domains requires nucleocapsid. J. Virol. 2012, 86, 4182–4193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottlinger, H.G. Influenza exits the cell without an ESCRT. Cell 2010, 142, 839–841. [Google Scholar] [CrossRef] [Green Version]
- Feng, Z.; Hirai-Yuki, A.; McKnight, K.L.; Lemon, S.M. Naked Viruses That Aren’t Always Naked: Quasi-Enveloped Agents of Acute Hepatitis. Ann. Rev. Virol. 2014, 1, 539–560. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Hensley, L.; McKnight, K.L.; Hu, F.; Madden, V.; Ping, L.; Jeong, S.H.; Walker, C.; Lanford, R.E.; Lemon, S.M. A pathogenic picornavirus acquires an envelope by hijacking cellular membranes. Nature 2013, 496, 367–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagashima, S.; Takahashi, M.; Jirintai, S.; Tanggis; Kobayashi, T.; Nishizawa, T.; Okamoto, H. The membrane on the surface of hepatitis E virus particles is derived from the intracellular membrane and contains trans-Golgi network protein 2. Arch. Virol 2014, 159, 979–991. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, S.; Takahashi, M.; Kobayashi, T.; Tanggis; Nishizawa, T.; Nishiyama, T.; Primadharsini, P.P.; Okamoto, H. Characterization of the Quasi-Enveloped Hepatitis E Virus Particles Released by the Cellular Exosomal Pathway. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izquierdo-Useros, N.; Naranjo-Gomez, M.; Erkizia, I.; Puertas, M.C.; Borras, F.E.; Blanco, J.; Martinez-Picado, J. HIV and mature dendritic cells: Trojan exosomes riding the Trojan horse? PLoS Pathog. 2010, 6, e1000740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramakrishnaiah, V.; Thumann, C.; Fofana, I.; Habersetzer, F.; Pan, Q.; de Ruiter, P.E.; Willemsen, R.; Demmers, J.A.; Stalin Raj, V.; Jenster, G.; et al. Exosome-mediated transmission of hepatitis C virus between human hepatoma Huh7.5 cells. Proc. Natl. Acad. Sci. USA 2013, 110, 13109–13113. [Google Scholar] [CrossRef] [Green Version]
- Giannessi, F.; Aiello, A.; Franchi, F.; Percario, Z.A.; Affabris, E. The Role of Extracellular Vesicles as Allies of HIV, HCV and SARS Viruses. Viruses 2020, 12, 571. [Google Scholar] [CrossRef]
- Longatti, A. The Dual Role of Exosomes in Hepatitis A and C Virus Transmission and Viral Immune Activation. Viruses 2015, 7, 6707–6715. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Woodson, M.; Sherman, M.B.; Neelakanta, G.; Sultana, H. Exosomes mediate Zika virus transmission through SMPD3 neutral Sphingomyelinase in cortical neurons. Emerg Microbes Infect. 2019, 8, 307–326. [Google Scholar] [CrossRef]
- Zhang, K.; Xu, S.; Shi, X.; Xu, G.; Shen, C.; Liu, X.; Zheng, H. Exosomes-mediated transmission of foot-and-mouth disease virus in vivo and in vitro. Vet. Microbiol. 2019, 233, 164–173. [Google Scholar] [CrossRef]
- Silvas, J.A.; Popov, V.L.; Paulucci-Holthauzen, A.; Aguilar, P.V. Extracellular Vesicles Mediate Receptor-Independent Transmission of Novel Tick-Borne Bunyavirus. J. Virol. 2016, 90, 873–886. [Google Scholar] [CrossRef] [Green Version]
- Reyes-Ruiz, J.M.; Osuna-Ramos, J.F.; De Jesus-Gonzalez, L.A.; Hurtado-Monzon, A.M.; Farfan-Morales, C.N.; Cervantes-Salazar, M.; Bolanos, J.; Cigarroa-Mayorga, O.E.; Martin-Martinez, E.S.; Medina, F.; et al. Isolation and characterization of exosomes released from mosquito cells infected with dengue virus. Virus Res. 2019, 266, 1–14. [Google Scholar] [CrossRef]
