
Multiple Mechanisms Regulate Eukaryotic Cytochrome C Oxidase

{kind=link}
Abstract
1. Introduction
2. Cytochrome C Oxidase (COX)
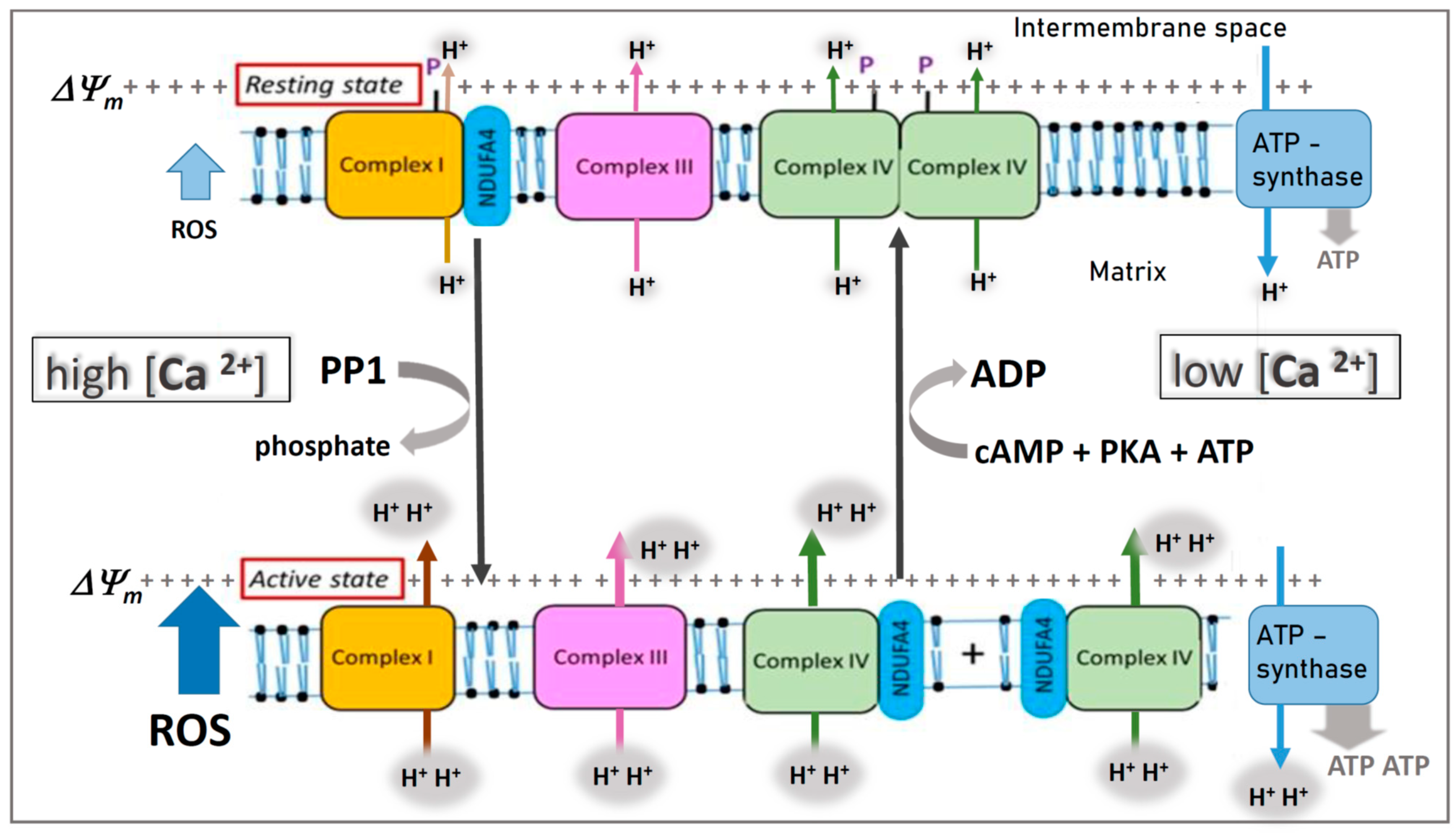
3. Regulation of COX Activity by “Allosteric ATP-Inhibition”
4. Role of Calcium in Generation of Increased ROS and ΔΨm
5. Regulation of COX via Reversible Phosphorylation
6. Regulation of COX via Expressing Supernumerary Subunit Isoforms
7. Regulation of COX via Binding Small Metabolites, Proteins, and Ligands and Deacetylation of Subunits
8. Overexpression of COX Subunits during Ischemic Injury, Cancerogenesis and Regulation via Forming Supercomplexes
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mitchell, P. Coupling of phosphorylation to electron and hydrogen transfer by a chemiosmotic type of mechanism. Nature 1961, 191, 144–148. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P. Chemiosmotic coupling in oxidative and photosynthetic phosphorylation. Biol. Rev. 1966, 41, 445–502. [Google Scholar] [CrossRef] [PubMed]
- Demine, S.; Renard, P.; Arnould, T. Mitochondrial uncoupling: A key controller of biological processes in physiology and diseases. Cells 2019, 8, 795. [Google Scholar] [CrossRef] [PubMed]
- Villani, G.; Attardi, G. In vivo control of respiration by cytochrome c oxidase in wildtype and mitochondrial DNA mutation-carrying human cells. Proc. Natl. Acad. Sci. USA 1997, 94, 1166–1171. [Google Scholar] [CrossRef] [PubMed]
- Villani, G.; Attardi, G. In vivo control of respiration by cytochrome c oxidase in human cells. Free Radic. Biol. Med. 2000, 29, 202–210. [Google Scholar] [CrossRef]
- Kacser, H.; Burns, J.A. The control of flux. Symp. Soc. Exp. Biol. 1973, 27, 65–104. [Google Scholar] [CrossRef]
- Heinrich, R.; Rapoport, T.A. A linear steady-state treatment of enzymatic chains. General properties, control and effector strength. Eur. J. Biochem. 1974, 42, 89–95. [Google Scholar] [CrossRef]
- Fell, D. Understanding the control of metabolism. In Frontiers in Metabolism; Portland Press: London, UK, 1997; Volume 2. [Google Scholar]
- Tager, J.M.; Wanders, R.J.A.; Groen, A.K.; Kunz, W.; Bohnensack, R.; Küster, U.; Letko, G.; Böhme, G.; Duszynski, J.; Woijtczak, L. Control of mitochondrial respiration. FEBS Lett. 1981, 1–9. [Google Scholar]
- Letellier, T.; Malgat, M.; Mazat, J.P. Control of oxidative phosphorylation in rat muscle mitochondria: Implications for mitochondrial myopathies. Biochim. Biophys. Acta 1993, 1141, 58–64. [Google Scholar] [CrossRef]
- Letellier, T.; Heinrich, R.; Malgat, M.; Mazat, J.P. The kinetic basis of threshold effects observed in mitochondrial diseases: A systemic approach. Biochem. J. 1994, 302, 171–174. [Google Scholar] [CrossRef] [PubMed]
- Pierron, D.; Wildman, D.E.; Hüttemann, M.; Markondapatnaikuni, G.C.; Aras, S.; Grossman, L.I. Cytochrome c oxidase: Evolution of control via nuclear subunit addition. Biochim. Biophys. Acta 2012, 1817, 590–597. [Google Scholar] [CrossRef] [PubMed]
- Kadenbach, B.; Jarausch, J.; Hartmann, R.; Merle, P. Separation of mammalian cytochrome c oxidase into 13 poly-peptides by a sodium dodecyl sulfate-gel electrophoretic procedure. Anal. Biochem. 1983, 129, 517–521. [Google Scholar] [CrossRef]
- Hundt, E.; Trapp, M.; Kadenbach, B. Biosynthesis of cytochrome c oxidase in isolated hepatocytes. FEBS Lett. 1980, 115, 95–99. [Google Scholar] [CrossRef][Green Version]
- Kang, Y.; Fielden, L.F.; Stojanovski, D. Mitochondrial protein transport in health and disease. Semin. Cell Dev. Biol. 2018, 76, 142–153. [Google Scholar] [CrossRef]
- Timón-Gómez, A.; Nývltová, E.; Abriata, L.A.; Vila, A.J.; Hosler, J.; Barrientos, A. Mitochondrial cytochrome c oxidase biogenesis: Recent developments. Semin. Cell Dev. Biol. 2018, 76, 163–178. [Google Scholar] [CrossRef] [PubMed]
- Arnold, S.; Kadenbach, B. Priority Paper. Cell respiration is controlled by ATP, an allosteric inhibitor of cytochrome c oxidase. Eur. J. Biochem. 1997, 249, 350–354. [Google Scholar] [CrossRef] [PubMed]
