
The Importance of Drosophila melanogaster Research to UnCover Cellular Pathways Underlying Parkinson’s Disease



Abstract
:1. Introduction
2. Drosophila melanogaster as Animal Model to Study Human Diseases
3. Drosophila melanogaster as Animal Model for Parkinson’s Disease
4. The Mitochondrial-PD Connection in Drosophila melanogaster
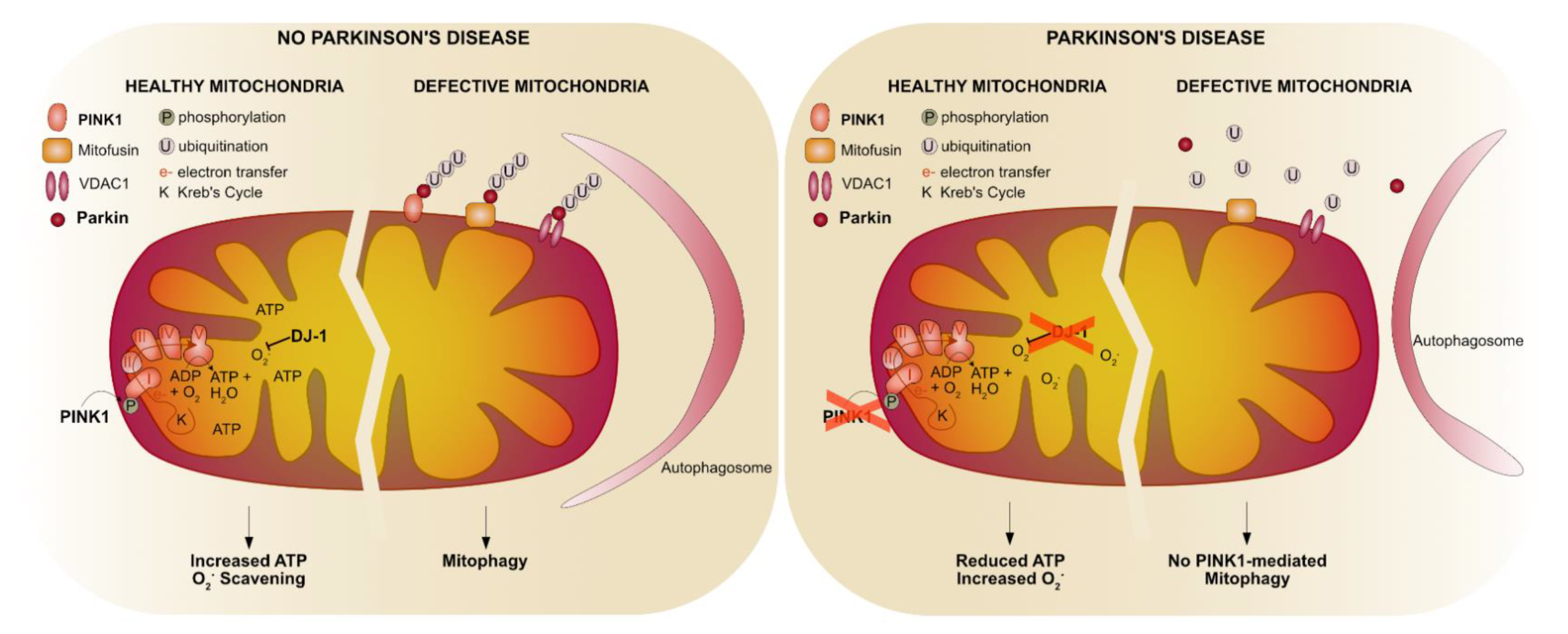
4.1. The Pink1-Parkin Pathway
4.1.1. Mitochondrial Fusion and Fission
4.1.2. Mitophagy
4.1.3. Mitochondrial Complex I
4.2. Parkin-Specific Functions
4.3. The Role of Oxidative Stress in PD
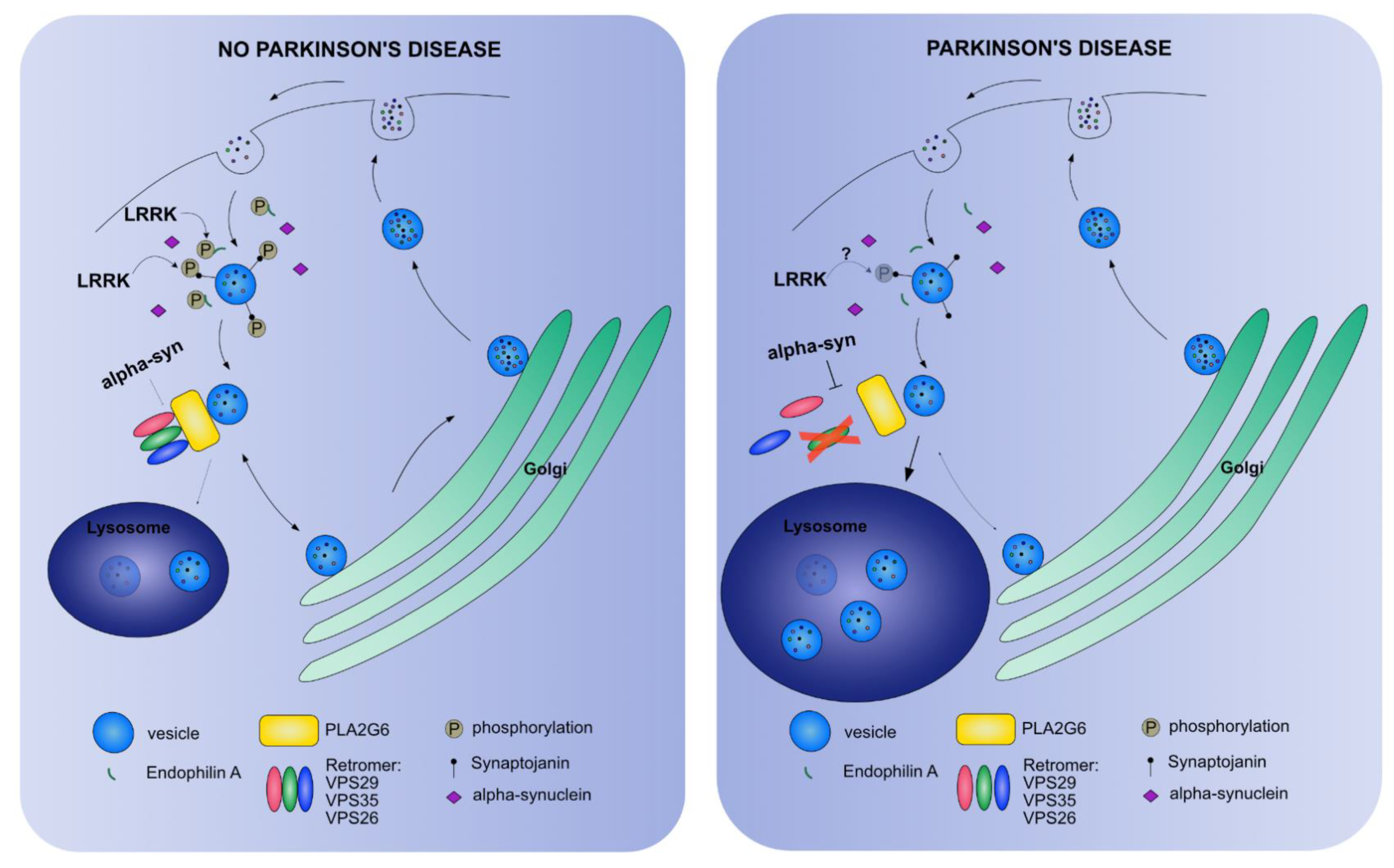
5. Endo-Lysosomal PD Connection in Drosophila melanogaster
5.1. Vesicle Trafficking Endocytosis
5.2. The Retromer Complex
5.3. The Autolysosome
6. Lipids as Connecting Factor between Mitochondrial Dysfunction and a Defective Endo-Lysosomal Pathway in Drosophila melanogaster
6.1. Glucorerebrosidase
6.2. Identification of Underlying Mechanisms in Non-Motor Symptoms in PD
7. Validation in a Mammalian System
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fahn, S.; Jankovic, J. Principles and Practice of Movement Disorders; Churchill Livingstone Elsevier: Philadelphia, PA, USA, 2007. [Google Scholar]
- Kasten, M.; Hartmann, C.; Hampf, J.; Schaake, S.; Westenberger, A.; Vollstedt, E.-J.; Balck, A.; Domingo, A.; Vulinovic, F.; Dulovic, M.; et al. Genotype-Phenotype Relations for the Parkinson’s Disease Genes Parkin, PINK1, DJ1: MDSGene Systematic Review. Mov. Disord. 2018, 33, 730–741. [Google Scholar] [CrossRef] [PubMed]
- Sulzer, D.; Surmeier, D.J. Neuronal vulnerability, pathogenesis, and Parkinson’s disease. Mov. Disord. 2013, 28, 715–724. [Google Scholar] [CrossRef]
- Shahmoradian, S.H.; Lewis, A.J.; Genoud, C.; Hench, J.; Moors, T.E.; Navarro, P.P.; Castaño-Díez, D.; Schweighauser, G.; Graff-Meyer, A.; Goldie, K.N.; et al. Lewy pathology in Parkinson’s disease consists of crowded organelles and lipid membranes. Nat. Neurosci. 2019, 22, 1099–1109. [Google Scholar] [CrossRef] [Green Version]
- Chaudhuri, K.R.; Healy, D.G.; Schapira, A.H. Non-motor symptoms of Parkinson’s disease: Diagnosis and management. Lancet Neurol. 2006, 5, 235–245. [Google Scholar] [CrossRef]
- Schapira, A.H.V.; Chaudhuri, K.R.; Jenner, P. Non-motor features of Parkinson disease. Nat. Rev. Neurosci. 2017, 18, 435. [Google Scholar] [CrossRef]
- Lee, H.; Koh, S. Many Faces of Parkinson’s Disease: Non-Motor Symptoms of Parkinson’s Disease. Mov. Disord. 2015, 8, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Kasten, M.; Marras, C.; Klein, C. Nonmotor Signs in Genetic Forms of Parkinson’s Disease. In International Review of Neurobiology; Elsevier: Amsterdam, The Netherlands, 2017; pp. 129–178. ISBN 9780128137086. [Google Scholar]
- Hermanowicz, N.; Jones, S.A.; Hauser, R.A. Impact of non-motor symptoms in Parkinson’s disease: A PMDAlliance survey. Neuropsychiatr. Dis. Treat. 2019, 15, 2205–2212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorsey, E.R.; Bloem, B.R. The Parkinson Pandemic-A Call to Action. JAMA Neurol. 2018, 75, 9–10. [Google Scholar] [CrossRef] [PubMed]
- GBD Global, regional, and national burden of neurological disorders, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 459–480. [CrossRef] [Green Version]
