1. Introduction
Intravital microscopic imaging (IVM) has proven to be a powerful imaging technique [
1,
2,
3,
4,
5,
6,
7,
8,
9,
10,
11], one of the few molecular imaging strategies that provides both spatial and temporal information regarding heterogeneous living systems in real-time at cellular resolution. In particular, this technology has become increasingly critical in studies of the tumor immune microenvironment, as it has become increasingly recognized that the immune system plays complex dual roles in cancer, both beneficially and adversely impacting tumorigenesis [
12,
13,
14,
15]. Thus, advances that allow for the study of interactions between immune cells and tumor cells in a dynamic, physiologically relevant system in vivo have become crucial. Enormous advances in immunology have been achieved though the analysis of tissues in bulk at different stages of tumor progression as well as a variety of in vitro and ex vivo methodologies. However, many of these findings provide only a snapshot in time of orchestrated molecular and cellular changes occurring over time. Analysis of tissue in bulk (e.g., Western blots, whole genome sequencing, PCR, RPPA) disregards spatial and temporal distributions and relationships during disease development and ignores the often massive heterogeneity of tumor cells themselves (genotype and phenotype), as well as heterogeneity within the immune cell microenvironment. Advances in single cell digital techniques (single cell RNAseq, CyTOF and multiplex imaging) have laid the foundational framework for advances in cancer immunology that account for cellular heterogeneity; however, these techniques are destructive and time-course dynamics require a large number of subjects for analysis of genetic, molecular and cellular changes that cannot be tracked or sampled temporally or spatially within the same individual. Only through non-invasive imaging can tumor progression and dynamic immune cell interaction in an intact in vivo system be monitored in real-time. However, few imaging modalities can achieve non-invasive cellular resolution sufficient to image the molecular mechanisms of cell trafficking, cell–cell interactions and the associated molecular signals that IVM provides.
Current IVM strategies primarily use fluorescence imaging [
1,
2,
3,
4,
5,
6,
7]. Unfortunately, fluorescence has several limitations, including high background (autofluorescence) that renders the quantification of fluorescence difficult, resulting in low signal-to-noise ratios, photo-bleaching, and expensive imaging systems [
1,
2,
3,
4,
5,
6,
7]. Although there are numerous fluorescent proteins and/or exogenous fluorochromes, current IVM microscopy systems are generally limited in the number of fluorophores that can be imaged simultaneously [
1,
2,
3,
4,
5,
6,
7]. In addition, many fluorescent proteins have long half-lives, e.g., green fluorescent protein (GFP) has a half-life of ~12–26 h [
16,
17,
18], which limits the capacity to quantify on–off dynamics in biological systems. In contrast, bioluminescence has become a standard preclinical macro-imaging tool for monitoring cancer cell fate and tumor growth [
11,
16,
17,
18,
19]. Traditionally, tumor cells are stably engineered to express constitutively active bioluminescent reporters, which directly measure live cell metabolism [
11,
16,
17,
19,
20,
21,
22]. Bioluminescence has several advantages over fluorescence, including low background signal, high signal-to-noise ratios, and modest cost [
23,
24,
25]. In addition, luciferases have a short half-life, e.g., North American
Photinus pyralis firefly and
Renilla luciferase have a half-life of ~ 3–5 h, where their rapid translation and maturation better reflects endogenous biological activation on-rates [
17]. With the development of new bioluminescent reporters with expanded emission spectra, advances in genomic techniques, and technical improvements in bioluminescence imaging and processing techniques [
11,
16,
17,
18,
19], the applications for bioluminescence imaging are increasing. Recent advances in microscopy technologies are extending bioluminescence applications to single cell imaging [
26,
27,
28,
29,
30].
A limitation of bioluminescence is the substrate dependence of luciferase enzymes where the pharmacokinetics of substrate delivery temporally impacts the bioluminescence signal in vivo. Unlike fluorophores, there are a limited number of bioluminescent proteins, although three luciferases have been spectrally unmixed macroscopically [
31,
32]. Luciferases, such as firefly luciferase and click beetle green luciferase, which both use the same D-luciferin substrate, can be imaged, and discriminated simultaneously [
11,
16,
17,
19,
31,
33,
34,
35]. Multi-color bioluminescence can be separated using appropriate emission filters and de-convoluted using spectral unmixing algorithms [
31,
32]. In addition, bioluminescent reporters that utilize different substrates are easily resolved in the same animal with separate sequential imaging sessions [
11,
16,
17,
19,
31,
32,
33,
34,
35]. Bioluminescent reporters have been developed that can image biological events at multiple levels; monitoring the regulation of specific genes, messenger RNA processing, signal transduction, protein processing and function, and protein–protein interactions are possible using transcriptional, translational and post-translational bioluminescent reporters [
11,
16,
17,
18,
19]. The combination of various reporters (genetic, protein and/or cellular) with different luciferases provides a powerful approach to study the temporal and spatial evolution of biological processes in vivo, wherein different molecular and/or cellular events can be monitored by bioluminescence simultaneously in a single imaging session. Herein, we describe a molecular imaging window chamber platform that uniquely combines bioluminescent and fluorescent reporters with intravital microscopy, providing a first-in-class advance in technology of intravital spectral unmixing in real-time concurrent with high resolution bioluminescence imaging of signaling dynamics applicable to heterogeneous living systems in vivo.
