
Multi-Modal Multi-Spectral Intravital Microscopic Imaging of Signaling Dynamics in Real-Time during Tumor–Immune Interactions


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Microscope Setup and Configuration
2.2. iKon Camera Characterization
2.3. Reagents
2.4. Cells
2.5. Plasmids
2.6. Generation of Stable Reporter Cells
2.7. Animals
2.8. Preparation for Window Chamber Implantation Surgery
2.9. Dorsal Skin Window Chamber Implantation Surgery
2.10. Abdominal Pancreas Window Chamber Implantation Surgery
2.11. Post Window Chamber Implantation Surgery
2.12. In Vivo Intravital Microscopy
2.13. Skin Window Chamber Intravital Microscopy
2.14. Photoswitching Dendra2 Fluorescent Protein
2.15. Spectral Unmixing
2.16. Abdominal Window Chamber Intravital Microscopy
2.17. Histological Analysis
2.18. Statistical Analyses
2.19. Data Availability
3. Results
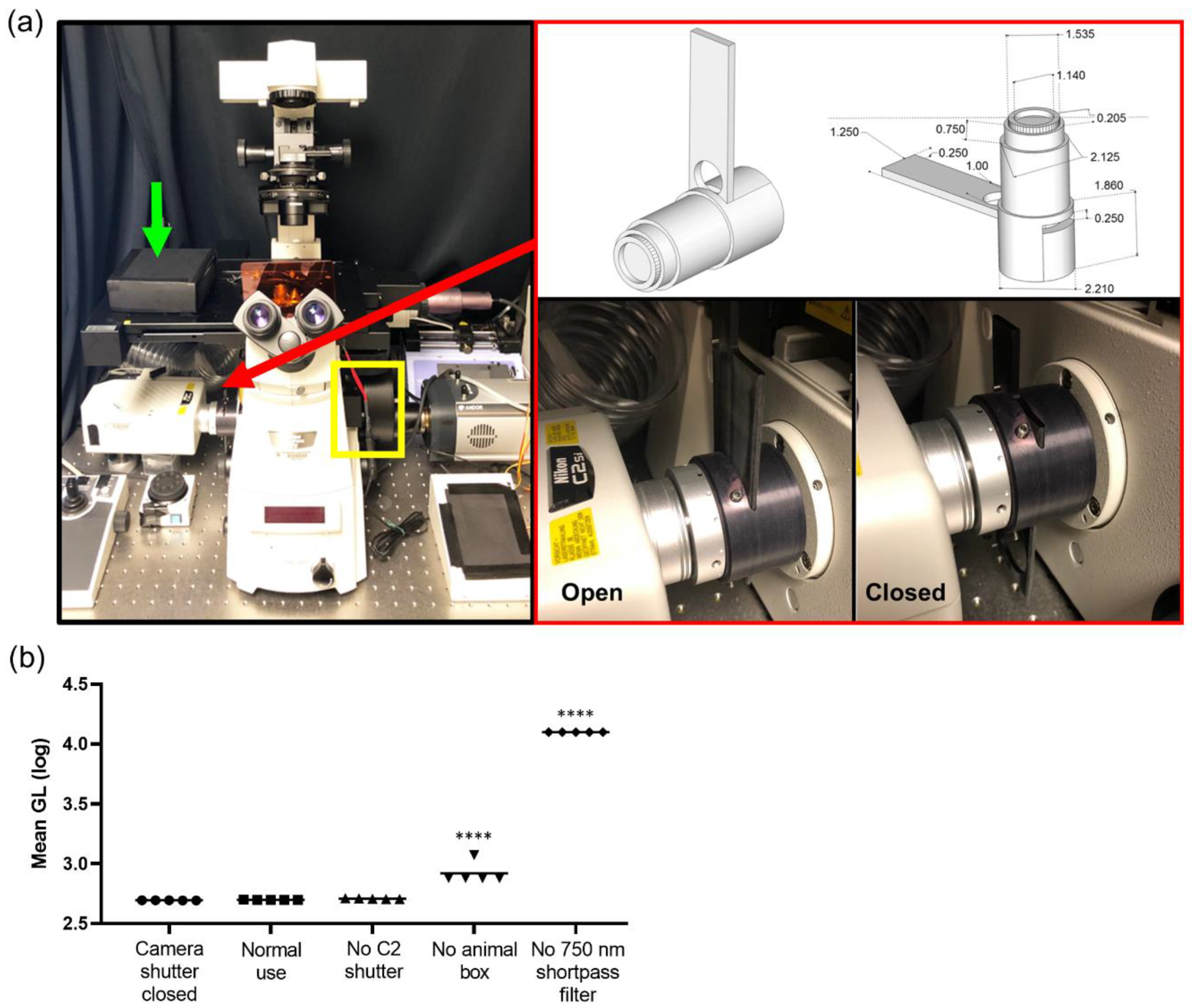
3.1. Elimination of Light Sources by Custom Modification of an off-the-Shelf Imaging System
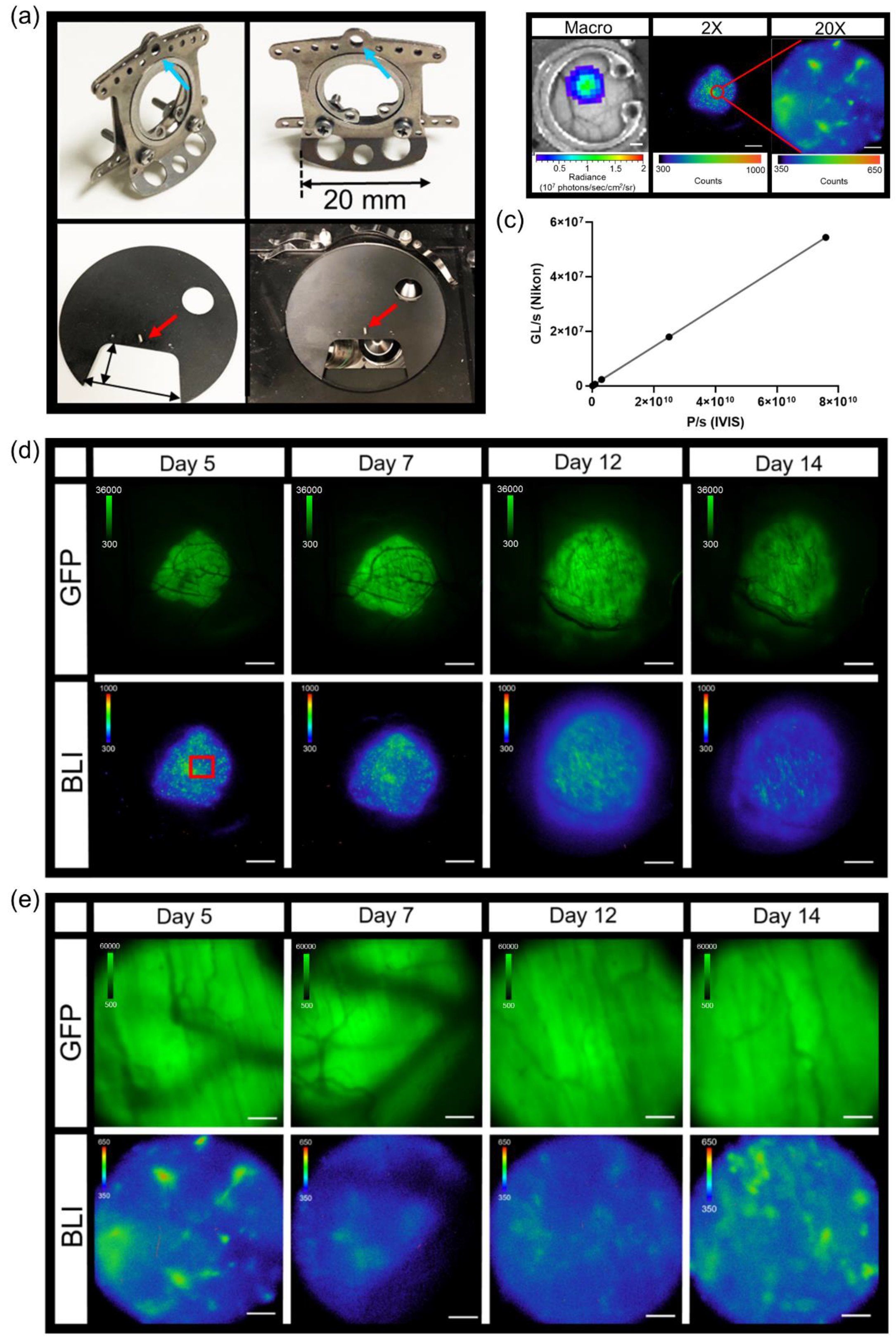
3.2. In Vivo Dorsal Skin Window Chamber Intravital Bioluminescence Imaging
3.3. Imaging and Quantification of Tumor Signaling Dynamics
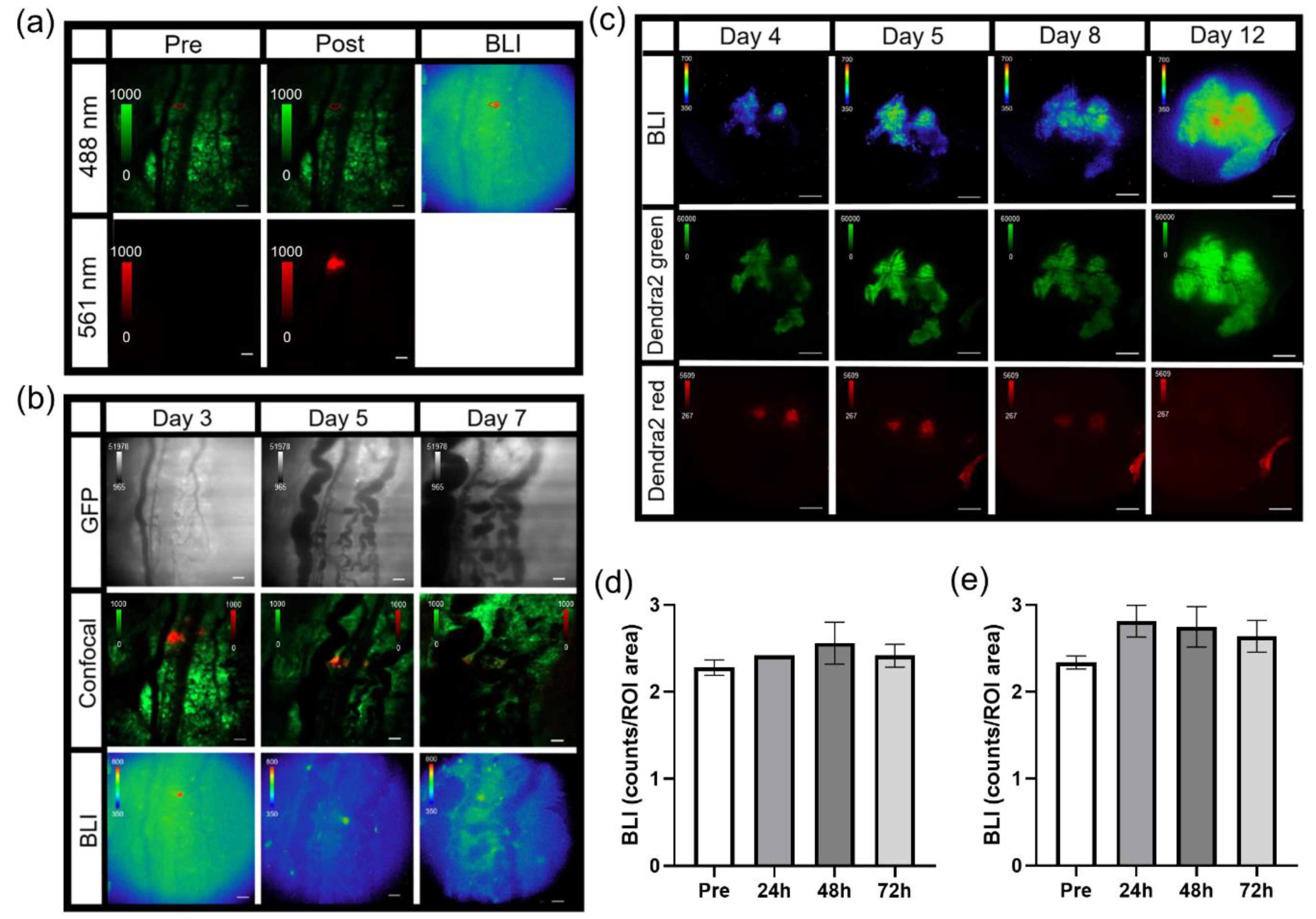
3.4. Photoswitchable IVM Imaging
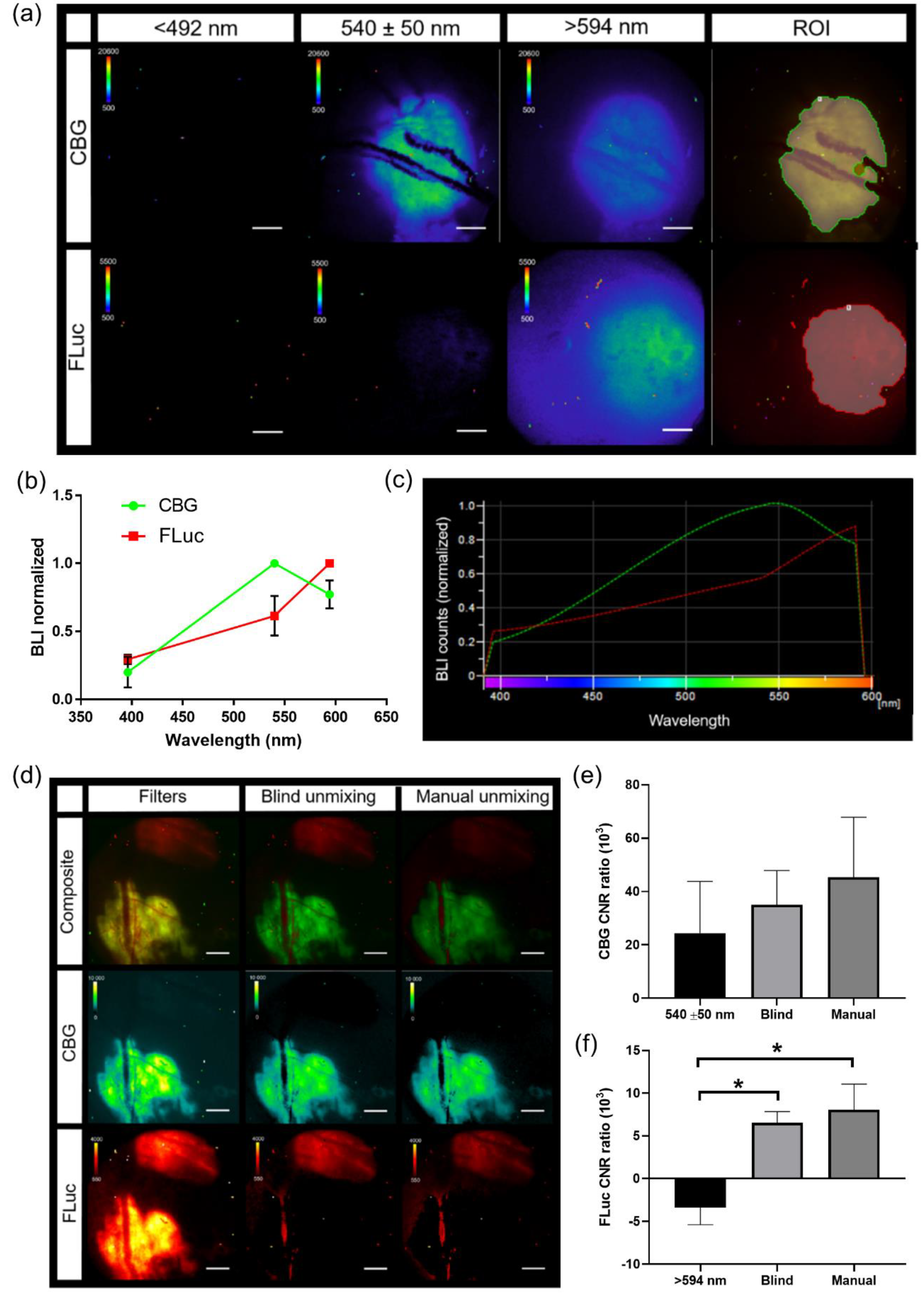
3.5. Generation of Spectra In Vivo for IVM
3.6. IVM of Abdominal Window Chamber Tumor-Bearing Animals
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pittet, M.J.; Weissleder, R. Intravital Imaging. Cell 2011, 147, 983–991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, G.M.; Fontanella, A.N.; Shan, S.; Hanna, G.; Zhang, G.; Fraser, C.L.; Dewhirst, M.W. In vivo optical molecular imaging and analysis in mice using dorsal window chamber models applied to hypoxia, vasculature and fluorescent reporters. Nat. Protoc. 2011, 6, 1355–1366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kircher, M.F.; Gambhir, S.S.; Grimm, J. Noninvasive cell-tracking methods. Nat. Rev. Clin. Oncol. 2011, 8, 677–688. [Google Scholar] [CrossRef]
- Entenberg, D.; Voiculescu, S.; Guo, P.; Borriello, L.; Wang, Y.; Karagiannis, G.S.; Jones, J.; Baccay, F.; Oktay, M.; Condeelis, J. A permanent window for the murine lung enables high-resolution imaging of cancer metastasis. Nat. Methods 2017, 15, 73–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suijkerbuijk, S.J.E.; van Rheenen, J. From good to bad: Intravital imaging of the hijack of physiological processes by cancer cells. Dev. Biol. 2017, 428, 328–337. [Google Scholar] [CrossRef]