- Dubrovsky, L.; Ward, A.; Choi, S.H.; Pushkarsky, T.; Brichacek, B.; Vanpouille, C.; Adzhubei, A.A.; Mukhamedova, N.; Sviridov, D.; Margolis, L.; et al. Inhibition of HIV Replication by Apolipoprotein A-I Binding Protein Targeting the Lipid Rafts. mBio 2020, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bello-Morales, R.; Lopez-Guerrero, J.A. Extracellular Vesicles in Herpes Viral Spread and Immune Evasion. Front. Microbiol. 2018, 9, 2572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bello-Morales, R.; Praena, B.; de la Nuez, C.; Rejas, M.T.; Guerra, M.; Galan-Ganga, M.; Izquierdo, M.; Calvo, V.; Krummenacher, C.; Lopez-Guerrero, J.A. Role of Microvesicles in the Spread of Herpes Simplex Virus 1 in Oligodendrocytic Cells. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mutsafi, Y.; Altan-Bonnet, N. Enterovirus Transmission by Secretory Autophagy. Viruses 2018, 10, 139. [Google Scholar] [CrossRef] [Green Version]
- Teo, Q.W.; van Leur, S.W.; Sanyal, S. Escaping the Lion’s Den: Redirecting autophagy for unconventional release and spread of viruses. FEBS J. 2020. [Google Scholar] [CrossRef] [PubMed]
- Hittelman, A.B.; Burakov, D.; Iniguez-Lluhi, J.A.; Freedman, L.P.; Garabedian, M.J. Differential regulation of glucocorticoid receptor transcriptional activation via AF-1-associated proteins. EMBO J. 1999, 18, 5380–5388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, M.; Yanagi, Y.; Masuhiro, Y.; Yano, T.; Yoshikawa, H.; Yanagisawa, J.; Kato, S. A putative tumor suppressor, TSG101, acts as a transcriptional suppressor through its coiled-coil domain. Biochem. Biophys. Res. Commun. 1998, 245, 900–905. [Google Scholar] [CrossRef] [PubMed]
- Chua, H.H.; Lee, H.H.; Chang, S.S.; Lu, C.C.; Yeh, T.H.; Hsu, T.Y.; Cheng, T.H.; Cheng, J.T.; Chen, M.R.; Tsai, C.H. Role of the TSG101 gene in Epstein-Barr virus late gene transcription. J. Virol. 2007, 81, 2459–2471. [Google Scholar] [CrossRef] [Green Version]
- Ruland, J.; Sirard, C.; Elia, A.; MacPherson, D.; Wakeham, A.; Li, L.; de la Pompa, J.L.; Cohen, S.N.; Mak, T.W. p53 accumulation, defective cell proliferation, and early embryonic lethality in mice lacking tsg101. Proc. Natl. Acad. Sci. USA 2001, 98, 1859–1864. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Liao, J.; Ruland, J.; Mak, T.W.; Cohen, S.N. A TSG101/MDM2 regulatory loop modulates MDM2 degradation and MDM2/p53 feedback control. Proc. Natl. Acad. Sci. USA 2001, 98, 1619–1624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, A.E.; Miller, T.; Schmidt, S.L.; Shiekhattar, R.; Shilatifard, A. Cloning and characterization of the EAP30 subunit of the ELL complex that confers derepression of transcription by RNA polymerase II. J. Biol. Chem. 1999, 274, 21981–21985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamura, T.; Burian, D.; Khalili, H.; Schmidt, S.L.; Sato, S.; Liu, W.J.; Conrad, M.N.; Conaway, R.C.; Conaway, J.W.; Shilatifard, A. Cloning and characterization of ELL-associated proteins EAP45 and EAP20. a role for yeast EAP-like proteins in regulation of gene expression by glucose. J. Biol. Chem. 2001, 276, 16528–16533. [Google Scholar] [CrossRef]
- Stauffer, D.R.; Howard, T.L.; Nyun, T.; Hollenberg, S.M. CHMP1 is a novel nuclear matrix protein affecting chromatin structure and cell-cycle progression. J. Cell Sci. 2001, 114, 2383–2393. [Google Scholar] [PubMed]