- De Lau, L.M.; Breteler, M.M. Epidemiology of Parkinson’s disease. Lancet Neurol. 2006, 5, 525–535. [Google Scholar] [CrossRef]
- Bloem, B.R.; Okun, M.S.; Klein, C. Parkinson’s disease. Lancet 2021, in press. [Google Scholar]
- You, H.; Mariani, L.; Mangone, G.; Le Febvre de Nailly, D.; Charbonnier-Beaupel, F.; Corvol, J. Molecular basis of dopamine replacement therapy and its side effects in Parkinson’s disease. Cell Tissue Res. Cell Tissue Res. 2018, 373, 111–135. [Google Scholar] [CrossRef]
- Neumann, M.; Kahle, P.J.; Giasson, B.I.; Ozmen, L.; Borroni, E.; Spooren, W.; Müller, V.; Odoy, S.; Fujiwara, H.; Hasegawa, M.; et al. Misfolded proteinase K-resistant hyperphosphorylated alpha-synuclein in aged transgenic mice with locomotor deterioration and in human alpha-synucleinopathies. J. Clin. Investig. 2002, 110, 1429–1439. [Google Scholar] [CrossRef]
- Giasson, B.I.; Duda, J.E.; Quinn, S.M.; Zhang, B.; Trojanowski, J.Q.; Lee, V.M.Y. Neuronal [alpha]-Synucleinopathy with Severe Movement Disorder in Mice Expressing A53T Human [alpha]-Synuclein. Neuron 2002, 34, 521–533. [Google Scholar] [CrossRef] [Green Version]
- Gispert, S.; Ricciardi, F.; Kurz, A.; Azizov, M.; Hoepken, H.-H.; Becker, D.; Voos, W.; Leuner, K.; Müller, W.E.; Kudin, A.P.; et al. Parkinson phenotype in aged PINK1-deficient mice is accompanied by progressive mitochondrial dysfunction in absence of neurodegeneration. PLoS ONE 2009, 4, e5777. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, M.S.; Fleming, S.M.; Palacino, J.J.; Cepeda, C.; Lam, H.A.; Bhatnagar, A.; Meloni, E.G.; Wu, N.; Ackerson, L.C.; Klapstein, G.J.; et al. Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J. Biol. Chem. 2003, 278, 43628–43635. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, S.; Seto, E. Dopamine dynamics and signaling in Drosophila: An overview of genes, drugs and behavioral paradigms. Exp. Anim. 2014, 63, 107–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagoshi, E. Drosophila Models of Sporadic Parkinson’s Disease. Int. J. Mol. Sci. 2018, 19, 3343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bier, E. Drosophila, the golden bug, emerges as a tool for human genetics. Nat. Rev. Genet. 2005, 6, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Zhang, L.; Huang, X. Genome modification by CRISPR/Cas9. FEBS J. 2014, 281, 5186–5193. [Google Scholar] [CrossRef]
- Gratz, S.; Cummings, A.; Nguyen, J.; Hamm, D.; Donohue, L.; Harrison, M.; Wildonger, J.; O’Connor-Giles, K. Genome engineering of Drosophila with the CRISPR RNA-guided Cas9 nuclease. Genetics. Genetics 2013, 194, 1029–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webster, N.; Jin, J.; Green, S.; Hollis, M.; Chambon, P. The yeast UASG is a transcriptional enhancer in human HeLa cells in the presence of the GAL4 trans-activator. Cell 1988, 52, 169–178. [Google Scholar] [CrossRef]
- Kakidani, H.; Ptashne, M. GAL4 activates gene expression in mammalian cells. Cell 1988, 52, 161–167. [Google Scholar] [CrossRef]
- Duffy JB GAL4 system in Drosophila: A fly geneticist’s Swiss army knife. Genes Brain Behav. 2002, 34, 1–15.
