
On Broken Ne(c)ks and Broken DNA: The Role of Human NEKs in the DNA Damage Response

 , , , and
, , , and
Abstract
:1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NEK Members | Gene Location (Chromosome) | Amino Acids; Molecular Weight | Functions | Subcellular Location | Protein Domains | 3D Structure; Method; PDB Entry | Ref. |
|---|---|---|---|---|---|---|---|
| NEK1 | 4q33 | 1258 aa, 143 kDa | Primary cilium formation, meiosis I spindle assembly, mitochondrial membrane permeability, cell cycle control, DNA damage response | Cytoplasm, cilia, centrosome, and nucleus upon DNA damage | Catalytic domain, coiled-coil, degradation motif (PEST sequence) | Yes; X-ray; PDB: 4APC | [3,11,36,44,45,46,47,48] |
| NEK2 | 1q32.3 | NEK2A: 445 aa, 48 kDa | Centrosome integrity and separation, cell cycle regulation, primary cilia, splicing | Centrosome, cytoplasm, nucleus | Catalytic domain, coiled-coil, degradation motif (PEST sequence) | Yes; X-ray and electron microscopy; PDB: 2W5H | [4,49,50,51,52,53,54,55,56,57,58,59] |
| NEK2B: 384 aa, 44 kDa | Centrosome, cytoplasm | Catalytic domain, coiled-coil | |||||
| NEK2C: 437 aa, 50 kDa | Centrosome, nucleus | Catalytic domain, coiled-coil, degradation motif (PEST sequence) | |||||
| NEK3 | 13q14.2 | 506 aa, 56 kDa | Cell cycle regulation | Cytoplasm | Catalytic domain, degradation motif (PEST sequence) | No | [60] |
| NEK4 | 3p21.1 | NEK4 I1: 841 aa, 94 kDa | Microtubule stabilization, primary cilia stabilization, DNA damage response, splicing | Cilia, basal bodies, nucleus, mitochondria | Catalytic domain | No | [13,61,62,63] |
| NEK4 I2: 781 aa, 88 kDa | |||||||
| NEK4 I3: 752 aa, 84 kDa | |||||||
| NEK5 | 13q14.3 | 708 aa, 81 kDa | Centrosome disjunction, DNA damage response, mitochondrial respiration, mtDNA maintenance | Cytoplasm, centrosome, mitochondria | Catalytic domain, dead-box helicase-like domain, coiled-coils | No | [14,64] |
| NEK6 | 9q33.3 | 313 aa, 35 kDa | Mitotic spindle and kinetochore fiber formation, metaphase-anaphase transition, cytokinesis, checkpoint regulation | Cytoplasm, nucleus, mitotic spindle, centrosome, central spindle, midbody | Short unfolded interaction region, catalytic domain | Yes; SAXS | [65,66,67,68,69,70] |
| NEK7 | 1q31.3 | 302 aa, 34 kDa | Mitotic spindle formation, centrosome separation, cytokinesis, NLRP3 inflammasome activation, DNA telomeric integrity | Centrosome, spindle midzone, midbody | Short unfolded interaction region, catalytic domain | Yes; X-ray and electron microscopy; PDB: 6S76 | [65,71,72,73,74,75,76,77] |
| NEK8 | 17q11.2 | 692 aa, 74 kDa | Stability and function of the primary cilium, DNA damage response | Cytoplasm, centrosome, cilia, nucleus, perinuclear compartments | Catalytic domain, Regulator of Chromosome Condensation (RCC1) domain | No | [78,79,80,81] |
| NEK9 | 14q24.3 | 979 aa, 120 kDa | Mitotic spindle formation, centrosome separation, replication stress response | Spindle poles, centrosome, cytoplasm, nucleus | Catalytic domain, Regulator of Chromosome Condensation (RCC1) domain, degradation motif (PEST sequence), coiled-coil | No | [82,83,84,85,86,87] |
| NEK10 | 3p24.1 | 1172 aa, 133 kDa | DNA damage response, mitochondrial metabolism | Associated with mitochondria | Armadillo repeats, coiled-coil, catalytic domain, degradation motif (PEST sequence) | No | [15,88] |
| NEK11 | 3q22.1 | NEK11 Long (L): 645 aa, 74 kDa | DNA replication and DNA damage response | Cytoplasm | Catalytic domain, coiled-coil, degradation motif (PEST sequence) | No | [89,90] |
| NEK11 Short (S): 470 aa, 54 kDa | Nucleus, cytoplasm | ||||||
| NEK11C: 482 aa, 56 kDa | Nucleus, cytoplasm | ||||||
| NEK11D: 599 aa, 69 kDa | Cytoplasm |
2. NEK1
3. NEK2
4. NEK3
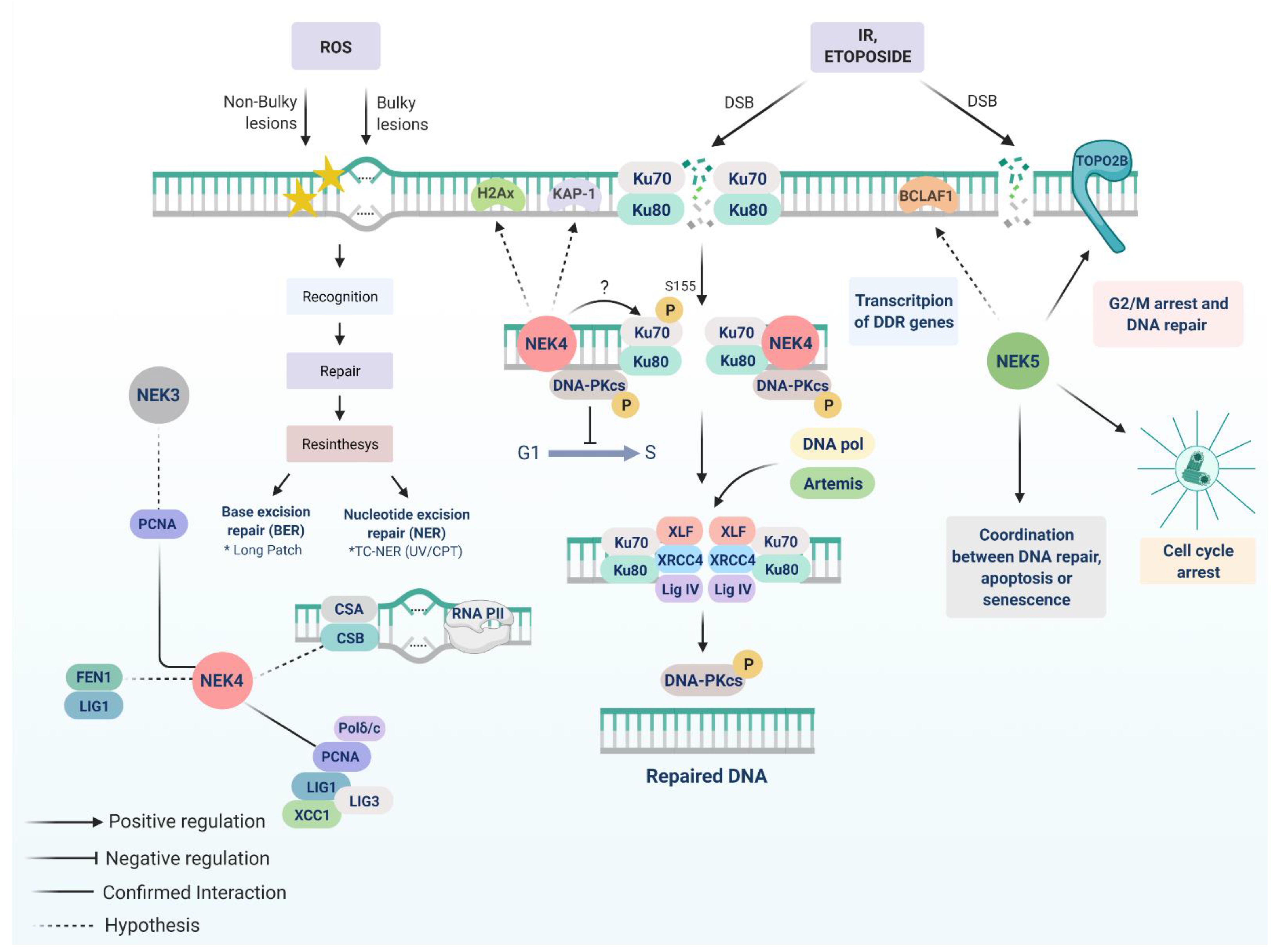
5. NEK4
6. NEK5
7. NEK6
8. NEK7
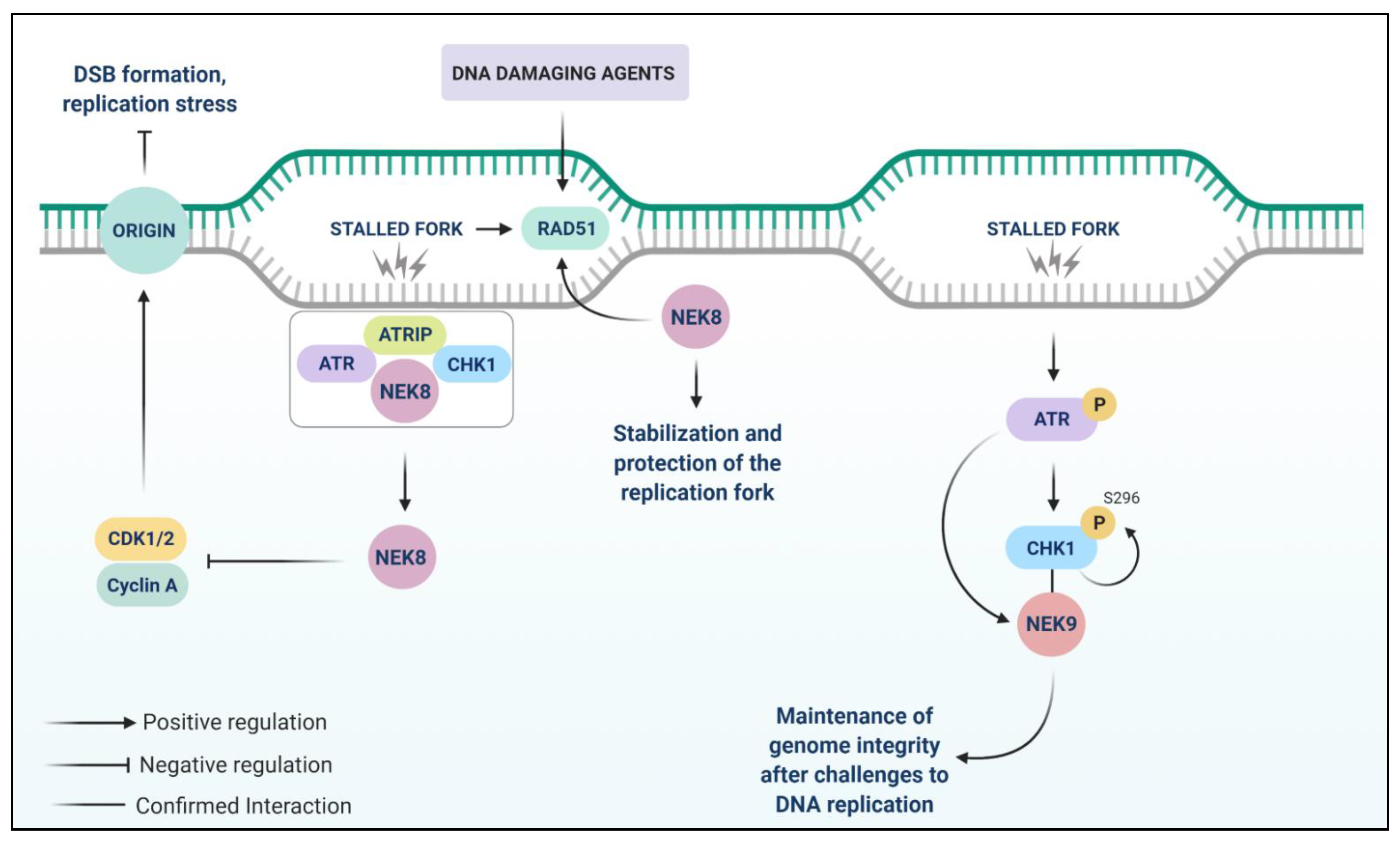
9. NEK8
10. NEK9
11. NEK10
12. NEK11
13. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Oakley, B.R.; Morris, N.R. A mutation in Aspergillus nidulans that blocks the transition from interphase to prophase. J. Cell Biol. 1983, 96, 1155–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meirelles, G.V.; Perez, A.M.; de Souza, E.E.; Basei, F.L.; Papa, P.F.; Melo Hanchuk, T.D.; Cardoso, V.B.; Kobarg, J. “Stop Ne(c)king around”: How interactomics contributes to functionally characterize Nek family kinases. World J. Biol. Chem. 2014, 5, 141–160. [Google Scholar] [CrossRef]
- Holloway, K.; Roberson, E.C.; Corbett, K.L.; Kolas, N.K.; Nieves, E.; Cohen, P.E. NEK1 Facilitates Cohesin Removal during Mammalian Spermatogenesis. Genes 2011, 2, 260–279. [Google Scholar] [CrossRef] [Green Version]
- Naro, C.; Barbagallo, F.; Chieffi, P.; Bourgeois, C.F.; Paronetto, M.P.; Sette, C. The centrosomal kinase NEK2 is a novel splicing factor kinase involved in cell survival. Nucleic Acids Res. 2014, 42, 3218–3227. [Google Scholar] [CrossRef] [Green Version]
- Gu, Z.; Xia, J.; Xu, H.; Frech, I.; Tricot, G.; Zhan, F. NEK2 Promotes Aerobic Glycolysis in Multiple Myeloma Through Regulating Splicing of Pyruvate Kinase. J. Hematol. Oncol. 2017, 10, 17. [Google Scholar] [CrossRef] [Green Version]
- Tadokoro, D.; Takahama, S.; Shimizu, K.; Hayashi, S.; Endo, Y.; Sawasaki, T. Characterization of a caspase-3-substrate kinome using an N- and C-terminally tagged protein kinase library produced by a cell-free system. Cell Death Dis. 2010, 1, e89. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.; Sawasaki, T. Nek5, a novel substrate for caspase-3, promotes skeletal muscle differentiation by up-regulating caspase activity. FEBS Lett. 2013, 587, 2219–2225. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Zeng, M.Y.; Yang, D.; Motro, B.; Núñez, G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 2016, 530, 354–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Oliveira, A.P.; Issayama, L.K.; Pavan, I.C.B.; Silva, F.R.; Melo-Hanchuk, T.D.; Simabuco, F.M.; Kobarg, J. Checking neks: Overcoming a bottleneck in human diseases. Molecules 2020, 25, 1778. [Google Scholar] [CrossRef]
