
Oxidized Phospholipids in Tumor Microenvironment Stimulate Tumor Metastasis via Regulation of Autophagy


 , ,
, ,  ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Reagents
2.3. Immunoblotting Analysis
2.4. Confocal Microscopy Analysis
2.5. Wound Healing Assay
2.6. Transwell Migration and Invasion Assay
2.7. Measurement of Autophagosomes
2.8. siRNA Transfection
2.9. LC-MS/MS Analysis for Oxidized Phosphatidylcholines
2.10. Statistical Analysis
3. Results
3.1. POVPC Increases the Expression of Epithelial-Mesenchymal Transition Markers in Cancer Cells
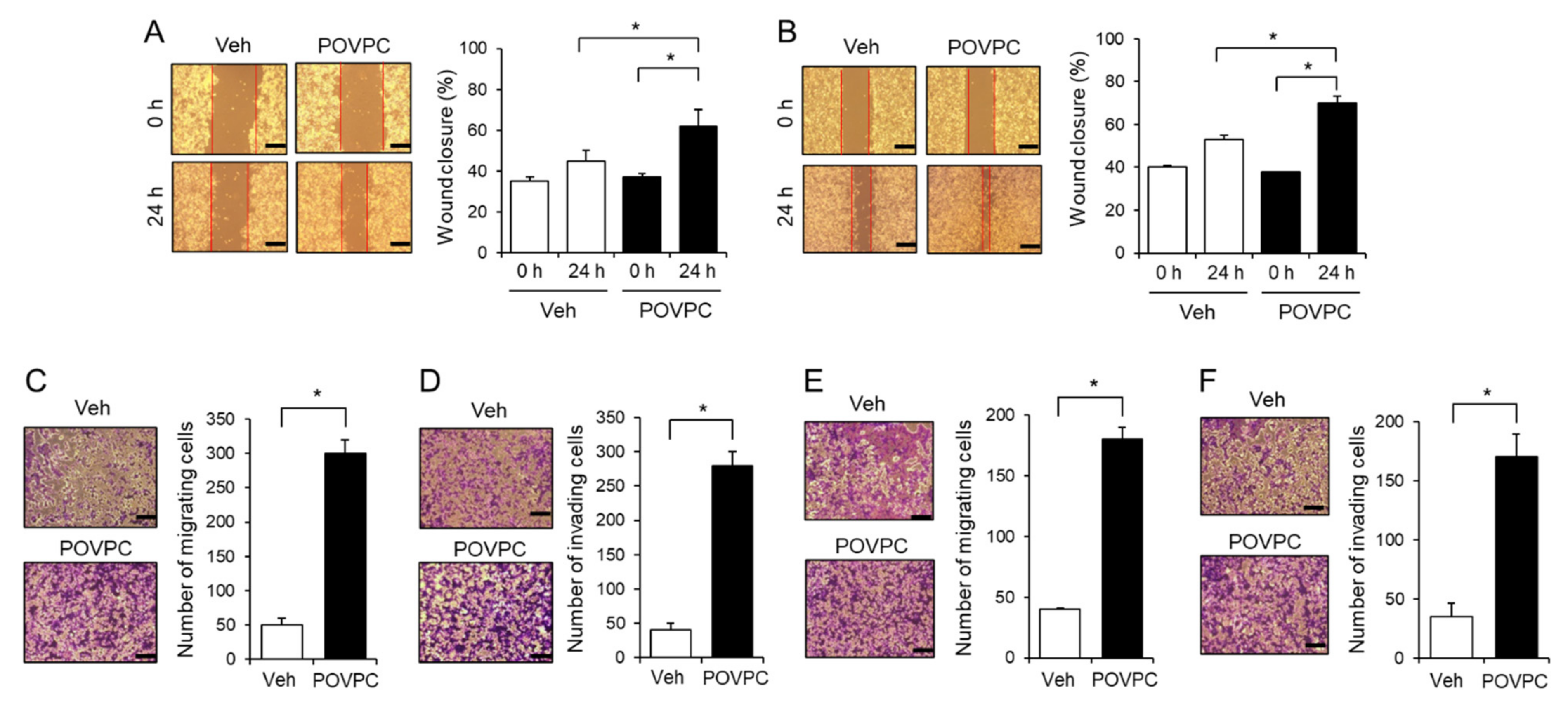
3.2. An Oxidized Phosphatidylcholine, POVPC, Increases the Metastatic Potential of Cancer Cells
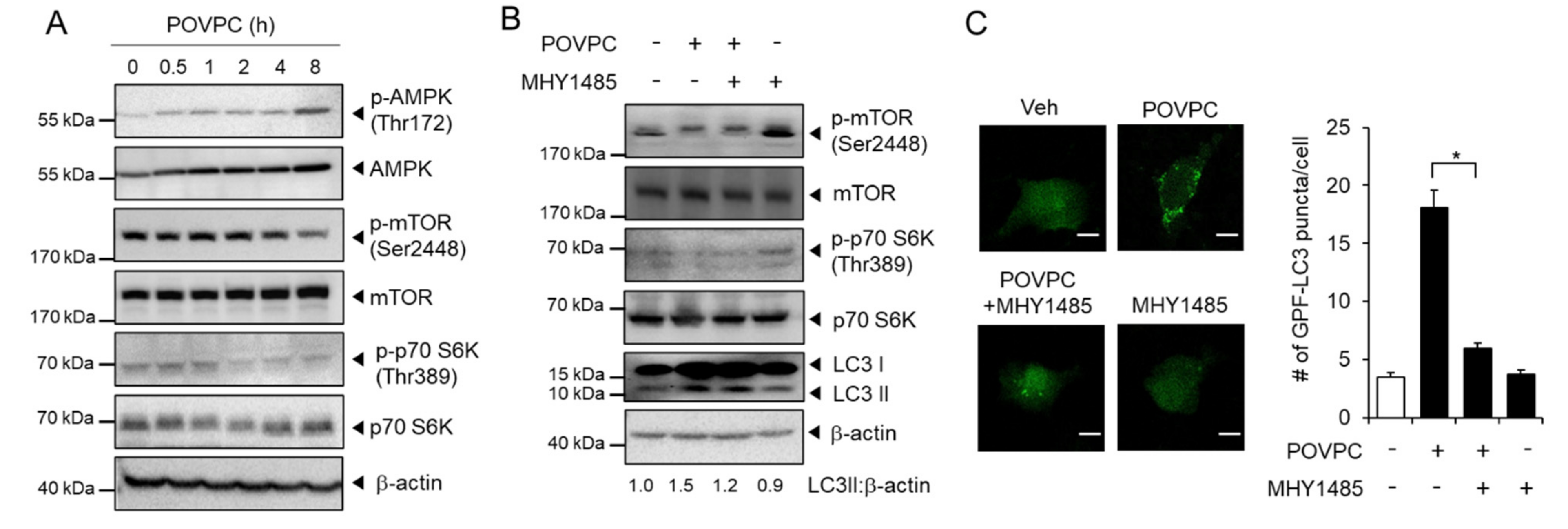
3.3. POVPC Upregulates Autophagic Flux in Cancer Cells
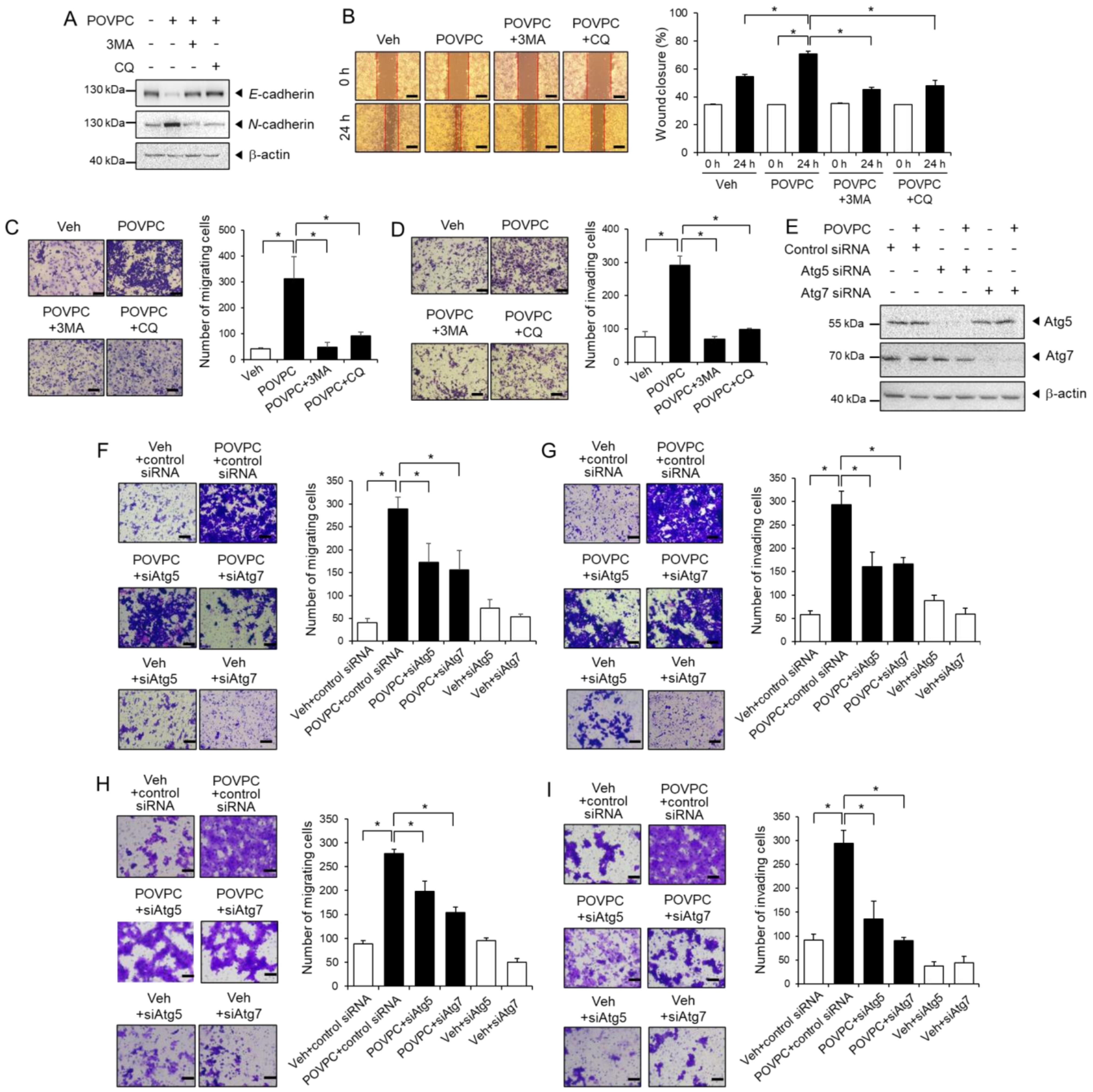
3.4. The Pro-Metastatic Effects of POVPC Are Mediated through the Autophagic Pathway
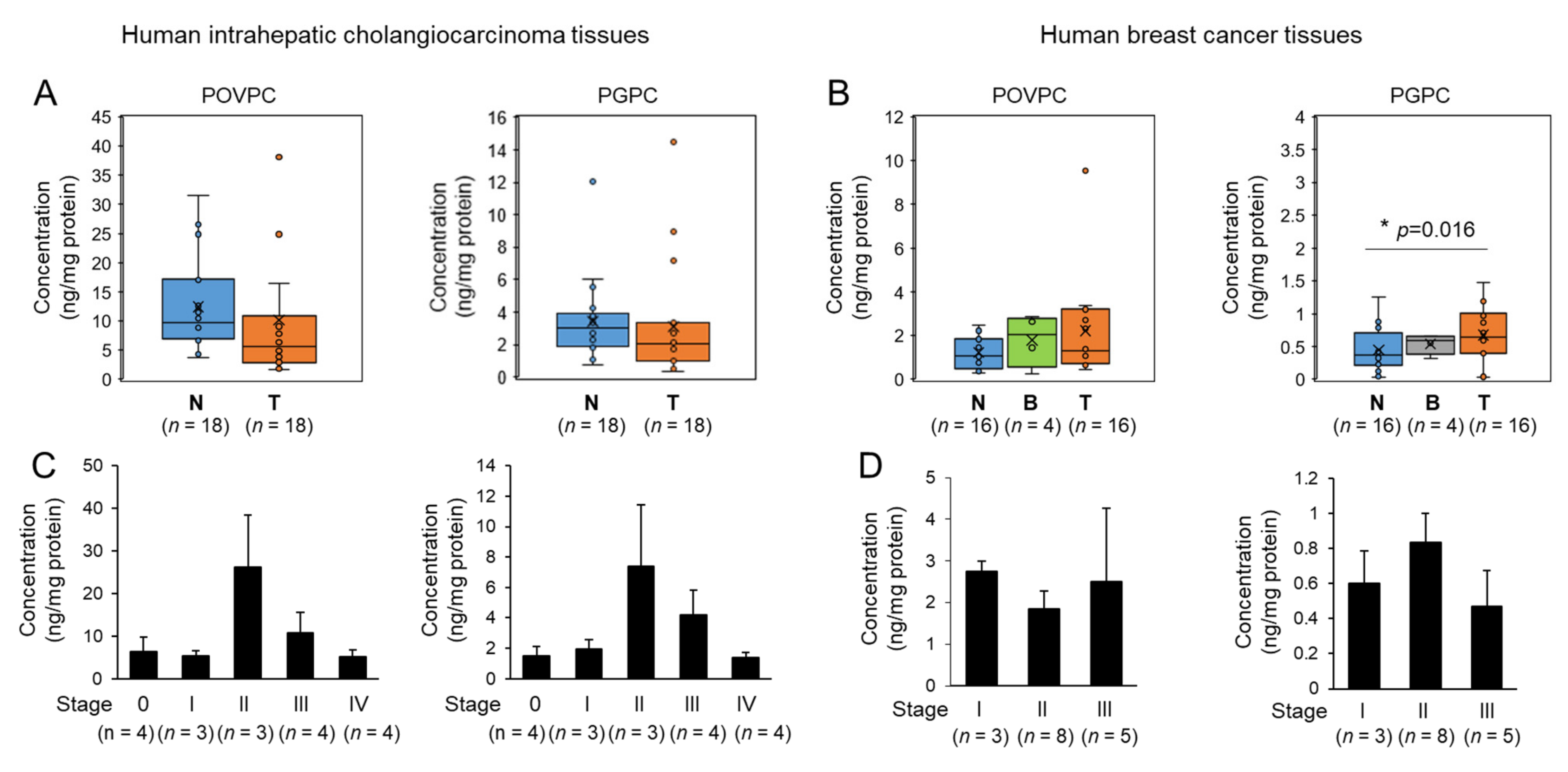
3.5. Changes in Oxidized Phospholipids Levels of Human Tumor Tissues
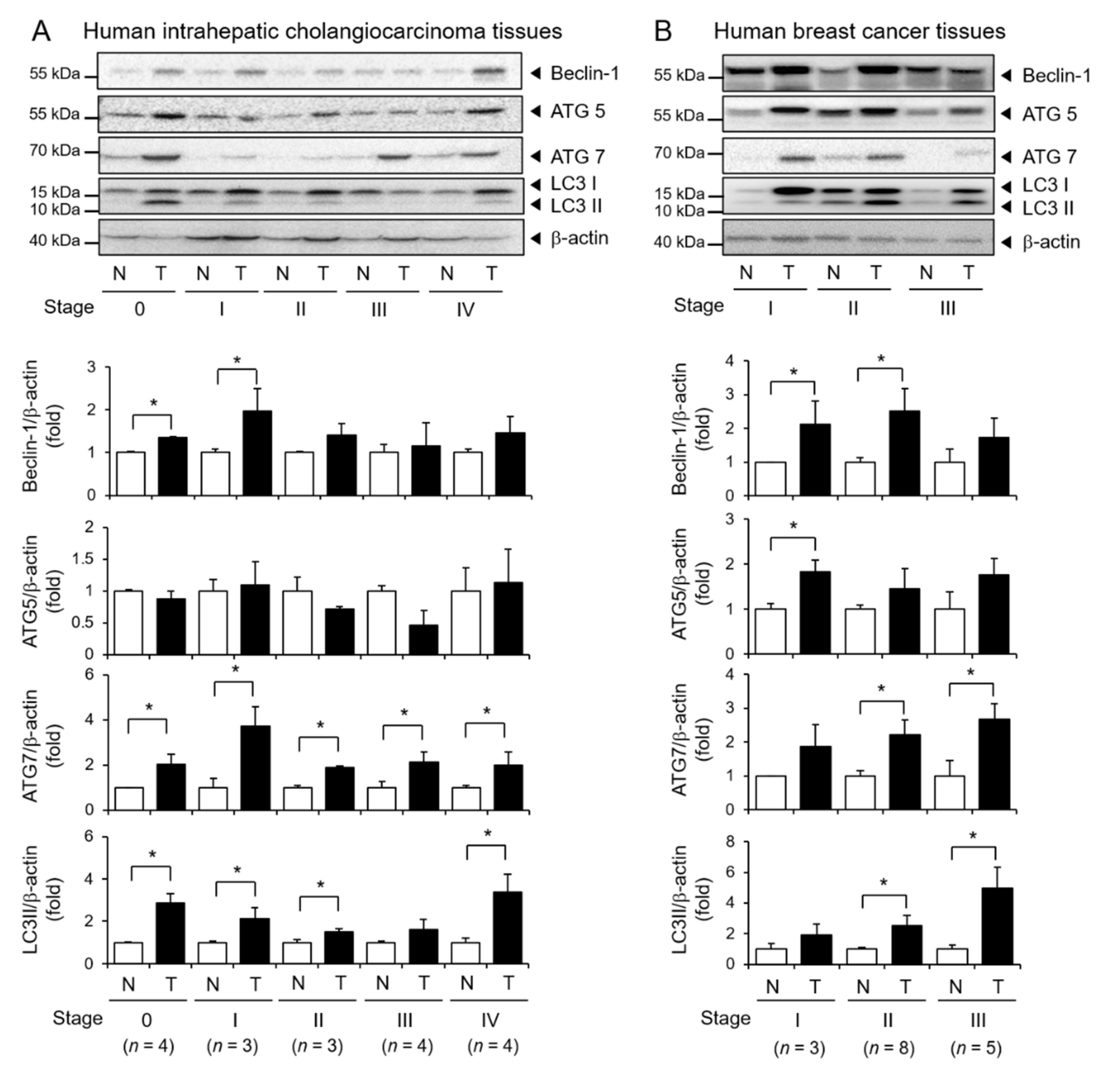
3.6. Elevated Expression of Autophagy Markers in Human Tumor Tissues
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guan, X. Cancer metastases: Challenges and opportunities. Acta Pharm. Sin. B 2015, 5, 402–418. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.; Weinberg, R.A. Epithelial–Mesenchymal Plasticity: A Central Regulator of Cancer Progression. Trends Cell Biol. 2015, 25, 675–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, J.; Debnath, J. Autophagy at the crossroads of catabolism and anabolism. Nat. Rev. Mol. Cell Biol. 2015, 16, 461–472. [Google Scholar] [CrossRef] [Green Version]
- Kenific, C.M.; Thorburn, A.; Debnath, J. Autophagy and metastasis: Another double-edged sword. Curr. Opin. Cell Biol. 2010, 22, 241–245. [Google Scholar] [CrossRef] [Green Version]