- Lohela, M.; Werb, Z. Intravital imaging of stromal cell dynamics in tumors. Curr. Opin. Genet. Dev. 2010, 20, 72–78. [Google Scholar] [CrossRef] [Green Version]
- Fein, M.R.; Egeblad, M. Caught in the act: Revealing the metastatic process by live imaging. Dis. Model. Mech. 2013, 6, 580–593. [Google Scholar] [CrossRef] [Green Version]
- Harney, A.S.; Arwert, E.N.; Entenberg, D.; Wang, Y.; Guo, P.; Qian, B.-Z.; Oktay, M.H.; Pollard, J.W.; Jones, J.G.; Condeelis, J.S. Real-Time Imaging Reveals Local, Transient Vascular Permeability, and Tumor Cell Intravasation Stimulated by TIE2hi Macrophage–Derived VEGFA. Cancer Discov. 2015, 5, 932–943. [Google Scholar] [CrossRef] [Green Version]
- Lohela, M.; Casbon, A.-J.; Olow, A.; Bonham, L.; Branstetter, D.; Weng, N.; Smith, J.; Werb, Z. Intravital imaging reveals distinct responses of depleting dynamic tumor-associated macrophage and dendritic cell subpopulations. Proc. Natl. Acad. Sci. USA 2014, 111, E5086–E5095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Wysocki, R.W.; Amoozgar, Z.; Maiorino, L.; Fein, M.R.; Jorns, J.; Schott, A.F.; Kinugasa-Katayama, Y.; Lee, Y.; Won, N.H.; et al. Cancer cells induce metastasis-supporting neutrophil extracellular DNA traps. Sci. Transl. Med. 2016, 8, 361ra138. [Google Scholar] [CrossRef] [Green Version]
- Prescher, J.A.; Contag, C.H. Guided by the light: Visualizing biomolecular processes in living animals with bioluminescence. Curr. Opin. Chem. Biol. 2010, 14, 80–89. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, S.; Piwnicaworms, D. Spying on cancerMolecular imaging in vivo with genetically encoded reporters. Cancer Cell 2005, 7, 5–15. [Google Scholar] [CrossRef]
- Kocher, B.; Piwnica-Worms, D. Illuminating Cancer Systems with Genetically Engineered Mouse Models and Coupled Luciferase Reporters In Vivo. Cancer Discov. 2013, 3, 616–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gammon, S.T.; Liu, T.W.; Piwnica-Worms, D. Interrogating Cellular Communication in Cancer with Genetically Encoded Imaging Reporters. Radiol. Imaging Cancer 2020, 2, e190053. [Google Scholar] [CrossRef]
