
Gamma-Chain Receptor Cytokines & PD-1 Manipulation to Restore HCV-Specific CD8+ T Cell Response during Chronic Hepatitis C

 ,
,
Abstract
:1. Introduction
2. Key Role of Specific CD8 T Cells in HCV Infection
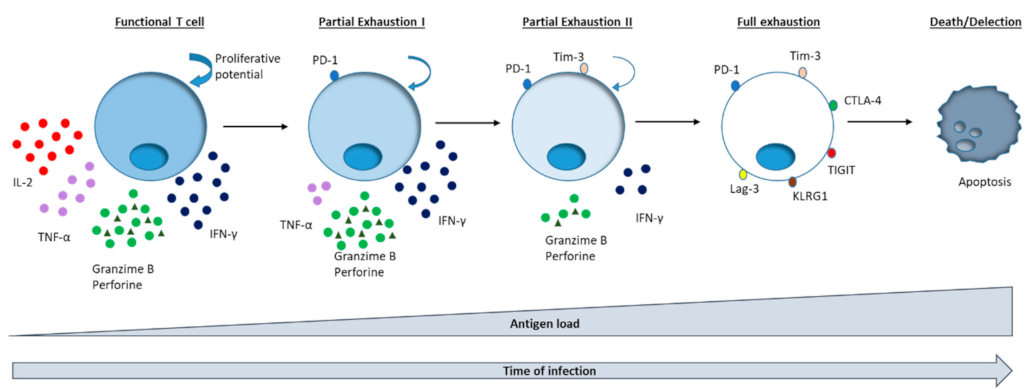
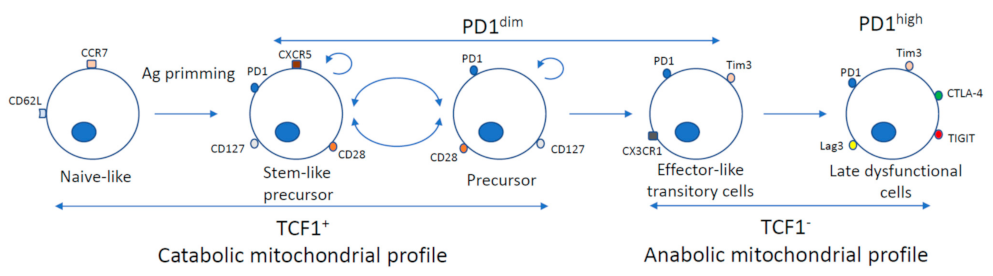
3. PD-1 Modulation for T Cell Exhaustion Reversion
4. Gamma (γ) Chain Cytokines for T Cell Exhaustion Reversion
4.1. Interleukin-2
4.2. Interleukin-7
4.3. Interleukin-15
4.4. Interleukin-21
5. Effect of PD-1 Modulation and γc-Cytokines on HCV-Specific CD8+ T Cell Response
6. Conclusions
Author Contributions
Funding
Institutional Review Board
Informed Consent
Data Availability
Conflicts of Interest
References
- Thrift, A.P.; El-Serag, H.B.; Kanwal, F. Global epidemiology and burden of HCV infection and HCV-related disease. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 122–132. [Google Scholar] [CrossRef]
- European Association for Study of, L. EASL Clinical Practice Guidelines: Management of hepatitis C virus infection. J. Hepatol. 2014, 60, 392–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Degasperi, E.; Spinetti, A.; Lombardi, A.; Landonio, S.; Rossi, M.C.; Pasulo, L.; Pozzoni, P.; Giorgini, A.; Fabris, P.; Romano, A.; et al. Real-life effectiveness and safety of sofosbuvir/velpatasvir/voxilaprevir in hepatitis C patients with previous DAA failure. J. Hepatol. 2019, 71, 1106–1115. [Google Scholar] [CrossRef]
- Cooper, S.; Erickson, A.L.; Adams, E.J.; Kansopon, J.; Weiner, A.J.; Chien, D.Y.; Houghton, M.; Parham, P.; Walker, C.M. Analysis of a successful immune response against hepatitis C virus. Immunity 1999, 10, 439–449. [Google Scholar] [CrossRef] [Green Version]
- Thimme, R.; Oldach, D.; Chang, K.M.; Steiger, C.; Ray, S.C.; Chisari, F.V. Determinants of viral clearance and persistence during acute hepatitis C virus infection. J. Exp. Med. 2001, 194, 1395–1406. [Google Scholar] [CrossRef] [PubMed]
- Klenerman, P.; Thimme, R. T cell responses in hepatitis C: The good, the bad and the unconventional. Gut 2012, 61, 1226–1234. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.L.; Mosbruger, T.; Mao, Q.; Liu, Z.; Wang, X.H.; Yang, H.C.; Sidney, J.; Sette, A.; Pardoll, D.; Thomas, D.L.; et al. Cellular immune selection with hepatitis C virus persistence in humans. J. Exp. Med. 2005, 201, 1741–1752. [Google Scholar] [CrossRef] [PubMed]
- Fitzmaurice, K.; Petrovic, D.; Ramamurthy, N.; Simmons, R.; Merani, S.; Gaudieri, S.; Sims, S.; Dempsey, E.; Freitas, E.; Lea, S.; et al. Molecular footprints reveal the impact of the protective HLA-A*03 allele in hepatitis C virus infection. Gut 2011, 60, 1563–1571. [Google Scholar] [CrossRef] [Green Version]
- Kim, A.Y.; Kuntzen, T.; Timm, J.; Nolan, B.E.; Baca, M.A.; Reyor, L.L.; Berical, A.C.; Feller, A.J.; Johnson, K.L.; Schulze zur Wiesch, J.; et al. Spontaneous control of HCV is associated with expression of HLA-B 57 and preservation of targeted epitopes. Gastroenterology 2011, 140, 686–696.e681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann-Haefelin, C.; McKiernan, S.; Ward, S.; Viazov, S.; Spangenberg, H.C.; Killinger, T.; Baumert, T.F.; Nazarova, N.; Sheridan, I.; Pybus, O.; et al. Dominant influence of an HLA-B27 restricted CD8+ T cell response in mediating HCV clearance and evolution. Hepatology 2006, 43, 563–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef]
- McLane, L.M.; Abdel-Hakeem, M.S.; Wherry, E.J. CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu. Rev. Immunol. 2019, 37, 457–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamphorst, A.O.; Wieland, A.; Nasti, T.; Yang, S.; Zhang, R.; Barber, D.L.; Konieczny, B.T.; Daugherty, C.Z.; Koenig, L.; Yu, K.; et al. Rescue of exhausted CD8 T cells by PD-1-targeted therapies is CD28-dependent. Science 2017, 355, 1423–1427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larrubia, J.R.; Lokhande, M.U.; Garcia-Garzon, S.; Miquel, J.; Gonzalez-Praetorius, A.; Parra-Cid, T.; Sanz-de-Villalobos, E. Persistent hepatitis C virus (HCV) infection impairs HCV-specific cytotoxic T cell reactivity through Mcl-1/Bim imbalance due to CD127 down-regulation. J. Viral. Hepat 2013, 20, 85–94. [Google Scholar] [CrossRef]
- Aregay, A.; Owusu Sekyere, S.; Deterding, K.; Port, K.; Dietz, J.; Berkowski, C.; Sarrazin, C.; Manns, M.P.; Cornberg, M.; Wedemeyer, H. Elimination of hepatitis C virus has limited impact on the functional and mitochondrial impairment of HCV-specific CD8+ T cell responses. J. Hepatol. 2019, 71, 889–899. [Google Scholar] [CrossRef]
