
Multi-Modal Multi-Spectral Intravital Macroscopic Imaging of Signaling Dynamics in Real Time during Tumor–Immune Interactions


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Plasmids
2.3. Cells
2.4. Generation of Stable Reporter Cells
2.5. Animals
2.6. Preparation for Window Chamber Implantation Surgery
2.7. Dorsal Skin Window Chamber Implantation Surgery
2.8. Abdominal Pancreas Window Chamber Implantation Surgery
2.9. Post Window Chamber Implantation Surgery
2.10. In Vivo Imaging
2.11. In Vivo Spectral Unmixing
2.12. Window Chamber Quantitative Analysis
2.13. Histological Analysis
2.14. Data Availability
3. Results
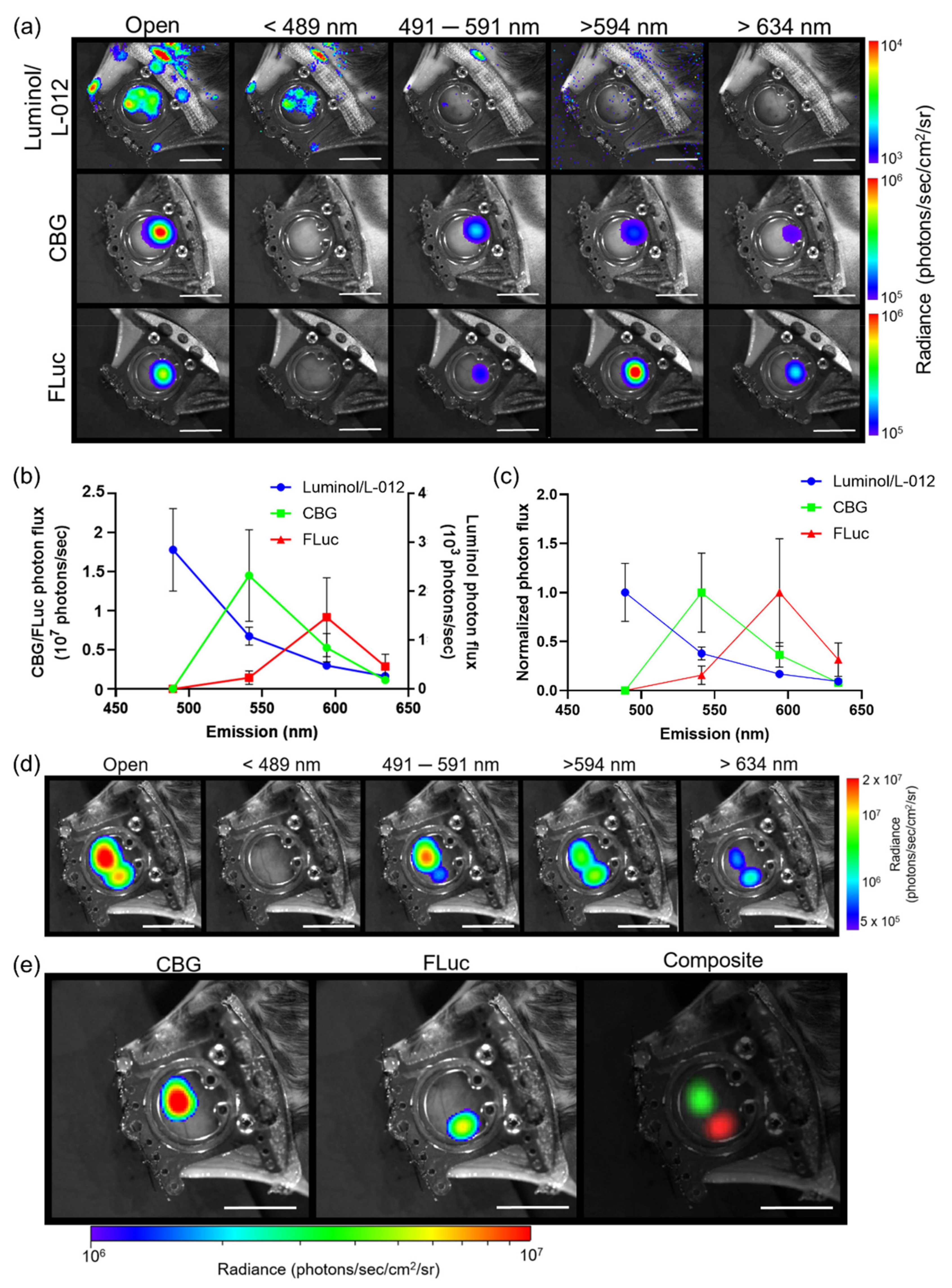
3.1. Generation of In Vivo Spectra
3.2. Imaging Reporters with Different Substrates
3.3. Limitations of Spectral Unmixing Luciferase–Luciferin Reporters In Vivo
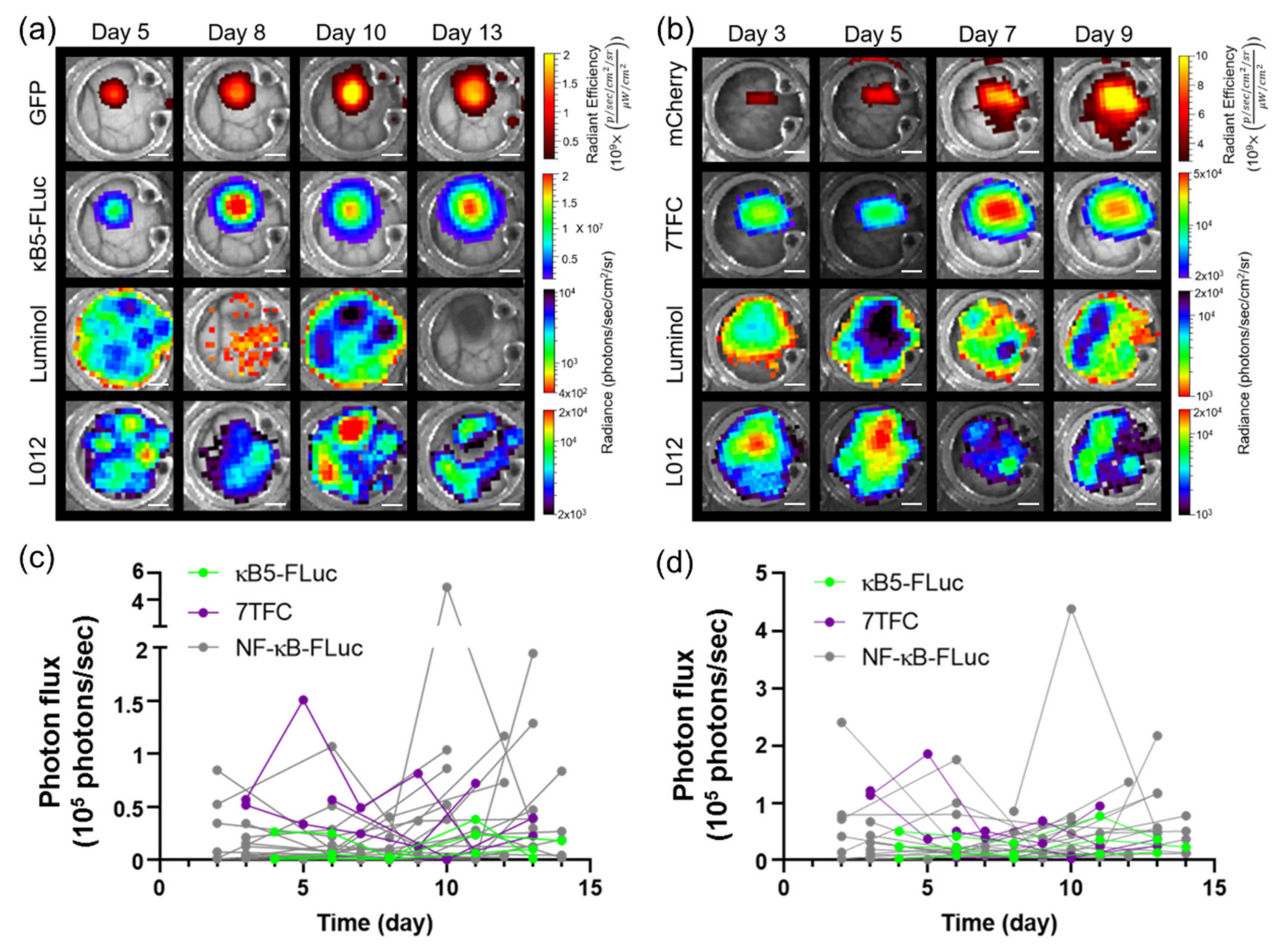
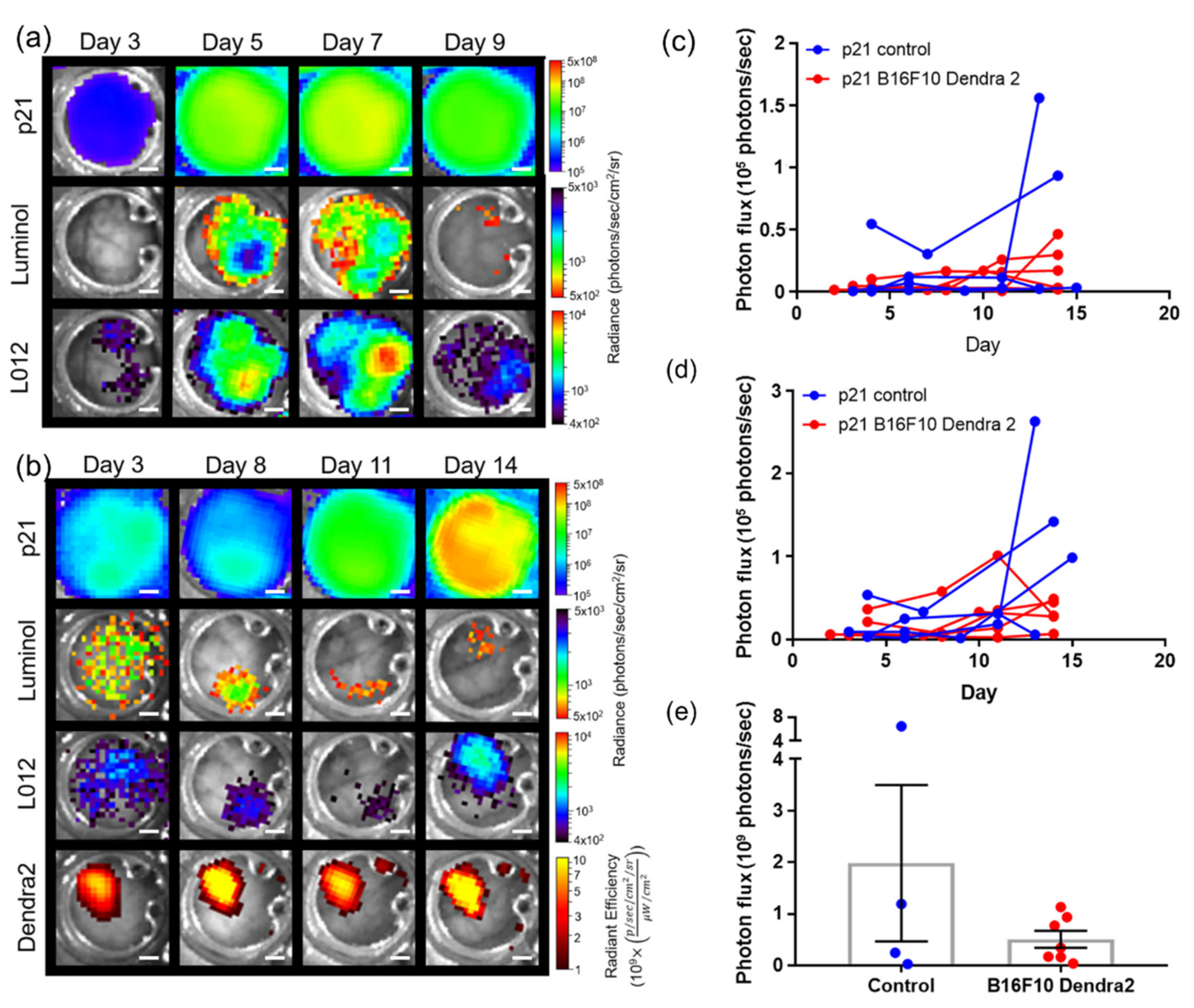
3.4. Imaging Using Transgenic Animals
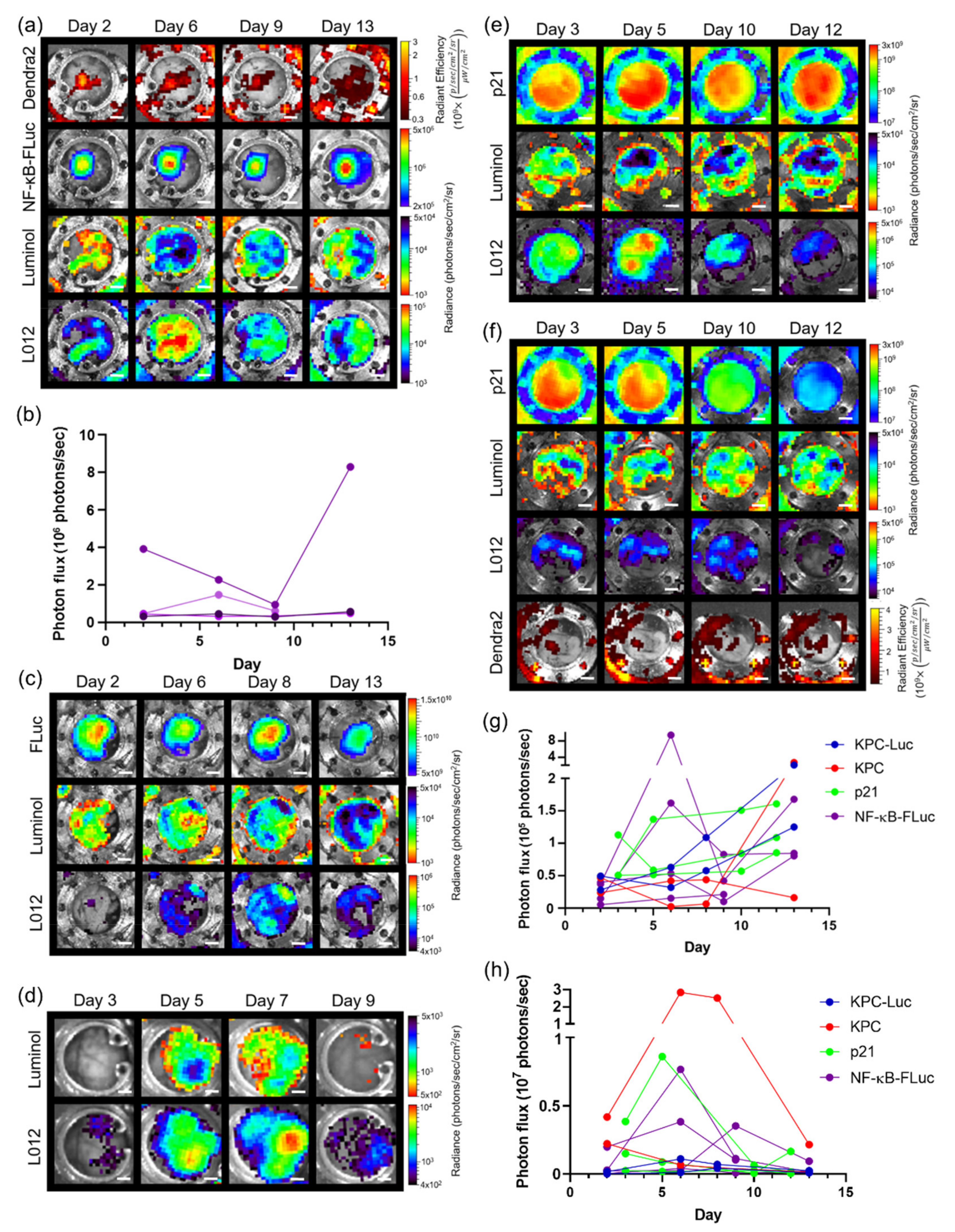
3.5. Imaging with Different Window Chambers
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Smyth, M.J.; Ngiow, S.F.; Ribas, A.; Teng, M.W.L. Combination cancer immunotherapies tailored to the tumour microenvironment. Nat. Rev. Clin. Oncol. 2016, 13, 143–158. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer Immunoediting: Integrating Immunity’s Roles in Cancer Suppression and Promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quail, D.F.; Joyce, J. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Gross, S.; Piwnicaworms, D. Spying on cancer: Molecular imaging in vivo with genetically encoded reporters. Cancer Cell 2005, 7, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Prescher, J.; Contag, C.H. Guided by the light: Visualizing