- Arnold, S.; Kadenbach, B. The intramitochondrial ATP/ADP-ratio controls cytochrome c oxidase activity allosterically. FEBS Lett. 1999, 443, 105–108. [Google Scholar] [CrossRef]
- Kadenbach, B.; Arnold, S. A second mechanism of respiratory control. FEBS Lett. 1999, 447, 131–134. [Google Scholar] [CrossRef]
- Tsukihara, T.; Aoyama, H.; Yamashita, E.; Tomizaki, T.; Yamaguchi, H.; Shinzawa-Itoh, K.; Nakashima, R.; Yaono, R.; Yoshikawa, S. The whole structure of the 13-subunit oxidized cytochrome c oxidase at 2.8 Å. Science 1996, 272, 1136–1144. [Google Scholar] [CrossRef]
- Osuda, Y.; Shinzawa-Itoh, K.; Tani, K.; Maeda, S.; Yoshikawa, S.; Tsukihara, T.; Gerle, C. Two dimensional crystallization of monomeric bovine cytochrome c oxidase with bound cytochrome c in reconstituted lipid membranes. Microscopy (Oxf.) 2016, 65, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Follmann, K.; Arnold, S.; Ferguson-Miller, S.; Kadenbach, B. Cytochrome c oxidase activity from eucaryotes but not from procaryotes is allosterically inhibited by ATP. Biochem. Mol. Biol. Intern. 1998, 45, 1047–1055. [Google Scholar]
- Alge, D.; Wastyn, M.; Mayer, C.; Jungwirth, C.; Zimmermann, U.; Zoder, R.; Fromwald, S.; Peschek., G.A. Allosteric properties of cyanobacterial cytochrome c oxidase: Inhibition of the coupled enzyme by ATP and stimulation by ADP. IUBMB Life 1999, 48, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Peschek, G.A. Cytochrome oxidase and cta operon of cyanobacteria. Biochim. Biophys. Acta 1996, 1275, 27–32. [Google Scholar] [CrossRef]
- Ramzan, R.; Rhiel, A.; Weber, P.; Kadenbach, B.; Vogt, S. Reversible dimerization of cytochrome c oxidase regulates mitochondrial respiration. Mitochondrion 2019, 49, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, B.; Bender, E.; Arnold, S.; Hüttemann, M.; Lee, I.; Kadenbach, B. Cytochrome c oxidase and the regulation of oxidative phosphorylation. ChemBioChem 2001, 2, 392–403. [Google Scholar] [CrossRef]
- Robb-Gaspers, L.D.; Rutter, G.A.; Denton, R.M.; Rizzuto, R.; Thomas, A.P. Integrating cytosolic calcium signals into mitochondrial metabolic responses. EMBO J. 1998, 17, 4987–5000. [Google Scholar] [CrossRef]
- Ramzan, R.; Vogt, S.; Kadenbach, B. Stress-mediated generation of deleterious ROS in healthy individuals—Role of cytochrome c oxidase. J. Mol. Med. (Berl.) 2020, 98, 651–657. [Google Scholar] [CrossRef]
- Kadenbach, B.; Hüttemann, M.; Arnold, S.; Lee, I.; Mühlenbein, N.; Bender, E. Mitochondrial energy metabolism is regulated via nuclear-coded subunits of cytochrome c oxidase. Free Radic. Biol. Med. 2000, 29, 211–221. [Google Scholar] [CrossRef]
- Kadenbach, B.; Ramzan, R.; Wen, L.; Vogt, S. New extension of the Mitchell Theory for oxidative phosphorylation in mitochondria of living organisms. Biochim. Biophys. Acta 2010, 1800, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive oxygen species in metabolic and inflammatory signaling. Circ. Res. 2018, 122, 877–902. [Google Scholar] [CrossRef]
- Kausar, S.; Wang, F.; Cui, H. The role of mitochondria in reactive oxygen species generation and its implications for neurodegenerative diseases. Cells 2018, 7, 274. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.-Z.; Jiang, S.; Zhang, L.; Yu, Z.-B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Hopper, R.K.; Carroll, S.; Aponte, A.M.; Johnson, D.T.; French, S.; Shen, R.F.; Witzmann, F.A.; Harris, R.A.; Balaban, R.S. Mitochondrial matrix phosphoproteome: Effect of extra mitochondrial calcium. Biochemistry 2006, 45, 2524–2536. [Google Scholar] [CrossRef] [PubMed]
- Sedlic, F.; Muravyeva, M.; Sepac, A.; Sedlic, M.; Williams, A.M.; Yang, M.; Bai, X.; Bosnjak, Z.J. Targeted modification of mitochondrial ROS production converts high glucose-induced cytotoxicity to cytoprotection: Effects on anesthetic preconditioning. J. Cell Physiol. 2017, 232, 216–224. [Google Scholar] [CrossRef]