- Pallett, L.J.; Schmidt, N.; Schurich, A. T cell metabolism in chronic viral infection. Clin. Exp. Immunol. 2019, 197, 143–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larrubia, J.R.; Calvino, M.; Benito, S.; Sanz-de-Villalobos, E.; Perna, C.; Perez-Hornedo, J.; Gonzalez-Mateos, F.; Garcia-Garzon, S.; Bienvenido, A.; Parra, T. The role of CCR5/CXCR3 expressing CD8+ cells in liver damage and viral control during persistent hepatitis C virus infection. J. Hepatol. 2007, 47, 632–641. [Google Scholar] [CrossRef] [PubMed]
- Bengsch, B.; Seigel, B.; Ruhl, M.; Timm, J.; Kuntz, M.; Blum, H.E.; Pircher, H.; Thimme, R. Coexpression of PD-1, 2B4, CD160 and KLRG1 on exhausted HCV-specific CD8+ T cells is linked to antigen recognition and T cell differentiation. PLoS Pathog. 2010, 6, e1000947. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, J.T.; Diepolder, H.M.; Jung, M.C.; Gruener, N.H.; Schraut, W.W.; Zachoval, R.; Hoffmann, R.; Schirren, C.A.; Santantonio, T.; Pape, G.R. Recurrence of hepatitis C virus after loss of virus-specific CD4(+) T-cell response in acute hepatitis C. Gastroenterology 1999, 117, 933–941. [Google Scholar] [CrossRef]
- Grakoui, A.; Shoukry, N.H.; Woollard, D.J.; Han, J.H.; Hanson, H.L.; Ghrayeb, J.; Murthy, K.K.; Rice, C.M.; Walker, C.M. HCV persistence and immune evasion in the absence of memory T cell help. Science 2003, 302, 659–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boettler, T.; Spangenberg, H.C.; Neumann-Haefelin, C.; Panther, E.; Urbani, S.; Ferrari, C.; Blum, H.E.; von Weizsacker, F.; Thimme, R. T cells with a CD4+CD25+ regulatory phenotype suppress in vitro proliferation of virus-specific CD8+ T cells during chronic hepatitis C virus infection. J. Virol. 2005, 79, 7860–7867. [Google Scholar] [CrossRef] [Green Version]
- Cabrera, R.; Tu, Z.; Xu, Y.; Firpi, R.J.; Rosen, H.R.; Liu, C.; Nelson, D.R. An immunomodulatory role for CD4(+)CD25(+) regulatory T lymphocytes in hepatitis C virus infection. Hepatology 2004, 40, 1062–1071. [Google Scholar] [CrossRef]
- Hui, E.; Cheung, J.; Zhu, J.; Su, X.; Taylor, M.J.; Wallweber, H.A.; Sasmal, D.K.; Huang, J.; Kim, J.M.; Mellman, I.; et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science 2017, 355, 1428–1433. [Google Scholar] [CrossRef]
- Lin, W.W.; Nish, S.A.; Yen, B.; Chen, Y.H.; Adams, W.C.; Kratchmarov, R.; Rothman, N.J.; Bhandoola, A.; Xue, H.H.; Reiner, S.L. CD8(+) T Lymphocyte Self-Renewal during Effector Cell Determination. Cell Rep. 2016, 17, 1773–1782. [Google Scholar] [CrossRef] [Green Version]
- Im, S.J.; Hashimoto, M.; Gerner, M.Y.; Lee, J.; Kissick, H.T.; Burger, M.C.; Shan, Q.; Hale, J.S.; Lee, J.; Nasti, T.H.; et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 2016, 537, 417–421. [Google Scholar] [CrossRef] [PubMed]
- Paley, M.A.; Kroy, D.C.; Odorizzi, P.M.; Johnnidis, J.B.; Dolfi, D.V.; Barnett, B.E.; Bikoff, E.K.; Robertson, E.J.; Lauer, G.M.; Reiner, S.L.; et al. Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science 2012, 338, 1220–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escobar, G.; Mangani, D.; Anderson, A.C. T cell factor 1: A master regulator of the T cell response in disease. Sci. Immunol. 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hu, J.; Li, Y.; Xiao, M.; Wang, H.; Tian, Q.; Li, Z.; Tang, J.; Hu, L.; Tan, Y.; et al. The Transcription Factor TCF1 Preserves the Effector Function of Exhausted CD8 T Cells During Chronic Viral Infection. Front. Immunol. 2019, 10, 169. [Google Scholar] [CrossRef]
- Utzschneider, D.T.; Charmoy, M.; Chennupati, V.; Pousse, L.; Ferreira, D.P.; Calderon-Copete, S.; Danilo, M.; Alfei, F.; Hofmann, M.; Wieland, D.; et al. T Cell Factor 1-Expressing Memory-like CD8(+) T Cells Sustain the Immune Response to Chronic Viral Infections. Immunity 2016, 45, 415–427. [Google Scholar] [CrossRef] [Green Version]
- Pellegrini, M.; Calzascia, T.; Toe, J.G.; Preston, S.P.; Lin, A.E.; Elford, A.R.; Shahinian, A.; Lang, P.A.; Lang, K.S.; Morre, M.; et al. IL-7 engages multiple mechanisms to overcome chronic viral infection and limit organ pathology. Cell 2011, 144, 601–613. [Google Scholar] [CrossRef] [Green Version]
- Nolz, J.C.; Richer, M.J. Control of memory CD8(+) T cell longevity and effector functions by IL-15. Mol. Immunol. 2020, 117, 180–188. [Google Scholar] [CrossRef]