- Kondo, Y.; Kanzawa, T.; Sawaya, R.; Kondo, S. The role of autophagy in cancer development and response to therapy. Nat. Rev. Cancer 2005, 5, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Beloribi-Djefaflia, S.; Vasseur, S.; Guillaumond, F. Lipid metabolic reprogramming in cancer cells. Oncogenesis 2016, 5, e189. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Wang, Y.; Zhou, D.; Li, Z. Significantly increased monounsaturated lipids relative to polyunsaturated lipids in six types of cancer microenvironment are observed by mass spectrometry imaging. Sci. Rep. 2015, 4, srep05959. [Google Scholar] [CrossRef] [Green Version]
- De Palma, M.; Biziato, D.; Petrova, T.V. Microenvironmental regulation of tumour angiogenesis. Nat. Rev. Cancer 2017, 17, 457–474. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Kurtova, A.V.; Xiao, J.; Mo, Q.; Pazhanisamy, S.K.; Krasnow, R.; Lerner, S.P.; Chen, F.; Roh, T.T.; Lay, E.; Ho, P.L.; et al. Blocking PGE2-induced tumour repopulation abrogates bladder cancer chemoresistance. Nature 2015, 517, 209–213. [Google Scholar] [CrossRef]
- Deigner, H.-P.; Hermetter, A. Oxidized phospholipids: Emerging lipid mediators in pathophysiology. Curr. Opin. Lipidol. 2008, 19, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Berliner, J.A.; Leitinger, N.; Tsimikas, S. The role of oxidized phospholipids in atherosclerosis. J. Lipid Res. 2009, 50, S207–S212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Gioia, M.; Spreafico, R.; Springstead, J.R.; Mendelson, M.M.; Joehanes, R.; Levy, D.; Zanoni, I. Endogenous oxidized phospholipids reprogram cellular metabolism and boost hyperinflammation. Nat. Immunol. 2020, 21, 42–53. [Google Scholar] [CrossRef]