- Tsang, H.T.; Connell, J.W.; Brown, S.E.; Thompson, A.; Reid, E.; Sanderson, C.M. A systematic analysis of human CHMP protein interactions: Additional MIT domain-containing proteins bind to multiple components of the human ESCRT III complex. Genomics 2006, 88, 333–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miranda-Saksena, M.; Denes, C.E.; Diefenbach, R.J.; Cunningham, A.L. Infection and Transport of Herpes Simplex Virus Type 1 in Neurons: Role of the Cytoskeleton. Viruses 2018, 10, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leuzinger, H.; Ziegler, U.; Schraner, E.M.; Fraefel, C.; Glauser, D.L.; Heid, I.; Ackermann, M.; Mueller, M.; Wild, P. Herpes simplex virus 1 envelopment follows two diverse pathways. J. Virol. 2005, 79, 13047–13059. [Google Scholar] [CrossRef] [Green Version]
- Draganova, E.B.; Zhang, J.; Zhou, Z.H.; Heldwein, E.E. Structural basis for capsid recruitment and coat formation during HSV-1 nuclear egress. Elife 2020, 9, e56627. [Google Scholar] [CrossRef]
- Klupp, B.G.; Granzow, H.; Mettenleiter, T.C. Nuclear envelope breakdown can substitute for primary envelopment-mediated nuclear egress of herpesviruses. J. Virol. 2011, 85, 8285–8292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lymberopoulos, M.H.; Bourget, A.; Ben Abdeljelil, N.; Pearson, A. Involvement of the UL24 protein in herpes simplex virus 1-induced dispersal of B23 and in nuclear egress. Virology 2011, 412, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Pan, S.; Zhang, L.; Baines, J.; Roller, R.; Ames, J.; Yang, M.; Wang, J.; Chen, D.; Liu, Y.; et al. Herpes Simplex Virus 1 Induces Phosphorylation and Reorganization of Lamin A/C through the gamma134.5 Protein That Facilitates Nuclear Egress. J. Virol. 2016, 90, 10414–10422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arii, J.; Watanabe, M.; Maeda, F.; Tokai-Nishizumi, N.; Chihara, T.; Miura, M.; Maruzuru, Y.; Koyanagi, N.; Kato, A.; Kawaguchi, Y. ESCRT-III mediates budding across the inner nuclear membrane and regulates its integrity. Nat. Commun. 2018, 9, 3379. [Google Scholar] [CrossRef] [Green Version]
- Crump, C.M.; Yates, C.; Minson, T. Herpes simplex virus type 1 cytoplasmic envelopment requires functional Vps4. J. Virol. 2007, 81, 7380–7387. [Google Scholar] [CrossRef] [Green Version]
- Kharkwal, H.; Smith, C.G.; Wilson, D.W. Blocking ESCRT-mediated envelopment inhibits microtubule-dependent trafficking of alphaherpesviruses in vitro. J. Virol. 2014, 88, 14467–14478. [Google Scholar] [CrossRef] [Green Version]
- Kharkwal, H.; Smith, C.G.; Wilson, D.W. Herpes Simplex Virus Capsid Localization to ESCRT-VPS4 Complexes in the Presence and Absence of the Large Tegument Protein UL36p. J. Virol. 2016, 90, 7257–7267. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, A.E.; Ryckman, B.J.; Baines, J.D.; Zhou, Y.; Liang, L.; Roller, R.J. U(L)31 and U(L)34 proteins of herpes simplex virus type 1 form a complex that accumulates at the nuclear rim and is required for envelopment of nucleocapsids. J. Virol. 2001, 75, 8803–8817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawliczek, T.; Crump, C.M. Herpes simplex virus type 1 production requires a functional ESCRT-III complex but is independent of TSG101 and ALIX expression. J. Virol. 2009, 83, 11254–11264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- English, L.; Chemali, M.; Duron, J.; Rondeau, C.; Laplante, A.; Gingras, D.; Alexander, D.; Leib, D.; Norbury, C.; Lippe, R.; et al. Autophagy enhances the presentation of endogenous viral antigens on MHC class I molecules during HSV-1 infection. Nat. Immunol. 2009, 10, 480–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radtke, K.; English, L.; Rondeau, C.; Leib, D.; Lippe, R.; Desjardins, M. Inhibition of the host translation shutoff response by herpes simplex virus 1 triggers nuclear envelope-derived autophagy. J. Virol. 2013, 87, 3990–3997. [Google Scholar] [CrossRef] [Green Version]