biomolecular processes in living animals with bioluminescence. Curr. Opin. Chem. Biol. 2010, 14, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Mezzanotte, L.; van’t Root, M.; Karatas, H.; Goun, E.A.; Löwik, C.W. In Vivo Molecular Bioluminescence Imaging: New Tools and Applications. Trends Biotechnol. 2017, 35, 640–652. [Google Scholar] [CrossRef] [PubMed]
- Kocher, B.; Piwnica-Worms, D. Illuminating Cancer Systems with Genetically Engineered Mouse Models and Coupled Luciferase Reporters In Vivo. Cancer Discov. 2013, 3, 616–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stacer, A.C.; Nyati, S.; Moudgil, P.; Iyengar, R.; Luker, K.E.; Rehemtulla, A.; Luker, G.D. NanoLuc reporter for dual luciferase imaging in living animals. Mol. Imaging 2013, 12, 1–13. [Google Scholar] [CrossRef]
- Daniel, C.; Poiret, S.; Dennin, V.; Boutillier, D.; Lacorre, D.A.; Foligné, B.; Pot, B. Dual-Color Bioluminescence Imaging for Simultaneous Monitoring of the Intestinal Persistence of Lactobacillus plantarum and Lactococcus lactis in Living Mice. Appl. Environ. Microbiol. 2015, 81, 5344–5349. [Google Scholar] [CrossRef] [Green Version]
- Mezzanotte, L.; Que, I.; Kaijzel, E.; Branchini, B.; Roda, A.; Löwik, C. Sensitive Dual Color In Vivo Bioluminescence Imaging Using a New Red Codon Optimized Firefly Luciferase and a Green Click Beetle Luciferase. PLoS ONE 2011, 6, e19277. [Google Scholar] [CrossRef] [PubMed]
- Gammon, S.T.; Leevy, W.M.; Gross, S.; Gokel, G.W.; Piwnica-Worms, D. Spectral Unmixing of Multicolored Bioluminescence Emitted from Heterogeneous Biological Sources. Anal. Chem. 2006, 78, 1520–1527. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.W.; Gammon, S.T.; Piwnica-Worms, D. Multi-modal multi-spectral intravital microscopic imaging of signaling dynamics in real-time during tumor-immune interactions. Cells 2021, 489. [Google Scholar] [CrossRef]