- Moreno-Cubero, E.; Subira, D.; Sanz-de-Villalobos, E.; Parra-Cid, T.; Madejon, A.; Miquel, J.; Olveira, A.; Gonzalez-Praetorius, A.; Garcia-Samaniego, J.; Larrubia, J.R. According to Hepatitis C Virus (HCV) Infection Stage, Interleukin-7 Plus 4-1BB Triggering Alone or Combined with PD-1 Blockade Increases TRAF1(low) HCV-Specific CD8(+) Cell Reactivity. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Barnaba, V. Hepatitis C virus infection: A “liaison a trois” amongst the virus, the host, and chronic low-level inflammation for human survival. J. Hepatol. 2010, 53, 752–761. [Google Scholar] [CrossRef] [Green Version]
- Larrubia, J.R.; Moreno-Cubero, E.; Lokhande, M.U.; Garcia-Garzon, S.; Lazaro, A.; Miquel, J.; Perna, C.; Sanz-de-Villalobos, E. Adaptive immune response during hepatitis C virus infection. World J. Gastroenterol. 2014, 20, 3418–3430. [Google Scholar] [CrossRef] [PubMed]
- Jo, J.; Aichele, U.; Kersting, N.; Klein, R.; Aichele, P.; Bisse, E.; Sewell, A.K.; Blum, H.E.; Bartenschlager, R.; Lohmann, V.; et al. Analysis of CD8+ T-cell-mediated inhibition of hepatitis C virus replication using a novel immunological model. Gastroenterology 2009, 136, 1391–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lechner, F.; Wong, D.K.; Dunbar, P.R.; Chapman, R.; Chung, R.T.; Dohrenwend, P.; Robbins, G.; Phillips, R.; Klenerman, P.; Walker, B.D. Analysis of successful immune responses in persons infected with hepatitis C virus. J. Exp. Med. 2000, 191, 1499–1512. [Google Scholar] [CrossRef] [Green Version]
- Thimme, R.; Bukh, J.; Spangenberg, H.C.; Wieland, S.; Pemberton, J.; Steiger, C.; Govindarajan, S.; Purcell, R.H.; Chisari, F.V. Viral and immunological determinants of hepatitis C virus clearance, persistence, and disease. Proc. Natl. Acad. Sci. USA 2002, 99, 15661–15668. [Google Scholar] [CrossRef] [Green Version]
- Shoukry, N.H.; Grakoui, A.; Houghton, M.; Chien, D.Y.; Ghrayeb, J.; Reimann, K.A.; Walker, C.M. Memory CD8+ T cells are required for protection from persistent hepatitis C virus infection. J. Exp. Med. 2003, 197, 1645–1655. [Google Scholar] [CrossRef]
- Uebelhoer, L.; Han, J.H.; Callendret, B.; Mateu, G.; Shoukry, N.H.; Hanson, H.L.; Rice, C.M.; Walker, C.M.; Grakoui, A. Stable cytotoxic T cell escape mutation in hepatitis C virus is linked to maintenance of viral fitness. PLoS Pathog. 2008, 4, e1000143. [Google Scholar] [CrossRef] [Green Version]
- Chang, K.M.; Rehermann, B.; McHutchison, J.G.; Pasquinelli, C.; Southwood, S.; Sette, A.; Chisari, F.V. Immunological significance of cytotoxic T lymphocyte epitope variants in patients chronically infected by the hepatitis C virus. J. Clin. Investig. 1997, 100, 2376–2385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann-Haefelin, C.; Killinger, T.; Timm, J.; Southwood, S.; McKinney, D.; Blum, H.E.; Thimme, R. Absence of viral escape within a frequently recognized HLA-A26-restricted CD8+ T-cell epitope targeting the functionally constrained hepatitis C virus NS5A/5B cleavage site. J. Gen. Virol. 2007, 88, 1986–1991. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, H.; Lauer, G.; Pybus, O.G.; Ouchi, K.; Wong, D.; Ward, S.; Walker, B.; Klenerman, P. Do antiviral CD8+ T cells select hepatitis C virus escape mutants? Analysis in diverse epitopes targeted by human intrahepatic CD8+ T lymphocytes. J. Viral. Hepat 2006, 13, 121–130. [Google Scholar] [CrossRef]
- Larrubia, J.R.; Benito-Martinez, S.; Miquel, J.; Calvino, M.; Sanz-de-Villalobos, E.; Gonzalez-Praetorius, A.; Albertos, S.; Garcia-Garzon, S.; Lokhande, M.; Parra-Cid, T. Bim-mediated apoptosis and PD-1/PD-L1 pathway impair reactivity of PD1(+)/CD127(-) HCV-specific CD8(+) cells targeting the virus in chronic hepatitis C virus infection. Cell Immunol. 2011, 269, 104–114. [Google Scholar] [CrossRef]
- Gruener, N.H.; Lechner, F.; Jung, M.C.; Diepolder, H.; Gerlach, T.; Lauer, G.; Walker, B.; Sullivan, J.; Phillips, R.; Pape, G.R.; et al. Sustained dysfunction of antiviral CD8+ T lymphocytes after infection with hepatitis C virus. J. Virol. 2001, 75, 5550–5558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamoto, N.; Kaplan, D.E.; Coleclough, J.; Li, Y.; Valiga, M.E.; Kaminski, M.; Shaked, A.; Olthoff, K.; Gostick, E.; Price, D.A.; et al. Functional restoration of HCV-specific CD8 T cells by PD-1 blockade is defined by PD-1 expression and compartmentalization. Gastroenterology 2008, 134, 1927–1937.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radziewicz, H.; Ibegbu, C.C.; Fernandez, M.L.; Workowski, K.A.; Obideen, K.; Wehbi, M.; Hanson, H.L.; Steinberg, J.P.; Masopust, D.; Wherry, E.J.; et al. Liver-infiltrating lymphocytes in chronic human hepatitis C virus infection display an exhausted phenotype with high levels of PD-1 and low levels of CD127 expression. J. Virol. 2007, 81, 2545–2553. [Google Scholar] [CrossRef] [Green Version]