- Miller, Y.I.; Shyy, J.Y.-J. Context-Dependent Role of Oxidized Lipids and Lipoproteins in Inflammation. Trends Endocrinol. Metab. 2017, 28, 143–152. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.J.; Choi, N.Y.; Koo, J.E.; Kim, S.Y.; Joung, S.M.; Jeong, E.; Lee, J.Y. Suppression of Toll-like receptor 4 activation by endogenous oxidized phosphatidylcholine, KOdiA-PC by inhibiting LPS binding to MD2. Inflamm. Res. 2013, 62, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Yeon, S.H.; Yang, G.; Lee, H.E.; Lee, J.Y. Oxidized phosphatidylcholine induces the activation of NLRP3 inflammasome in macrophages. J. Leukoc. Biol. 2016, 101, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Lee, H.E.; Yeon, S.H.; Kang, H.C.; Cho, Y.-Y.; Zouboulis, C.C.; Han, S.-H.; Lee, J.-H.; Lee, J.Y. Licochalcone A attenuates acne symptoms mediated by suppression of NLRP3 inflammasome. Phytother. Res. 2018, 32, 2551–2559. [Google Scholar] [CrossRef]
- Yang, G.; Lee, H.E.; Moon, S.; Ko, K.M.; Koh, J.H.; Seok, J.K.; Min, J.; Heo, T.; Kang, H.C.; Cho, Y.; et al. Direct Binding to NLRP3 Pyrin Domain as a Novel Strategy to Prevent NLRP3-Driven Inflammation and Gouty Arthritis. Arthritis Rheumatol. 2020, 72, 1192–1202. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.E.; Lee, J.Y.; Yang, G.; Kang, H.C.; Cho, Y.-Y.; Lee, J.Y. Inhibition of NLRP3 inflammasome in tumor microenvironment leads to suppression of metastatic potential of cancer cells. Sci. Rep. 2019, 9, 12277. [Google Scholar] [CrossRef] [Green Version]
- Koo, J.E.; Shin, S.W.; Um, S.H.; Lee, J.Y. X-shaped DNA potentiates therapeutic efficacy in colitis-associated colon cancer through dual activation of TLR9 and inflammasomes. Mol. Cancer 2015, 14, 104. [Google Scholar] [CrossRef] [Green Version]