- Gammon, S.T.; Liu, T.W.; Piwnica-Worms, D. Interrogating Cellular Communication in Cancer with Genetically Encoded Imaging Reporters. Radiol. Imaging Cancer 2020, 2, e190053. [Google Scholar] [CrossRef] [PubMed]
- Pittet, M.J.; Weissleder, R. Intravital Imaging. Cell 2011, 147, 983–991. [Google Scholar] [CrossRef] [Green Version]
- Palmer, G.M.; Fontanella, A.N.; Shan, S.; Hanna, G.; Zhang, G.; Fraser, C.L.; Dewhirst, M.W. In vivo optical molecular imaging and analysis in mice using dorsal window chamber models applied to hypoxia, vasculature and fluorescent reporters. Nat. Protoc. 2011, 6, 1355–1366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kircher, M.F.; Gambhir, S.S.; Grimm, J. Noninvasive cell-tracking methods. Nat. Rev. Clin. Oncol. 2011, 8, 677–688. [Google Scholar] [CrossRef] [PubMed]
- Entenberg, D.; Voiculescu, S.; Guo, P.; Borriello, L.; Wang, Y.; Karagiannis, G.S.; Jones, J.; Baccay, F.; Oktay, M.; Condeelis, J. A permanent window for the murine lung enables high-resolution imaging of cancer metastasis. Nat. Methods 2018, 15, 73–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harney, A.S.; Arwert, E.N.; Entenberg, D.; Wang, Y.; Guo, P.; Qian, B.-Z.; Oktay, M.H.; Pollard, J.W.; Jones, J.G.; Condeelis, J.S. Real-Time Imaging Reveals Local, Transient Vascular Permeability, and Tumor Cell Intravasation Stimulated by TIE2hi Macrophage–Derived VEGFA. Cancer Discov. 2015, 5, 932–943. [Google Scholar] [CrossRef] [Green Version]