- Chen, Y.; Gaczynska, M.; Osmulski, P.; Polci, R.; Riley, D.J. Phosphorylation by Nek1 regulates opening and closing of voltage dependent anion channel 1. Biochem. Biophys. Res. Commun. 2010, 394, 798–803. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Craigen, W.J.; Riley, D.J. Nek1 regulates cell death and mitochondrial membrane permeability through phosphorylation of VDAC1. Cell Cycle 2009, 8, 257–267. [Google Scholar] [CrossRef] [Green Version]
- Singh, V.; Khalil, M.I.; De Benedetti, A. The TLK1/Nek1 axis contributes to mitochondrial integrity and apoptosis prevention via phosphorylation of VDAC1. Cell Cycle 2020, 19, 363–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basei, F.L.; Meirelles, G.V.; Righetto, G.L.; Dos Santos Migueleti, D.L.; Smetana, J.H.C.; Kobarg, J. New interaction partners for Nek4.1 and Nek4.2 isoforms: From the DNA damage response to RNA splicing. Proteome Sci. 2015, 13, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanchuk, T.D.M.; Papa, P.F.; La Guardia, P.G.; Vercesi, A.E.; Kobarg, J. Nek5 interacts with mitochondrial proteins and interferes negatively in mitochondrial mediated cell death and respiration. Cell. Signal. 2015, 27, 1168–1177. [Google Scholar] [CrossRef]
- De Oliveira, A.P.; Basei, F.L.; Slepicka, P.F.; de Castro Ferezin, C.; Melo-Hanchuk, T.D.; de Souza, E.E.; Lima, T.I.; dos Santos, V.T.; Mendes, D.; Silveira, L.R.; et al. NEK10 interactome and depletion reveal new roles in mitochondria. Proteome Sci. 2020, 18, 4. [Google Scholar] [CrossRef]
- Fry, A.M.; O’Regan, L.; Sabir, S.R.; Bayliss, R. Cell cycle regulation by the NEK family of protein kinases. J. Cell Sci. 2012, 125, 4423–4433. [Google Scholar] [CrossRef] [Green Version]
- Quarmby, L.M.; Mahjoub, M.R. Caught Nek-ing: Cilia and centrioles. J. Cell Sci. 2005, 118, 5161–5169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fry, A.M.; Bayliss, R.; Roig, J. Mitotic Regulation by NEK Kinase Networks. Front. Cell Dev. Biol. 2017, 5, 102. [Google Scholar] [CrossRef] [Green Version]
- Giglia-Mari, G.; Zotter, A.; Vermeulen, W. DNA Damage Response. Cold Spring Harb. Perspect. Biol. 2011, 3, a000745. [Google Scholar] [CrossRef]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef] [Green Version]
- Xiao, W.; Samson, L. In vivo evidence for endogenous DNA alkylation damage as a source of spontaneous mutation in eukaryotic cells. Proc. Natl. Acad. Sci. USA 1993, 90, 2117–2121. [Google Scholar] [CrossRef] [Green Version]
- Mullins, E.A.; Rodriguez, A.A.; Bradley, N.P.; Eichman, B.F. Emerging Roles of DNA Glycosylases and the Base Excision Repair Pathway. Trends Biochem. Sci. 2019, 44, 765–781. [Google Scholar] [CrossRef] [PubMed]
- Costa, R.M.A.; Chiganças, V.; Galhardo, R.D.S.; Carvalho, H.; Menck, C.F.M. The eukaryotic nucleotide excision repair pathway. Biochimie 2003, 85, 1083–1099. [Google Scholar] [CrossRef]
- Jiricny, J. The multifaceted mismatch-repair system. Nat. Rev. Mol. Cell Biol. 2006, 7, 335–346. [Google Scholar] [CrossRef]
- Iyama, T.; Wilson, D.M. DNA repair mechanisms in dividing and non-dividing cells. DNA Repair Amst. 2013, 12, 620–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnum, K.J.; O’Connell, M.J. Cell cycle regulation by checkpoints. Methods Mol. Biol. 2014, 1170, 29–40. [Google Scholar] [CrossRef] [Green Version]
- Smith, H.L.; Southgate, H.; Tweddle, D.A.; Curtin, N.J. DNA damage checkpoint kinases in cancer. Expert Rev. Mol. Med. 2020, 22, e2. [Google Scholar] [CrossRef]
- Vera, J.; Raatz, Y.; Wolkenhauer, O.; Kottek, T.; Bhattacharya, A.; Simon, J.C.; Kunz, M. Chk1 and Wee1 control genotoxic-stress induced G2-M arrest in melanoma cells. Cell. Signal. 2015, 27, 951–960. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yu, Y.; Hamrick, H.E.; Duerksen-Hughes, P.J. ATM, ATR and DNA-PK: Initiators of the cellular genotoxic stress responses. Carcinogenesis 2003, 24, 1571–1580. [Google Scholar] [CrossRef] [Green Version]
- Stiff, T.; O’Driscoll, M.; Rief, N.; Iwabuchi, K.; Löbrich, M.; Jeggo, P.A. ATM and DNA-PK Function Redundantly to Phosphorylate H2AX after Exposure to Ionizing Radiation. Cancer Res. 2004, 64, 2390–2396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, I.M.; Chen, J. Histone H2AX Is Phosphorylated in an ATR-dependent Manner in Response to Replicational Stress. J. Biol. Chem. 2001, 276, 47759–47762. [Google Scholar] [CrossRef] [Green Version]
- Kopp, B.; Khoury, L.; Audebert, M. Validation of the γH2AX biomarker for genotoxicity assessment: A review. Arch. Toxicol. 2019, 93, 2103–2114. [Google Scholar] [CrossRef]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef] [Green Version]
- Hafner, A.; Bulyk, M.L.; Jambhekar, A.; Lahav, G. The multiple mechanisms that regulate p53 activity and cell fate. Nat. Rev. Mol. Cell Biol. 2019, 20, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Simabuco, F.M.; Morale, M.G.; Pavan, I.C.B.; Morelli, A.P.; Silva, F.R.; Tamura, R.E. p53 and metabolism: From mechanism to therapeutics. Oncotarget 2018, 9, 23780–23823. [Google Scholar] [CrossRef] [Green Version]
- Letwin, K.; Mizzen, L.; Motro, B.; Ben-David, Y.; Bernstein, A.; Pawson, T. A mammalian dual specificity protein kinase, Nek1, is related to the NIMA cell cycle regulator and highly expressed in meiotic germ cells. EMBO J. 1992, 11, 3521–3531. [Google Scholar] [CrossRef]
- Naro, C.; Bielli, P.; Pagliarini, V.; Sette, C. The interplay between DNA damage response and RNA processing: The unexpected role of splicing factors as gatekeepers of genome stability. Front. Genet. 2015, 6, 142. [Google Scholar] [CrossRef] [Green Version]
- Kai, M. Roles of RNA-binding proteins in DNA damage response. Int. J. Mol. Sci. 2016, 17, 310. [Google Scholar] [CrossRef] [Green Version]
- Shkreta, L.; Chabot, B. The RNA Splicing Response to DNA Damage. Biomolecules 2015, 5, 2935–2977. [Google Scholar] [CrossRef] [PubMed]