- Vande Walle, L.; Stowe, I.B.; Sacha, P.; Lee, B.L.; Demon, D.; Fossoul, A.; Van Hauwermeiren, F.; Saavedra, P.H.V.; Simon, P.; Subrt, V.; et al. MCC950/CRID3 potently targets the NACHT domain of wild-type NLRP3 but not disease-associated mutants for inflammasome inhibition. PLoS Biol. 2019, 17, e3000354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeon, S.H.; Song, M.J.; Kang, H.-R.; Lee, J.Y. Phosphatidylinositol-3-kinase and Akt are required for RIG-I-mediated anti-viral signalling through cross-talk with IPS-1. Immunology 2015, 144, 312–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolós, V.; Peinado, H.; Pérez-Moreno, M.A.; Fraga, M.F.; Esteller, M.; Cano, A. The transcription factor Slug represses E-cadherin expression and induces epithelial to mesenchymal transitions: A comparison with Snail and E47 repressors. J. Cell Sci. 2002, 116, 499–511. [Google Scholar] [CrossRef] [Green Version]
- Hajra, K.M.; Chen, D.Y.; Fearon, E.R. The SLUG zinc-finger protein represses E-cadherin in breast cancer. Cancer Res. 2002, 62, 1613–1618. [Google Scholar] [PubMed]
- Martin, T.A.; Goyal, A.; Watkins, G.; Jiang, W.G. Expression of the Transcription Factors Snail, Slug, and Twist and Their Clinical Significance in Human Breast Cancer. Ann. Surg. Oncol. 2005, 12, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Lorin, S.; Hamaï, A.; Mehrpour, M.; Codogno, P. Autophagy regulation and its role in cancer. Semin. Cancer Biol. 2013, 23, 361–379. [Google Scholar] [CrossRef]
- McGowan, P.M.; Kirstein, J.M.; Chambers, A.F. Micrometastatic disease and metastatic outgrowth: Clinical issues and experimental approaches. Future Oncol. 2009, 5, 1083–1098. [Google Scholar] [CrossRef] [PubMed]