- Gerencser, A.A. Metabolic activation-driven mitochondrial hyperpolarization predicts insulin secretion in human pancreatic beta-cells. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 817–828. [Google Scholar] [CrossRef]
- Capellino, S.; Claus, M.; Watzl, C. Regulation of natural killer cell activity by glucocorticoids, serotonin, dopamine, and epinephrine. Cell. Mol. Immunol. 2020, 17, 705–711. [Google Scholar] [CrossRef]
- Vincent, A.M.; Olzmann, J.A.; Brownlee, M.; Sivitz, W.I.; Russell, J.W. Uncoupling proteins prevent glucose-induced neuronal oxidative stress and programmed cell death. Diabetes 2004, 53, 726–734. [Google Scholar] [CrossRef]
- Dorn, G.W.; Force, T. Protein kinase cascades in the regulation of cardiac hypertrophy. J. Clin. Investig. 2005, 115, 527–537. [Google Scholar] [CrossRef]
- Kass, G.E.; Orenius, S. Calcium signaling and cytotoxicity. Environ. Health Perspect. 1999, 107, 25–35. [Google Scholar]
- Sher, L.D.; Geddie, H.V.; Olivier, L.; Cairns, M.; Truter, N.; Beselaar, L.; Essop, M.F. Chronic stress and endothelial dysfunction: Mechanisms, experimental challenges and way ahead. Am. J. Physiol. Heart Circ. Physiol. 2020, 319, H488–H506. [Google Scholar] [CrossRef]
- Barbiero, S.; Aimo, A.; Castiglione, V.; Giannoni, A.; Vergaro, G.; Passino, C.; Emdin, M. Healthy hearts at hectic pace: From daily life stress to abnormal cardiomyocyte function and arrhythmias. Eur. J. Prev. Cardiol. 2018, 25, 1419–1430. [Google Scholar] [CrossRef] [PubMed]
- Kullmann, F.A.; McDonnell, B.M.; Wolf-Johnston, A.S.; Kanai, A.J.; Shiva, S.; Chelimsky, T.; Rodriguez, L.; Birder, L.A. Stress-induced autonomic dysregulation of mitochondrial function in the rat urothelium. Neurourol. Urodyn. 2019, 38, 572–581. [Google Scholar] [CrossRef] [PubMed]
- Matsuhisa, F.; Kitamura, N.; Satoh, E. Effects of acute and chronic psychological stress on platelet aggregation in mice. Stress 2014, 17, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Solanki, N.; Salvi, A.; Patki, G.; Salim, S. Modulating oxidative stress relieves stress-induced behavioral and cognitive impairments in rats. Int. J. Neuropsychopharmacol. 2017, 20, 550–561. [Google Scholar] [CrossRef] [PubMed]
- Turdi, S.; Yuan, M.; Leedy, G.M.; Wu, Z.; Ren, J. Chronic social stress induces cardiomyocyte contractile dysfunction and intracellular Ca2+ derangement in rats. Physiol. Behav. 2012, 105, 498–509. [Google Scholar] [CrossRef]
- Kumari, S.; Mehta, S.L.; Li, P.A. Glutamate induces mitochondrial dynamic imbalance and autophagy activation: Preventive effects of selenium. PLoS ONE 2012, 7, e39382. [Google Scholar] [CrossRef]
- Napiwotzki, J.; Shinzawa-Itoh, K.; Yoshikawa, S.; Kadenbach, B. ATP and ADP bind to cytochrome c oxidase and regulate its activity. Biol. Chem. 1997, 378, 1013–1021. [Google Scholar] [CrossRef]
- Jastroch, M.; Divakaruni, A.S.; Mookerjee, S.; Treberg, J.R.; Brand, M.D. Mitochondrial proton and electron leaks. Essays Biochem. 2010, 47, 53–67. [Google Scholar]
- O’Shea, P.S.; Petrone, G.; Casey, R.P.; Azzi, A. The current– voltage relationships of liposomes and mitochondria. Biochem. J. 1984, 219, 719–726. [Google Scholar] [CrossRef]
- Steverding, D.; Kadenbach, B. Influence of N-ethoxycarbonyl-2-ethoxy-1,2-dihydroquinoline modification on proton translocation and membrane potential of reconstituted cytochrome c oxidase support “proton slippage”. J. Biol. Chem. 1991, 266, 8097–8101. [Google Scholar] [CrossRef]
- Babcock, G.T.; Wikström, M. Oxygen activation and the conservation of energy in cell respiration. Nature 1992, 356, 301–309. [Google Scholar] [CrossRef]
- Frank, V.; Kadenbach, B. Regulation of the H+/e--stoichiometry of cytochrome c oxidase from bovine heart by intraliposomal ATP/ADP ratios. FEBS Lett. 1996, 382, 121–124. [Google Scholar] [CrossRef]
- Yoshikawa, S. A cytochrome c oxidase proton pumping mechanism that excludes the O2 reduction site. FEBS Lett. 2003, 555, 8–12. [Google Scholar] [CrossRef]