- Turan, A.; Grosche, L.; Krawczyk, A.; Muhl-Zurbes, P.; Drassner, C.; Duthorn, A.; Kummer, M.; Hasenberg, M.; Voortmann, S.; Jastrow, H.; et al. Autophagic degradation of lamins facilitates the nuclear egress of herpes simplex virus type 1. J. Cell Biol. 2019, 218, 508–523. [Google Scholar] [CrossRef]
- Owen, D.J.; Crump, C.M.; Graham, S.C. Tegument Assembly and Secondary Envelopment of Alphaherpesviruses. Viruses 2015, 7, 5084–5114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ventimiglia, L.N.; Martin-Serrano, J. ESCRT machinery: Damage control at the nuclear membrane. Cell Res. 2016, 26, 641–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.P.; Liu, P.T.; Kung, H.N.; Su, M.T.; Chua, H.H.; Chang, Y.H.; Chang, C.W.; Tsai, C.H.; Liu, F.T.; Chen, M.R. The ESCRT machinery is recruited by the viral BFRF1 protein to the nucleus-associated membrane for the maturation of Epstein-Barr Virus. PLoS Pathog. 2012, 8, e1002904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.P.; Liu, G.T.; Kung, H.N.; Liu, P.T.; Liao, Y.T.; Chow, L.P.; Chang, L.S.; Chang, Y.H.; Chang, C.W.; Shu, W.C.; et al. The Ubiquitin Ligase Itch and Ubiquitination Regulate BFRF1-Mediated Nuclear Envelope Modification for Epstein-Barr Virus Maturation. J. Virol. 2016, 90, 8994–9007. [Google Scholar] [CrossRef] [Green Version]
- Granzow, H.; Klupp, B.G.; Fuchs, W.; Veits, J.; Osterrieder, N.; Mettenleiter, T.C. Egress of alphaherpesviruses: Comparative ultrastructural study. J. Virol. 2001, 75, 3675–3684. [Google Scholar] [CrossRef] [Green Version]
- van Genderen, I.L.; Brandimarti, R.; Torrisi, M.R.; Campadelli, G.; van Meer, G. The phospholipid composition of extracellular herpes simplex virions differs from that of host cell nuclei. Virology 1994, 200, 831–836. [Google Scholar] [CrossRef]
- Hollinshead, M.; Johns, H.L.; Sayers, C.L.; Gonzalez-Lopez, C.; Smith, G.L.; Elliott, G. Endocytic tubules regulated by Rab GTPases 5 and 11 are used for envelopment of herpes simplex virus. EMBO J. 2012, 31, 4204–4220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schipke, J.; Pohlmann, A.; Diestel, R.; Binz, A.; Rudolph, K.; Nagel, C.H.; Bauerfeind, R.; Sodeik, B. The C terminus of the large tegument protein pUL36 contains multiple capsid binding sites that function differently during assembly and cell entry of herpes simplex virus. J. Virol. 2012, 86, 3682–3700. [Google Scholar] [CrossRef] [Green Version]
- Leis, J.; Luan, C.H.; Audia, J.E.; Dunne, S.F.; Heath, C.M. Ilaprazole and other novel prazole-based compounds that bind Tsg101 inhibit viral budding of HSV-1/2 and HIV from cells. BioRxiv 2020. [Google Scholar] [CrossRef]
- Kattenhorn, L.M.; Korbel, G.A.; Kessler, B.M.; Spooner, E.; Ploegh, H.L. A deubiquitinating enzyme encoded by HSV-1 belongs to a family of cysteine proteases that is conserved across the family Herpesviridae. Mol. Cell 2005, 19, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Schlieker, C.; Korbel, G.A.; Kattenhorn, L.M.; Ploegh, H.L. A deubiquitinating activity is conserved in the large tegument protein of the herpesviridae. J. Virol. 2005, 79, 15582–15585. [Google Scholar] [CrossRef] [Green Version]