- Chaffer, C.L.; Weinberg, R.A. A Perspective on Cancer Cell Metastasis. Science 2011, 331, 1559–1564. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Wang, H.; Guan, H.; Shi, S.; Li, Y.; Wu, X.; Li, N.; Yang, C.; Bai, X.; Cai, W.; et al. IL10 inhibits starvation-induced autophagy in hypertrophic scar fibroblasts via cross talk between the IL10-IL10R-STAT3 and IL10-AKT-mTOR pathways. Cell Death Dis. 2016, 7, e2133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, M.-K.; Binder, C.J.; Torzewski, M.; Witztum, J.L. C-reactive protein binds to both oxidized LDL and apoptotic cells through recognition of a common ligand: Phosphorylcholine of oxidized phospholipids. Proc. Natl. Acad. Sci. USA 2002, 99, 13043–13048. [Google Scholar] [CrossRef] [Green Version]
- Bochkov, V.N.; Oskolkova, O.V.; Birukov, K.G.; Levonen, A.-L.; Binder, C.J.; Stöckl, J. Generation and Biological Activities of Oxidized Phospholipids. Antioxid. Redox Signal. 2010, 12, 1009–1059. [Google Scholar] [CrossRef] [Green Version]
- Berliner, J.A.; Territo, M.C.; Sevanian, A.; Ramin, S.; Kim, J.A.; Bamshad, B.; Esterson, M.; Fogelman, A.M. Minimally modified low density lipoprotein stimulates monocyte endothelial interactions. J. Clin. Investig. 1990, 85, 1260–1266. [Google Scholar] [CrossRef] [Green Version]
- Weismann, D.; Binder, C.J. The innate immune response to products of phospholipid peroxidation. Biochim. Biophys. Acta Biomembr. 2012, 1818, 2465–2475. [Google Scholar] [CrossRef] [Green Version]
- Binder, C.J.; Papac-Milicevic, C.J.B.N.; Witztum, J.L. Innate sensing of oxidation-specific epitopes in health and disease. Nat. Rev. Immunol. 2016, 16, 485–497. [Google Scholar] [CrossRef]