- Rodell, C.B.; Arlauckas, S.P.; Cuccarese, M.F.; Garris, C.S.; Li, R.; Ahmed, M.S.; Kohler, R.H.; Pittet, M.J.; Weissleder, R. TLR7/8-agonist-loaded nanoparticles promote the polarization of tumour-associated macrophages to enhance cancer immunotherapy. Nat. Biomed. Eng. 2018, 2, 578–588. [Google Scholar] [CrossRef] [PubMed]
- Suijkerbuijk, S.J.; Van Rheenen, J. From good to bad: Intravital imaging of the hijack of physiological processes by cancer cells. Dev. Biol. 2017, 428, 328–337. [Google Scholar] [CrossRef]
- Lohela, M.; Casbon, A.-J.; Olow, A.; Bonham, L.; Branstetter, D.; Weng, N.; Smith, J.; Werb, Z. Intravital imaging reveals distinct responses of depleting dynamic tumor-associated macrophage and dendritic cell subpopulations. Proc. Natl. Acad. Sci. USA 2014, 111, E5086–E5095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lohela, M.; Werb, Z. Intravital imaging of stromal cell dynamics in tumors. Curr. Opin. Genet. Dev. 2010, 20, 72–78. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Wysocki, R.W.; Amoozgar, Z.; Maiorino, L.; Fein, M.R.; Jorns, J.; Schott, A.F.; Kinugasa-Katayama, Y.; Lee, Y.; Won, N.H.; et al. Cancer cells induce metastasis-supporting neutrophil extracellular DNA traps. Sci. Transl. Med. 2016, 8, 361ra138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fein, M.R.; Egeblad, M. Caught in the act: Revealing the metastatic process by live imaging. Dis. Model. Mech. 2013, 6, 580–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Figley, S.A.; Chen, Y.; Maeda, A.; Conroy, L.; McMullen, J.D.; Silver, J.I.; Stapleton, S.; Vitkin, A.; Lindsay, P.; Burrell, K.; et al. A Spinal Cord Window Chamber Model for In Vivo Longitudinal Multimodal Optical and Acoustic Imaging in a Murine Model. PLoS ONE 2013, 8, e58081. [Google Scholar] [CrossRef] [PubMed]