2. Materials and Methods
2.1. Microscope Setup and Configuration
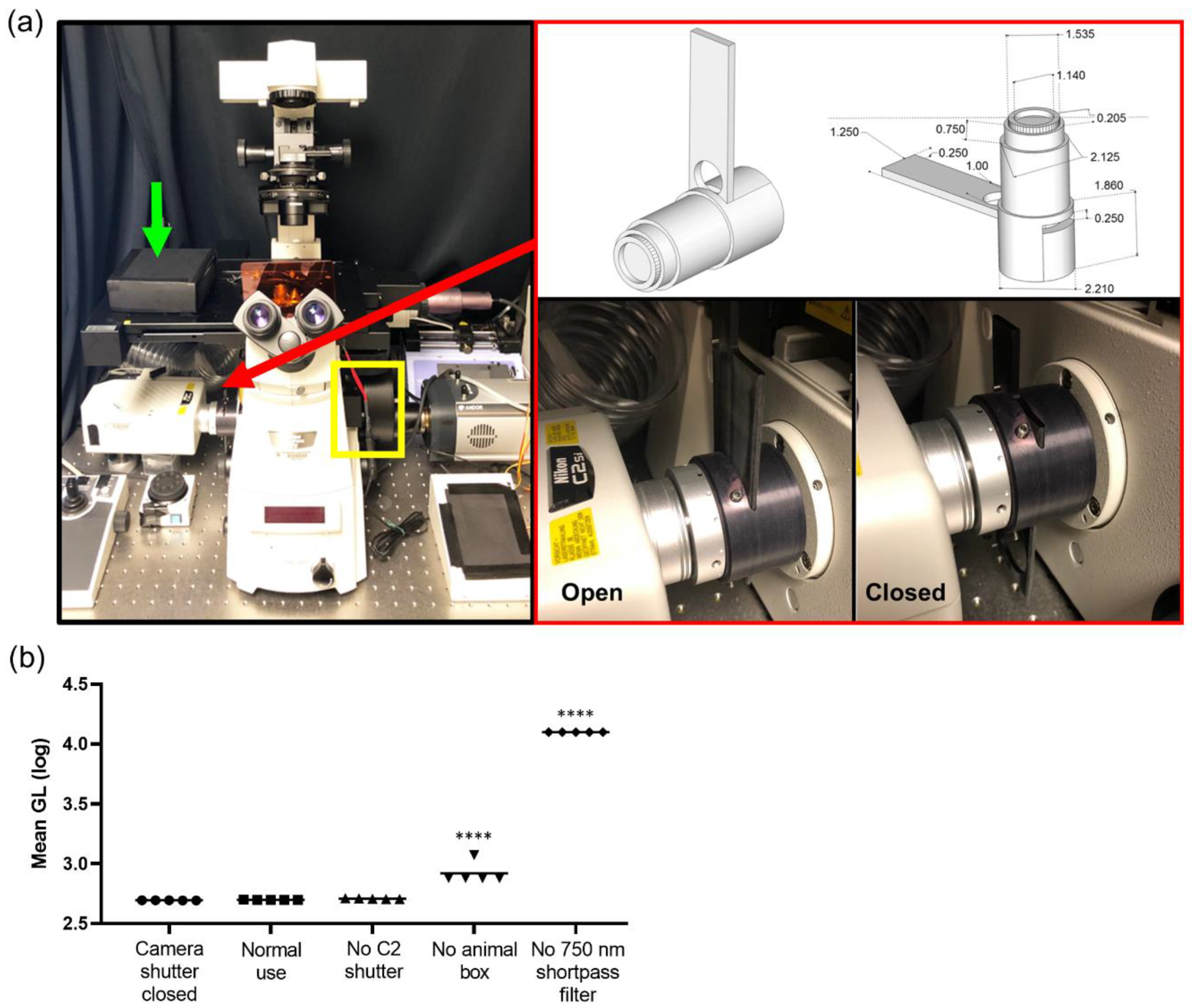
A Nikon TiE inverted microscope (Nikon Instruments, Melville, NY, USA) provided the core scaffold for the custom imaging system and was used for all intravital imaging studies. The microscope was equipped with the following objectives: 2X NA 0.1 8.5 mm WD (CFI Plan Apochromat Lambda 2X), 10X NA 0.45 (Plan Apo) and 20X NA 0.45 (s Plan Fluor extra-long working distance) (Nikon Instruments, Melville, NY, USA). The internal reflection of NIR light from the real time focal adjustment system (Perfect Focus, Nikon Instruments) was blocked by placing a 25 mm 750 nm short pass filter with >6 OD stopping power and >90% transmittance (ET750sp-2p8, Chroma Technology Corp, Bellows Falls, VT, USA) in the filter wheel (FLBW-E, Nikon Instrument) either alone or in series with 25 mm short pass, band pass, and long pass filters (BrightLine FF01-492/SP-25, Brightline FF01-540/50, Edgebasic BLP01-635R, Semrock Corporation Rochester, NY, USA). An open slot was maintained for use during fluorescence imaging (including NIR). The filter wheel was placed directly in line with the camera system, enabling both the collection of single reporter bioluminescence imaging (750 nm short pass), or multispectral bioluminescence imaging (750 nm short pass in series with the appropriate visible filter). This filter wheel was under the automated control of the microscope for the facile programming of single acquisition and multiple mixed fluorescence and bioluminescence protocols. For bioluminescence and epifluorescence imaging, a back-illuminated 1024 × 1024 pixel CCD with a 13 µm pixel pitch (iKon-M 934; DU934P-BEX2-DD, Andor Inc/Oxford Instruments Belfast, Northern Ireland) was air cooled to −85 °C during normal operations with deep depletion fringe suppression and anti-reflective coating. When acquiring bioluminescence images, the camera was read out at 50 kHz, 4X gain, and binning 2 to minimize read noise and maximize sensitivity. In the epifluorescence mode, the readout speed was 1 MHz. Confocal imaging was integrated into the platform using the LSM C2 Confocal system (Nikon Instruments, Melville, NY, USA). The isoflurane vaporizer and induction chamber were placed on a separate table adjacent to the microscope table to minimize vibrations during imaging. An isoflurane vaporizer (XGI-8, Perkin Elmer, Waltham, MA, USA) supplied isoflurane and air plus a vacuum to anaesthetize animals. The induction chamber was placed onto a custom 24′′ × 24′′ perforated downdraft table (Biomedical Solutions Inc, Stafford, TX, USA) to further minimize isoflurane exposure to the operator. Furthermore, a continuous low flow “snorkel” was placed near the nose cone on the stage to further minimize exposure of isoflurane. A single beam infrared spectrophotometer (MIRAN SapphIRe gas analyzer, Thermo Environmental Instruments, Franklin, MA, USA) was utilized to measure the instantaneous isoflurane exposure rates at the microscope, near the nose cone, and at the isoflurane vaporizer, induction box, induction box connectors, filters, and breathing zone above the mouse. A custom heated stage top (Oko Lab, NA, Italy) was designed for intravital imaging with a stage heating system (h401-t-single-bl, Oko Lab, NA, Italy) to ensure animals were maintained at 37 °C during image acquisition.
2.2. iKon Camera Characterization
The camera was tested to independently validate the read noise both globally and on a per pixel basis. In total, 300 short (100 µsec) images were acquired with the camera shutter closed. A bias image was generated by calculating the median value at each pixel to reject the effects of cosmic ray strikes. Both a median absolute deviation image and a 5-sigma filtered standard deviation image were generated. Read noise was calculated via 5-sigma filtering of all pixels in all 300 images, and then calculating the global standard deviation. Read noise was well controlled (3 Gl), with very small but detectable top to bottom and left to right gradients in read noise: 4.9 × 10−4 Gl/pixel and 1.8 × 10−6 Gl/pixel, respectively. The number of electrons was calculated from vendor-supplied electron-Gl conversion values for a 50 kHz read and 4X gain (1.2) and the number of read photons required by accounting for 0.95 quantum yield. Test–retest data and correlation with a macroscopic imaging system (IVIS Spectrum) were conducted with an NIST traceable luminometer microplate reference standard (LRM 168-96 622 nm emission, HARTA Instruments Gaithersburg, MD, USA).
2.3. Reagents
D-Luciferin, potassium salt, was purchased from Gold Biotechnology, Inc.® (St. Louis, MO, USA). TNFα was purchased from R&D Systems, Inc. (Minneapolis, MN, USA). L-012 sodium salt was purchased from Wako Chemicals USA (Richmond, VA, USA). L-012 powder was dissolved in sterile double distilled water (ddH20) to a final concentration of 20 mM and stored at −20 °C. Puromycin was purchased from Thermo Fisher Scientific (Waltham, MA, USA).