- Lenzken, S.C.; Loffreda, A.; Barabino, S.M.L. RNA Splicing: A New Player in the DNA Damage Response. Int. J. Cell Biol. 2013, 2013, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Pabla, N.; Bhatt, K.; Dong, Z. Checkpoint kinase 1 (Chk1)-short is a splice variant and endogenous inhibitor of Chk1 that regulates cell cycle and DNA damage checkpoints. Proc. Natl. Acad. Sci. USA 2012, 109, 197–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandler, D.S.; Singh, R.K.; Caldwell, L.C.; Bitler, J.L.; Lozano, G. Genotoxic Stress Induces Coordinately Regulated Alternative Splicing of the p53 Modulators MDM2 and MDM4. Cancer Res. 2006, 66, 9502–9508. [Google Scholar] [CrossRef] [Green Version]
- Albert, H.; Battaglia, E.; Monteiro, C.; Bagrel, D. Genotoxic stress modulates CDC25C phosphatase alternative splicing in human breast cancer cell lines. Mol. Oncol. 2012, 6, 542–552. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Ho, C.K.; Ouyang, J.; Zou, L. Nek1 kinase associates with ATR-ATRIP and primes ATR for efficient DNA damage signaling. Proc. Natl. Acad. Sci. USA 2013, 110, 2175–2180. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Chen, P.-L.; Chen, C.-F.; Jiang, X.; Riley, D.J. Never-in-mitosis related Kinase 1 functions in DNA damage response and checkpoint control. Cell Cycle 2008, 7, 3194–3201. [Google Scholar] [CrossRef]
- Melo-Hanchuk, T.D.; Slepicka, P.F.; Meirelles, G.V.; Basei, F.L.; Lovato, D.V.; Granato, D.C.; Pauletti, B.A.; Domingues, R.R.; Leme, A.F.P.; Pelegrini, A.L.; et al. NEK1 kinase domain structure and its dynamic protein interactome after exposure to Cisplatin. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Brieño-Enríquez, M.A.; Moak, S.L.; Holloway, J.K.; Cohen, P.E. NIMA-related kinase 1 (NEK1) regulates meiosis I spindle assembly by altering the balance between α-Adducin and Myosin X. PLoS ONE 2017, 12, e0185780. [Google Scholar] [CrossRef] [Green Version]
- Shalom, O.; Shalva, N.; Altschuler, Y.; Motro, B. The mammalian Nek1 kinase is involved in primary cilium formation. FEBS Lett. 2008, 582, 1465–1470. [Google Scholar] [CrossRef] [Green Version]
- Fry, A.M.; Meraldi, P.; Nigg, E.A. A centrosomal function for the human Nek2 protein kinase, a member of the NIMA family of cell cycle regulators. EMBO J. 1998, 17, 470–481. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Yang, Y.; Xia, J.; Wang, H.; Salama, M.E.; Xiong, W.; Xu, H.; Shetty, S.; Chen, T.; Zeng, Z.; et al. NEK2 induces drug resistance mainly through activation of efflux drug pumps and is associated with poor prognosis in myeloma and other cancers. Cancer Cell 2013, 23, 48–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uto, K.; Nakajo, N.; Sagata, N. Two Structural Variants of Nek2 Kinase, Termed Nek2A and Nek2B, Are Differentially Expressed inXenopusTissues and Development. Dev. Biol. 1999, 208, 456–464. [Google Scholar] [CrossRef] [Green Version]
- Rellos, P.; Ivins, F.J.; Baxter, J.E.; Pike, A.; Nott, T.J.; Parkinson, D.-M.; Das, S.; Howell, S.; Fedorov, O.; Shen, Q.Y.; et al. Structure and regulation of the human Nek2 centrosomal kinase. J. Biol. Chem. 2007, 282, 6833–6842. [Google Scholar] [CrossRef] [Green Version]
- Alfieri, C.; Tischer, T.; Barford, D. A unique binding mode of Nek2A to the APC/C allows its ubiquitination during prometaphase. EMBO Rep. 2020, 21, e49831. [Google Scholar] [CrossRef] [PubMed]
- Fry, A.M. The Nek2 protein kinase: A novel regulator of centrosome structure. Oncogene 2002, 21, 6184–6194. [Google Scholar] [CrossRef] [Green Version]
- Fry, A.M.; Schultz, S.J.; Bartek, J.; Nigg, E.A. Substrate specificity and cell cycle regulation of the Nek2 protein kinase, a potential human homolog of the mitotic regulator NIMA of Aspergillus nidulans. J. Biol. Chem. 1995, 270, 12899–12905. [Google Scholar] [CrossRef] [Green Version]
- Jeong, A.L.; Ka, H.I.; Han, S.; Lee, S.; Lee, E.; Soh, S.J.; Joo, H.J.; Sumiyasuren, B.; Park, J.Y.; Lim, J.; et al. Oncoprotein CIP 2A promotes the disassembly of primary cilia and inhibits glycolytic metabolism. EMBO Rep. 2018, 19. [Google Scholar] [CrossRef]
- Wu, W.; Baxter, J.E.; Wattam, S.L.; Hayward, D.G.; Fardilha, M.; Knebel, A.; Ford, E.M.; da Cruz e Silva, E.F.; Fry, A.M. Alternative splicing controls nuclear translocation of the cell cycle-regulated Nek2 kinase. J. Biol. Chem. 2007, 282, 26431–26440. [Google Scholar] [CrossRef] [Green Version]
- HAMES, R.S.; FRY, A.M. Alternative splice variants of the human centrosome kinase Nek2 exhibit distinct patterns of expression in mitosis. Biochem. J. 2002, 361, 77. [Google Scholar] [CrossRef]
- Fletcher, L.; Cerniglia, G.J.; Nigg, E.A.; Yend, T.J.; Muschel, R.J. Inhibition of centrosome separation after DNA damage: A role for Nek2. Radiat. Res. 2004, 162, 128–135. [Google Scholar] [CrossRef]
- Kimura, M.; Okano, Y. Molecular cloning and characterization of the human NIMA-related protein kinase 3 gene (NEK3). Cytogenet. Cell Genet. 2001, 95, 177–182. [Google Scholar] [CrossRef]
- Coene, K.L.M.; Mans, D.A.; Boldt, K.; Gloeckner, C.J.; van Reeuwijk, J.; Bolat, E.; Roosing, S.; Letteboer, S.J.F.; Peters, T.A.; Cremers, F.P.M.; et al. The ciliopathy-associated protein homologs RPGRIP1 and RPGRIP1L are linked to cilium integrity through interaction with Nek4 serine/threonine kinase. Hum. Mol. Genet. 2011, 20, 3592–3605. [Google Scholar] [CrossRef] [Green Version]
- Doles, J.; Hemann, M.T. Nek4 status differentially alters sensitivity to distinct microtubule poisons. Cancer Res. 2010, 70, 1033–1041. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, C.L.; Possemato, R.; Bauerlein, E.L.; Xie, A.; Scully, R.; Hahn, W.C. Nek4 Regulates Entry into Replicative Senescence and the Response to DNA Damage in Human Fibroblasts. Mol. Cell. Biol. 2012, 32, 3963–3977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prosser, S.L.; Sahota, N.K.; Pelletier, L.; Morrison, C.G.; Fry, A.M. Nek5 promotes centrosome integrity in interphase and loss of centrosome cohesion in mitosis. J. Cell Biol. 2015, 209, 339–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Regan, L.; Fry, A.M. The Nek6 and Nek7 protein kinases are required for robust mitotic spindle formation and cytokinesis. Mol. Cell. Biol. 2009, 29, 3975–3990. [Google Scholar] [CrossRef] [Green Version]