- Rouffiac, V.; Roux, K.S.; Salomé-Desnoulez, S.; Leguerney, I.; Ginefri, J.; Sébrié, C.; Jourdain, L.; Lécluse, Y.; Laplace-Builhé, C. Multimodal imaging for tumour characterization from micro- to macroscopic level using a newly developed dorsal chamber designed for long-term follow-up. J. Biophotonics 2020, 13, e201900217. [Google Scholar] [CrossRef]
- Souris, J.S.; Hickson, J.; Msezane, L.; Rinker-Schaeffer, C.W.; Chen, C.-T. Flexible peritoneal windows for quantitative fluorescence and bioluminescence preclinical imaging. Mol. Imaging 2013, 12, 28–38. [Google Scholar]
- Moss, B.L.; Gross, S.; Gammon, S.T.; Vinjamoori, A.; Piwnica-Worms, D. Identification of a ligand-induced transient refractory period in nuclear factor-kappaB signaling. J. Biol. Chem. 2008, 283, 8687–8698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moss, B.L.; Elhammali, A.; Fowlkes, T.; Gross, S.; Vinjamoori, A.; Contag, C.H.; Piwnica-Worms, D. Interrogation of Inhibitor of Nuclear Factor κB α/Nuclear Factor κB (IκBα/NF-κB) Negative Feedback Loop Dynamics. J. Biol. Chem. 2012, 287, 31359–31370. [Google Scholar] [CrossRef] [Green Version]
- Warrington, N.M.; Gianino, S.M.; Jackson, E.; Goldhoff, P.; Garbow, J.R.; Piwnica-Worms, D.; Gutmann, D.H.; Rubin, J.B. Cyclic AMP Suppression Is Sufficient to Induce Gliomagenesis in a Mouse Model of Neurofibromatosis-1. Cancer Res. 2010, 70, 5717–5727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, S.; Piwnica-Worms, D. Real-time imaging of ligand-induced IKK activation in intact cells and in living mice. Nat. Chem. Biol. 2005, 2, 607–614. [Google Scholar] [CrossRef]
- Fuerer, C.; Nusse, R. Lentiviral Vectors to Probe and Manipulate the Wnt Signaling Pathway. PLoS ONE 2010, 5, e9370. [Google Scholar] [CrossRef]