2.4. Cells
B16F10 melanoma cells were purchased from the MD Anderson Cancer Center Cell core (originally from the American Type Culture Collection, ATCC, Manassas, VA, USA). B16F10 cells were cultured in DMEM supplemented with 10% heat-inactivated fetal bovine serum (FBS). Pan02 cells were purchased from the DCTD Tumor Repository, National Cancer Institute (Frederick, Maryland). Pan02 cells were cultured in RPMI 1640 supplemented with 10% heat-inactivated FBS and 2 mM glutamine. Cell cultures were grown at 37 °C in a humidified 5% CO2 atmosphere.
2.5. Plasmids
The κB5→IκBα-FLuc and κB5→FLuc plasmid were previously described [
30,
36]. The FUW-FLG plasmid encoding a fusion of CBG and EGFP proteins driven by the human ubiquitin C promoter within an established lentiviral backbone has been previously described [
37]. The pFLuc control plasmid was generated as previously described [
25]. The GFP-IRES-GFP lentivirus was a gift from Ron DePinho (UT MD Anderson Cancer Center, TX, USA). The pDendra2-N and pLVX-IRES-Puro were purchased from Clontech (Clontech, Moutain View, CA, USA). A Cignal lenti NFκB→FLuc reporter was purchased from Qiagen (SABiosceinces, Frederick, MD, USA).
2.6. Generation of Stable Reporter Cells
The generation of a B16F10 cell line stably expressing GFP: B16F10 cells at 50% confluence were infected with the second generation GFP-IRES-GFP, CBG or FLuc lentiviruses. GFP-IRES-GFP and CBG infected B16F10 cells were selected for GFP expression by FACS sorting. FLuc infected B16F10 cells were selected by bioluminescence imaging; after two additional weeks, bioluminescence images of isolated cell colonies using phenol free DMEM media with 10% FBS and 150 µg/mL D-Luciferin were acquired to assess reporter gene expression, upon which bioluminescent colonies were harvested and expanded. Cells were transduced at a multiplicity of infection (MOI) of 10. The generation of the B16F10 GFP-IRES-GFP cell line stably expressing pκB5→FLuc: cells at 95% confluence was co-transfected with 10 µg of pκB5→FLuc plasmid DNA and 3 µg of pLVX-IRES-Puro plasmid DNA using Fugene6 transfection reagent (Promega, Madison, WI, USA) in a 6-well plate. At 24 h post transfection, media were replaced with fresh cell media, and at 48 h post transfection, cells were trypsinized and plated at a variety of densities (1:2, 1:5, 1:10) into media containing 2 µg/mL puromycin to select for stable transformants. After two weeks, bioluminescence images of isolated cell colonies using phenol free DMEM media with 10% FBS and 150 µg/mL D-Luciferin were acquired to assess reporter gene expression, upon which bioluminescent colonies were harvested and expanded. Cells were continuously cultured in the presence of 2 µg/mL puromycin to maintain expression of the reporter plasmid. The generation of a B16F10 and Pan02 cell line stably expressing Dendra2: cells at 95% confluence were co-transfected with 10 µg of pDendra2-N plasmid DNA using Fugene6 in a 6-well plate. At 24 h post transfection, media were replaced with fresh cell media, and at 48 h post transfection, cells were trypsinized and plated in media containing 1.5 mg/mL G418 for B16F10 cells and 150 µg/mL G418 for Pan02 cells to select for stable transformants. Flow cytometry was used to isolate Dendra2-positive cells; cells were sorted twice for Dendra2 fluorescence (λex = 409 nm, λem = 507 nm). The generation of a B16F10 Dendra2 and Pan02 Dendra2 cell stably expressing NF-κB→FLuc: B16F10 Dendra2 or Pan02 Dendra2 cells at 50% confluence were transduced with the NF-κB→FLuc lentivirus and selected using 2 µg/mL puromycin. Cells were transduced at a MOI of 10. All cell lines tested negative for mycoplasma.
2.7. Animals
The Institutional Animal Care and Use Committee at the University of Texas MD Anderson Cancer Center approve all animal protocols. The following animals were used for window chamber experiments: wild type C57BL/6 animals (Taconic Biosciences, Rensselaer, NY, USA); syngeneic C57BL/6 myeloperoxidase-deficient
MPO−/− (
MPOtm1Lus, The Jackson Laboratory, Bar Harbor, ME, USA) mice;
p21-
FLuc reporter mice [
26]; and
LSL-KrasG12D/+;LSL-p53T172H/+;Pdx-1-Cre (
KPC) animals were kindly provided by Dr. Chun Li [
38].
2.8. Preparation for Window Chamber Implantation Surgery
Animals weighed at least 20 g prior to window chamber implantation surgery. Surgical kits with chlorhexidine, ophthalmic ointment, buprenorphine and sterile materials were provided from the Department of Veterinary Services at MDACC. Animals were anaesthetized using 2% isoflurane in oxygen (2 mL/min flow rate) and the fur was removed from the dorsum. Animals received a single injection of 0.1 mg/kg of buprenorphine prior to beginning the surgery. All surgical procedures were performed in aseptic conditions while maintaining body temperature at physiological levels using a heating pad. The skin was prepared by three alternating washes of chlorhexidine and 70% ethanol and ophthalmic ointment applied to the animal’s eyes. Before any incisions were made, the toe pinch reflex test was used to determine if the animal had attained a surgical level of anesthesia.