- White, E. Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer 2012, 12, 401–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhi, X.; Zhong, Q. Autophagy in cancer. F1000Prime Rep. 2015, 7, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimmelman, A.C.; White, E. Autophagy and Tumor Metabolism. Cell Metab. 2017, 25, 1037–1043. [Google Scholar] [CrossRef]
- Fung, C.; Lock, R.; Gao, S.; Salas, E.; Debnath, J. Induction of Autophagy during Extracellular Matrix Detachment Promotes Cell Survival. Mol. Biol. Cell 2008, 19, 797–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrero-Martín, G.; Høyer-Hansen, M.; García-García, C.; Fumarola, C.; Farkas, T.; López-Rivas, A.; Jäättelä, M. TAK1 activates AMPK-dependent cytoprotective autophagy in TRAIL-treated epithelial cells. EMBO J. 2009, 28, 677–685. [Google Scholar] [CrossRef] [Green Version]
- Jung, C.H.; Ro, S.-H.; Cao, J.; Otto, N.M.; Kim, D.-H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef] [Green Version]
- Hill, B.G.; Haberzettl, P.; Ahmed, Y.; Srivastava, S.; Bhatnagar, A. Unsaturated lipid peroxidation-derived aldehydes activate autophagy in vascular smooth-muscle cells. Biochem. J. 2008, 410, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Ni, C.; Narzt, M.-S.; Nagelreiter, I.-M.; Zhang, C.F.; Larue, L.; Rossiter, H.; Grillari, J.; Tschachler, E.; Gruber, F. Autophagy deficient melanocytes display a senescence associated secretory phenotype that includes oxidized lipid mediators. Int. J. Biochem. Cell Biol. 2016, 81, 375–382. [Google Scholar] [CrossRef]
- Galluzzi, L.; Pedro, J.M.B.-S.; Levine, B.; Green, D.R.; Kroemer, G. Pharmacological modulation of autophagy: Therapeutic potential and persisting obstacles. Nat. Rev. Drug Discov. 2017, 16, 487–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solomon, V.R.; Lee, H. Chloroquine and its analogs: A new promise of an old drug for effective and safe cancer therapies. Eur. J. Pharmacol. 2009, 625, 220–233. [Google Scholar] [CrossRef]