- Mezzanotte, L.; Root, M.V.; Karatas, H.; Goun, E.A.; Löwik, C.W. In Vivo Molecular Bioluminescence Imaging: New Tools and Applications. Trends Biotechnol. 2017, 35, 640–652. [Google Scholar] [CrossRef]
- Souris, J.S.; Hickson, J.A.; Msezane, L.; Rinker-Schaeffer, C.W.; Chen, C.-T. Flexible peritoneal windows for quantitative fluorescence and bioluminescence preclinical imaging. Mol. Imaging 2013, 12, 28–38. [Google Scholar]
- Figley, S.A.; Chen, Y.; Maeda, A.; Conroy, L.; McMullen, J.D.; Silver, J.I.; Stapleton, S.; Vitkin, A.; Lindsay, P.; Burrell, K.; et al. A Spinal Cord Window Chamber Model for In Vivo Longitudinal Multimodal Optical and Acoustic Imaging in a Murine Model. PLoS ONE 2013, 8, e58081. [Google Scholar] [CrossRef]
- Rouffiac, V.; Roux, K.S.; Salomé-Desnoulez, S.; Leguerney, I.; Ginefri, J.; Sébrié, C.; Jourdain, L.; Lécluse, Y.; Laplace-Builhé, C. Multimodal imaging for tumour characterization from micro- to macroscopic level using a newly developed dorsal chamber designed for long-term follow-up. J. Biophotonics 2019, 13, e201900217. [Google Scholar] [CrossRef] [PubMed]
- Singer, R.H.; Lawrence, D.S.; Ovryn, B.; Condeelis, J. Imaging of gene expression in living cells and tissues. J. Biomed. Opt. 2005, 10, 051406. [Google Scholar] [CrossRef] [PubMed]
- Dothager, R.S.; Flentie, K.; Moss, B.; Pan, M.-H.; Kesarwala, A.; Piwnica-Worms, D. Advances in bioluminescence imaging of live animal models. Curr. Opin. Biotechnol. 2009, 20, 45–53. [Google Scholar] [CrossRef] [Green Version]
- Gross, S.; Piwnica-Worms, D. Real-time imaging of ligand-induced IKK activation in intact cells and in living mice. Nat. Chem. Biol. 2005, 2, 607–614. [Google Scholar] [CrossRef]
- Tinkum, K.L.; Marpegan, L.; White, L.S.; Sun, J.; Herzog, E.D.; Piwnica-Worms, D. Bioluminescence Imaging Captures the Expression and Dynamics of Endogenous p21 Promoter Activity in Living Mice and Intact Cells. Mol. Cell. Biol. 2011, 31, 3759–3772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilagan, M.X.G.; Lim, S.; Fulbright, M.; Piwnica-Worms, D.; Kopan, R. Real-Time Imaging of Notch Activation with a Luciferase Complementation-Based Reporter. Sci. Signal. 2011, 4, rs7. [Google Scholar] [CrossRef] [Green Version]
- Spiller, D.G.; Wood, C.D.; Rand, D.A.; White, M.R.H. Measurement of single-cell dynamics. Nature 2010, 465, 736–745. [Google Scholar] [CrossRef] [PubMed]