- Vos, M.; Esposito, G.; Edirisinghe, J.N.; Vilain, S.; Haddad, D.M.; Slabbaert, J.R.; Van Meensel, S.; Schaap, O.; De Strooper, B.; Meganathan, R.; et al. Vitamin K2 is a mitochondrial electron carrier that rescues pink1 deficiency. Science 2012, 336, 1306–1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esposito, G.; Vos, M.; Vilain, S.; Swerts, J.; De Sousa Valadas, J.; Van Meensel, S.; Schaap, O.; Verstreken, P. Van Aconitase Causes Iron Toxicity in Drosophila pink1 Mutants. PLoS Genet. 2013, 9, e1003478. [Google Scholar] [CrossRef] [Green Version]
- Ivatt, R.; Sanchez-Martinez, A.; Godena, V.; Brown, S.; Ziviani, E.; Whitworth, A. Genome-wide RNAi screen identifies the Parkinson disease GWAS risk locus SREBF1 as a regulator of mitophagy. Proc. Natl. Acad. Sci. USA 2014, 111, 8494–8499. [Google Scholar] [CrossRef] [Green Version]
- Dulovic-Mahlow, M.; Trinh, J.; Kandaswamy, K.K.; Braathen, G.J.; Di Donato, N.; Rahikkala, E.; Beblo, S.; Werber, M.; Krajka, V.; Busk, Ø.L.; et al. De Novo Variants in TAOK1 Cause Neurodevelopmental Disorders. Am. J. Hum. Genet. 2019, 105, 213–220. [Google Scholar] [CrossRef] [Green Version]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [Green Version]
- Marras, C.; Lang, A.; van de Warrenburg, B.; Sue, C.; Tabrizi, S.; Bertram, L.; Mercimek-Mahmutoglu, S.; Ebrahimi-Fakhari, D.; Warner, T.; Durr, A.; et al. Nomenclature of genetic movement disorders: Recommendations of the international Parkinson and movement disorder society task force. Mov. Disord. 2016, 31, 436–457. [Google Scholar] [CrossRef] [PubMed]
- Marras, C.; Lohmann, K.; Lang, A.; Klein, C. Fixing the broken system of genetic locus symbols: Parkinson disease and dystonia as examples. Neurology 2012, 78, 1016–1024. [Google Scholar] [CrossRef] [Green Version]
- Feany, M.B.; Bender, W.W. A Drosophila model of Parkinson’s disease. Nature 2000, 404, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Stefanis, L. α-Synuclein in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009399. [Google Scholar] [CrossRef] [Green Version]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.-E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Prim. 2017, 3, 17013. [Google Scholar] [CrossRef] [PubMed]
- Klein, A.; Mazzulli, J. Is Parkinson’s disease a lysosomal disorder? Brain 2018, 141, 2255–2262. [Google Scholar] [CrossRef] [Green Version]
- Maor, G.; Rencus-Lazar, S.; Filocamo, M.; Steller, H.; Segal, D.; Horowitz, M. Unfolded protein response in Gaucher disease: From human to Drosophila. Orphanet J. Rare Dis. 2013, 8, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, M.; Trinh, K.; Thomas, R.; Yu, S.; Germanos, A.; Whitley, B.; Sardi, S.; Montine, T.; Pallanck, L. Glucocerebrosidase Deficiency in Drosophila Results in α-Synuclein-Independent Protein Aggregation and Neurodegeneration. PLoS Genet. 2016, 12, e1005944. [Google Scholar] [CrossRef] [Green Version]
- Maor, G.; Rapaport, D.; Horowitz, M. The effect of mutant GBA1 on accumulation and aggregation of α-synuclein. Hum. Mol. Genet. 2019, 28, 1768–1781. [Google Scholar] [CrossRef]
- Chai, C.; Lim, K.-L. Genetic insights into sporadic Parkinson’s disease pathogenesis. Curr. Genomics 2013, 14, 486–501. [Google Scholar] [CrossRef] [Green Version]
- Lesage, S.; Brice, A. Role of Mendelian genes in “sporadic” Parkinson’s disease. Parkinsonism Relat. Disord. 2012, 18, S66–S70. [Google Scholar] [CrossRef] [Green Version]
- Lesage, S.; Brice, A. Parkinson’s disease: From monogenic forms to genetic susceptibility factors. Hum. Mol. Genet. 2009, 18, R48–R59. [Google Scholar] [CrossRef] [PubMed]
- Baba, Y.; Markopoulou, K.; Putzke, J.; Whaley, N.; Farrer, M.; Wszolek, Z.; Uitti, R. Phenotypic commonalities in familial and sporadic Parkinson disease. Arch. Neurol. 2006, 63, 579–583. [Google Scholar] [CrossRef] [Green Version]
- Papapetropoulos, S.; Adi, N.; Ellul, J.; Argyriou, A.; Chroni, E. A prospective study of familial versus sporadic Parkinson’s disease. Neurodegener. Dis. 2007, 4, 424–427. [Google Scholar] [CrossRef]
- Schapira, A.H.; Cooper, J.M.; Dexter, D.; Jenner, P.; Clark, J.B.; Marsden, C.D. Mitochondrial complex I deficiency in Parkinson’s disease. Lancet 1989, 1, 1269. [Google Scholar] [CrossRef]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.K.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary Early-Onset Parkinson’s Disease Caused by Mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonifati, V.; Rizzu, P.; van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.; Squitieri, F.; Ibanez, P.; Joosse, M.; et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003, 299, 256–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Lee, S.B.; Lee, S.; Kim, Y.; Song, S.; Kim, S.; Bae, E.; Kim, J.; Shong, M.; Kim, J.-M.; et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 2006, 441, 1157–1161. [Google Scholar] [CrossRef] [PubMed]
- Clark, I.E.; Dodson, M.W.; Jiang, C.; Cao, J.H.; Huh, J.R.; Seol, J.H.; Yoo, S.J.; Hay, B.A.; Guo, M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 2006, 441, 1162–1166. [Google Scholar] [CrossRef] [PubMed]
- Gautier, C.A.; Kitada, T.; Shen, J. Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc. Natl. Acad. Sci. USA 2008, 105, 11364–11369. [Google Scholar] [CrossRef] [Green Version]