- Larrubia, J.R.; Benito-Martinez, S.; Calvino, M.; Sanz-de-Villalobos, E.; Parra-Cid, T. Role of chemokines and their receptors in viral persistence and liver damage during chronic hepatitis C virus infection. World J. Gastroenterol. 2008, 14, 7149–7159. [Google Scholar] [CrossRef]
- Hajarizadeh, B.; Grebely, J.; Dore, G.J. Epidemiology and natural history of HCV infection. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Golden-Mason, L.; Palmer, B.E.; Kassam, N.; Townshend-Bulson, L.; Livingston, S.; McMahon, B.J.; Castelblanco, N.; Kuchroo, V.; Gretch, D.R.; Rosen, H.R. Negative immune regulator Tim-3 is overexpressed on T cells in hepatitis C virus infection and its blockade rescues dysfunctional CD4+ and CD8+ T cells. J. Virol. 2009, 83, 9122–9130. [Google Scholar] [CrossRef] [Green Version]
- Adams, W.C.; Chen, Y.H.; Kratchmarov, R.; Yen, B.; Nish, S.A.; Lin, W.W.; Rothman, N.J.; Luchsinger, L.L.; Klein, U.; Busslinger, M.; et al. Anabolism-Associated Mitochondrial Stasis Driving Lymphocyte Differentiation over Self-Renewal. Cell Rep. 2016, 17, 3142–3152. [Google Scholar] [CrossRef] [PubMed]
- Seo, H.; Chen, J.; Gonzalez-Avalos, E.; Samaniego-Castruita, D.; Das, A.; Wang, Y.H.; Lopez-Moyado, I.F.; Georges, R.O.; Zhang, W.; Onodera, A.; et al. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8(+) T cell exhaustion. Proc. Natl. Acad. Sci. USA 2019, 116, 12410–12415. [Google Scholar] [CrossRef] [Green Version]
- Zander, R.; Schauder, D.; Xin, G.; Nguyen, C.; Wu, X.; Zajac, A.; Cui, W. CD4(+) T Cell Help Is Required for the Formation of a Cytolytic CD8(+) T Cell Subset that Protects against Chronic Infection and Cancer. Immunity 2019, 51, 1028–1042.e4. [Google Scholar] [CrossRef] [PubMed]
- Johnnidis, J.B.; Muroyama, Y.; Ngiow, S.F.; Chen, Z.; Manne, S.; Cai, Z.; Song, S.; Platt, J.M.; Schenkel, J.M.; Abdel-Hakeem, M.; et al. Inhibitory signaling sustains a distinct early memory CD8(+) T cell precursor that is resistant to DNA damage. Sci. Immunol. 2021, 6. [Google Scholar] [CrossRef]
- Gattinoni, L.; Lugli, E.; Ji, Y.; Pos, Z.; Paulos, C.M.; Quigley, M.F.; Almeida, J.R.; Gostick, E.; Yu, Z.; Carpenito, C.; et al. A human memory T cell subset with stem cell-like properties. Nat. Med. 2011, 17, 1290–1297. [Google Scholar] [CrossRef]
- Cieri, N.; Camisa, B.; Cocchiarella, F.; Forcato, M.; Oliveira, G.; Provasi, E.; Bondanza, A.; Bordignon, C.; Peccatori, J.; Ciceri, F.; et al. IL-7 and IL-15 instruct the generation of human memory stem T cells from naive precursors. Blood 2013, 121, 573–584. [Google Scholar] [CrossRef]
- Nakano, S.; Eso, Y.; Okada, H.; Takai, A.; Takahashi, K.; Seno, H. Recent Advances in Immunotherapy for Hepatocellular Carcinoma. Cancers 2020, 12, 775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pauken, K.E.; Sammons, M.A.; Odorizzi, P.M.; Manne, S.; Godec, J.; Khan, O.; Drake, A.M.; Chen, Z.; Sen, D.R.; Kurachi, M.; et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 2016, 354, 1160–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sen, D.R.; Kaminski, J.; Barnitz, R.A.; Kurachi, M.; Gerdemann, U.; Yates, K.B.; Tsao, H.W.; Godec, J.; LaFleur, M.W.; Brown, F.D.; et al. The epigenetic landscape of T cell exhaustion. Science 2016, 354, 1165–1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shourian, M.; Beltra, J.C.; Bourdin, B.; Decaluwe, H. Common gamma chain cytokines and CD8 T cells in cancer. Semin Immunol. 2019, 42, 101307. [Google Scholar] [CrossRef]
- Fukuda, R.; Sugawara, S.; Kondo, Y. Immune Checkpoint Inhibitor Can Reduce HCV-RNA without Liver Damage. Intern. Med. 2020, 59, 2245–2248. [Google Scholar] [CrossRef]
- Kasprowicz, V.; Schulze Zur Wiesch, J.; Kuntzen, T.; Nolan, B.E.; Longworth, S.; Berical, A.; Blum, J.; McMahon, C.; Reyor, L.L.; Elias, N.; et al. High level of PD-1 expression on hepatitis C virus (HCV)-specific CD8+ and CD4+ T cells during acute HCV infection, irrespective of clinical outcome. J. Virol. 2008, 82, 3154–3160. [Google Scholar] [CrossRef] [Green Version]
- Gong, J.; Chehrazi-Raffle, A.; Reddi, S.; Salgia, R. Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: A comprehensive review of registration trials and future considerations. J. Immunother Cancer 2018, 6, 8. [Google Scholar] [CrossRef]