- Negrin, R.S.; Contag, C.H. In vivo imaging using bioluminescence: A tool for probing graft-versus-host disease. Nat. Rev. Immunol. 2006, 6, 484–490. [Google Scholar] [CrossRef]
- Moss, B.L.; Elhammali, A.; Fowlkes, T.; Gross, S.; Vinjamoori, A.; Contag, C.H.; Piwnica-Worms, D. Interrogation of inhibitor of nuclear factor kappaB alpha/nuclear factor kappaB (IkappaBalpha/NF-kappaB) negative feedback loop dynamics: From single cells to live animals in vivo. J. Biol. Chem. 2012, 287, 31359–31370. [Google Scholar] [CrossRef] [Green Version]
- Gammon, S.T.; Leevy, W.M.; Gross, S.; Gokel, G.W.; Piwnica-Worms, D. Spectral Unmixing of Multicolored Bioluminescence Emitted from Heterogeneous Biological Sources. Anal. Chem. 2006, 78, 1520–1527. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.W.; Gammon, S.T.; Fuentes, D.; Piwnica-Worms, D. Multi-modal multi-spectral intravital macroscopic imaging of signaling dynamics in real-time during tumor-immune interactions. Cells 2021. Accepted. Companion paper 2. [Google Scholar]
- Stacer, A.C.; Nyati, S.; Moudgil, P.; Iyengar, R.; Luker, K.E.; Rehemtulla, A.; Luker, G.D. NanoLuc reporter for dual luciferase imaging in living animals. Mol. Imaging 2013, 12, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Daniel, C.; Poiret, S.; Dennin, V.; Boutillier, D.; Lacorre, D.A.; Foligné, B.; Pot, B. Dual-Color Bioluminescence Imaging for Simultaneous Monitoring of the Intestinal Persistence of Lactobacillus plantarum and Lactococcus lactis in Living Mice. Appl. Environ. Microbiol. 2015, 81, 5344–5349. [Google Scholar] [CrossRef] [Green Version]
- Mezzanotte, L.; Que, I.; Kaijzel, E.; Branchini, B.; Roda, A.; Löwik, C. Sensitive Dual Color In Vivo Bioluminescence Imaging Using a New Red Codon Optimized Firefly Luciferase and a Green Click Beetle Luciferase. PLoS ONE 2011, 6, e19277. [Google Scholar] [CrossRef]
- Moss, B.L.; Gross, S.; Gammon, S.T.; Vinjamoori, A.; Piwnica-Worms, D. Identification of a ligand-induced transient refractory period in nuclear factor-kappaB signaling. J. Biol. Chem. 2008, 283, 8687–8698. [Google Scholar] [CrossRef] [Green Version]