- Morais, V.A.; Verstreken, P.; Roethig, A.; Smet, J.; Snellinx, A.; Vanbrabant, M.; Haddad, D.; Frezza, C.; Mandemakers, W.; Vogt-Weisenhorn, D.; et al. Parkinson’s disease mutations in PINK1 result in decreased Complex I activity and deficient synaptic function. EMBO Mol. Med. 2009, 1, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Qian, L.; Xiong, H.; Liu, J.; Neckameyer, W.S.; Oldham, S.; Xia, K.; Wang, J.; Bodmer, R.; Zhang, Z. Antioxidants protect PINK1-dependent dopaminergic neurons in Drosophila. Proc. Natl. Acad. Sci. USA 2006, 103, 13520–13525. [Google Scholar] [CrossRef] [Green Version]
- Tetrud, J.W.; Langston, J.W.; Garbe, P.L.; Ruttenber, A.J. Mild parkinsonism in persons exposed to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Neurology 1989, 39, 1483–1487. [Google Scholar] [CrossRef]
- Tetrud, J.W.; Langston, J.W. MPTP-induced parkinsonism as a model for Parkinson’s disease. Acta Neurol. Scand. Suppl. 1989, 126, 35–40. [Google Scholar] [CrossRef]
- Alam, M.; Schmidt, W.J. Rotenone destroys dopaminergic neurons and induces parkinsonian symptoms in rats. Behav. Brain Res. 2002, 136, 317–324. [Google Scholar] [CrossRef]
- Inden, M.; Kitamura, Y.; Takeuchi, H.; Yanagida, T.; Takata, K.; Kobayashi, Y.; Taniguchi, T.; Yoshimoto, K.; Kaneko, M.; Okuma, Y.; et al. Neurodegeneration of mouse nigrostriatal dopaminergic system induced by repeated oral administration of rotenone is prevented by 4-phenylbutyrate, a chemical chaperone. J. Neurochem. 2007, 101, 1491–1504. [Google Scholar] [CrossRef] [PubMed]
- Tanner, C.M.; Kamel, F.; Ross, G.W.; Hoppin, J.A.; Goldman, S.M.; Korell, M.; Marras, C.; Bhudhikanok, G.S.; Kasten, M.; Chade, A.R.; et al. Rotenone, paraquat, and Parkinson’s disease. Environ. Health Perspect. 2011, 119, 866–872. [Google Scholar] [CrossRef] [Green Version]
- Coulom, H.; Birman, S. Chronic Exposure to Rotenone Models Sporadic Parkinson’s Disease in Drosophila melanogaster. J. Neurosci. 2004, 24, 10993–10998. [Google Scholar] [CrossRef] [Green Version]
- Trevor, A.J.; Castagnoli, N.; Singer, T.P. The formation of reactive intermediates in the MAO-catalyzed oxidation of the nigrostriatal toxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Toxicology 1988, 49, 513–519. [Google Scholar] [CrossRef]
- Esteve-Rudd, J.; Fernandez-Sanchez, L.; Lax, P.; De Juan, E.; Martin-Nieto, J.; Cuenca, N. Rotenone induces degeneration of photoreceptors and impairs the dopaminergic system in the rat retina. Neurobiol. Dis. 2011, 44, 102–115. [Google Scholar] [CrossRef]
- Simola, N.; Morelli, M.; Carta, A. The 6-hydroxydopamine model of Parkinson’s disease. Neurotox. Res. 2007, 11, 151–167. [Google Scholar] [CrossRef] [PubMed]
- Langston, J.W.; Ballard, P.; Tetrud, J.W.; Irwin, I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 1983, 219, 979–980. [Google Scholar] [CrossRef] [Green Version]
- Schapira, A.H.; Cooper, J.M.; Dexter, D.; Jenner, P.; Clark, J.B.; Marsden, C.D. Mitochondrial complex I deficiency in Parkinson’s disease. J. Neurochem. 1990, 1, 1269. [Google Scholar] [CrossRef] [PubMed]
- Przedborski, S.; Jackson-Lewis, V. Mechanisms of MPTP toxicity. Mov. Disord. 1998, 13, 35–38. [Google Scholar]
- Xiong, N.; Long, X.; Xiong, J.; Jia, M.; Chen, C.; Huang, J.; Ghoorah, D.; Kong, X.; Lin, Z.; Wang, T. Mitochondrial complex I inhibitor rotenone-induced toxicity and its potential mechanisms in Parkinson’s disease models. Crit. Rev. Toxicol. 2012, 42, 613–632. [Google Scholar] [CrossRef]
- Manneschi, L.; Dotti, M.T.; Battisti, C.; de Stefano, N.; Federico, A. Muscle respiratory chain enzyme activities in parkinson’s disease and in multisystem extrapyramidal disorders with parkinsonism as the main clinical feature. Arch. Gerontol. Geriatr. 1994, 19, 155–161. [Google Scholar] [CrossRef]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin-induced mitophagy in the pathogenesis of Parkinson disease. Autophagy 2009, 5, 706–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [Green Version]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, I.; Youle, R.J. Mitochondrial fission and fusion. Essays Biochem. 2010, 47, 85–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rube, D.; van der Bliek, A. Mitochondrial morphology is dynamic and varied. Mol. Cell. Biochem. 2004, 256, 331–339. [Google Scholar] [CrossRef]
- Poole, A.C.; Thomas, R.E.; Andrews, L.A.; McBride, H.M.; Whitworth, A.J.; Pallanck, L.J. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc. Natl. Acad. Sci. USA 2008, 105, 1638–1643. [Google Scholar] [CrossRef] [Green Version]
- Deng, H.; Dodson, M.W.; Huang, H.; Guo, M. The Parkinson’s disease genes pink1 and parkin promote mitochondrial fission and/or inhibit fusion in Drosophila. Proc. Natl. Acad. Sci. USA 2008, 105, 14503–14508. [Google Scholar] [CrossRef] [Green Version]
- Han, H.; Tan, J.; Wang, R.; Wan, H.; He, Y.; Yan, X.; Guo, J.; Gao, Q.; Li, J.; Shang, S.; et al. PINK1 phosphorylates Drp1S616 to regulate mitophagy-independent mitochondrial dynamics. EMBO Rep. 2020, 21, e48686. [Google Scholar] [CrossRef]
- Verstreken, P.; Ly, C.V.; Venken, K.J.T.; Koh, T.-W.; Zhou, Y.; Bellen, H.J. Synaptic Mitochondria Are Critical for Mobilization of Reserve Pool Vesicles at Drosophila Neuromuscular Junctions. Neuron 2005, 47, 365–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greene, J.C.; Whitworth, A.J.; Kuo, I.; Andrews, L.A.; Feany, M.B.; Pallanck, L.J. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc. Natl. Acad. Sci. USA 2003, 100, 4078–4083. [Google Scholar] [CrossRef] [Green Version]