2.9. Dorsal Skin Window Chamber Implantation Surgery
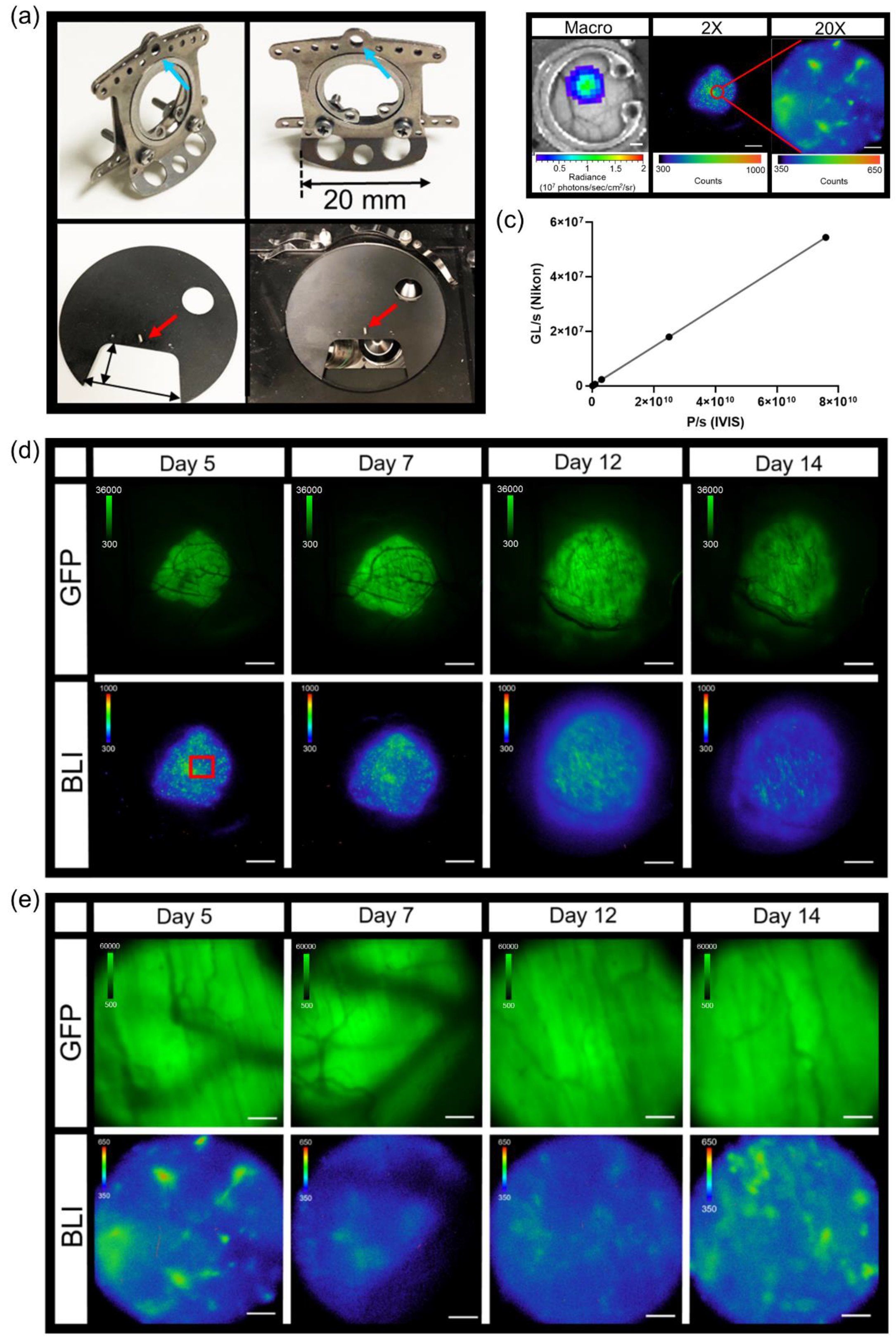
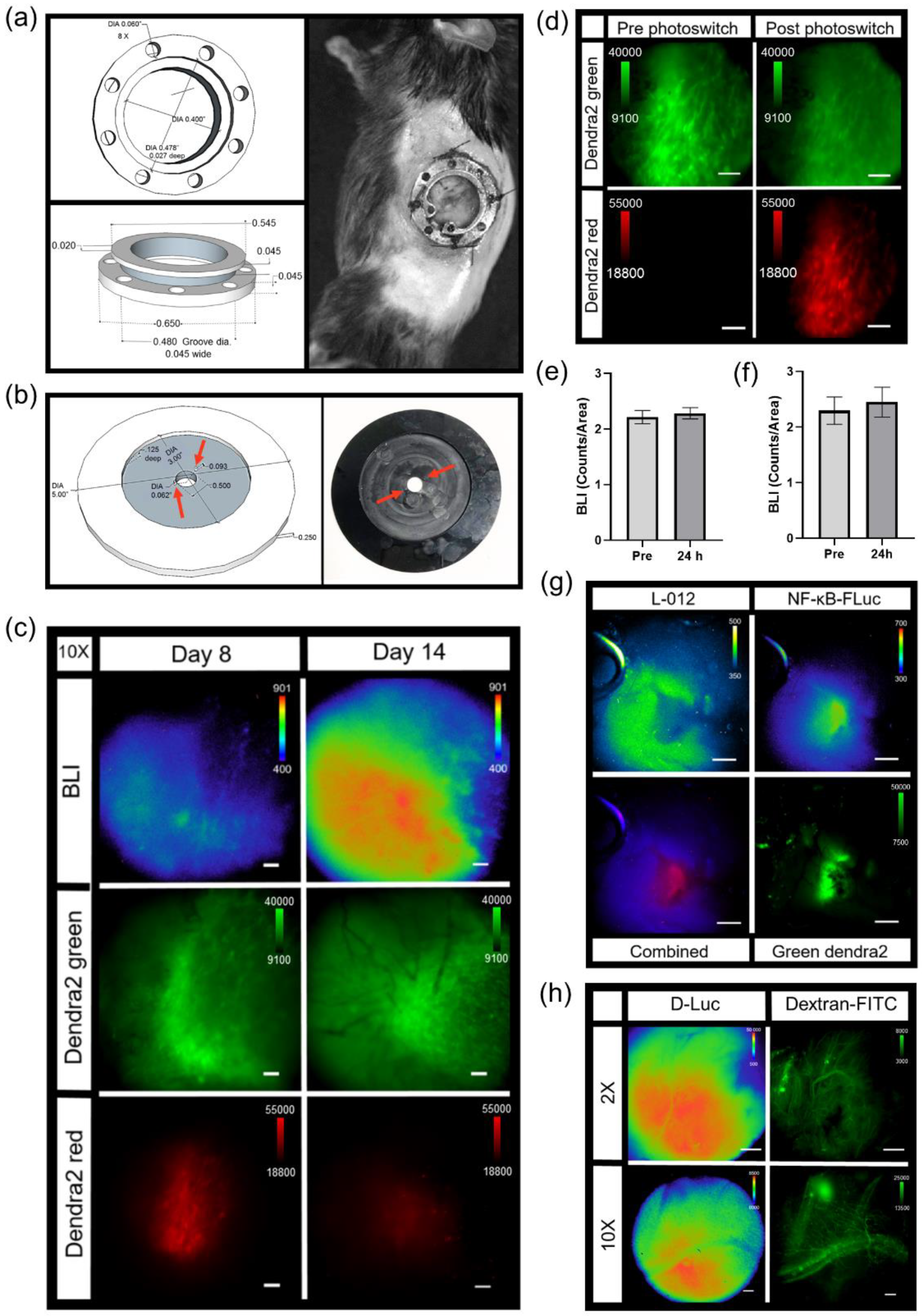
Titanium dorsal skin window chambers were purchased from APJ Trading (APJ Trading Co, Inc, Ventura, CA, USA). The skin window chambers were modified by the UT MD Anderson Cancer Center Radiation Physics machine shop. The dorsal skin was drawn up into a longitudinal fold using a straight suture needle and sterile silk. The skinfold was trans-illuminated so that the symmetrical pattern of blood vessels on either side of the dorsal midline were matched for an even skinfold. The flat side of one of the window chamber pieces was held against the skin fold and the positions of the screw holes were marked. A hole was punched through both sides of the skin at each screw location using an 18 G needle. The front window chamber frame was then screwed into the rear frame and the top of the skinfold sutured along the edge of the frame through the skin to hold the window chamber in place. Antibiotic ointment was applied to all sites where screws and sutures passed through the skin. A 1.2 cm diameter circle in the top layer of the skin was cut with scissors along the circumference of the window chamber adjacent to the frame. The exposed dermis was washed with saline prior to an injection of 0.5 – 1 × 106 tumor reporter cells between the exposed fascial plane and dermis. The exposed window area was then filled with saline until a meniscus formed; a glass cover slip was placed over the window, and secured with a retaining ring.