- Warrington, N.M.; Gianino, S.M.; Jackson, E.; Goldhoff, P.; Garbow, J.R.; Piwnica-Worms, D.; Gutmann, D.H.; Rubin, J.B. Cyclic AMP Suppression Is Sufficient to Induce Gliomagenesis in a Mouse Model of Neurofibromatosis-1. Cancer Res. 2010, 70, 5717–5727. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Wang, H.; Hsiao, C.-H.; Chow, D.S.-L.; Koay, E.J.; Kang, Y.; Wen, X.; Huang, Q.; Ma, Y.; Bankson, J.A.; et al. Simultaneous inhibition of hedgehog signaling and tumor proliferation remodels stroma and enhances pancreatic cancer therapy. Biomaterials 2018, 159, 215–228. [Google Scholar] [CrossRef]
- Ritsma, L.; Steller, E.J.A.; Ellenbroek, S.I.J.; Kranenburg, O.; Rinkes, I.H.M.B.; Van Rheenen, J. Surgical implantation of an abdominal imaging window for intravital microscopy. Nat. Protoc. 2013, 8, 583–594. [Google Scholar] [CrossRef] [Green Version]
- Egeblad, M.; Ewald, A.J.; Askautrud, H.A.; Truitt, M.L.; Welm, B.E.; Bainbridge, E.; Peeters, G.; Krummel, M.F.; Werb, Z. Visualizing stromal cell dynamics in different tumor microenvironments by spinning disk confocal microscopy. Dis. Model. Mech. 2008, 1, 155–167. [Google Scholar] [CrossRef] [Green Version]
- Devi, S.; Li, A.; Westhorpe, C.L.V.; Lo, C.Y.; Abeynaike, L.D.; Snelgrove, S.L.; Hall, P.; Ooi, J.D.; Sobey, C.G.; Kitching, A.R.; et al. Multiphoton imaging reveals a new leukocyte recruitment paradigm in the glomerulus. Nat. Med. 2012, 19, 107–112. [Google Scholar] [CrossRef]
- Kolaczkowska, E.; Jenne, C.N.; Surewaard, B.G.; Thanabalasuriar, A.; Lee, W.Y.; Sanz, M.J.; Mowen, K.; Opdenakker, G.; Kubes, P. Molecular mechanisms of NET formation and degradation revealed by intravital imaging in the liver vasculature. Nat. Commun. 2015, 6, 6673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, C.A.; Kim, E.Y.; Marangoni, F.; Carrizosa, E.; Claudio, N.M.; Mempel, T.R. Dynamic Treg interactions with intratumoral APCs promote local CTL dysfunction. J. Clin. Investig. 2014, 124, 2425–2440. [Google Scholar] [CrossRef] [PubMed]
- Roh-Johnson, M.; Shah, A.N.; Stonick, J.A.; Poudel, K.R.; Kargl, J.; Yang, G.H.; Di Martino, J.; Hernandez, R.E.; Gast, C.E.; Zarour, L.R.; et al. Macrophage-Dependent Cytoplasmic Transfer during Melanoma Invasion In Vivo. Dev. Cell 2017, 43, 549–562.e6. [Google Scholar] [CrossRef]
- Laviron, M.; Combadière, C.; Boissonnas, A. Tracking Monocytes and Macrophages in Tumors with Live Imaging. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Fiole, D.; Deman, P.; Trescos, Y.; Mayol, J.F.; Mathieu, J.; Vial, J.C.; Douady, J.; Tournier, J.N. Two-photon intravital imaging of lungs during anthrax infection reveals long-lasting macrophage-dendritic cell contacts. Infect. Immun. 2014, 82, 864–872. [Google Scholar] [CrossRef] [Green Version]