- Rapley, J.; Nicolàs, M.; Groen, A.; Regué, L.; Bertran, M.T.; Caelles, C.; Avruch, J.; Roig, J. The NIMA-family kinase Nek6 phosphorylates the kinesin Eg5 at a novel site necessary for mitotic spindle formation. J. Cell Sci. 2008, 121, 3912–3921. [Google Scholar] [CrossRef] [Green Version]
- Meirelles, G.V.; Silva, J.C.; Mendonça, Y.D.A.; Ramos, C.H.I.; Torriani, I.L.; Kobarg, J. Human Nek6 is a monomeric mostly globular kinase with an unfolded short N-terminal domain. BMC Struct. Biol. 2011, 11, 12. [Google Scholar] [CrossRef] [Green Version]
- O’Regan, L.; Sampson, J.; Richards, M.W.; Knebel, A.; Roth, D.; Hood, F.E.; Straube, A.; Royle, S.J.; Bayliss, R.; Fry, A.M.; et al. Hsp72 is targeted to the mitotic spindle by Nek6 to promote K-fiber assembly and mitotic progression. J. Cell Biol. 2015, 209, 349–358. [Google Scholar] [CrossRef] [Green Version]
- Yin, M.-J.J.; Shao, L.; Voehringer, D.; Smeal, T.; Jallal, B. The Serine/Threonine Kinase Nek6 Is Required for Cell Cycle Progression through Mitosis. J. Biol. Chem. 2003, 278, 52454–52460. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.Y.; Kim, H.J.; Kim, M.A.; Hye, J.J.; Ae, J.K.; Bae, Y.S.; Park, J.I.; Chung, J.H.; Yun, J. Nek6 is involved in G2/M phase cell cycle arrest through DNA damage-induced phosphorylation. Cell Cycle 2008, 7, 2705–2709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Lee, K.; Rhee, K. NEK7 is a centrosomal kinase critical for microtubule nucleation. Biochem. Biophys. Res. Commun. 2007, 360, 56–62. [Google Scholar] [CrossRef]
- Haq, T.; Richards, M.W.; Burgess, S.G.; Gallego, P.; Yeoh, S.; O’Regan, L.; Reverter, D.; Roig, J.; Fry, A.M.; Bayliss, R. Mechanistic basis of Nek7 activation through Nek9 binding and induced dimerization. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byrne, M.J.; Nasir, N.; Basmadjian, C.; Bhatia, C.; Cunnison, R.F.; Carr, K.H.; Mas-Droux, C.; Yeoh, S.; Cano, C.; Bayliss, R. Nek7 conformational flexibility and inhibitor binding probed through protein engineering of the R-spine. Biochem. J. 2020, 477, 1525–1539. [Google Scholar] [CrossRef] [Green Version]
- Yissachar, N.; Salem, H.; Tennenbaum, T.; Motro, B. Nek7 kinase is enriched at the centrosome, and is required for proper spindle assembly and mitotic progression. FEBS Lett. 2006, 580, 6489–6495. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Meng, J.; Bi, F.; Li, H.; Chang, C.; Ji, C.; Liu, W. NEK7 Regulates NLRP3 Inflammasome Activation and Neuroinflammation Post-traumatic Brain Injury. Front. Mol. Neurosci. 2019, 12, 202. [Google Scholar] [CrossRef] [Green Version]
- Schmacke, N.A.; Gaidt, M.M.; Szymanska, I.; O’Duill, F.; Stafford, C.A.; Chauhan, D.; Fröhlich, A.L.; Nagl, D.; Pinci, F.; Schmid-Burgk, J.L.; et al. Priming enables a NEK7-independent route of NLRP3 activation. bioRxiv 2019, 799320. [Google Scholar] [CrossRef] [Green Version]
- Tan, R.; Nakajima, S.; Wang, Q.; Levine, A.S.; Su, B.; Lan, L.; Tan, R.; Nakajima, S.; Wang, Q.; Sun, H.; et al. Nek7 Protects Telomeres from Oxidative DNA Damage by Phosphorylation and Stabilization Article Nek7 Protects Telomeres from Oxidative DNA Damage by Phosphorylation and Stabilization of TRF1. Mol. Cell 2017, 65, 818–831.e5. [Google Scholar] [CrossRef] [Green Version]
- Otto, E.A.; Trapp, M.L.; Schultheiss, U.T.; Helou, J.; Quarmby, L.M.; Hildebrandt, F. NEK8 mutations affect ciliary and centrosomal localization and may cause nephronophthisis. J. Am. Soc. Nephrol. 2008, 19, 587–592. [Google Scholar] [CrossRef] [Green Version]
- Zalli, D.; Bayliss, R.; Fry, A.M. The Nek8 protein kinase, mutated in the human cystic kidney disease nephronophthisis, is both activated and degraded during ciliogenesis. Hum. Mol. Genet. 2012, 21, 1155–1171. [Google Scholar] [CrossRef] [Green Version]
- Sohara, E.; Luo, Y.; Zhang, J.; Manning, D.K.; Beier, D.R.; Zhou, J. Nek8 regulates the expression and localization of polycystin-1 and polycystin-2. J. Am. Soc. Nephrol. 2008, 19, 469–476. [Google Scholar] [CrossRef] [Green Version]
- Choi, H.J.C.; Lin, J.R.; Vannier, J.B.; Slaats, G.G.; Kile, A.C.; Paulsen, R.D.; Manning, D.K.; Beier, D.R.; Giles, R.H.; Boulton, S.J.; et al. NEK8 links the ATR-regulated replication stress response and S phase CDK activity to renal ciliopathies. Mol. Cell 2013, 51, 423–439. [Google Scholar] [CrossRef] [Green Version]
- Tan, B.C.M.; Lee, S.C. Nek9, a Novel FACT-associated Protein, Modulates Interphase Progression. J. Biol. Chem. 2004, 279, 9321–9330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belham, C.; Roig, J.; Caldwell, J.A.; Aoyama, Y.; Kemp, B.E.; Comb, M.; Avruch, J. A mitotic cascade of NIMA family kinases: Nercc1/Nek9 activates the Nek6 and Nek7 kinases. J. Biol. Chem. 2003, 278, 34897–34909. [Google Scholar] [CrossRef] [Green Version]
- Roig, J.; Groen, A.; Caldwell, J.; Avruch, J. Active Nercc1 Protein Kinase Concentrates at Centrosomes Early in Mitosis and Is Necessary for Proper Spindle Assembly. Mol. Biol. Cell 2005, 16, 4827–4840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sdelci, S.; Schütz, M.; Pinyol, R.; Bertran, M.T.; Regué, L.; Caelles, C.; Vernos, I.; Roig, J. Nek9 phosphorylation of NEDD1/GCP-WD contributes to Plk1 control of γ-tubulin recruitment to the mitotic centrosome. Curr. Biol. 2012, 22, 1516–1523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaneta, Y.; Ullrich, A. NEK9 depletion induces catastrophic mitosis by impairment of mitotic checkpoint control and spindle dynamics. Biochem. Biophys. Res. Commun. 2013, 442, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.C.; Petrova, A.V.; Madden, M.Z.; Wang, H.; Pan, Y.; Warren, M.D.; Hardy, C.W.; Liang, D.; Liu, E.A.; Robinson, M.H.; et al. A gemcitabine sensitivity screen identifies a role for NEK9 in the replication stress response. Nucleic Acids Res. 2014, 42, 11517–11527. [Google Scholar] [CrossRef] [Green Version]
- Moniz, L.S.; Stambolic, V. Nek10 mediates G2/M cell cycle arrest and MEK autoactivation in response to UV irradiation. Mol. Cell. Biol. 2011, 31, 30–42. [Google Scholar] [CrossRef] [Green Version]
- Noguchi, K.; Fukazawa, H.; Murakami, Y.; Uehara, Y. Nek11, a new member of the NIMA family of kinases, involved in DNA replication and genotoxic stress responses. J. Biol. Chem. 2002, 277, 39655–39665. [Google Scholar] [CrossRef] [Green Version]