- Twig, G.; Hyde, B.; Shirihai, O. Mitochondrial fusion, fission and autophagy as a quality control axis: The bioenergetic view. Biochim. Biophys. Acta 2008, 1777, 1092–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Twig, G.; Elorza, A.; Molina, A.; Mohamed, H.; Wikstrom, J.; Walzer, G.; Stiles, L.; Haigh, S.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatsuta, T.; Langer, T. Quality control of mitochondria: Protection against neurodegeneration and ageing. EMBO J. 2008, 27, 306–314. [Google Scholar] [CrossRef] [Green Version]
- Shimura, H.; Hattori, N.; Kubo, S.; Mizuno, Y.; Asakawa, S.; Minoshima, S.; Shimizu, N.; Iwai, K.; Chiba, T.; Tanaka, K.; et al. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat. Genet. 2000, 25, 302–305. [Google Scholar] [CrossRef]
- Scarffe, L.; Stevens, D.; Dawson, V.; Dawson, T. Parkin and PINK1: Much more than mitophagy. Trends Neurosci. 2014, 37, 315–324. [Google Scholar] [CrossRef] [Green Version]
- Winklhofer, K. Parkin and mitochondrial quality control: Toward assembling the puzzle. Trends Cell Biol. 2014, 24, 332–341. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y. PINK1-Parkin signaling in Parkinson’s disease: Lessons from Drosophila. Neurosci. Res. 2020, 159, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Ziviani, E.; Tao, R.N.; Whitworth, A.J. Drosophila parkin requires PINK1 for mitochondrial translocation and ubiquitinates mitofusin. Proc. Natl. Acad. Sci. USA 2010, 107, 5018–5023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziviani, E.; Whitworth, A.J. How could Parkin-mediated ubiquitination of mitofusin promote mitophagy? Autophagy 2010, 6, 660–662. [Google Scholar] [CrossRef] [Green Version]
- Burchell, V.; Nelson, D.; Sanchez-Martinez, A.; Delgado-Camprubi, M.; Ivatt, R.; Pogson, J.; Randle, S.; Wray, S.; Lewis, P.; Houlden, H.; et al. The Parkinson’s disease-linked proteins Fbxo7 and Parkin interact to mediate mitophagy. Nat. Neurosci. 2013, 16, 1257–1265. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Lee, J.; Tan, E. Pathophysiological mechanisms linking F-box only protein 7 (FBXO7) and Parkinson’s disease (PD). Mutat. Res. 2018, 778, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Yun, J.; Liu, J.; Malide, D.; Liu, C.; Rovira, I.I.; Holmström, K.M.; Fergusson, M.M.; Yoo, Y.H.; Combs, C.A.; et al. Measuring In Vivo Mitophagy. Mol. Cell 2015, 60, 685–696. [Google Scholar] [CrossRef] [Green Version]
- Cornelissen, T.; Vilain, S.; Vints, K.; Gounko, N.; Verstreken, P.; Vandenberghe, W. Deficiency of parkin and PINK1 impairs age-dependent mitophagy in Drosophila. Elife 2018, 7, e35878. [Google Scholar] [CrossRef]
- Lee, J.J.; Sanchez-Martinez, A.; Zarate, A.M.; Benincá, C.; Mayor, U.; Clague, M.J.; Whitworth, A.J. Basal mitophagy is widespread in Drosophila but minimally affected by loss of Pink1 or parkin. J. Cell Biol. 2018, 217, 1613–1622. [Google Scholar] [CrossRef] [Green Version]
- Vilain, S.; Esposito, G.; Haddad, D.; Schaap, O.; Dobreva, M.P.; Vos, M.; Van Meensel, S.; Morais, V.A.; De Strooper, B.; Verstreken, P. The yeast complex I equivalent NADH dehydrogenase rescues pink1 mutants. PLoS Genet. 2012, 8, e1002456. [Google Scholar] [CrossRef]
- Liu, W.; Acín-Peréz, R.; Geghman, K.D.; Manfredi, G.; Lu, B.; Li, C. Pink1 regulates the oxidative phosphorylation machinery via mitochondrial fission. Proc. Natl. Acad. Sci. USA 2011, 108, 12920–12924. [Google Scholar] [CrossRef] [Green Version]
- Morais, V.A.; Haddad, D.; Craessaerts, K.; De Bock, P.-J.; Swerts, J.; Vilain, S.; Aerts, L.; Overbergh, L.; Grünewald, A.; Seibler, P.; et al. PINK1 Loss-of-Function Mutations Affect Mitochondrial Complex I Activity via NdufA10 Ubiquinone Uncoupling. Science 2014, 344, 203–207. [Google Scholar] [CrossRef]
- Vos, M.; Lovisa, B.; Geens, A.; Morais, V.A.; Wagnières, G.; Van Den Bergh, H.; Ginggen, A.; De Strooper, B.; Tardy, Y.; Verstreken, P. Near-infrared 808 nm light boosts complex IV-dependent respiration and rescues a Parkinson-related pink1 model. PLoS ONE 2013, 8, e78562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oueslati, A.; Lovisa, B.; Perrin, J.; Wagnières, G.; van den Bergh, H.; Tardy, Y.; Lashuel, H. Photobiomodulation Suppresses Alpha-Synuclein-Induced Toxicity in an AAV-Based Rat Genetic Model of Parkinson’s Disease. PLoS ONE 2015, 10, e0140880. [Google Scholar]
- Prasuhn, J.; Brüggemann, N.; Hessler, N.; Berg, D.; Gasser, T.; Brockmann, K.; Olbrich, D.; Ziegler, A.; König, I.; Klein, C.; et al. An omics-based strategy using coenzyme Q10 in patients with Parkinson’s disease: Concept evaluation in a double-blind randomized placebo-controlled parallel group trial. Neurol. Res. Pract. 2019, 1, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasuhn, J.; Kasten, M.; Vos, M.; König, I.R.; Schmid, S.M.; Wilms, B.; Klein, C.; Brüggemann, N. The Use of Vitamin K2 in Patients With Parkinson’s Disease and Mitochondrial Dysfunction (PD-K2): A Theranostic Pilot Study in a Placebo-Controlled Parallel Group Design. Front. Neurol. 2021, 11, 592104. [Google Scholar] [CrossRef]
- Sofic, E.; Paulus, W.; Jellinger, K.; Riederer, P.; Youdim, M.B. Selective increase of iron in substantia nigra zona compacta of parkinsonian brains. J. Neurochem. 1991, 56, 978–982. [Google Scholar] [CrossRef]
- Beinert, H.; Kennedy, M.C. Aconitase, a two-faced protein: Enzyme and iron regulatory factor. FASEB J. 1993, 7, 1442–1449. [Google Scholar] [CrossRef] [PubMed]