- Yoshikawa, S.; Muramoto, K.; Sinzawa-Itoh, K.; Aoyama, H.; Tsukihara, T.; Shimokata, K.; Katayama, Y.; Shimada, H.H. Proton pumping mechanism of bovine heart cytochrome c oxidase. Biochim. Biophys. Acta 2006, 1757, 1110–1116. [Google Scholar] [CrossRef][Green Version]
- Yoshikawa, S.; Muramoto, K.; Shinzawa-Itoh, K. The O(2) reduction and proton pumping gate mechanism of bovine heart cytochrome c oxidase. Biochim. Biophys. Acta 2011, 1807, 1279–1286. [Google Scholar] [CrossRef]
- Salje, J.; Ludwig, B.; Richter, O.M. Is a third proton-conducting pathway operative in bacterial cytochrome c oxidase? Biochem. Soc. Trans. 2005, 33, 829–831. [Google Scholar] [CrossRef] [PubMed]
- Costa, L.E.; Reynafarje, B.; Lehninger, A.L. Stoichiometry of mitochondrial H+ translocation coupled to succinate oxidation at level flow. J. Biol. Chem. 1984, 259, 4802–4811. [Google Scholar] [CrossRef]
- Reynafarje, B.; Alexandre, A.; Davies, P.; Lehninger, A.L. Proton translocation stoichiometry of cytochrome oxidase: Use of a fast-responding oxygen electrode. Proc. Natl. Acad. Sci. USA 1982, 79, 7218–7222. [Google Scholar] [CrossRef] [PubMed]
- Reynafarje, B.; Costa, L.E.; Lehninger, A.L. Upper and lower limits of the proton stoichiometry of cytochrome c oxidation in rat liver mitoplasts. J. Biol. Chem. 1986, 261, 8254–8262. [Google Scholar] [CrossRef]
- Setty, O.H.; Shrager, R.I.; Bunow, B.; Reynafarje, B.; Lehninger, A.L.; Hendler, R.W. Direct measurement of the initial proton extrusion to oxygen uptake ratio accompanying succinate oxidation by rat liver mitochondria. Biophys. J. 1986, 50, 391–404. [Google Scholar] [CrossRef]
- Zong, S.; Wu, M.; Gu, J.; Liu, T.; Guo, R.; Yang, M. Structure of the intact 14-subunit human cytochrome c oxidase. Cell Res. 2018, 28, 1026–1034. [Google Scholar] [CrossRef]
- Weishaupt, A.; Kadenbach, B. Selective removal of subunit VIb increases the activity of cytochrome c oxidase. Biochemistry 1992, 31, 11477–11481. [Google Scholar] [CrossRef]
- Nesterov, S.; Chesnokov, Y.; Kamyshinsky, R.; Panteleeva, A.; Lyamzaev, K.; Vasilov, R.; Yaguzhinsky, L. Ordered clusters of the complete oxidative phosphorylation system in cardiac mitochondria. Int. J. Mol. Sci. 2021, 22, 1462. [Google Scholar] [CrossRef] [PubMed]
- Chance, B.; Sies, H.; Boveris, A. Hydroperoxide metabolism in mammalian organs. Physiol. Rev. 1979, 59, 527–605. [Google Scholar] [CrossRef] [PubMed]
- Dalle-Donne, I.; Rossi, R.; Colombo, R.; Giustarini, D.; Milzani, A. Biomarkers of oxidative damage in human disease. Clin. Chem. 2006, 52, 601–623. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Leibfritz, D.; Moncola, J.; Cronin, M.T.D.; Mazura, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Trachootham, D.; Zhang, H.; Zhang, W.; Feng, L.; Du, M.; Zhou, Y.; Chen, Z.; Pelicano, H.; Plunkett, W.; Wierda, W.G.; et al. Redox regulation of cell survival. Antioxi. Redox Signal. 2008, 10, 1343–1374. [Google Scholar] [CrossRef] [PubMed]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef]
- Sreedhar, A.; Aguilera-Aguirre, L.; Singh, K.K. Mitochondria in skin health, aging, and disease. Cell. Death Dis. 2020, 11, 444. [Google Scholar] [CrossRef]
- Go, Y.M.; Chandler, J.D.; Jones, D.P. The cysteine proteome. Free Radic. Biol. Med. 2015, 84, 227–245. [Google Scholar] [CrossRef]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-mediated cellular signaling. Oxidative Med. Cell. Longev. 2016, 4350965. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.S. Generating, partitioning, targeting and functioning of superoxide in mitochondria. Biosci. Rep. 1997, 17, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Korshunov, S.S.; Skulachev, V.P.; Starkov, A.A. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997, 416, 15–18. [Google Scholar] [CrossRef]
- Starkov, A.A.; Fiskum, G. Regulation of brain mitochondrial H2O2 production by membrane potential and NAD(P)H redox state. J. Neurochem. 2003, 86, 1101–1107. [Google Scholar] [CrossRef] [PubMed]