- Bolstad, M.; Abaitua, F.; Crump, C.M.; O’Hare, P. Autocatalytic activity of the ubiquitin-specific protease domain of herpes simplex virus 1 VP1-2. J. Virol. 2011, 85, 8738–8751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butt, B.G.; Owen, D.J.; Jeffries, C.M.; Ivanova, L.; Hill, C.H.; Houghton, J.W.; Ahmed, M.F.; Antrobus, R.; Svergun, D.I.; Welch, J.J.; et al. Insights into herpesvirus assembly from the structure of the pUL7:pUL51 complex. Elife 2020, 9, e53789. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Cellular Process | Key Player/s | Effect of Silencing | References |
|---|---|---|---|
| MVB Biogenesis | ESCRT-0 (HRS) ESCRT-I (TSG101) ESCRT-III/VPS4 | HRS depletion: enlarged MVBs with few ILVs TSG101 depletion: MVB formation strongly reduced ESCRT-III/Vps24 depletion: smaller MVBs in clusters HRS TSG101, Vps22 and Vps24 co-depletion: MVBs and ILVs still formed | [53,128,129,130] |
| Autophagy | ESCRT-I (VPS37A) ESCRT-III (CHMP2A)/VPS4 | VPS37A depletion: accumulation of phagophores due to defects in autophagosome completion CHMP2A depletion: accumulation of immature autophagosomal structures; impairment of autophagic flux; inhibition of phagophore sealing during mitophagy CHMP2A, CHMP3, CHMP7 depletion: increase in immature autophagosomal membranes under starvation CHMP2A, CHMP4B and VPS4 depletion: inhibition of mitophagy | [66,67] |
| Cytokinesis | ESCRT-I (TSG101)/ESCRT-II Alix ESCRT-III (CHMP-6,CHMP4B,CHMP4C)/VPS4 | Alix depletion: an increase in multinuclear cells; furrow regression; a failure in CHMP4C recruitment to the midbody; CHMP4B still recruited TSG101 and Alix co-depletion: failure in CHMP4B recruitment to the midbody; multinucleation non aggravated Alix, VPS22, and CHMP6 co-depletion: CHMP4B is not recruited to the intercellular bridge CHMP4C depletion: altered cytokinetic arrest in the presence of chromosomal problems; furrow regression and binucleation | [73,74,80,88,131] |
| Cell Membrane Repair | ESCRT-I (TSG101) Alix ESCRT-III (CHMP4B)/VPS4 | Alix, CHMP2B VPS4 depletion: failure of the repairing process followed by cell death (CHMP4B and VPS4 silencing) CHMP2A depletion: impairment of the repairing process CHMP3 depletion: no significant effect | [99,101] |
| Nuclear Membrane Repair | ESCRT-III (CHMP4B, CHMP7)/VPS4 | Alix, HD-PTP, HRS, TSG101 depletion: no effects on CHMP4B recruitment to the site of ruptures CHMP7depletion: failure of CHMP4B recruitment to the nuclear envelope | [107,108] |
| Lysosomal Membrane Repair | ESCRT-I (TSG101) Alix ESCRT-III (CHMP2A, CHMP4B)/VPS4 | HRS depletion: no effect on CHMP4B recruitment to lysosomes TSG101 depletion: consistent delay in CHMP4B recruitment CHMP2A depletion: increased accumulation of CHMP4B on damaged lysosomes Alix depletion: no detectable effect on CHM4B recruitment TSG101 and Alix co-depletion: almost complete abrogation of CHMP4B recruitment; failure of recovering of damaged lysosomes | [118,119] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Calistri, A.; Reale, A.; Palù, G.; Parolin, C. Why Cells and Viruses Cannot Survive without an ESCRT. Cells 2021, 10, 483. https://doi.org/10.3390/cells10030483
Calistri A, Reale A, Palù G, Parolin C. Why Cells and Viruses Cannot Survive without an ESCRT. Cells. 2021; 10(3):483. https://doi.org/10.3390/cells10030483
Chicago/Turabian StyleCalistri, Arianna, Alberto Reale, Giorgio Palù, and Cristina Parolin. 2021. "Why Cells and Viruses Cannot Survive without an ESCRT" Cells 10, no. 3: 483. https://doi.org/10.3390/cells10030483
APA StyleCalistri, A., Reale, A., Palù, G., & Parolin, C. (2021). Why Cells and Viruses Cannot Survive without an ESCRT. Cells, 10(3), 483. https://doi.org/10.3390/cells10030483