- Wan, Z.; Xu, J.; Huang, Y.; Zhai, Y.; Ma, Z.; Zhou, B.; Cao, Z. Elevating bioavailable iron levels in mitochondria suppresses the defective phenotypes caused by PINK1 loss-of-function in Drosophila melanogaster. Biochem. Biophys. Res. Commun. 2020, 532, 285–291. [Google Scholar] [CrossRef]
- Pickart, C.; Eddins, M. Ubiquitin: Structures, functions, mechanisms. Biochim. Biophys. Acta 2004, 1695, 55–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haddad, D.M.; Vilain, S.; Vos, M.; Esposito, G.; Matta, S.; Kalscheuer, V.M.; Craessaerts, K.; Leysen, M.; Nascimento, R.; Vianna-Morgante, A.M.; et al. Mutations in the intellectual disability gene Ube2a cause neuronal dysfunction and impair parkin-dependent mitophagy. Mol. Cell 2013, 50, 831–843. [Google Scholar] [CrossRef] [Green Version]
- Reyes-Turcu, F.E.; Ventii, K.H.; Wilkinson, K.D. Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu. Rev. Biochem. 2009, 78, 363–397. [Google Scholar] [CrossRef] [Green Version]
- Cornelissen, T.; Haddad, D.; Wauters, F.; Van Humbeeck, C.; Mandemakers, W.; Koentjoro, B.; Sue, C.; Gevaert, K.; De Strooper, B.; Verstreken, P.; et al. The deubiquitinase USP15 antagonizes Parkin-mediated mitochondrial ubiquitination and mitophagy. Hum. Mol. Genet. 2014, 23, 5227–5242. [Google Scholar] [CrossRef] [Green Version]
- Zhu, M.; Li, X.; Tian, X.; Wu, C. Mask loss-of-function rescues mitochondrial impairment and muscle degeneration of Drosophila pink1 and parkin mutants. Hum. Mol. Genet. 2015, 24, 3272–3285. [Google Scholar] [CrossRef] [Green Version]
- Geisler, S.; Holmstrom, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell. Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Ham, S.; Lee, D.; Yoo, H.; Jun, K.; Shin, H.; Chung, J. Decision between mitophagy and apoptosis by Parkin via VDAC1 ubiquitination. Proc. Natl. Acad. Sci. USA 2020, 117, 4281–4291. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; De Pinto, V.; Zweckstetter, M.; Raviv, Z.; Keinan, N.; Arbel, N. VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol. Asp. Med. 2010, 31, 227–285. [Google Scholar] [CrossRef]
- Shin, J.; Ko, H.; Kang, H.; Lee, Y.; Lee, Y.; Pletinkova, O.; Troconso, J.; Dawson, V.; Dawson, T. PARIS (ZNF746) repression of PGC-1α contributes to neurodegeneration in Parkinson’s disease. Cell 2011, 144, 689–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pirooznia, S.; Yuan, C.; Khan, M.; Karuppagounder, S.; Wang, L.; Xiong, Y.; Kang, S.; Lee, Y.; Dawson, V.; Dawson, T. PARIS induced defects in mitochondrial biogenesis drive dopamine neuron loss under conditions of parkin or PINK1 deficiency. Mol. Neurodegener. 2020, 15, 17. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Kim, S.; Cha, G.; Lee, S.; Kim, S.; Chung, J. Drosophila DJ-1 mutants show oxidative stress-sensitive locomotive dysfunction. Gene 2005, 361, 133–139. [Google Scholar] [CrossRef]
- Lavara-Culebras, E.; Paricio, N. Drosophila DJ-1 mutants are sensitive to oxidative stress and show reduced lifespan and motor deficits. Gene 2007, 400, 158–165. [Google Scholar] [CrossRef]
- Meulener, M.C.; Xu, K.; Thomson, L.; Ischiropoulos, H.; Bonini, N.M. Mutational analysis of DJ-1 in Drosophila implicates functional inactivation by oxidative damage and aging. Proc. Natl. Acad. Sci. USA 2006, 103, 12517–12522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shendelman, S.; Jonason, A.; Martinat, C.; Leete, T.; Abeliovich, A. DJ-1 is a redox-dependent molecular chaperone that inhibits alpha-synuclein aggregate formation. PLoS Biol. 2004, 2, e362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martín-Nieto, J.; Uribe, M.; Esteve-Rudd, J.; Herrero, M.; Campello, L. A role for DJ-1 against oxidative stress in the mammalian retina. Neurosci. Lett. 2019, 708, 134361. [Google Scholar] [CrossRef]
- Hao, L.Y.; Giasson, B.I.; Bonini, N.M. DJ-1 is critical for mitochondrial function and rescues PINK1 loss of function. Proc. Natl. Acad. Sci. USA 2010, 107, 9747–9752. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Kim, M.; Yoon, W.; Kim, E.; Kim, H.; Lee, Y.; Min, B.; Kang, K.; Son, J.; Park, H.; et al. Isocitrate protects DJ-1 null dopaminergic cells from oxidative stress through NADP+-dependent isocitrate dehydrogenase (IDH). PLoS Genet. 2017, 13, e1006975. [Google Scholar] [CrossRef]
- Smolders, S.; Van Broeckhoven, C. Genetic perspective on the synergistic connection between vesicular transport, lysosomal and mitochondrial pathways associated with Parkinson’s disease pathogenesis. Acta Neuropathol. Commun. 2020, 8, 63. [Google Scholar] [CrossRef] [PubMed]
- Malik, B.; Maddison, D.; Smith, G.; Peters, O. Autophagic and endo-lysosomal dysfunction in neurodegenerative disease. Mol. Brain 2019, 12, 100. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.; Wang, L.; Marcogliese, P.; Bellen, H. Sphingolipids in the Pathogenesis of Parkinson’s Disease and Parkinsonism. Trends Endocrinol. Metab. 2019, 30, 106–117. [Google Scholar] [CrossRef]
- Kuijpers, M.; Azarnia Tehran, D.; Haucke, V.; Soykan, T. The axonal endolysosomal and autophagic systems. J. Neurochem. 2020. [Google Scholar] [CrossRef]
- Andres-Alonso, M.; Kreutz, M.; Karpova, A. Autophagy and the endolysosomal system in presynaptic function. Cell Mol. Life Sci. 2020. [Google Scholar] [CrossRef]
- Giovedì, S.; Ravanelli, M.; Parisi, B.; Bettegazzi, B.; Guarnieri, F. Dysfunctional Autophagy and Endolysosomal System in Neurodegenerative Diseases: Relevance and Therapeutic Options. Front. Cell Neurosci. 2020, 14, 602116. [Google Scholar] [CrossRef] [PubMed]