- Tomiyama, H.; Matsuda, T.; Takiguchi, M. Differentiation of human CD8(+) T cells from a memory to memory/effector phenotype. J. Immunol. 2002, 168, 5538–5550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizuno, R.; Sugiura, D.; Shimizu, K.; Maruhashi, T.; Watada, M.; Okazaki, I.M.; Okazaki, T. PD-1 Primarily Targets TCR Signal in the Inhibition of Functional T Cell Activation. Front. Immunol. 2019, 10, 630. [Google Scholar] [CrossRef] [Green Version]
- Ebinuma, H.; Nakamoto, N.; Li, Y.; Price, D.A.; Gostick, E.; Levine, B.L.; Tobias, J.; Kwok, W.W.; Chang, K.M. Identification and in vitro expansion of functional antigen-specific CD25+ FoxP3+ regulatory T cells in hepatitis C virus infection. J. Virol. 2008, 82, 5043–5053. [Google Scholar] [CrossRef] [Green Version]
- Qureshi, O.S.; Zheng, Y.; Nakamura, K.; Attridge, K.; Manzotti, C.; Schmidt, E.M.; Baker, J.; Jeffery, L.E.; Kaur, S.; Briggs, Z.; et al. Trans-endocytosis of CD80 and CD86: A molecular basis for the cell-extrinsic function of CTLA-4. Science 2011, 332, 600–603. [Google Scholar] [CrossRef] [Green Version]
- Nakamoto, N.; Cho, H.; Shaked, A.; Olthoff, K.; Valiga, M.E.; Kaminski, M.; Gostick, E.; Price, D.A.; Freeman, G.J.; Wherry, E.J.; et al. Synergistic reversal of intrahepatic HCV-specific CD8 T cell exhaustion by combined PD-1/CTLA-4 blockade. PLoS Pathog. 2009, 5, e1000313. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; McPherson, A.J.; Jones, R.B.; Kawamura, K.S.; Lin, G.H.; Lang, P.A.; Ambagala, T.; Pellegrini, M.; Calzascia, T.; Aidarus, N.; et al. Loss of the signaling adaptor TRAF1 causes CD8+ T cell dysregulation during human and murine chronic infection. J. Exp. Med. 2012, 209, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Smith-Garvin, J.E.; Koretzky, G.A.; Jordan, M.S. T cell activation. Annu Rev. Immunol. 2009, 27, 591–619. [Google Scholar] [CrossRef] [PubMed]
- Urbani, S.; Amadei, B.; Tola, D.; Massari, M.; Schivazappa, S.; Missale, G.; Ferrari, C. PD-1 expression in acute hepatitis C virus (HCV) infection is associated with HCV-specific CD8 exhaustion. J. Virol. 2006, 80, 11398–11403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutebemberwa, A.; Ray, S.C.; Astemborski, J.; Levine, J.; Liu, L.; Dowd, K.A.; Clute, S.; Wang, C.; Korman, A.; Sette, A.; et al. High-programmed death-1 levels on hepatitis C virus-specific T cells during acute infection are associated with viral persistence and require preservation of cognate antigen during chronic infection. J. Immunol. 2008, 181, 8215–8225. [Google Scholar] [CrossRef] [Green Version]
- Penna, A.; Pilli, M.; Zerbini, A.; Orlandini, A.; Mezzadri, S.; Sacchelli, L.; Missale, G.; Ferrari, C. Dysfunction and functional restoration of HCV-specific CD8 responses in chronic hepatitis C virus infection. Hepatology 2007, 45, 588–601. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, D.; Lalezari, J.; Lawitz, E.; DiMicco, M.; Ghalib, R.; Reddy, K.R.; Chang, K.M.; Sulkowski, M.; Marro, S.O.; Anderson, J.; et al. A randomized, double-blind, placebo-controlled assessment of BMS-936558, a fully human monoclonal antibody to programmed death-1 (PD-1), in patients with chronic hepatitis C virus infection. PLoS ONE 2013, 8, e63818. [Google Scholar] [CrossRef]
- Owusu Sekyere, S.; Suneetha, P.V.; Kraft, A.R.; Zhang, S.; Dietz, J.; Sarrazin, C.; Manns, M.P.; Schlaphoff, V.; Cornberg, M.; Wedemeyer, H. A heterogeneous hierarchy of co-regulatory receptors regulates exhaustion of HCV-specific CD8 T cells in patients with chronic hepatitis C. J. Hepatol. 2015, 62, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Fisicaro, P.; Valdatta, C.; Massari, M.; Loggi, E.; Ravanetti, L.; Urbani, S.; Giuberti, T.; Cavalli, A.; Vandelli, C.; Andreone, P.; et al. Combined blockade of programmed death-1 and activation of CD137 increase responses of human liver T cells against HBV, but not HCV. Gastroenterology 2012, 143, 1576–1585. [Google Scholar] [CrossRef]
- Li, Y.; Liu, S.; Hernandez, J.; Vence, L.; Hwu, P.; Radvanyi, L. MART-1-specific melanoma tumor-infiltrating lymphocytes maintaining CD28 expression have improved survival and expansion capability following antigenic restimulation in vitro. J. Immunol. 2010, 184, 452–465. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, M.; Im, S.J.; Araki, K.; Ahmed, R. Cytokine-Mediated Regulation of CD8 T-Cell Responses During Acute and Chronic Viral Infection. Cold Spring Harb Perspect Biol. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, D.M.; Ravkov, E.V.; Williams, M.A. Distinct roles for IL-2 and IL-15 in the differentiation and survival of CD8+ effector and memory T cells. J. Immunol. 2010, 184, 6719–6730. [Google Scholar] [CrossRef] [Green Version]