- Lim, B.; Woodward, W.A.; Wang, X.; Reuben, J.M.; Ueno, N.T. Inflammatory breast cancer biology: The tumour microenvironment is key. Nat. Rev. Cancer 2018, 18, 485–499. [Google Scholar] [CrossRef] [PubMed]
- Valenta, T.; Hausmann, G.; Basler, K. The many faces and functions of beta-catenin. EMBO J. 2012, 31, 2714–2736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, A.H.; Weis, W.I. The structure of the beta-catenin/E-cadherin complex and the molecular basis of diverse ligand recognition by beta-catenin. Cell 2001, 105, 391–402. [Google Scholar] [CrossRef] [Green Version]
- Birukova, A.A.; Malyukova, I.; Poroyko, V.; Birukov, K.G. Paxillin-beta-catenin interactions are involved in Rac/Cdc42-mediated endothelial barrier-protective response to oxidized phospholipids. Am. J. Physiol. Lung. Cell Mol. Physiol. 2007, 293, L199–L211. [Google Scholar] [CrossRef] [PubMed]
- Karki, P.; Meliton, A.; Shah, A.; Tian, Y.; Ohmura, T.; Sarich, N.; Birukova, A.A.; Birukov, K.G. Role of truncated oxidized phospholipids in acute endothelial barrier dysfunction caused by particulate matter. PLoS ONE 2018, 13, e0206251. [Google Scholar] [CrossRef] [PubMed]
- Yeh, M.; Gharavi, N.M.; Choi, J.; Hsieh, X.; Reed, E.; Mouillesseaux, K.P.; Cole, A.L.; Reddy, S.T.; Berliner, J.A. Oxidized Phospholipids Increase Interleukin 8 (IL-8) Synthesis by Activation of the c-src/Signal Transducers and Activators of Transcription (STAT)3 Pathway. J. Biol. Chem. 2004, 279, 30175–30181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, W. Role of JAK/STAT3 Signaling in the Regulation of Metastasis, the Transition of Cancer Stem Cells, and Chemoresistance of Cancer by Epithelial–Mesenchymal Transition. Cells 2020, 9, 217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freigang, S.B. The regulation of inflammation by oxidized phospholipids. Eur. J. Immunol. 