- Afonso, V.; Champy, R.; Mitrovic, D.; Collin, P.; Lomri, A. Reactive oxygen species and superoxide dismutases: Role in joint diseases. Jt. Bone Spine 2007, 74, 324–329. [Google Scholar] [CrossRef]
- Kaim, G.; Dimroth, P. ATP synthesis by F-type ATP synthase is obligatorily dependent on the transmembrane voltage. EMBO J. 1999, 18, 4118–4127. [Google Scholar] [CrossRef]
- Hüttemann, M.; Lee, I.; Pecinova, A.; Pecina, P.; Przyklenk, K.; Doan, J.W. Regulation of oxidative phosphorylation, the mitochondrial membrane potential, and their role in human disease. J. Bioenerg. Biomembr. 2008, 40, 445–456. [Google Scholar] [CrossRef]
- Kadenbach, B.; Arnold, S.; Lee, I.; Hüttemann, M. The possible role of cytochrome c oxidase in stress-induced apoptosis and degenerative diseases. Biochim. Biophys. Acta 2004, 1655, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Skulachev, V.P. Bioenergetic aspects of apoptosis, necrosis and mitoptosis. Apoptosis 2006, 11, 473–485. [Google Scholar] [CrossRef] [PubMed]
- Ramzan, R.; Staniek, K.; Kadenbach, B.; Vogt, S. Mitochondrial respiration and membrane potential are regulated by the allosteric ATP-inhibition of cytochrome c oxidase. Biochim. Biophys. Acta 2010, 1797, 1672–1680. [Google Scholar] [CrossRef] [PubMed]
- Kalpage, H.A.; Wan, J.; Morse, P.T.; Zurek, M.P.; Turner, A.A.; Khobeir, A.; Yazdi, N.; Hakim, L.; Liu, J.; Vaishnav, A.; et al. Cytochrome c phosphorylation: Control of mitochondrial electron transport chain flux and apoptosis. Intern. J. Biochem. Cell Biol. 2020, 121, 105704. [Google Scholar] [CrossRef] [PubMed]
- Yegorov, Y.E.; Poznyak, A.; Nikiforov, N.G.; Sobenin, I.A.; Orekhov, A.N. The link between chronic stress and accelerated aging. Biomedicines 2020, 8, 198. [Google Scholar] [CrossRef] [PubMed]
- Musatov, A.; Ortega-Lopez, J.; Robinson, N.C. Detergent-solubilized bovine cytochrome c oxidase: Dimerization depends on the amphiphilic environment. Biochemistry 2000, 39, 12996–13004. [Google Scholar] [CrossRef] [PubMed]
- Schägger, H.; Pfeiffer, K. The ratio of oxidative phosphorylation complexes I–V in bovine heart mitochondria and the composition of respiratory chain supercomplexes. J. Biol. Chem. 2001, 276, 37861–37867. [Google Scholar] [CrossRef]
- Musatov, A.; Robinson, N.C. Cholate-induced dimerization of detergent- or phospholipid-solubilized bovine cytochrome c oxidase. Biochemistry 2002, 41, 4371–4376. [Google Scholar] [CrossRef]
- Lee, I.; Bender, E.; Arnold, S.; Kadenbach, B. Minireview-Hypothesis. New control of mitochondrial membrane potential and ROS-formation. Biol. Chem. 2001, 382, 1629–1633. [Google Scholar] [CrossRef]
- Hüttemann, M.; Lee, I.; Grossman, L.I.; Doan, J.W.; Sanderson, T.H. Phosphorylation of mammalian cytochrome c and cytochrome c oxidase in the regulation of cell destiny: Rion, apoptosis, and human disease. Adv. Exp. Med. Biol. Chapter X 2012, 748, 237–264. [Google Scholar]
- Kadenbach, B. Complex IV—The regulatory center of mitochondrial oxidative phosphorylation. Mitochondrion 2021, 56, S1567–S7249. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.; Bender, E.; Kadenbach, B. Control of mitochondrial membrane potential and ROS formation by reversible phosphorylation of cytochrome c oxidase. Mol. Cell. Biochem. 2002, 234–235, 63–70. [Google Scholar] [CrossRef]
- Covian, R.; Balaban, R.S. Cardiac mitochondrial matrix and respiratory complex protein phosphorylation. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H940–H966. [Google Scholar] [CrossRef] [PubMed]
- Helling, S.; Hüttemann, M.; Ramzan, R.; Kim, S.H.; Lee, I.; Müller, T.; Langenfeld, E.; Meyer, H.E.; Kadenbach, B.; Vogt, S.; et al. Multiple phosphorylations of cytochrome c oxidase and their functions. Proteomics 2012, 12, 950–959. [Google Scholar] [CrossRef]
- Fang, J.K.; Prabu, S.K.; Sepuri, N.B.; Raza, H.; Anandatheerthavarada, H.K.; Galati, D.; Spear, J.; Avadhani, N.G. Site specific phosphorylation of cytochrome c oxidase subunits I, IVi1 and Vb in rabbit hearts subjected to ischemia/reperfusion. FEBS Lett. 2007, 581, 1302–1310. [Google Scholar] [CrossRef] [PubMed]