- Obar, J.J.; Molloy, M.J.; Jellison, E.R.; Stoklasek, T.A.; Zhang, W.; Usherwood, E.J.; Lefrancois, L. CD4+ T cell regulation of CD25 expression controls development of short-lived effector CD8+ T cells in primary and secondary responses. Proc. Natl. Acad. Sci. USA 2010, 107, 193–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pipkin, M.E.; Sacks, J.A.; Cruz-Guilloty, F.; Lichtenheld, M.G.; Bevan, M.J.; Rao, A. Interleukin-2 and inflammation induce distinct transcriptional programs that promote the differentiation of effector cytolytic T cells. Immunity 2010, 32, 79–90. [Google Scholar] [CrossRef] [Green Version]
- Feau, S.; Arens, R.; Togher, S.; Schoenberger, S.P. Autocrine IL-2 is required for secondary population expansion of CD8(+) memory T cells. Nat. Immunol. 2011, 12, 908–913. [Google Scholar] [CrossRef] [Green Version]
- West, E.E.; Jin, H.T.; Rasheed, A.U.; Penaloza-Macmaster, P.; Ha, S.J.; Tan, W.G.; Youngblood, B.; Freeman, G.J.; Smith, K.A.; Ahmed, R. PD-L1 blockade synergizes with IL-2 therapy in reinvigorating exhausted T cells. J. Clin. Investig. 2013, 123, 2604–2615. [Google Scholar] [CrossRef] [PubMed]
- Kaech, S.M.; Tan, J.T.; Wherry, E.J.; Konieczny, B.T.; Surh, C.D.; Ahmed, R. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat. Immunol. 2003, 4, 1191–1198. [Google Scholar] [CrossRef]
- Opferman, J.T.; Letai, A.; Beard, C.; Sorcinelli, M.D.; Ong, C.C.; Korsmeyer, S.J. Development and maintenance of B and T lymphocytes requires antiapoptotic MCL-1. Nature 2003, 426, 671–676. [Google Scholar] [CrossRef] [PubMed]
- Schluns, K.S.; Kieper, W.C.; Jameson, S.C.; Lefrancois, L. Interleukin-7 mediates the homeostasis of naive and memory CD8 T cells in vivo. Nat. Immunol. 2000, 1, 426–432. [Google Scholar] [CrossRef]
- Lang, K.S.; Recher, M.; Navarini, A.A.; Harris, N.L.; Lohning, M.; Junt, T.; Probst, H.C.; Hengartner, H.; Zinkernagel, R.M. Inverse correlation between IL-7 receptor expression and CD8 T cell exhaustion during persistent antigen stimulation. Eur. J. Immunol. 2005, 35, 738–745. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, M.; Calzascia, T.; Elford, A.R.; Shahinian, A.; Lin, A.E.; Dissanayake, D.; Dhanji, S.; Nguyen, L.T.; Gronski, M.A.; Morre, M.; et al. Adjuvant IL-7 antagonizes multiple cellular and molecular inhibitory networks to enhance immunotherapies. Nat. Med. 2009, 15, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Jabri, B.; Abadie, V. IL-15 functions as a danger signal to regulate tissue-resident T cells and tissue destruction. Nat. Rev. Immunol. 2015, 15, 771–783. [Google Scholar] [CrossRef]
- Becker, T.C.; Wherry, E.J.; Boone, D.; Murali-Krishna, K.; Antia, R.; Ma, A.; Ahmed, R. Interleukin 15 is required for proliferative renewal of virus-specific memory CD8 T cells. J. Exp. Med. 2002, 195, 1541–1548. [Google Scholar] [CrossRef] [Green Version]
- Sandau, M.M.; Kohlmeier, J.E.; Woodland, D.L.; Jameson, S.C. IL-15 regulates both quantitative and qualitative features of the memory CD8 T cell pool. J. Immunol. 2010, 184, 35–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wherry, E.J.; Blattman, J.N.; Murali-Krishna, K.; van der Most, R.; Ahmed, R. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J. Virol. 2003, 77, 4911–4927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teague, R.M.; Sather, B.D.; Sacks, J.A.; Huang, M.Z.; Dossett, M.L.; Morimoto, J.; Tan, X.; Sutton, S.E.; Cooke, M.P.; Ohlen, C.; et al. Interleukin-15 rescues tolerant CD8+ T cells for use in adoptive immunotherapy of established tumors. Nat. Med. 2006, 12, 335–341. [Google Scholar] [CrossRef]
- Buck, M.D.; O’Sullivan, D.; Klein Geltink, R.I.; Curtis, J.D.; Chang, C.H.; Sanin, D.E.; Qiu, J.; Kretz, O.; Braas, D.; van der Windt, G.J.; et al. Mitochondrial Dynamics Controls T Cell Fate through Metabolic Programming. Cell 2016, 166, 63–76. [Google Scholar] [CrossRef] [Green Version]
- van der Windt, G.J.; Everts, B.; Chang, C.H.; Curtis, J.D.; Freitas, T.C.; Amiel, E.; Pearce, E.J.; Pearce, E.L. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity 2012, 36, 68–78. [Google Scholar] [CrossRef] [Green Version]