- Sabir, S.R.; Sahota, N.K.; Jones, G.D.D.; Fry, A.M. Loss of Nek11 Prevents G2/M Arrest and Promotes Cell Death in HCT116 Colorectal Cancer Cells Exposed to Therapeutic DNA Damaging Agents. PLoS ONE 2015, 10, e0140975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surpili, M.J.; Delben, T.M.; Kobarg, J. Identification of proteins that interact with the central coiled-coil region of the human protein kinase NEK1. Biochemistry 2003, 42, 15369–15376. [Google Scholar] [CrossRef]
- Polci, R.; Peng, A.; Chen, P.-L.; Riley, D.J.; Chen, Y. NIMA-Related Protein Kinase 1 Is Involved Early in the Ionizing Radiation-Induced DNA Damage Response. Cancer Res. 2004, 64, 8800–8803. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Chen, C.F.; Chiang, H.C.; Pena, M.; Polci, R.; Wei, R.L.; Edwards, R.A.; Hansel, D.E.; Chen, P.L.; Riley, D.J. Mutation of NIMA-related kinase 1 (NEK1) leads to chromosome instability. Mol. Cancer 2011, 10, 5. [Google Scholar] [CrossRef] [Green Version]
- Patil, M.; Pabla, N.; Ding, H.-F.; Dong, Z. Nek1 interacts with Ku80 to assist chromatin loading of replication factors and S-phase progression. Cell Cycle 2013, 12, 2608–2616. [Google Scholar] [CrossRef] [Green Version]
- Spies, J.; Waizenegger, A.; Barton, O.; Sürder, M.; Wright, W.D.; Heyer, W.D.; Löbrich, M. Nek1 Regulates Rad54 to Orchestrate Homologous Recombination and Replication Fork Stability. Mol. Cell 2016, 62, 903–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciccia, A.; Elledge, S.J. The DNA Damage Response: Making It Safe to Play with Knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Chen, C.-F.; Riley, D.J.; Chen, P.-L. Nek1 kinase functions in DNA damage response and checkpoint control through a pathway independent of ATM and ATR. Cell Cycle 2011, 10, 655–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, E.J.; Baltimore, D. ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev. 2000, 14, 397–402. [Google Scholar] [PubMed]
- Upadhya, P.; Birkenmeier, E.H.; Birkenmeier, C.S.; Barker, J.E. Mutations in a NIMA-related kinase gene, Nek1, cause pleiotropic effects including a progressive polycystic kidney disease in mice. Proc. Natl. Acad. Sci. USA 2000, 97, 217–221. [Google Scholar] [CrossRef] [Green Version]
- Singh, V.; Connelly, Z.M.; Shen, X.; De Benedetti, A. Identification of the proteome complement of humanTLK1 reveals it binds and phosphorylates NEK1 regulating its activity. Cell Cycle 2017, 16, 915–926. [Google Scholar] [CrossRef] [Green Version]
- Singh, V.; Jaiswal, P.K.; Ghosh, I.; Koul, H.K.; Yu, X.; De Benedetti, A. The TLK1-Nek1 axis promotes prostate cancer progression. Cancer Lett. 2019, 453, 131–141. [Google Scholar] [CrossRef]
- Singh, V.; Jaiswal, P.K.; Ghosh, I.; Koul, H.K.; Yu, X.; De Benedetti, A. Targeting the TLK1/NEK1 DDR axis with Thioridazine suppresses outgrowth of androgen independent prostate tumors. Int. J. Cancer 2019, 145, 1055–1067. [Google Scholar] [CrossRef] [Green Version]
- De Benedetti, A. The Tousled-Like Kinases as Guardians of Genome Integrity. ISRN Mol. Biol. 2012, 2012, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Cirulli, E.T.; Lasseigne, B.N.; Petrovski, S.; Sapp, P.C.; Dion, P.A.; Leblond, C.S.; Couthouis, J.; Lu, Y.-F.; Wang, Q.; Krueger, B.J.; et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 2015, 347, 1436–1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gratten, J.; Zhao, Q.; Benyamin, B.; Garton, F.; He, J.; Leo, P.J.; Mangelsdorf, M.; Anderson, L.; Zhang, Z.; Chen, L.; et al. Whole-exome sequencing in amyotrophic lateral sclerosis suggests NEK1 is a risk gene in Chinese. Genome Med. 2017, 1–9. [Google Scholar] [CrossRef]
- Kenna, K.P.; Van Doormaal, P.T.C.; Dekker, A.M.; Ticozzi, N.; Kenna, B.J.; Diekstra, F.P.; Van Rheenen, W.; Van Eijk, K.R.; Jones, A.R.; Keagle, P.; et al. NEK1 variants confer susceptibility to amyotrophic lateral sclerosis. Nat. Genet. 2016, 48, 1037–1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higelin, J.; Catanese, A.; Semelink-Sedlacek, L.L.; Oeztuerk, S.; Lutz, A.-K.; Bausinger, J.; Barbi, G.; Speit, G.; Andersen, P.M.; Ludolph, A.C.; et al. NEK1 loss-of-function mutation induces DNA damage accumulation in ALS patient-derived motoneurons. Stem Cell Res. 2018, 30, 150–162. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Lin, H.; Wang, X.; Zuo, Q.; Qin, J.; Zhang, P. The NEK1 interactor, C21ORF2, is required for efficient DNA damage repair. Acta Biochim. Biophys. Sin. (Shanghai) 2015, 47, 834–841. [Google Scholar] [CrossRef] [Green Version]
- Van Rheenen, W.; Shatunov, A.; Dekker, A.M.; McLaughlin, R.L.; Diekstra, F.P.; Pulit, S.L.; van der Spek, R.A.A.; Võsa, U.; de Jong, S.; Robinson, M.R.; et al. Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat. Genet. 2016, 48, 1043–1048. [Google Scholar] [CrossRef] [Green Version]
- Walworth, N.C. DNA damage: Chk1 and Cdc25, more than meets the eye. Curr. Opin. Genet. Dev. 2001, 11, 78–82. [Google Scholar] [CrossRef]
- Smits, V.A.; Medema, R.H. Checking out the G(2)/M transition. Biochim. Biophys. Acta 2001, 1519, 1–12. [Google Scholar] [CrossRef]
- Andreassen, P.R.; Lacroix, F.B.; Villa-Moruzzi, E.; Margolis, R.L. Differential subcellular localization of protein phosphatase-1 alpha, gamma1, and delta isoforms during both interphase and mitosis in mammalian cells. J. Cell Biol. 1998, 141, 1207–1215. [Google Scholar] [CrossRef] [Green Version]
- Mi, J.; Guo, C.; Brautigan, D.L.; Larner, J.M. Protein phosphatase-1alpha regulates centrosome splitting through Nek2. Cancer Res. 2007, 67, 1082–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meraldi, P.; Nigg, E.A. Centrosome cohesion is regulated by a balance of kinase and phosphatase activities. J. Cell Sci. 2001, 114, 3749–3757. [Google Scholar]
- Chong, L.; van Steensel, B.; Broccoli, D.; Erdjument-Bromage, H.; Hanish, J.; Tempst, P.; de Lange, T. A human telomeric protein. Science 1995, 270, 1663–1667. [Google Scholar] [CrossRef]