- Rua, R.; McGavern, D.B. Elucidation of monocyte/macrophage dynamics and function by intravital imaging. J. Leukoc. Biol. 2015, 98, 319–332. [Google Scholar] [CrossRef]
- Hickman, H.D.; Li, L.; Reynoso, G.V.; Rubin, E.J.; Skon, C.N.; Mays, J.W.; Gibbs, J.; Schwartz, O.; Bennink, J.R.; Yewdell, J.W. Chemokines control naive CD8+ T cell selection of optimal lymph node antigen presenting cells. J. Exp. Med. 2011, 208, 2511–2524. [Google Scholar] [CrossRef]
- Evans, T.A.; Barkauskas, D.S.; Myers, J.T.; Hare, E.G.; You, J.Q.; Ransohoff, R.M.; Huang, A.Y.; Silver, J. High-resolution intravital imaging reveals that blood-derived macrophages but not resident microglia facilitate secondary axonal dieback in traumatic spinal cord injury. Exp. Neurol. 2014, 254, 109–120. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.W.; Gammon, S.T.; Yang, P.; Fuentes, D.; Piwnica-Worms, D. Myeloid cell-derived HOCl is a paracrine effector that trans-inhibits IKK/NF-κB in melanoma cells and limits early tumor progression. Sci. Signal 2021. Accepted. [Google Scholar]
- Gurskaya, N.G.; Verkhusha, V.V.; Shcheglov, A.S.; Staroverov, D.B.; Chepurnykh, T.V.; Fradkov, A.F.; Lukyanov, S.; Lukyanov, K.A. Engineering of a monomeric green-to-red photoactivatable fluorescent protein induced by blue light. Nat. Biotechnol. 2006, 24, 461–465. [Google Scholar] [CrossRef] [PubMed]
- Goiffon, R.J.; Martinez, S.C.; Piwnica-Worms, D. A rapid bioluminescence assay for measuring myeloperoxidase activity in human plasma. Nat. Commun. 2015, 6, 6271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Askoxylakis, V.; Badeaux, M.; Roberge, S.; Batista, A.; Kirkpatrick, N.; Snuderl, M.; Amoozgar, Z.; Seano, G.; Ferraro, G.B.; Chatterjee, S.; et al. A cerebellar window for intravital imaging of normal and disease states in mice. Nat. Protoc. 2017, 12, 2251–2262. [Google Scholar] [CrossRef] [PubMed]
- Bousso, P. T-cell activation by dendritic cells in the lymph node: Lessons from the movies. Nat. Rev. Immunol. 2008, 8, 675–684. [Google Scholar] [CrossRef]
- Celli, S.; Albert, M.L.; Bousso, P. Visualizing the innate and adaptive immune responses underlying allograft rejection by two-photon microscopy. Nat. Med. 2011, 17, 744–749. [Google Scholar] [CrossRef] [PubMed]