- Bengsch, B.; Johnson, A.L.; Kurachi, M.; Odorizzi, P.M.; Pauken, K.E.; Attanasio, J.; Stelekati, E.; McLane, L.M.; Paley, M.A.; Delgoffe, G.M.; et al. Bioenergetic Insufficiencies Due to Metabolic Alterations Regulated by the Inhibitory Receptor PD-1 Are an Early Driver of CD8(+) T Cell Exhaustion. Immunity 2016, 45, 358–373. [Google Scholar] [CrossRef] [Green Version]
- Shenoy, A.R.; Kirschnek, S.; Hacker, G. IL-15 regulates Bcl-2 family members Bim and Mcl-1 through JAK/STAT and PI3K/AKT pathways in T cells. Eur J. Immunol. 2014, 44, 2500–2507. [Google Scholar] [CrossRef]
- Goshu, B.A.; Chen, H.; Moussa, M.; Cheng, J.; Catalfamo, M. Combination rhIL-15 and Anti-PD-L1 (Avelumab) Enhances HIVGag-Specific CD8 T-Cell Function. J. Infect. Dis. 2020, 222, 1540–1549. [Google Scholar] [CrossRef]
- Chen, P.; Chen, H.; Moussa, M.; Cheng, J.; Li, T.; Qin, J.; Lifson, J.D.; Sneller, M.C.; Krymskaya, L.; Godin, S.; et al. Recombinant Human Interleukin-15 and Anti-PD-L1 Combination Therapy Expands a CXCR3+PD1-/low CD8 T-Cell Subset in Simian Immunodeficiency Virus-Infected Rhesus Macaques. J. Infect. Dis. 2020, 221, 523–533. [Google Scholar] [CrossRef]
- Kim, K.H.; Kim, H.K.; Kim, H.D.; Kim, C.G.; Lee, H.; Han, J.W.; Choi, S.J.; Jeong, S.; Jeon, M.; Kim, H.; et al. PD-1 blockade-unresponsive human tumor-infiltrating CD8(+) T cells are marked by loss of CD28 expression and rescued by IL-15. Cell Mol. Immunol. 2020. [Google Scholar] [CrossRef]
- Giuffrida, L.; Sek, K.; Henderson, M.A.; House, I.G.; Lai, J.; Chen, A.X.Y.; Todd, K.L.; Petley, E.V.; Mardiana, S.; Todorovski, I.; et al. IL-15 Preconditioning Augments CAR T Cell Responses to Checkpoint Blockade for Improved Treatment of Solid Tumors. Mol. Ther. 2020, 28, 2379–2393. [Google Scholar] [CrossRef]
- Tian, Y.; Zajac, A.J. IL-21 and T Cell Differentiation: Consider the Context. Trends Immunol. 2016, 37, 557–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spolski, R.; Leonard, W.J. Interleukin-21: A double-edged sword with therapeutic potential. Nat. Rev. Drug Discov. 2014, 13, 379–395. [Google Scholar] [CrossRef] [PubMed]
- Boni, A.; Muranski, P.; Cassard, L.; Wrzesinski, C.; Paulos, C.M.; Palmer, D.C.; Gattinoni, L.; Hinrichs, C.S.; Chan, C.C.; Rosenberg, S.A.; et al. Adoptive transfer of allogeneic tumor-specific T cells mediates effective regression of large tumors across major histocompatibility barriers. Blood 2008, 112, 4746–4754. [Google Scholar] [CrossRef]
- Loschinski, R.; Bottcher, M.; Stoll, A.; Bruns, H.; Mackensen, A.; Mougiakakos, D. IL-21 modulates memory and exhaustion phenotype of T-cells in a fatty acid oxidation-dependent manner. Oncotarget 2018, 9, 13125–13138. [Google Scholar] [CrossRef] [Green Version]
- Pearce, E.L.; Walsh, M.C.; Cejas, P.J.; Harms, G.M.; Shen, H.; Wang, L.S.; Jones, R.G.; Choi, Y. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature 2009, 460, 103–107. [Google Scholar] [CrossRef]
- Kwon, H.; Thierry-Mieg, D.; Thierry-Mieg, J.; Kim, H.P.; Oh, J.; Tunyaplin, C.; Carotta, S.; Donovan, C.E.; Goldman, M.L.; Tailor, P.; et al. Analysis of interleukin-21-induced Prdm1 gene regulation reveals functional cooperation of STAT3 and IRF4 transcription factors. Immunity 2009, 31, 941–952. [Google Scholar] [CrossRef] [Green Version]
- Moroz, A.; Eppolito, C.; Li, Q.; Tao, J.; Clegg, C.H.; Shrikant, P.A. IL-21 enhances and sustains CD8+ T cell responses to achieve durable tumor immunity: Comparative evaluation of IL-2, IL-15, and IL-21. J. Immunol. 2004, 173, 900–909. [Google Scholar] [CrossRef] [Green Version]
- Zeng, R.; Spolski, R.; Finkelstein, S.E.; Oh, S.; Kovanen, P.E.; Hinrichs, C.S.; Pise-Masison, C.A.; Radonovich, M.F.; Brady, J.N.; Restifo, N.P.; et al. Synergy of IL-21 and IL-15 in regulating CD8+ T cell expansion and function. J. Exp. Med. 2005, 201, 139–148. [Google Scholar] [CrossRef] [Green Version]
- Lewis, K.E.; Selby, M.J.; Masters, G.; Valle, J.; Dito, G.; Curtis, W.R.; Garcia, R.; Mink, K.A.; Waggie, K.S.; Holdren, M.S.; et al. Interleukin-21 combined with PD-1 or CTLA-4 blockade enhances antitumor immunity in mouse tumor models. Oncoimmunology 2017, 7, e1377873. [Google Scholar] [CrossRef]
- Wieland, D.; Kemming, J.; Schuch, A.; Emmerich, F.; Knolle, P.; Neumann-Haefelin, C.; Held, W.; Zehn, D.; Hofmann, M.; Thimme, R. TCF1(+) hepatitis C virus-specific CD8(+) T cells are maintained after cessation of chronic antigen stimulation. Nat. Commun. 2017, 8, 15050. [Google Scholar] [CrossRef] [Green Version]