2.10. Abdominal Pancreas Window Chamber Implantation Surgery
The abdominal window chamber was produced by the UT MD Anderson Cancer Center Radiation Physics machine shop, custom modified using the specifications from Ritsma et al. [
39] After window chamber preparation, described above, a sterile drape was placed over the animal with the surgery area remaining uncovered. Using a scalpel, a 15 mm incision through the skin on the dorsal left of the spine was made and the skin retracted. A similar length incision through the muscular layer was then made and the muscular tissue retracted, exposing the abdominal organs. The abdominal window chamber was held in position using a purse-string non-resorbable suture, which was concealed within the groove of the abdominal window chamber ring. A purse-string suture in the muscular layer was first made, followed by a purse-string suture through the skin. Concealing the suture within the groove of the ring prevented mice from biting or pulling the sutures. Once the abdominal window chamber was sutured in place, the pancreas was located and secured by gluing the organ to the interior inner side of the abdominal window chamber using high viscosity cyanoacrylates (Sigma-Aldrich, St. Louis, MO, USA). Once the pancreas was positioned within the abdominal window chamber, an orthotopic injection of 1 × 10
6 reporter tumor cells was performed; note that no tumor cell injections were required for
KPC or
KPC-Luc animals. To further hold the organ in place, an optically translucent silicone seal (Kwik-Sil
TM, World Precision Instruments, Sarasota, FL, USA) was placed around the pancreas. A 1.2 cm diameter glass cover slip was then placed on top of the window chamber and held in place by either glue or a retaining ring.
2.11. Post Window Chamber Implantation Surgery
After each window chamber implantation, the animal was placed on another heating pad and gently warmed during recovery; animals were injected subcutaneously with 1 mL of saline to maintain hydration and 0.1 mg/kg buprenorphine SR. Animals were monitored until fully recovered from the surgical procedure. Antibiotic ointment was applied along the tissue–implant interface. All animals were housed separately after window chamber implantation and the wire bar lid removed from the microisolator cages. A 2 mg Rimadyl tablet (Bio-Serv, Flemington, NJ, USA) was given orally in hydrogel and placed in the animal’s cage.
2.12. In Vivo Intravital Microscopy
To limit motion during intravital imaging, the UT MD Anderson Cancer Center Radiation Physics machine shop custom built window chamber imaging stage inserts made of aluminum and painted with black enamel that fit in the custom-crafted heated stage surface recess. The stage temperature was set to 37 °C to continuously warm animals during acquisition. Mice were kept under isoflurane anesthesia throughout the entire imaging session. To further minimize stray light during acquisitions, a small black box was placed over the animals with a notch cut out for the anesthetic nose cone. Intravital imaging generally occurred as follows: epifluorescence imaging of GFP or Dendra2 was performed (GFP cube) to locate and center the tumor. Next, confocal imaging was performed where indicated in which the manual shutter was placed in the open position to acquire confocal images. An i.p. injection of 300 mg/kg D-Luciferin was used for FLuc microscopy for single imaging sessions. Immediately following D-Luciferin administration, an epifluorescence image of GFP or Dendra2 occurred to ensure the tumor was still in focus. The confocal manual shutter was then pushed into the closed position and a bioluminescence image was taken 5 min post administration of D-Luciferin. Based on the 2X epifluorescence and bioluminescence images, an area of interest was magnified using either the 10X or 20X objective by epifluorescence or confocal imaging. When using the 10X and 20X objectives, image focus occurred using perfect focus to further prevent any drift from the focal plane. If an acquisition time of 10 min or less was used at 2X, a bioluminescence image at the higher magnification immediately followed. If an acquisition time of greater than 10 min occurred at 2X, a bioluminescence image occurred 5 min following a second i.p. injection of 300 mg/kg D-Luciferin.
2.13. Skin Window Chamber Intravital Microscopy
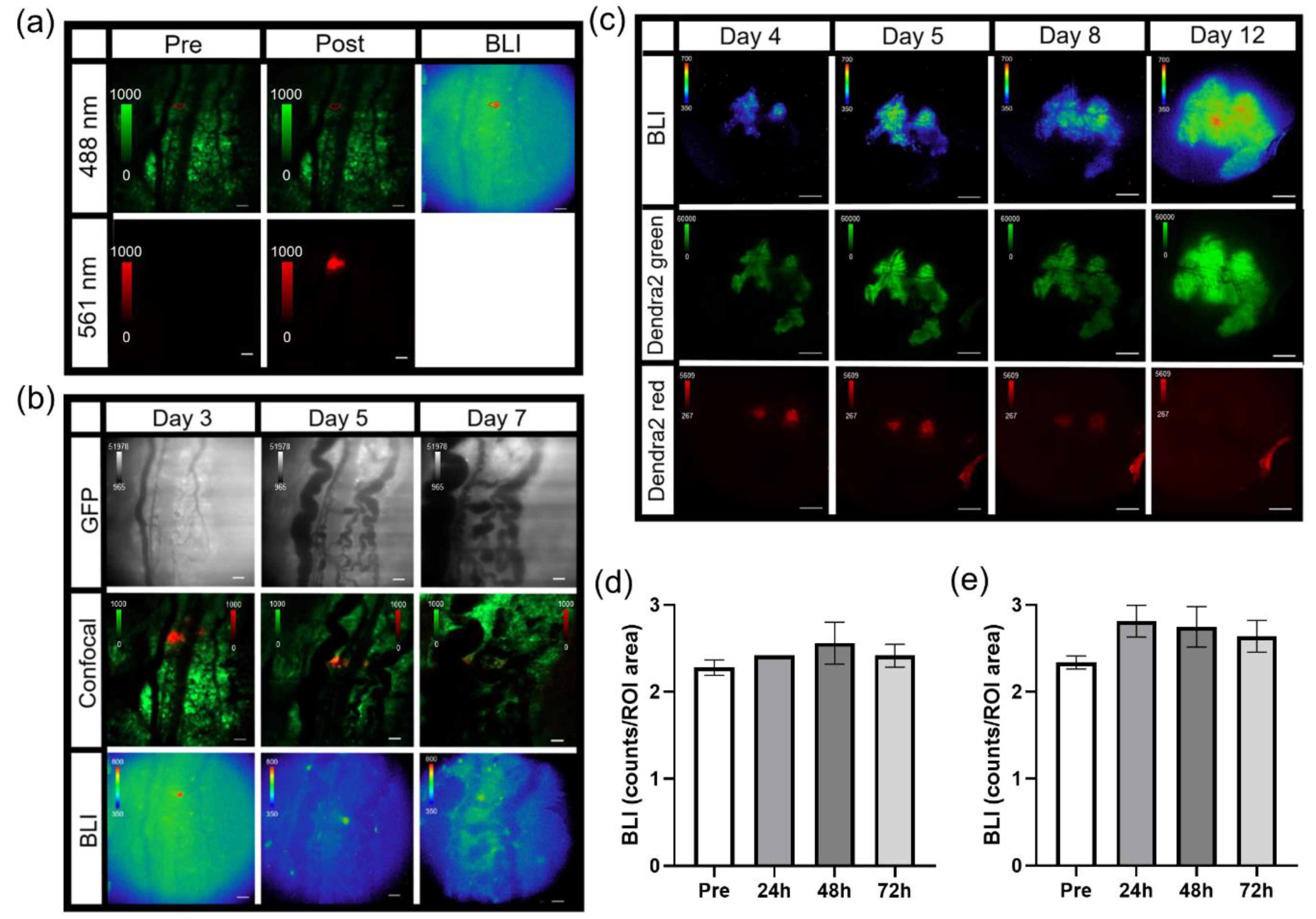