- Kadenbach, B.; Hartmann, R.; Glanville, R.; Buse, G. Tissue-specific genes code for polypeptide VIa of beef liver and heart cytochrome c oxidase. FEBS Lett. 1982, 138, 236–238. [Google Scholar] [CrossRef]
- Anthony, G.; Stroh, A.; Lottspeich, F.; Kadenbach, B. Different isozymes of cytochrome c oxidase are expressed in bovine smooth muscle and skeletal or heart muscle. FEBS Lett. 1990, 277, 97–100. [Google Scholar] [CrossRef]
- Schlerf, A.; Droste, M.; Winter, M.; Kadenbach, B. Characterization of two different genes (cDNA) for cytochrome c oxidase subunit VIa from heart and liver of the rat. EMBO J. 1988, 7, 2387–2391. [Google Scholar] [CrossRef] [PubMed]
- Sinkler, C.A.; Kalpage, H.; Shay, J.; Lee, I.; Malek, M.H.; Grossman, L.I.; Hüttemann, M. Tissue- and condition-specific isoforms of mammalian cytochrome c oxidase subunits: From function to human disease. Oxid Med. Cell Longev. 2017, 2017, 1534056. [Google Scholar] [CrossRef] [PubMed]
- Hüttemann, M.; Kadenbach, B.; Grossman, L.I. Mammalian subunit IV isoforms of cytochrome c oxidase. Gene 2001, 267, 111–123. [Google Scholar] [CrossRef]
- Endou, M.; Yoshida, K.; Hirota, M.; Nakajima, C.; Sakaguchi, A.; Kurihara, Y. Coxfa4l3, a novel mitochondrial electron transport chain Complex 4 subunit protein, switches from Coxfa4 during spermatogenesis. Mitochondrion 2020, 52, 1–7. [Google Scholar] [CrossRef]
- Fukuda, R.; Zhang, H.; Kim, J.W.; Shimoda, L.; Dang, C.V.; Semenza, G.L. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 2007, 129, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Horvat, S.; Beyer, C.; Arnold, S. Effect of hypoxia on the transcription pattern of subunit isoforms and the kinetics of cytochrome c oxidase in cortical astrocytes and cerebellar neurons. J. Neurochem. 2006, 99, 937–951. [Google Scholar] [CrossRef] [PubMed]
- Hüttemann, M.; Jaradat, S.; Grossman, L.I. Cytochrome c oxidase of mammals contains a testes-specific isoform of subunit VIb—the counterpart to testes-specific Cytochrome c? Mol. Reprod. Dev. 2003, 66, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Segade, F.; Hurle, B.; Claudio, E.; Ramos, S.; Lazo, P.S. Identification of an additional member of the cytochrome c oxidase subunit VIIa family of proteins. J. Biol. Chem. 1996, 271, 12343–12349. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, K.; Wang, G.; Zhang, X.; Hüttemann, P.P.; Qiu, Y.; Liu, J.; Mitchell, A.; Lee, I.; Zhang, C.; Lee, J.S.; et al. COX7AR is a Stress-inducible Mitochondrial COX Subunit that Promotes Breast Cancer Malignancy. Sci. Rep. 2016, 6, 31742. [Google Scholar] [CrossRef]
- Indrieri, A.; van Rahden, V.A.; Tiranti, V.; Morleo, M.; Iaconis, D.; Tammaro, R.; D’Amato, I.; Conte, I.; Maystadt, I.; Demuth, S.; et al. Mutations in COX7B cause microphthalmia with linear skin lesions, an unconventional mitochondrial disease. Am. J. Hum. Genet. 2012, 91, 942–949. [Google Scholar] [CrossRef]
- Hüttemann, M.; Schmidt, T.R.; Grossman, L.I. A third isoform of cytochrome c oxidase subunit VIII is present in mammals. Gene 2003, 312, 95–102. [Google Scholar] [CrossRef]
- Bonne, G.; Seibel, P.; Possekel, S.; Marsac, C.; Kadenbach, B. Expression of human cytochrome c oxidase subunits during fetal development. Eur. J. Biochem. 1993, 217, 1099–1107. [Google Scholar] [CrossRef]
- Ewart, G.D.; Zhang, Y.Z.; Capaldi, R.A. Switching of bovine cytochrome c oxidase subunit VIa isoforms in skeletal muscle during development. FEBS Lett. 1991, 292, 79–84. [Google Scholar]
- Ogórek, M.; Gąsior, Ł.; Pierzchała, O.; Daszkiewicz, R.; Lenartowicz, M. Role of copper in the process of spermatogenesis. Postepy Hig Med. Dosw (Online) 2017, 71, 663–683. [Google Scholar] [CrossRef] [PubMed]