- Seigel, B.; Bengsch, B.; Lohmann, V.; Bartenschlager, R.; Blum, H.E.; Thimme, R. Factors that determine the antiviral efficacy of HCV-specific CD8(+) T cells ex vivo. Gastroenterology 2013, 144, 426–436. [Google Scholar] [CrossRef] [PubMed]
- Larrea, E.; Riezu-Boj, J.I.; Aldabe, R.; Guembe, L.; Echeverria, I.; Balasiddaiah, A.; Gastaminza, P.; Civeira, M.P.; Sarobe, P.; Prieto, J. Dysregulation of interferon regulatory factors impairs the expression of immunostimulatory molecules in hepatitis C virus genotype 1-infected hepatocytes. Gut 2014, 63, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Kared, H.; Fabre, T.; Bedard, N.; Bruneau, J.; Shoukry, N.H. Galectin-9 and IL-21 mediate cross-regulation between Th17 and Treg cells during acute hepatitis C. PLoS Pathog. 2013, 9, e1003422. [Google Scholar] [CrossRef] [Green Version]
- Yu, P.; Steel, J.C.; Zhang, M.; Morris, J.C.; Waitz, R.; Fasso, M.; Allison, J.P.; Waldmann, T.A. Simultaneous inhibition of two regulatory T-cell subsets enhanced Interleukin-15 efficacy in a prostate tumor model. Proc. Natl. Acad. Sci. USA 2012, 109, 6187–6192. [Google Scholar] [CrossRef] [Green Version]
- Yu, P.; Steel, J.C.; Zhang, M.; Morris, J.C.; Waldmann, T.A. Simultaneous blockade of multiple immune system inhibitory checkpoints enhances antitumor activity mediated by interleukin-15 in a murine metastatic colon carcinoma model. Clin. Cancer Res. 2010, 16, 6019–6028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfannenstiel, L.W.; Diaz-Montero, C.M.; Tian, Y.F.; Scharpf, J.; Ko, J.S.; Gastman, B.R. Immune-Checkpoint Blockade Opposes CD8(+) T-cell Suppression in Human and Murine Cancer. Cancer Immunol. Res. 2019, 7, 510–525. [Google Scholar] [CrossRef] [Green Version]
- Naidoo, J.; Page, D.B.; Li, B.T.; Connell, L.C.; Schindler, K.; Lacouture, M.E.; Postow, M.A.; Wolchok, J.D. Toxicities of the anti-PD-1 and anti-PD-L1 immune checkpoint antibodies. Ann. Oncol. 2015, 26, 2375–2391. [Google Scholar] [CrossRef]
- Waldmann, T.A. The shared and contrasting roles of IL2 and IL15 in the life and death of normal and neoplastic lymphocytes: Implications for cancer therapy. Cancer Immunol. Res. 2015, 3, 219–227. [Google Scholar] [CrossRef] [Green Version]
- Kammula, U.S.; White, D.E.; Rosenberg, S.A. Trends in the safety of high dose bolus interleukin-2 administration in patients with metastatic cancer. Cancer 1998, 83, 797–805. [Google Scholar] [CrossRef]
- Sportes, C.; Babb, R.R.; Krumlauf, M.C.; Hakim, F.T.; Steinberg, S.M.; Chow, C.K.; Brown, M.R.; Fleisher, T.A.; Noel, P.; Maric, I.; et al. Phase I study of recombinant human interleukin-7 administration in subjects with refractory malignancy. Clin. Cancer Res. 2010, 16, 727–735. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IL-2 | IL-7 | IL-15 | IL-21 | |
|---|---|---|---|---|
| Survival | X | X | ||
| Memory differentiation: | ||||
| Central memory | X † | X | ||
| Effector memory | X § | X | X | |
| Homeostatic proliferation | X | X | ||
| Ag-specific proliferation | X | X | X | |
| Decrease of negative IC | X | X | X | |
| Long-lasting stem-like cells | X | X | ||
| Self-renewal | X | |||
| Catabolic mitochondrial reprogramming | X | X |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peña-Asensio, J.; Calvo, H.; Torralba, M.; Miquel, J.; Sanz-de-Villalobos, E.; Larrubia, J.-R. Gamma-Chain Receptor Cytokines & PD-1 Manipulation to Restore HCV-Specific CD8+ T Cell Response during Chronic Hepatitis C. Cells 2021, 10, 538. https://doi.org/10.3390/cells10030538
Peña-Asensio J, Calvo H, Torralba M, Miquel J, Sanz-de-Villalobos E, Larrubia J-R. Gamma-Chain Receptor Cytokines & PD-1 Manipulation to Restore HCV-Specific CD8+ T Cell Response during Chronic Hepatitis C. Cells. 2021; 10(3):538. https://doi.org/10.3390/cells10030538
Chicago/Turabian StylePeña-Asensio, Julia, Henar Calvo, Miguel Torralba, Joaquín Miquel, Eduardo Sanz-de-Villalobos, and Juan-Ramón Larrubia. 2021. "Gamma-Chain Receptor Cytokines & PD-1 Manipulation to Restore HCV-Specific CD8+ T Cell Response during Chronic Hepatitis C" Cells 10, no. 3: 538. https://doi.org/10.3390/cells10030538
APA StylePeña-Asensio, J., Calvo, H., Torralba, M., Miquel, J., Sanz-de-Villalobos, E., & Larrubia, J.-R. (2021). Gamma-Chain Receptor Cytokines & PD-1 Manipulation to Restore HCV-Specific CD8+ T Cell Response during Chronic Hepatitis C. Cells, 10(3), 538. https://doi.org/10.3390/cells10030538