For imaging tumor signaling dynamics, an i.v. tail vein catheter was inserted and taped to the animal prior to being positioned on the microscope. The animal was placed on the microscope where the window chamber was locked in place using the custom stage insert. For imaging B16F10 κB-FLuc GFP reporter cells, an i.p. catheter was placed in the abdomen. Using the 2X objective, the tumor was located and centered using epifluorescence (GFP cube) and confocal (laser 488 nm) imaging. An i.p. injection of 300 mg/kg D-Luciferin occurred, followed by a steady infusion of D-Luciferin using a syringe pump at 8 µL/min to maintain a steady bioluminescence signal [
25]. A basal bioluminescence image occurred at 5 min post i.p. injection of D-Luciferin at 2X (5 min exposure, open filter). Then, 10X epifluorescence (GFP cube) and confocal (4.8 speed, laser lines: 488 nm (Dendra2 excitation) and 561 nm (Texas Red excitation)) imaging occurred based on an area of interest chosen from the 2X bioluminescence image. A basal 10X bioluminescence image (10 min exposure, open filter) was taken. Following the basal bioluminescence image at 10X, live sequential C2 confocal imaging acquisition occurred and continued during the i.v. injection of dextran-Texas Red (10,000 MW, 4 mg/mL, ThermoFisher Scientific, Waltham, MA) to confirm a successful i.v. injection (
n = 3 control animals,
n = 4 TNFα treated animals). Animals were injected with dextran alone or a combination of dextran and TNFα. The total sequential C2 confocal acquisition time was 5 min. Following the successful i.v. injection of dextran with or without TNFα, sequential bioluminescence imaging (10X, 10 min exposure, open filter) occurred for 45 min. A final confocal image was taken following sequential bioluminescence imaging. The quantification of signaling dynamics occurred as follows: using the highest 10X bioluminescence image from the bioluminescence time series, multiple auto-detect ROIs were created around single cells or clusters of cells within the tumor. These ROIs were copied and pasted onto every bioluminescence image and counts were measured. All bioluminescence counts were normalized to the ROI area. Imaging
MPO+/+ (
n = 14) and
MPO−/− (
n = 9) animals with B16F10 NF-κB-FLuc tumors occurred as follows: a Dendra2 fluorescence image (GFP cube) at 2X was acquired to locate the tumor, and then a bioluminescence image (20 min acquisition, open filter) occurred 5 min post i.p. injection of 300 mg/kg D-luciferin. The quantification of NF-κB transcriptional activation occurred as follows: an auto ROI was drawn around the tumor using the Dendra2 fluorescence image and copied to the corresponding bioluminescence image. Total counts were measured and normalized to ROI area. For vasculature leakage, live sequential confocal imaging acquisition (4.8 speed, laser lines: 488 nm (Dendra2 excitation) and 561 nm (Texas Red excitation)) occurred and continued during an i.v. administration of dextran-Texas Red (70,000 MW, 4 mg/mL, ThermoFisher Scientific, Waltham, MA) using the tail vein catheter. The total sequential C2 confocal acquisition time was 10 min. Quantification of dextran leakage occurred by drawing an ROI on a vessel and dextran fluorescence was measured as a function of time. The first image with measurable dextran fluorescence was set as time = 0, and the fluorescence intensity normalized to fluorescence intensity at time = 0 for all measured time points that followed to account for dextran delivery variability between animals (
n = 4 animal per group). Normalized dextran leakage curves were fit using non-linear regression, least square fit. For in vivo imaging of myeloid cells, dextran-Texas Red (10,000 MW, 4 mg/mL, ThermoFisher Scientific, Waltham, MA) and αGr-1 Alexafluor647 (10 μg, BioLegend, San Diego, CA) were intravenously administered to label myeloid cells [
9,
40,
41,
42,
43,
44,
45,
46,
47,
48,
49]. Live sequential confocal imaging acquisition (4.8 speed, laser lines: 488 nm (Dendra2 excitation), 561 nm (Texas Red excitation) and 640 nm (Alexfluor647 excitation)) occurred and continued during the intravenous injection of dextran and αGr-1 antibody. The total sequential C2 confocal acquisition time was 10 min. A region of interest was determined from the 2X confocal images to then image using the 10X objective. Five minutes post i.p. injection of 300 mg/kg D-Luciferin, a bioluminescence image was taken at 10X objective using a 20 min exposure (
n = 4
MPO+/+ animals). Following a 10X objective bioluminescence acquisition, a C2 confocal acquisition of the same region of interest at 10X occurred using the same acquisition parameters described above using a Z stack acquisition of 110 slices at 1 μm steps. Following intravital imaging, animals were given a bolus injection of 1 mL saline and recovered in their cage on a heating pad.