- Condeelis, J.; Segall, J.E. Intravital imaging of cell movement in tumours. Nat. Rev. Cancer 2003, 3, 921–930. [Google Scholar] [CrossRef]
- Conway, J.R.; Carragher, N.O.; Timpson, P. Developments in preclinical cancer imaging: Innovating the discovery of thera-peutics. Nat. Rev. Cancer 2014, 14, 314–328. [Google Scholar] [CrossRef]
- Entenberg, D.; Wyckoff, J.; Gligorijevic, B.; Roussos, E.T.; Verkhusha, V.V.; Pollard, J.W.; Condeelis, J. Setup and use of a two-laser multiphoton microscope for multichannel intravital fluorescence imaging. Nat. Protoc. 2011, 6, 1500–1520. [Google Scholar] [CrossRef] [Green Version]
- Germain, R.N.; Robey, E.A.; Cahalan, M.D. A Decade of Imaging Cellular Motility and Interaction Dynamics in the Immune System. Science 2012, 336, 1676–1681. [Google Scholar] [CrossRef] [Green Version]
- Liew, P.X.; Kubes, P. Intravital imaging—Dynamic insights into natural killer T cell biology. Front. Immunol. 2015, 6, 240. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.Z.; McKeown, M.R.; Ng, H.-L.; Aguilera, T.A.; Shaner, N.C.; Campbell, R.E.; Adams, S.R.; Gross, L.A.; Ma, W.; Alber, T.; et al. Autofluorescent Proteins with Excitation in the Optical Window for Intravital Imaging in Mammals. Chem. Biol. 2009, 16, 1169–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo Celso, C.; Wu, J.W.; Lin, C.P. In vivo imaging of hematopoietic stem cells and their microenvironment. J. Biophotonics 2009, 2, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Karin, M. Nuclear factor-kappaB in cancer development and progression. Nature 2006, 441, 431–436. [Google Scholar] [CrossRef]
- Oeckinghaus, A.; Hayden, M.S.; Ghosh, S. Crosstalk in NF-kappaB signaling pathways. Nat. Immunol. 2011, 12, 695–708. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.D. The diverse and complex roles of NF-kappaB subunits in cancer. Nat. Rev. Cancer 2012, 12, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Karin, M. NF-κB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef]
- Yang, G.; Pan, F.; Parkhurst, C.N.; Grutzendler, J.; Gan, W.-B. Thinned-skull cranial window technique for long-term imaging of the cortex in live mice. Nat. Protoc. 2010, 5, 201–208. [Google Scholar] [CrossRef] [Green Version]