- Tinkum, K.L.; Marpegan, L.; White, L.S.; Sun, J.; Herzog, E.D.; Piwnica-Worms, D. Bioluminescence Imaging Captures the Expression and Dynamics of Endogenous p21 Promoter Activity in Living Mice and Intact Cells. Mol. Cell. Biol. 2011, 31, 3759–3772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Wang, H.; Hsiao, C.-H.; Chow, D.S.-L.; Koay, E.J.; Kang, Y.; Wen, X.; Huang, Q.; Ma, Y.; Bankson, J.A.; et al. Simultaneous inhibition of hedgehog signaling and tumor proliferation remodels stroma and enhances pancreatic cancer therapy. Biomaterials 2018, 159, 215–228. [Google Scholar] [CrossRef]
- Ritsma, L.; Steller, E.J.; Ellenbroek, S.I.J.; Kranenburg, O.; Rinkes, I.H.M.B.; Van Rheenen, J. Surgical implantation of an abdominal imaging window for intravital microscopy. Nat. Protoc. 2013, 8, 583–594. [Google Scholar] [CrossRef] [Green Version]
- Gandelman, O.; Brovko, L.; Ugarova, N.; Chikishev, A.; Shkurimov, A. Oxyluciferin fluorescence is a model of native bioluminescence in the firefly luciferin—luciferase system. J. Photochem. Photobiol. B Biol. 1993, 19, 187–191. [Google Scholar] [CrossRef]
- Gross, S.; Gammon, S.T.; Moss, B.L.; Rauch, D.; Harding, J.J.; Heinecke, J.W.; Ratner, L.; Piwnica-Worms, D. Bioluminescence imaging of myeloperoxidase activity in vivo. Nat. Med. 2009, 15, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goiffon, R.J.; Martinez, S.C.; Piwnica-Worms, D. A rapid bioluminescence assay for measuring myeloperoxidase activity in human plasma. Nat. Commun. 2015, 6, 6271. [Google Scholar] [CrossRef] [Green Version]
- Kielland, A.; Blom, T.; Nandakumar, K.S.; Holmdahl, R.; Blomhoff, R.; Carlsen, H. In vivo imaging of reactive oxygen and nitrogen species in inflammation using the luminescent probe L-012. Free Radic. Biol. Med. 2009, 47, 760–766. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.W.; Gammon, S.T.; Yang, P.; Fuentes, D.; Piwnica-Worms, D. HOCl is a paracrine effector linking myeloid cells to NF-kB signaling in melanoma by trans-inhibition of IKK. Sci. Signal. 2021. Accepted. [Google Scholar]