2.14. Photoswitching Dendra2 Fluorescent Protein
Single cell photoswitching in vivo was performed using the 405 nm laser on the confocal imaging system. Imaging acquisition occurred as follows: epifluorescence imaging (GFP cube) of Dendra2 located and centered the tumor at 2X. A bioluminescence image was acquired (2X, 10 min acquisition, open filter) 5 min post an i.p. injection of 300 mg/kg D-Luciferin. A bioluminescence image (10X, 20 min acquisition, open filter) followed, magnifying an area of interest chosen from the 2X bioluminescence image. Confocal imaging (4.8 speed, laser lines: 488 nm (green Dendra2 excitation) and 561 nm (red photoswitched Dendra2 excitation)) acquired a pre photoconversion image. An ROI was drawn around a cell to be photomarked. A live cell real-time sequential confocal sequence (4.8 speed, laser lines: 488nm and 561 nm) was acquired as follows: 5 confocal images prior to photoswitching, 20 confocal images acquired during stimulation with the 405 nm laser, and 20 confocal images post photoconversion. The 405 nm laser (3 stimulations, 20% power) photoswitched Dendra2 from green to red. Dendra2 photoswitching of larger tumor areas occurred as follows: epifluorescence imaging (GFP cube) of Dendra2 located and centered the tumor at 2X. A bioluminescence image was acquired (2X, 20 min acquisition, open filter) 5 min post an i.p. injection of 300 mg/kg D-Luciferin. Epifluorecence imaging (GFP and DsRed cubes) were performed at 20X; regions of interest were chosen based on the 2X bioluminescence image. The area of illumination for photoconversion was decreased to smaller than the field of view using the field stop slider. The target photoconversion area was exposed to the DAPI cube excitation light for 10 min to photoconvert Dendra2 at 20X. Epifluorescence imaging (GFP and DsRed cubes) at 20X and 2X occurred to confirm photoconversion. Imaging acquisition following single cell or tumor area Dendra2 photoconversion followed the general intravital imaging protocol described above at least 24 h later. For the quantification of Dendra2 photoconversion effects on bioluminescence reporter imaging, an auto ROI was drawn around the tumor using the Dendra2 epifluorescence image at 2X and copied to the corresponding bioluminescence image. Fluorescence and bioluminescence intensities were quantified on the day of, 24 h, and 48 h post photoconversion. Total quantified counts were normalized to the ROI area. Bioluminescence counts/area as well as bioluminescence counts/area normalized to fluorescence counts/area were calculated.
2.15. Spectral Unmixing
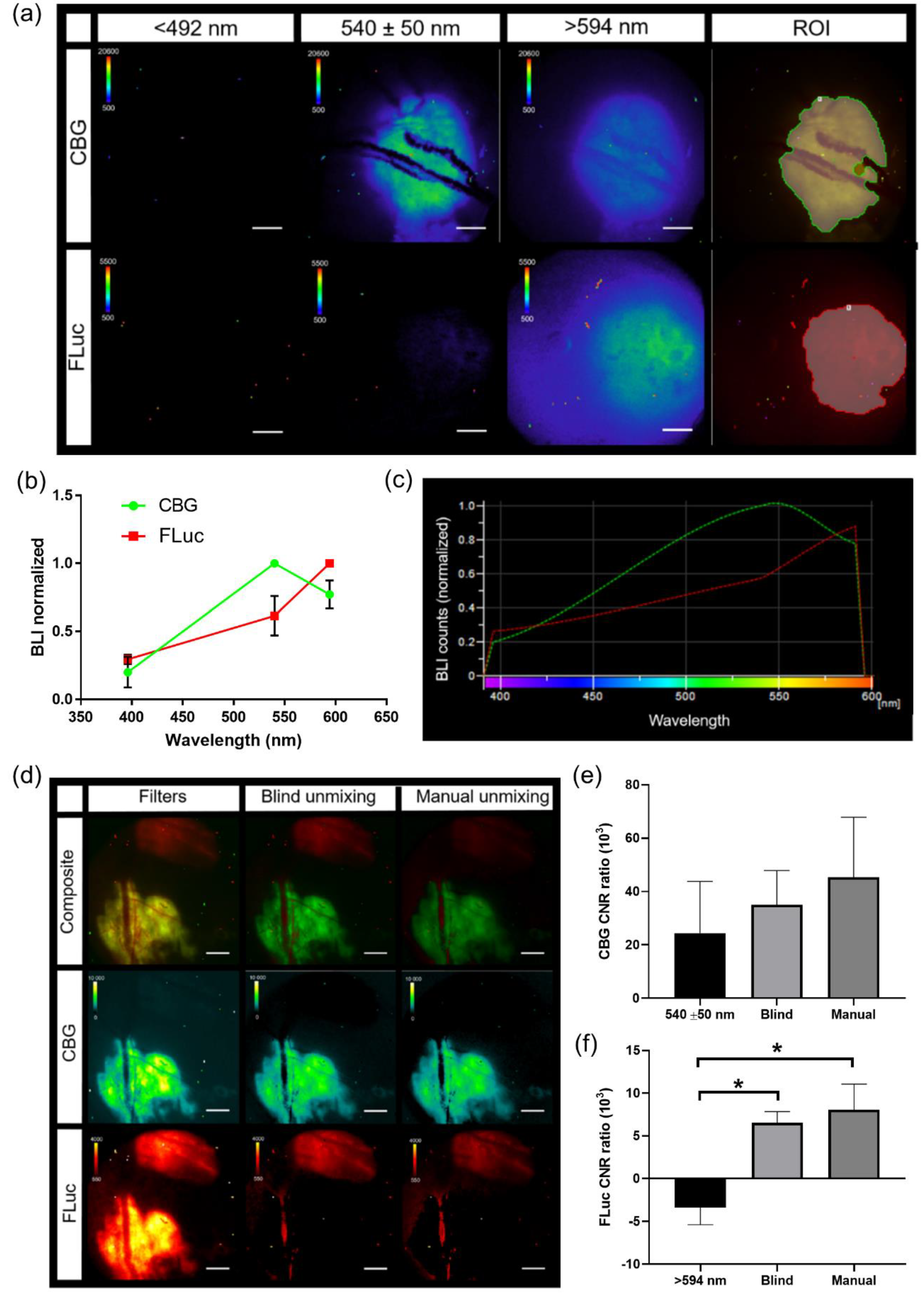
In vivo bioluminescence spectra were created using window chamber models with either B16F0 CBG or B16F10 FLuc tumors. Bioluminescence imaging occurred 5 min following a 150 mg/kg of body weight D-Luciferin using all emission filters (<492 nm, 540 ± 50 nm, and >594 nm filters, 5 min acquisition at all filters, 2X objective). Spectra for CBG and FLuc were generated using the analysis software, NIS-Elements AR Analysis (Nikon Instruments, Melville, NY, USA). Using all bioluminescence images with each emission filter, an auto threshold ROI was drawn around the tumor and a spectrum for each reporter was generated in each animal. These spectra were exported into excel where the mean spectrum was calculated (CBG,
n = 5 images; FLuc,
n = 4 images). The mean spectrum for each wavelength was saved as a csv file and uploaded into NIS-Elements software. Pure spectra for each reporter were found under a user-defined library when performing spectral unmixing. When spectral unmixing images, only the spectra for reporters that needed to be visually separated were selected. When quantifying the contrast to noise ratio (CNR), an auto threshold ROI was created on each spectrally unmixed BLI image. This ROI was copied onto each BLI image to quantify CNR. CNR was calculated using the following equation:
2.16. Abdominal Window Chamber Intravital Microscopy
Bioluminescence imaging of the abdominal window chamber was described above in in vivo intravital microscopy. Vasculature imaging of abdominal window chamber animals occurred following an i.v. injection of dextran-FITC (200,000 MW, 4 mg/mL). The photoconversion of Dendra2 in abdominal window chambers occurred as described above in the photoswitching Dendra2 fluorescent protein methods section but using the Dendra2 photoswitching of larger tumor areas protocol.