- Heo, C.; Park, H.; Kim, Y.-T.; Baeg, E.; Kim, S.-G.; Suh, M. A soft, transparent, freely accessible cranial window for chronic imaging and electrophysiology. Sci. Rep. 2016, 6, 27818. [Google Scholar] [CrossRef] [Green Version]
- Hansen-Algenstaedt, N.; Schaefer, C.; Wolfram, L.; Joscheck, C.; Schroeder, M.; Algenstaedt, P.; Ruther, W. Femur window—A new approach to microcirculation of living bone in situ. J. Orthop. Res. 2005, 23, 1073–1082. [Google Scholar] [CrossRef]
- Kedrin, D.; Gligorijevic, B.; Wyckoff, J.; Verkhusha, V.V.; Condeelis, J.; Segall, J.E.; Van Rheenen, J. Intravital imaging of metastatic behavior through a mammary imaging window. Nat. Methods 2008, 5, 1019–1021. [Google Scholar] [CrossRef]
- Ito, K.; Smith, B.R.; Parashurama, N.; Yoon, J.-K.; Song, S.Y.; Miething, C.; Mallick, P.; Lowe, S.; Gambhir, S.S. Unexpected Dissemination Patterns in Lymphoma Progression Revealed by Serial Imaging within a Murine Lymph Node. Cancer Res. 2012, 72, 6111–6118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrar, M.J.; Bernstein, I.M.; Schlafer, D.H.; Cleland, T.A.; Fetcho, J.R.; Schaffer, C.B. Chronic in vivo imaging in the mouse spinal cord using an implanted chamber. Nat. Methods 2012, 9, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, T.; Pollard, J.W.; Vendrell, M. Optical Windows for Imaging the Metastatic Tumour Microenvironment in vivo. Trends Biotechnol. 2017, 35, 5–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, T.W.; Gammon, S.T.; Piwnica-Worms, D. Multi-Modal Multi-Spectral Intravital Microscopic Imaging of Signaling Dynamics in Real-Time during Tumor–Immune Interactions. Cells 2021, 10, 499. https://doi.org/10.3390/cells10030499
Liu TW, Gammon ST, Piwnica-Worms D. Multi-Modal Multi-Spectral Intravital Microscopic Imaging of Signaling Dynamics in Real-Time during Tumor–Immune Interactions. Cells. 2021; 10(3):499. https://doi.org/10.3390/cells10030499
Chicago/Turabian StyleLiu, Tracy W., Seth T. Gammon, and David Piwnica-Worms. 2021. "Multi-Modal Multi-Spectral Intravital Microscopic Imaging of Signaling Dynamics in Real-Time during Tumor–Immune Interactions" Cells 10, no. 3: 499. https://doi.org/10.3390/cells10030499
APA StyleLiu, T. W., Gammon, S. T., & Piwnica-Worms, D. (2021). Multi-Modal Multi-Spectral Intravital Microscopic Imaging of Signaling Dynamics in Real-Time during Tumor–Immune Interactions. Cells, 10(3), 499. https://doi.org/10.3390/cells10030499