- Cottin, S.C.; Roussel, G.; Gambling, L.; Hayes, H.E.; Currie, V.J.; McArdle, H.J. The effect of maternal iron deficiency on zinc and copper levels and on genes of zinc and copper metabolism during pregnancy in the rat. Br. J. Nutr. 2019, 121, 121–129. [Google Scholar] [CrossRef]
- Sharpe, M.A.; Krzyaniak, M.D.; Xu, S.; McCracken, J.; Ferguson-Miller, S. EPR evidence of cyanide binding to the Mn(Mg) center of cytochrome c oxidase: Support for Cu(A)-Mg involvement in proton pumping. Biochemistry 2009, 48, 328–335. [Google Scholar] [CrossRef][Green Version]
- Florens, L.; Schmidt, B.; McCracken, J.; Ferguson-Miller, S. Fast deuterium access to the buried magnesium/manganese site in cytochrome c oxidase. Biochemistry 2001, 40, 7491–7497. [Google Scholar] [CrossRef] [PubMed]
- Szabo, C. Hydrogen sulfide, an endogenous stimulator of mitochondrial function in cancer cells. Cells 2021, 10, 220. [Google Scholar] [CrossRef]
- Buhrow, L.; Hiser, C.; Van Voorst, J.R.; Ferguson-Miller, S.; Kuhn, L.A. Computational prediction and in vitro analysis of potential physiological ligands of the bile acid binding site in cytochrome c oxidase. Biochemistry 2013, 52, 6995–7006. [Google Scholar] [CrossRef][Green Version]
- Qin, L.; Mills, D.A.; Buhrow, L.; Hiser, C.; Ferguson-Miller, S. A conserved steroid binding site in cytochrome C oxidase. Biochemistry 2008, 47, 9931–9933. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shoji, K.; Giuffro, A.; D’Itri, E.; Hagiwara, K.; Yamanaka, T.; Brunori, M.; Sarti, P. The ratio between the fast and slow forms of bovine cytochrome c oxidase is changed by cholate or nucleotides bound to the cholate-binding site close to the cytochrome a3/CuB binuclear centre. Cell Mol. Life Sci. 2000, 57, 1482–1487. [Google Scholar] [CrossRef]
- Arnold, S.; Goglia, F.; Kadenbach, B. 3,5-Diiodothyronine binds to subunit Va of cytochrome c oxidase and abolishes the allosteric inhibition of respiration by ATP. Eur. J. Biochem. 1998, 252, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.; Kadenbach, B. Palmitate decreases proton pumping of liver-type cytochrome c oxidase. Eur. J. Biochem. 2001, 268, 6329–6334. [Google Scholar] [CrossRef]
- Tu, L.F.; Cao, L.F.; Zhang, Y.H.; Guo, Y.L.; Zhou, Y.F.; Lu, W.Q.; Zhang, T.Z.; Zhang, T.; Zhang, G.X.; Kurihara, H.; et al. Sirt3-dependent deacetylation of COX-1 counteracts oxidative stress-induced cell apoptosis. FASEB J. 2019, 33, 14118–14128. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, Y.; Tang, R.; Jiang, X.; Wang, Y.; Gu, T. Effect of TFAM on ATP content in tachypacing primary cultured cardiomyocytes and atrial fibrillation patients. Mol. Med. Rep. 2020, 22, 5105–5112. [Google Scholar] [CrossRef] [PubMed]
- Xiyang, Y.B.; Liu, R.; Wang, X.Y.; Li, S.; Zhao, Y.; Lu, B.T.; Xiao, Z.C.; Zhang, L.F.; Wang, T.H.; Zhang, J. COX5A Plays a vital role in memory impairment associated with brain aging via the BDNF/ERK1/2 signaling pathway. Front. Aging Neurosci. 2020, 12, 215. [Google Scholar] [CrossRef]
- Jiang, Y.; Bai, X.; Li, T.T.; Al-Hawwas, M.; Jin, Y.; Zou, Y.; Hu, Y.; Liu, L.Y.; Zhang, Y.; Liu, Q.; et al. COX5A over-expression protects cortical neurons from hypoxic ischemic injury in neonatal rats associated with TPI up-regulation. BMC Neurosci. 2020, 21, 18. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Shu, L.; Zhang, W.; Wang, Z. Cca-miR398 increases copper sulfate stress sensitivity via the regulation of CSD mRNA transcription levels in transgenic Arabidopsis thaliana. PeerJ 2020, 8, e9105. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, X.; Chen, Y.; Wu, Z.; Xu, Q.; Chen, M.; Shao, M.; Cao, X.; Zhou, Y.; Xie, M.; Shi, Y.; et al. Mitochondrial miR-181a-5p promotes glucose metabolism reprogramming in liver cancer by regulating the electron transport chain. Carcinogenesis 2020, 41, 972–983. [Google Scholar] [CrossRef]