- Nakamura, M.; Zhou, X.Z.; Kishi, S.; Kosugi, I.; Tsutsui, Y.; Lu, K.P. A specific interaction between the telomeric protein Pin2/TRF1 and the mitotic spindle. Curr. Biol. 2001, 11, 1512–1516. [Google Scholar] [CrossRef] [Green Version]
- Shen, M.; Haggblom, C.; Vogt, M.; Hunter, T.; Lu, K.P. Characterization and cell cycle regulation of the related human telomeric proteins Pin2 and TRF1 suggest a role in mitosis. Proc. Natl. Acad. Sci. USA 1997, 94, 13618–13623. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Gollahon, L. Mitotic perturbations induced by Nek2 overexpression require interaction with TRF1 in breast cancer cells. Cell Cycle 2013, 12, 3599–3614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, J.; Franqui Machin, R.; Gu, Z.; Zhan, F. Role of NEK2A in human cancer and its therapeutic potentials. BioMed. Res. Int. 2015, 2015, 862461. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.; Zhang, X. Targeting NEK2 as a promising therapeutic approach for cancer treatment. Cell Cycle 2016, 15, 895–907. [Google Scholar] [CrossRef] [Green Version]
- Kokuryo, T.; Yokoyama, Y.; Yamaguchi, J.; Tsunoda, N.; Ebata, T.; Nagino, M. NEK2 Is an Effective Target for Cancer Therapy With Potential to Induce Regression of Multiple Human Malignancies. Anticancer Res. 2019, 39, 2251–2258. [Google Scholar] [CrossRef]
- Choi, B.-K.; Dayaram, T.; Parikh, N.; Wilkins, A.D.; Nagarajan, M.; Novikov, I.B.; Bachman, B.J.; Jung, S.Y.; Haas, P.J.; Labrie, J.L.; et al. Literature-based automated discovery of tumor suppressor p53 phosphorylation and inhibition by NEK2. Proc. Natl. Acad. Sci. USA 2018, 115, 10666–10671. [Google Scholar] [CrossRef] [Green Version]
- Franqui-Machin, R.; Hao, M.; Bai, H.; Gu, Z.; Zhan, X.; Habelhah, H.; Jethava, Y.; Qiu, L.; Frech, I.; Tricot, G.; et al. Destabilizing NEK2 overcomes resistance to proteasome inhibition in multiple myeloma. J. Clin. Investig. 2018, 128, 2877–2893. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Zeng, Y.; Shi, L.; Yang, Q.; Chen, Y.; Wu, G.; Li, G.; Xu, S. Targeting NEK2 impairs oncogenesis and radioresistance via inhibiting the Wnt1/β-catenin signaling pathway in cervical cancer. J. Exp. Clin. Cancer Res. 2020, 39, 183. [Google Scholar] [CrossRef] [PubMed]
- Bindra, R.S.; Schaffer, P.J.; Meng, A.; Woo, J.; Måseide, K.; Roth, M.E.; Lizardi, P.; Hedley, D.W.; Bristow, R.G.; Glazer, P.M. Down-regulation of Rad51 and decreased homologous recombination in hypoxic cancer cells. Mol. Cell. Biol. 2004, 24, 8504–8518. [Google Scholar] [CrossRef] [Green Version]
- Dutertre, M.; Sanchez, G.; Barbier, J.; Corcos, L.; Auboeuf, D. The emerging role of pre-messenger RNA splicing in stress responses: Sending alternative messages and silent messengers. RNA Biol. 2011, 8, 740–747. [Google Scholar] [CrossRef] [Green Version]
- Cardoso, V.B.; Hanchuk, T.; De Souza, E.; Papa, P.; Meirelles, G.; Kobarg, J. Identification of NEK3 interacting proteins and functional characterization of its signaling mechanisms. J. Integr. OMICS 2017, 7. [Google Scholar] [CrossRef]
- Mailand, N.; Gibbs-Seymour, I.; Bekker-Jensen, S. Regulation of PCNA–protein interactions for genome stability. Nat. Rev. Mol. Cell Biol. 2013, 14, 269–282. [Google Scholar] [CrossRef] [PubMed]
- Fell, V.L.; Walden, E.A.; Hoffer, S.M.; Rogers, S.R.; Aitken, A.S.; Salemi, L.M.; Schild-Poulter, C. Ku70 Serine 155 mediates Aurora B inhibition and activation of the DNA damage response. Sci. Rep. 2016, 6, 37194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Shao, Y.; Wang, Q.; Zhai, Y.; Li, X. Genotoxic stress causes the accumulation of DNA-dependent protein kinase catalytic subunit phosphorylated at serine 2056 at nuclear speckles and alters pre-mRNA alternative splicing. FEBS Open Bio. 2019, 9, 304–314. [Google Scholar] [CrossRef]
- Melo-Hanchuk, T.D.; Slepicka, P.F.; Pelegrini, A.L.; Menck, C.F.M.; Kobarg, J. NEK5 interacts with topoisomerase IIβ and is involved in the DNA damage response induced by etoposide. J. Cell. Biochem. 2019, 120, 16853–16866. [Google Scholar] [CrossRef]
- Sciascia, N.; Wu, W.; Zong, D.; Sun, Y.; Wong, N.; John, S.; Wangsa, D.; Ried, T.; Bunting, S.F.; Pommier, Y.; et al. Suppressing proteasome mediated processing of topoisomerase II DNA-protein complexes preserves genome integrity. Elife 2020, 9. [Google Scholar] [CrossRef]
- Nitiss, J.L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 2009, 9, 338–350. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Berger, J.M. Cell Cycle-Dependent Control and Roles of DNA Topoisomerase II. Genes 2019, 10, 859. [Google Scholar] [CrossRef] [Green Version]
- Kasof, G.M.; Goyal, L.; White, E. Btf, a Novel Death-Promoting Transcriptional Repressor That Interacts with Bcl-2-Related Proteins. Mol. Cell. Biol. 1999, 19, 4390–4404. [Google Scholar] [CrossRef] [Green Version]
- Sarras, H.; Alizadeh Azami, S.; McPherson, J.P. In Search of a Function for BCLAF1. Sci. World J. 2010, 10, 1450–1461. [Google Scholar] [CrossRef]
- Lee, Y.Y.; Yu, Y.B.; Gunawardena, H.P.; Xie, L.; Chen, X. BCLAF1 is a radiation-induced H2AX-interacting partner involved in cH2AX-mediated regulation of apoptosis and DNA repair. Cell Death Dis. 2012, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vohhodina, J.; Barros, E.M.; Savage, A.L.; Liberante, F.G.; Manti, L.; Bankhead, P.; Cosgrove, N.; Madden, A.F.; Harkin, D.P.; Savage, K.I. The RNA processing factors THRAP3 and BCLAF1 promote the DNA damage response through selective mRNA splicing and nuclear export. Nucleic Acids Res. 2017, 45, 12816–12833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, J.; Zhang, J.; Yang, X.; Wu, Z.; Sun, C.; Wang, Z.; Wang, B. NEK5 promotes breast cancer cell proliferation through up-regulation of Cyclin A2. Mol. Carcinog. 2019, 58, 933–943. [Google Scholar] [CrossRef] [PubMed]
- Gong, D.; Ferrell, J.E. The roles of cyclin A2, B1, and B2 in early and late mitotic events. Mol. Biol. Cell 2010, 21, 3149–3161. [Google Scholar] [CrossRef] [Green Version]
- Sarkaria, J.N.; Busby, E.C.; Tibbetts, R.S.; Roos, P.; Taya, Y.; Karnitz, L.M.; Abraham, R.T. Inhibition of ATM and ATR kinase activities by the radiosensitizing agent, caffeine. Cancer Res. 1999, 59, 4375–4382. [Google Scholar] [PubMed]