2016, 46, 1818–1825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karki, P.; Birukov, K.G. Oxidized Phospholipids in Healthy and Diseased Lung Endothelium. Cells 2020, 9, 981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, G.; Batra, S.; Shukla, N.K.; Deo, S.; Raina, V.; Ashok, S.; Husain, S.A. Lipid peroxidation, free radical production and antioxidant status in breast cancer. Breast Cancer Res. Treat. 2000, 59, 163–170. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seok, J.K.; Hong, E.-H.; Yang, G.; Lee, H.E.; Kim, S.-E.; Liu, K.-H.; Kang, H.C.; Cho, Y.-Y.; Lee, H.S.; Lee, J.Y. Oxidized Phospholipids in Tumor Microenvironment Stimulate Tumor Metastasis via Regulation of Autophagy. Cells 2021, 10, 558. https://doi.org/10.3390/cells10030558
Seok JK, Hong E-H, Yang G, Lee HE, Kim S-E, Liu K-H, Kang HC, Cho Y-Y, Lee HS, Lee JY. Oxidized Phospholipids in Tumor Microenvironment Stimulate Tumor Metastasis via Regulation of Autophagy. Cells. 2021; 10(3):558. https://doi.org/10.3390/cells10030558
Chicago/Turabian StyleSeok, Jin Kyung, Eun-Hee Hong, Gabsik Yang, Hye Eun Lee, Sin-Eun Kim, Kwang-Hyeon Liu, Han Chang Kang, Yong-Yeon Cho, Hye Suk Lee, and Joo Young Lee. 2021. "Oxidized Phospholipids in Tumor Microenvironment Stimulate Tumor Metastasis via Regulation of Autophagy" Cells 10, no. 3: 558. https://doi.org/10.3390/cells10030558
APA StyleSeok, J. K., Hong, E.-H., Yang, G., Lee, H. E., Kim, S.-E., Liu, K.-H., Kang, H. C., Cho, Y.-Y., Lee, H. S., & Lee, J. Y. (2021). Oxidized Phospholipids in Tumor Microenvironment Stimulate Tumor Metastasis via Regulation of Autophagy. Cells, 10(3), 558. https://doi.org/10.3390/cells10030558