- Singh, R.K.; Saini, S.K.; Prakasam, G.; Kalairasan, P.; Bamezai, R.N.K. Role of ectopically expressed mtDNA encoded cytochrome c oxidase subunit I (MT-COI) in tumorigenesis. Mitochondrion 2019, 49, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Wu, P.; Xiao, J.; Jiang, L. Overexpression of COX6B1 protects against I/R-induced neuronal injury in rat hippocampal neurons. Mol. Med. Rep. 2019, 19, 4852–4862. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, Y.; Wan, J.; Zhang, P.; Pei, F. COX6B1 relieves hypoxia/reoxygenation injury of neonatal rat cardiomyocytes by regulating mitochondrial function. Biotechnol. Lett. 2019, 41, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Chen, X.; Feng, Y.; Wang, G.; Nawaz, I.; Hu, L.; Liu, P. COX7A1 suppresses the viability of human non-small cell lung cancer cells via regulating autophagy. Cancer Med. 2019, 8, 7762–7773. [Google Scholar] [CrossRef]
- Chen, Z.X.; Pervaiz, S. Involvement of cytochrome c oxidase subunits Va and Vb in the regulation of cancer cell metabolism by Bcl-2. Cell Death Differ. 2010, 17, 408–420. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.P.; Sun, H.F.; Jiang, H.L.; Li, L.D.; Hu, X.; Xu, X.E.; Jin, W. Loss of COX5B inhibits, and promotes senescence via mitochondrial dysfunction in breast cancer. Oncotarget 2015, 6, 43363–43374. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Xi, J. Identification of COX5B as a novel biomarker in high-grade glioma patients. Onco Targets Ther. 2017, 10, 5463–5470. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.P.; Sun, H.F.; Fu, W.Y.; Li, L.D.; Zhao, Y.; Chen, M.T.; Jin, W. High expression of COX5B is associated with poor prognosis in breast cancer. Future Oncol. 2017, 13, 1711–1719. [Google Scholar] [CrossRef] [PubMed]
- Krupar, R.; Hautmann, M.G.; Pathak, R.R.; Varier, I.; McLaren, C.; Gaag, D.; Hellerbrand, C.; Evert, M.; Laban, S.; Idel, C.; et al. Immunometabolic determinants of chemoradiotherapy response and survival in head and neck squamous cell carcinoma. Am. J. Pathol. 2018, 188, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Stein, J.; Tenbrock, J.; Kristiansen, G.; Müller, S.C.; Ellinger, J. Systematic expression analysis of the mitochondrial respiratory chain protein subunits identifies COX5B as a prognostic marker in clear cell renal cell carcinoma. J. Urol. 2019, 26, 910–916. [Google Scholar] [CrossRef]
- Chu, Y.D.; Lin, W.R.; Lin, Y.H.; Kuo, W.H.; Tseng, C.J.; Lim, S.N.; Huang, Y.L.; Huang, S.C.; Wu, T.J.; Lin, K.H.; et al. COX5B-mediated bioenergetic alteration regulates tumor growth and migration by modulating AMPK-UHMK1-ERK cascade in hepatoma. Cancers 2020, 12, 1646. [Google Scholar] [CrossRef]
- Signes, A.; Fernandez-Vizarra, E. Assembly of mammalian oxidative phosphorylation complexes I-V and supercomplexes. Essays Biochem. 2018, 62, 255–270. [Google Scholar] [PubMed]
- Letts, J.A.; Sazanov, L.A. Clarifying the supercomplex: The higher-order organization of the mitochondrial electron transport chain. Nat. Struct. Mol. Biol. 2017, 24, 800–808. [Google Scholar] [CrossRef]
- Baker, N.; Patel, J.; Khacho, M. Linking mitochondrial dynamics, cristae remodeling and supercomplex formation: How mitochondrial structure can regulate bioenergetics. Mitochondrion 2019, 49, 259–268. [Google Scholar] [CrossRef]
- Genova, M.L.; Lenaz, G. A critical appraisal of the role of respiratory supercomplexes in mitochondria. Biol. Chem. 2013, 394, 631–639. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramzan, R.; Kadenbach, B.; Vogt, S. Multiple Mechanisms Regulate Eukaryotic Cytochrome C Oxidase. Cells 2021, 10, 514. https://doi.org/10.3390/cells10030514
Ramzan R, Kadenbach B, Vogt S. Multiple Mechanisms Regulate Eukaryotic Cytochrome C Oxidase. Cells. 2021; 10(3):514. https://doi.org/10.3390/cells10030514
Chicago/Turabian StyleRamzan, Rabia, Bernhard Kadenbach, and Sebastian Vogt. 2021. "Multiple Mechanisms Regulate Eukaryotic Cytochrome C Oxidase" Cells 10, no. 3: 514. https://doi.org/10.3390/cells10030514
APA StyleRamzan, R., Kadenbach, B., & Vogt, S. (2021). Multiple Mechanisms Regulate Eukaryotic Cytochrome C Oxidase. Cells, 10(3), 514. https://doi.org/10.3390/cells10030514