- Blajeski, A.L.; Phan, V.A.; Kottke, T.J.; Kaufmann, S.H. G1 and G2 cell-cycle arrest following microtubule depolymerization in human breast cancer cells. J. Clin. Investig. 2002, 110, 91–99. [Google Scholar] [CrossRef]
- Minoguchi, S.; Minoguchi, M.; Yoshimura, A. Differential control of the NIMA-related kinases, Nek6 and Nek7, by serum stimulation. Biochem. Biophys. Res. Commun. 2003, 301, 899–906. [Google Scholar] [CrossRef]
- Zhang, B.; Zhang, H.A.I.; Wang, D.; Han, S.; Wang, K.E.; Yao, A.; Li, X. Never in mitosis gene A-related kinase 6 promotes cell proliferation of hepatocellular carcinoma via cyclin B modulation. Oncol. Lett. 2014, 1163–1168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jee, H.J.; Kim, H.J.; Kim, A.J.; Song, N.; Kim, M.; Yun, J. Nek6 suppresses the premature senescence of human cancer cells induced by camptothecin and doxorubicin treatment. Biochem. Biophys. Res. Commun. 2011, 408, 669–673. [Google Scholar] [CrossRef]
- Jee, H.J.; Kim, A.J.; Song, N.; Kim, H.J.; Kim, M.; Koh, H.; Yun, J. Nek6 overexpression antagonizes p53-induced senescence in human cancer cells. Cell Cycle 2010, 9, 4703–4710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, N.; Zhu, M.; Wang, X.; Tan, H.Y.; Tsao, S.W.; Feng, Y. Berberine-induced tumor suppressor p53 up-regulation gets involved in the regulatory network of MIR-23a in hepatocellular carcinoma. Biochim. Biophys. Acta Gene Regul. Mech. 2014, 1839, 849–857. [Google Scholar] [CrossRef]
- Guo, Z.; Zhou, B.; Liu, W.; Xu, Y.; Wu, D.; Yin, Z.; Permatasari, F.; Luo, D. MiR-23a regulates DNA damage repair and apoptosis in UVB-irradiated HaCaT cells. J. Dermatol. Sci. 2013, 69, 68–76. [Google Scholar] [CrossRef]
- Zhu, Y.; Ma, N.; Li, H.X.; Tian, L.; Ba, Y.F.; Hao, B. Berberine induces apoptosis and DNA damage in MG-63 human osteosarcoma cells. Mol. Med. Rep. 2014, 10, 1734–1738. [Google Scholar] [CrossRef] [Green Version]
- Zhu, M.; Wang, N.; Tsao, S.W.; Yuen, M.F.; Feng, Y.; Wan, T.S.K.; Man, K.; Feng, Y. Up-regulation of microRNAs, miR21 and miR23a in human liver cancer cells treated with Coptidis rhizoma aqueous extract. Exp. Ther. Med. 2011, 2, 27–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaz Meirelles, G.; Ferreira Lanza, D.C.; Da Silva, J.C.; Santana Bernachi, J.; Paes Leme, A.F.; Kobarg, J. Characterization of hNek6 interactome reveals an important role for its short N-terminal domain and colocalization with proteins at the centrosome. J. Proteome Res. 2010, 9, 6298–6316. [Google Scholar] [CrossRef] [PubMed]
- Zuo, J.; Ma, H.; Cai, H.; Wu, Y.; Jiang, W.; Yu, L. An inhibitory role of NEK6 in TGFß/Smad signaling pathway. BMB Rep. 2015, 48, 473–478. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.; Jarvis, I.W.H.; Bottai, M.; Dreij, K.; Stenius, U. TGF beta promotes repair of bulky DNA damage through increased ERCC1/XPF and ERCC1/XPA interaction. Carcinogenesis 2019, 40, 580–591. [Google Scholar] [CrossRef]
- Mitra, D.; Fernandez, P.; Bian, L.; Song, N.; Li, F.; Han, G.; Wang, X.J. Smad4 loss in mouse keratinocytes leads to increased susceptibility to UV carcinogenesis with reduced ercc1-mediated DNA repair. J. Invest. Dermatol. 2013, 133, 2609–2616. [Google Scholar] [CrossRef] [Green Version]
- Levy, L.; Hill, C.S. Smad4 Dependency Defines Two Classes of Transforming Growth Factor β (TGF-β) Target Genes and Distinguishes TGF-β-Induced Epithelial-Mesenchymal Transition from Its Antiproliferative and Migratory Responses. Mol. Cell. Biol. 2005, 25, 8108–8125. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.-Y.; Nie, J.; Huang, J.-P.; Zheng, G.-J.; Feng, B. Targeting STAT3 inhibition to reverse cisplatin resistance. Biomed. Pharmacother. 2019, 117, 109135. [Google Scholar] [CrossRef] [PubMed]
- De Donato, M.; Fanelli, M.; Mariani, M.; Raspaglio, G.; Pandya, D.; He, S.; Fiedler, P.; Petrillo, M.; Scambia, G.; Ferlini, C. Nek6 and Hif-1α cooperate with the cytoskeletal gateway of drug resistance to drive outcome in serous ovarian cancer. Am. J. Cancer Res. 2015, 5, 1862–1877. [Google Scholar]
- De Donato, M.; Righino, B.; Filippetti, F.; Battaglia, A.; Petrillo, M.; Pirolli, D.; Scambia, G.; De Rosa, M.C.; Gallo, D. Identification and antitumor activity of a novel inhibitor of the NIMA-related kinase NEK6. Sci. Rep. 2018, 8, 16047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeon, Y.J.; Lee, K.Y.; Cho, Y.Y.; Pugliese, A.; Kim, H.G.; Jeong, C.H.; Bode, A.M.; Dong, Z. Role of NEK6 in tumor promoter-induced transformation in JB6 C141 mouse skin epidermal cells. J. Biol. Chem. 2010, 285, 28126–28133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koganti, S.; Hui-Yuen, J.; McAllister, S.; Gardner, B.; Grasser, F.; Palendira, U.; Tangye, S.G.; Freeman, A.F.; Bhaduri-McIntosh, S. STAT3 interrupts ATR-Chk1 signaling to allow oncovirus-mediated cell proliferation. Proc. Natl. Acad. Sci. USA 2014, 111, 4946–4951. [Google Scholar] [CrossRef] [Green Version]
- Fu, B.; Xue, W.; Zhang, H.; Zhang, R.; Feldman, K.; Zhao, Q.; Zhang, S.; Shi, L.; Pavani, K.C.; Nian, W.; et al. Microrna-325-3p facilitates immune escape of mycobacterium tuberculosis through targeting lnx1 via nek6 accumulation to promote anti-apoptotic stat3 signaling. MBio 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Barry, S.P.; Townsend, P.A.; Knight, R.A.; Scarabelli, T.M.; Latchman, D.S.; Stephanou, A. STAT3 modulates the DNA damage response pathway. Int. J. Exp. Pathol. 2010, 91, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Lin, S.; Xu, L.; Lin, J.; Zhao, C.; Huang, X. Novel activators and small-molecule inhibitors of STAT3 in cancer. Cytokine Growth Factor Rev. 2019, 49, 10–22. [Google Scholar] [CrossRef]
- Donate, L.E.; Blasco, M.A. Telomeres in cancer and ageing. Philos. Trans. R. Soc. B Biol. Sci. 2011, 366, 76–84. [Google Scholar] [CrossRef]
- Smith, S. Telomerase can’t handle the stress. Genes Dev. 2018, 32, 597–599. [Google Scholar] [CrossRef] [PubMed]
- Diotti, R.; Loayza, D. Shelterin complex and associated factors at human telomeres. Nucleus 2011, 2, 119–135. [Google Scholar] [CrossRef] [Green Version]