2.17. Histological Analysis
Necropsy of animals occurred after 14 days, wherein the titanium window chamber was removed, skin excised and tissue frozen in OCT media. Prior to histological analysis, tumor samples were formalin fixed for 48 h and stored in 70% ethanol until histological staining. All tumor samples were sent to the Research Histology, Pathology and Imaging Core at UT MD Anderson Cancer Center where they were sectioned, stained and analyzed.
2.18. Statistical Analyses
Graphs were generated and statistical analyses were performed using GraphPad Prism (GraphPad Software, Inc, La Jolla, CA, USA). Data were expressed as mean ± s.e.m. Analysis of differences between two normally distributed paired test groups was performed using a Student’s t-test. Analysis of differences between unpaired groups was performed using an unpaired two-tail Mann–Whitney t test. p values were considered statistically significant if p < 0.05. For analysis of three or more groups, mixed-effects analysis were performed with Dunnett’s multiple comparisons test.
2.19. Data Availability
All relevant data are within the manuscript and the supplementary information files.
4. Discussion
A variety of custom-built intravital microscopy systems have been reported by expert laboratories [
1,
2,
3,
4,
5,
6,
7,
40,
41,
42,
53,
54,
55,
56,
57,
58,
59,
60,
61,
62]. Our goal was to expand the utility of the intravital imaging strategy by first starting with an off-the-shelf commercial system, and by modifying this to achieve high quality imaging within the reach of any biology laboratory. This intravital microscopic imaging platform, which combines both bioluminescent and fluorescent genetically encoded reporters, as well as exogenous reporters, provides a powerful multi-plex strategy to study molecular and cellular processes in real-time in intact living systems at single cell resolution. The combination of bioluminescence and fluorescence capacity with intravital microscopy all in a single system provided a first-in-class advance in technology for the high-resolution multi-modal imaging of single cells, signaling dynamics, and real-time spectral unmixing of heterogeneous living systems in vivo. The addition of bioluminescence imaging to a fluorescence microscope was achieved by the addition of a high quality, cooled, back-illuminated CCD camera. These cameras also are often “plug and play” ready, with well-controlled background, and validated linear response, all critical for not only detecting but measuring changes in emitted light. As new detector schemes become integrated into vendor software, further integrations are expected to accommodate EMCCD, ICCD, cooled scientific CMOS and other emergent camera technologies. While minimizing stray external light was readily achieved, significant effort was expended in identifying key internal sources of stray light, many of which may be vendor specific. Nonetheless, careful assessment and strategies to block stray photons can be achieved in low-noise systems.
Intravital bioluminescence microscopy demonstrated the heterogeneous activation of NF-κB within melanoma tumors, which was not captured by a matched macroscopic imaging system [
32]. Baseline heterogeneous NF-κB signaling levels within individual cells of a tumor and between individual tumors were confirmed using two different bioluminescence-based NF-κB transcriptional activation reporters at single cell resolution, as well as in two different tumor models (melanoma and pancreas). Real-time changes in NF-κB cell signaling dynamics within individual cells of a tumor were demonstrated using TNFα-induced activation during bioluminescence microscopy in vivo. NF-κB signaling was examined due to the biological significance of this pathway; NF-κB is considered a central coordinator of the immune system and implicated during cancer development [
63,
64,
65,
66]. The visualization of bioluminescence micro-structures by microscopy, but not by macro-imaging, was the result of the change in sensitivity, pixel size and effective f stop between the systems. Photon-emitting sources arising from single cells or a cluster of cells that were smaller than the macro-imaging pixel size often were undetectable by macro-imaging due to partial volume effects, which were not a limitation of the higher resolution microscopy system. However, by leveraging the strengths of both systems, multi-modal, multi-spectral whole body (and whole window chamber) imaging [
32] could be combined with dynamic molecular signaling and cellular imaging, providing multi-scale information of disease progression in an individual animal. Furthermore, with the high correlation between the widely utilized macro-imaging systems (IVIS) and the 2X objective on the custom microscopy system, global trends could be interrogated and cross-validated on both imaging platforms.
Herein, we also demonstrated that bioluminescent reporters were complimentary to fluorescent reporters. The inclusion of bioluminescent reporters did not affect fluorescence imaging and vice versa, demonstrated by using genetically encoded reporters expressed in both implanted tumor cells and transgenic reporter animals. Photomarking tumor cells using a photoswitchable fluorophore, Dendra2, was also not affected by the presence of bioluminescent reporters, nor did photoconversion affect bioluminescence signals. The inclusion of a photoswitchable fluorophore allowed cells, or areas of interest, to be marked, and changes in signaling dynamics, or cell–cell interactions could be followed for many days throughout disease progression. However, when combining a bioluminescent signaling reporter with a constitutively active photoswitchable fluorophore, if photoconversion occurs, challenges can arise in the normalization of the bioluminescence signal to tumor mass fluorescence due to difficulties in controlling the quantity of photoconversion, the differences in the fluorescence intensity between unphotoswitched and photoswitched signals, and the loss of photon converted signal over time. When using photoconverted fluorescent proteins, the normalization of bioluminescence signaling dynamics to initial bioluminescence may be better suited for quantification purposes when comparing between different animals. The capability to spectrally resolve different luciferases that use the same substrate, as well as bioluminescent reporters that use different substrates, was also achieved on a microscopic level in vivo.
As we have demonstrated, this platform could be used to image, in real time, the interaction between cancer cells and immune cells and could be further extended to other diseases in vivo. With the continued development of genetically encoded imaging reporters, different signaling networks, as well as changes in transcription, translation, protein folding, protein–protein interactions, protein degradation and second messengers could be studied. We demonstrated the spectral resolution of up to four imaging reporters (two bioluminescent and two fluorescent reporters). However, in principle, our current intravital microscopy system has the capability to resolve three bioluminescence and four fluorescence channels. Herein, we demonstrated two different permanent window chamber models—skin and abdomen—but numerous other tissue areas have also been imaged by fluorescence window chamber imaging, including the liver, spleen, small intestine, brain, lung, femur, mammary fat pad, lymphodes, and spine [
4,
39,
53,
54,
67,
68,
69,
70,
71,
72,
73]; these window chamber models could easily be extended to include in vivo bioluminescence. The use of our intravital microscopy imaging platform provides a strategy to begin to understand how cells communicate as well as changes in signaling networks, both spatially and temporally, in a quantitative manner in a heterogeneous living system at cellular resolution. With the paradoxical dual role that the immune microenvironment plays during cancer progression, where it can restrain or promote cancer progression [
12,
13,
14,
15], it is essential to develop strategies to better understand the interaction networks between tumor cells and immune cells to improve disease detection and monitor therapeutic response.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}