- Conroy, E.; Aviello, G. Imaging Intestinal ROS in Homeostatic Conditions Using L-012. Breast Cancer 2019, 1982, 313–327. [Google Scholar] [CrossRef]
- Emani, R.; Asghar, M.N.; Toivonen, R.; Laurén, L.; Soderstrom, M.; Toivola, D.M.; Van Tol, E.A.F.; Hänninen, A. Casein hydrolysate diet controls intestinal T cell activation, free radical production and microbial colonisation in NOD mice. Diabetologia 2013, 56, 1781–1791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, M.P.; Woodroofe, C.C.; Wood, M.G.; Que, I.; Root, M.V.; Ridwan, Y.; Shi, C.; Kirkland, T.A.; Encell, L.P.; Wood, K.V.; et al. Click beetle luciferase mutant and near infrared naphthyl-luciferins for improved bioluminescence imaging. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, T.W.; Gammon, S.T.; Fuentes, D.; Piwnica-Worms, D. Multi-Modal Multi-Spectral Intravital Macroscopic Imaging of Signaling Dynamics in Real Time during Tumor–Immune Interactions. Cells 2021, 10, 489. https://doi.org/10.3390/cells10030489
Liu TW, Gammon ST, Fuentes D, Piwnica-Worms D. Multi-Modal Multi-Spectral Intravital Macroscopic Imaging of Signaling Dynamics in Real Time during Tumor–Immune Interactions. Cells. 2021; 10(3):489. https://doi.org/10.3390/cells10030489
Chicago/Turabian StyleLiu, Tracy W., Seth T. Gammon, David Fuentes, and David Piwnica-Worms. 2021. "Multi-Modal Multi-Spectral Intravital Macroscopic Imaging of Signaling Dynamics in Real Time during Tumor–Immune Interactions" Cells 10, no. 3: 489. https://doi.org/10.3390/cells10030489