- Outeiro, T.; Lindquist, S. Yeast cells provide insight into alpha-synuclein biology and pathobiology. Science 2003, 302, 1772–1775. [Google Scholar] [CrossRef] [Green Version]
- Devi, L.; Raghavendran, V.; Prabhu, B.M.; Avadhani, N.G.; Anandatheerthavarada, H.K. Mitochondrial Import and Accumulation of α-Synuclein Impair Complex I in Human Dopaminergic Neuronal Cultures and Parkinson Disease Brain. J. Biol. Chem. 2008, 283, 9089–9100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, Y.; Yu, J. Modeling Parkinson’s Disease in Drosophila: What Have We Learned for Dominant Traits? Front. Neurol. 2018, 9, 228. [Google Scholar] [CrossRef] [Green Version]
- Mohite, G.; Dwivedi, S.; Das, S.; Kumar, R.; Paluri, S.; Mehra, S.; Ruhela, N.; Arunima, S.; Jha, N.; Maji, S. Parkinson’s Disease Associated α-Synuclein Familial Mutants Promote Dopaminergic Neuronal Death in Drosophila melanogaster. ACS Chem. Neurosci. 2018, 9, 2628–2638. [Google Scholar] [CrossRef]
- Takahashi, M.; Kanuka, H.; Fujiwara, H.; Koyama, A.; Hasegawa, M.; Miura, M.; Iwatsubo, T. Phosphorylation of alpha-synuclein characteristic of synucleinopathy lesions is recapitulated in alpha-synuclein transgenic Drosophila. Neurosci. Lett. 2003, 336, 155–158. [Google Scholar] [CrossRef]
- Fujiwara, H.; Hasegawa, M.; Dohmae, N.; Kawashima, A.; Masliah, E.; Goldberg, M.; Shen, J.; Takio, K.; Iwatsubo, T. alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 2002, 4, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Kawashima, A.; Ruberu, N.; Fujiwara, H.; Koyama, S.; Sawabe, M.; Arai, T.; Nagura, H.; Yamanouchi, H.; Hasegawa, M.; et al. Accumulation of phosphorylated alpha-synuclein in aging human brain. J. Neuropathol. Exp. Neurol. 2003, 62, 644–654. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Feany, M.B. Alpha-synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson disease. Nat. Neurosci. 2005, 8, 657–663. [Google Scholar] [CrossRef]
- Cooper, A.A.; Gitler, A.D.; Cashikar, A.; Haynes, C.M.; Hill, K.J.; Bhullar, B.; Liu, K.; Xu, K.; Strathearn, K.E.; Liu, F.; et al. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science 2006, 313, 324–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stenmark, H.; Olkkonen, V.M. The Rab GTPase family. Genome Biol. 2001, 2, reviews3007.1. [Google Scholar] [CrossRef] [Green Version]
- Breda, C.; Nugent, M.L.; Estranero, J.G.; Kyriacou, C.P.; Outeiro, T.F.; Steinert, J.R.; Giorgini, F. Rab11 modulates α-synuclein-mediated defects in synaptic transmission and behaviour. Hum. Mol. Genet. 2015, 24, 1077–1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinter, E.; Saridaki, T.; Nippold, M.; Plum, S.; Diederichs, L.; Komnig, D.; Fensky, L.; May, C.; Marcus, K.; Voigt, A.; et al. Rab7 induces clearance of α-synuclein aggregates. J. Neurochem. 2016, 138, 758–774. [Google Scholar] [CrossRef]
- Yin, G.; Lopes da Fonseca, T.; Eisbach, S.; Anduaga, A.; Breda, C.; Orcellet, M.; Szegő, É.; Guerreiro, P.; Lázaro, D.; Braus, G.; et al. α-Synuclein interacts with the switch region of Rab8a in a Ser129 phosphorylation-dependent manner. Neurobiol. Dis. 2014, 70, 149–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodson, M.W.; Zhang, T.; Jiang, C.; Chen, S.; Guo, M. Roles of the Drosophila LRRK2 homolog in Rab7-dependent lysosomal positioning. Hum. Mol. Genet. 2012, 21, 1350–1363. [Google Scholar] [CrossRef]
- Matta, S.; Van Kolen, K.; da Cunha, R.; van den Bogaart, G.; Mandemakers, W.; Miskiewicz, K.; De Bock, P.-J.; Morais, V.A.; Vilain, S.; Haddad, D.; et al. LRRK2 Controls an EndoA Phosphorylation Cycle in Synaptic Endocytosis. Neuron 2012, 75, 1008–1021. [Google Scholar] [CrossRef] [Green Version]
- Soukup, S.; Kuenen, S.; Vanhauwaert, R.; Manetsberger, J.; Hernández-Díaz, S.; Swerts, J.; Schoovaerts, N.; Vilain, S.; Gounko, N.; Vints, K.; et al. A LRRK2-Dependent EndophilinA Phosphoswitch Is Critical for Macroautophagy at Presynaptic Terminals. Neuron 2016, 92, 829–844. [Google Scholar] [CrossRef] [Green Version]
- Huttner, W.; Schmidt, A. Lipids, lipid modification and lipid-protein interaction in membrane budding and fission—Insights from the roles of endophilin A1 and synaptophysin in synaptic vesicle endocytosis. Curr. Opin. Neurol. 2000, 10, 543–551. [Google Scholar] [CrossRef]
- Vanhauwaert, R.; Kuenen, S.; Masius, R.; Bademosi, A.; Manetsberger, J.; Schoovaerts, N.; Bounti, L.; Gontcharenko, S.; Swerts, J.; Vilain, S.; et al. The SAC1 domain in synaptojanin is required for~autophagosome maturation at presynaptic~terminals. EMBO J. 2017, 36, 1392–1411. [Google Scholar] [CrossRef]
- Song, W.; Zinsmaier, K. Endophilin and synaptojanin hook up to promote synaptic vesicle endocytosis. Neuron 2003, 40, 665–667. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.B.; Kim, W.; Lee, S.; Chung, J. Loss of LRRK2/PARK8 induces degeneration of dopaminergic neurons in Drosophila. Biochem. Biophys. Res. Commun. 2007, 358, 534–539. [Google Scholar] [CrossRef]
- Dodson, M.W.; Leung, L.K.; Lone, M.; Lizzio, M.A.; Guo, M. Novel ethyl methanesulfonate (EMS)-induced null alleles of the Drosophila homolog of LRRK2 reveal a crucial role in endolysosomal functions and autophagy in vivo. Dis. Model. Mech. 2014, 7, 1351–1363. [Google Scholar] [CrossRef]
- Wang, D.; Tang, B.; Zhao, G.; Pan, Q.; Xia, K.; Bodmer, R.; Zhang, Z. Dispensable role of Drosophila ortholog of LRRK2 kinase activity in survival of dopaminergic neurons. Mol. Neurodegener. 2008, 3, 3. [Google Scholar] [CrossRef] [Green Version]