- De Souza, E.E.; Meirelles, G.V.; Godoy, B.B.; Perez, A.M.; Smetana, J.H.C.; Doxsey, S.J.; McComb, M.E.; Costello, C.E.; Whelan, S.A.; Kobarg, J. Characterization of the human NEK7 interactome suggests catalytic and regulatory properties distinct from those of NEK6. J. Proteome Res. 2014, 13, 4074–4090. [Google Scholar] [CrossRef] [Green Version]
- Carney, J.P.; Maser, R.S.; Olivares, H.; Davis, E.M.; Le Beau, M.; Yates, J.R.; Hays, L.; Morgan, W.F.; Petrini, J.H. The hMre11/hRad50 Protein Complex and Nijmegen Breakage Syndrome: Linkage of Double-Strand Break Repair to the Cellular DNA Damage Response. Cell 1998, 93, 477–486. [Google Scholar] [CrossRef] [Green Version]
- Limbo, O.; Yamada, Y.; Russell, P. Mre11-Rad50–dependent activity of ATM/Tel1 at DNA breaks and telomeres in the absence of Nbs1. Mol. Biol. Cell 2018, 29, 1389–1399. [Google Scholar] [CrossRef]
- Baranovskiy, A.G.; Babayeva, N.D.; Suwa, Y.; Gu, J.; Pavlov, Y.I.; Tahirov, T.H. Structural basis for inhibition of DNA replication by aphidicolin. Nucleic Acids Res. 2014, 42, 14013–14021. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Lu, W.; Obara, T.; Kuida, S.; Lehoczky, J.; Dewar, K.; Drummond, I.A.; Beier, D.R. A defect in a novel Nek-family kinase causes cystic kidney disease in the mouse and in zebrafish. Development 2002, 129, 5839–5846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Heyer, W.-D. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008, 18, 99–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krejci, L.; Altmannova, V.; Spirek, M.; Zhao, X. Homologous recombination and its regulation. Nucleic Acids Res. 2012, 40, 5795–5818. [Google Scholar] [CrossRef] [PubMed]
- Abeyta, A.; Castella, M.; Jacquemont, C.; Taniguchi, T. NEK8 regulates DNA damage-induced RAD51 foci formation and replication fork protection. Cell Cycle 2017, 16, 335–347. [Google Scholar] [CrossRef]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R.; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR Substrate Analysis Reveals Extensive Protein Networks Responsive to DNA Damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Behrends, C.; Sowa, M.E.; Gygi, S.P.; Harper, J.W. Network organization of the human autophagy system. Nature 2010, 466, 68–76. [Google Scholar] [CrossRef] [Green Version]
- Schäfer, A.; Schomacher, L.; Barreto, G.; Döderlein, G.; Niehrs, C. Gemcitabine Functions Epigenetically by Inhibiting Repair Mediated DNA Demethylation. PLoS ONE 2010, 5, e14060. [Google Scholar] [CrossRef]
- Parker, J.D.K.; Bradley, B.A.; Mooers, A.O.; Quarmby, L.M. Phylogenetic Analysis of the Neks Reveals Early Diversification of Ciliary-Cell Cycle Kinases. PLoS ONE 2007, 2, e1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lippke, J.A.; Gordon, L.K.; Brash, D.E.; Haseltine, W.A. Distribution of UV light-induced damage in a defined sequence of human DNA: Detection of alkaline-sensitive lesions at pyrimidine nucleoside-cytidine sequences. Proc. Natl. Acad. Sci. USA 1981, 78, 3388–3392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, D.L.; Nairn, R.S. The biology of the (6–4) photoproduct. Photochem. Photobiol. 1989, 49, 805–819. [Google Scholar] [CrossRef]
- De Lima-Bessa, K.M.; Armelini, M.G.; Chiganças, V.; Jacysyn, J.F.; Amarante-Mendes, G.P.; Sarasin, A.; Menck, C.F.M. CPDs and 6-4PPs play different roles in UV-induced cell death in normal and NER-deficient human cells. DNA Repair Amst. 2008, 7, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Roberts, P.J.; Der, C.J. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 2007, 26, 3291–3310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haider, N.; Dutt, P.; van de Kooij, B.; Ho, J.; Palomero, L.; Pujana, M.A.; Yaffe, M.; Stambolic, V. NEK10 tyrosine phosphorylates p53 and controls its transcriptional activity. Oncogene 2020, 39, 5252–5266. [Google Scholar] [CrossRef]
- Kumari, R.; Kohli, S.; Das, S. p53 regulation upon genotoxic stress: Intricacies and complexities. Mol. Cell. Oncol. 2014, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toufektchan, E.; Toledo, F. The Guardian of the Genome Revisited: p53 Downregulates Genes Required for Telomere Maintenance, DNA Repair, and Centromere Structure. Cancers 2018, 10, 135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boutros, R.; Lobjois, V.; Ducommun, B. CDC25 phosphatases in cancer cells: Key players? Good targets? Nat. Rev. Cancer 2007, 7, 495–507. [Google Scholar] [CrossRef] [PubMed]
- Goto, H.; Natsume, T.; Kanemaki, M.T.; Kaito, A.; Wang, S.; Gabazza, E.C.; Inagaki, M.; Mizoguchi, A. Chk1-mediated Cdc25A degradation as a critical mechanism for normal cell cycle progression. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef] [Green Version]
- De Man, F.M.; Goey, A.K.L.; van Schaik, R.H.N.; Mathijssen, R.H.J.; Bins, S. Individualization of Irinotecan Treatment: A Review of Pharmacokinetics, Pharmacodynamics, and Pharmacogenetics. Clin. Pharmacokinet. 2018, 57, 1229–1254. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pavan, I.C.B.; Peres de Oliveira, A.; Dias, P.R.F.; Basei, F.L.; Issayama, L.K.; Ferezin, C.d.C.; Silva, F.R.; Rodrigues de Oliveira, A.L.; Alves dos Reis Moura, L.; Martins, M.B.; et al. On Broken Ne(c)ks and Broken DNA: The Role of Human NEKs in the DNA Damage Response. Cells 2021, 10, 507. https://doi.org/10.3390/cells10030507
Pavan ICB, Peres de Oliveira A, Dias PRF, Basei FL, Issayama LK, Ferezin CdC, Silva FR, Rodrigues de Oliveira AL, Alves dos Reis Moura L, Martins MB, et al. On Broken Ne(c)ks and Broken DNA: The Role of Human NEKs in the DNA Damage Response. Cells. 2021; 10(3):507. https://doi.org/10.3390/cells10030507
Chicago/Turabian StylePavan, Isadora Carolina Betim, Andressa Peres de Oliveira, Pedro Rafael Firmino Dias, Fernanda Luisa Basei, Luidy Kazuo Issayama, Camila de Castro Ferezin, Fernando Riback Silva, Ana Luisa Rodrigues de Oliveira, Lívia Alves dos Reis Moura, Mariana Bonjiorno Martins, and et al. 2021. "On Broken Ne(c)ks and Broken DNA: The Role of Human NEKs in the DNA Damage Response" Cells 10, no. 3: 507. https://doi.org/10.3390/cells10030507