- Inoshita, T.; Arano, T.; Hosaka, Y.; Meng, H.; Umezaki, Y.; Kosugi, S.; Morimoto, T.; Koike, M.; Chang, H.; Imai, Y.; et al. Vps35 in cooperation with LRRK2 regulates synaptic vesicle endocytosis through the endosomal pathway in Drosophila. Hum. Mol. Genet. 2017, 26, 2933–2948. [Google Scholar] [CrossRef] [Green Version]
- Pfeffer, S. Membrane transport: Retromer to the rescue. Curr. Biol. 2001, 11, R109–R111. [Google Scholar] [CrossRef] [Green Version]
- Seaman, M. Recycle your receptors with retromer. Trends Cell. Biol. 2005, 15, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Burd, C.; Cullen, P. Retromer: A master conductor of endosome sorting. Cold Spring Harb. Perpect. Biol. 2014, 6, a016774. [Google Scholar] [CrossRef]
- Gallon, M.; Cullen, P. Retromer and sorting nexins in endosomal sorting. Biochem. Soc. Trans. 2015, 43, 33–47. [Google Scholar] [CrossRef]
- Williams, E.T.; Chen, X.; Moore, D.J. VPS35, the Retromer Complex and Parkinson’s Disease. J. Parkinsons Dis. 2017, 7, 219–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojas, R.; van Vlijmen, T.; Mardones, G.A.; Prabhu, Y.; Rojas, A.L.; Mohammed, S.; Heck, A.J.R.; Raposo, G.; van der Sluijs, P.; Bonifacino, J.S. Regulation of retromer recruitment to endosomes by sequential action of Rab5 and Rab7. J. Cell Biol. 2008, 183, 513–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Toh, J.; Ho, P.; Tio, M.; Zhao, Y.; Tan, E.-K. In vivo evidence of pathogenicity of VPS35 mutations in the Drosophila. Mol. Brain 2014, 7, 73. [Google Scholar] [CrossRef]
- Malik, B.R.; Godena, V.K.; Whitworth, A.J. VPS35 pathogenic mutations confer no dominant toxicity but partial loss of function in Drosophila and genetically interact with parkin. Hum. Mol. Genet. 2015, 24, 6106–6117. [Google Scholar] [CrossRef] [Green Version]
- Ishizu, N.; Yui, D.; Hebisawa, A.; Aizawa, H.; Cui, W.; Fujita, Y.; Hashimoto, K.; Ajioka, I.; Mizusawa, H.; Yokota, T.; et al. Impaired striatal dopamine release in homozygous Vps35 D620N knock-in mice. Hum. Mol. Genet. 2016, 25, 4507–4517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munsie, L.N.; Milnerwood, A.J.; Seibler, P.; Beccano-Kelly, D.A.; Tatarnikov, I.; Khinda, J.; Volta, M.; Kadgien, C.; Cao, L.P.; Tapia, L.; et al. Retromer-dependent neurotransmitter receptor trafficking to synapses is altered by the Parkinson’s disease VPS35 mutation p.D620N. Hum. Mol. Genet. 2015, 24, 1691–1703. [Google Scholar] [CrossRef] [Green Version]
- Linhart, R.; Wong, S.A.; Cao, J.; Tran, M.; Huynh, A.; Ardrey, C.; Park, J.M.; Hsu, C.; Taha, S.; Peterson, R.; et al. Vacuolar protein sorting 35 (Vps35) rescues locomotor deficits and shortened lifespan in Drosophila expressing a Parkinson’s disease mutant of Leucine-Rich Repeat Kinase 2 (LRRK2). Mol. Neurodegener. 2014, 9, 23. [Google Scholar] [CrossRef] [Green Version]
- MacLeod, D.A.; Rhinn, H.; Kuwahara, T.; Zolin, A.; Di Paolo, G.; McCabe, B.D.; Marder, K.S.; Honig, L.S.; Clark, L.N.; Small, S.A.; et al. RAB7L1 interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson’s disease risk. Neuron 2013, 77, 425–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miura, E.; Hasegawa, T.; Konno, M.; Suzuki, M.; Sugeno, N.; Fujikake, N.; Geisler, S.; Tabuchi, M.; Oshima, R.; Kikuchi, A.; et al. VPS35 dysfunction impairs lysosomal degradation of α-synuclein and exacerbates neurotoxicity in a Drosophila model of Parkinson’s disease. Neurobiol. Dis. 2014, 71, 1–13. [Google Scholar] [CrossRef]
- Lin, G.; Lee, P.-T.; Chen, K.; Mao, D.; Tan, K.L.; Zuo, Z.; Lin, W.-W.; Wang, L.; Bellen, H.J. Phospholipase PLA2G6, a Parkinsonism-Associated Gene, Affects Vps26 and Vps35, Retromer Function, and Ceramide Levels, Similar to α-Synuclein Gain. Cell Metab. 2018, 28, 605–618.e6. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.-P.; Tang, B.-S.; Guo, J.-F. PLA2G6-Associated Neurodegeneration (PLAN): Review of Clinical Phenotypes and Genotypes. Front. Neurol. 2018, 9, 1100. [Google Scholar] [CrossRef] [Green Version]
- Kinghorn, K.J.; Castillo-Quan, J.I.; Bartolome, F.; Angelova, P.R.; Li, L.; Pope, S.; Cochemé, H.M.; Khan, S.; Asghari, S.; Bhatia, K.P.; et al. Loss of PLA2G6 leads to elevated mitochondrial lipid peroxidation and mitochondrial dysfunction. Brain 2015, 138, 1801–1816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, A.; Hatano, T.; Inoshita, T.; Shiba-Fukushima, K.; Koinuma, T.; Meng, H.; Kubo, S.-I.; Spratt, S.; Cui, C.; Yamashita, C.; et al. Parkinson’s disease-associated iPLA2-VIA/PLA2G6 regulates neuronal functions and α-synuclein stability through membrane remodeling. Proc. Natl. Acad. Sci. USA 2019, 116, 20689–20699. [Google Scholar] [CrossRef] [Green Version]
- Iliadi, K.G.; Gluscencova, O.B.; Iliadi, N.; Boulianne, G.L. Mutations in the Drosophila homolog of human PLA2G6 give rise to age-dependent loss of psychomotor activity and neurodegeneration. Sci. Rep. 2018, 8, 2939. [Google Scholar] [CrossRef] [Green Version]
- Panevska, A.; Skočaj, M.; Križaj, I.; Maček, P.; Sepčić, K. Ceramide phosphoethanolamine, an enigmatic cellular membrane sphingolipid. Biochim. Biophys. Acta Biomembr. 2019, 1861, 1284–1292. [Google Scholar] [CrossRef]
- Taniguchi, M.; Okazaki, T. The role of sphingomyelin and sphingomyelin synthases in cell death, proliferation and migration—from cell and animal models to human disorders. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2014, 1841, 692–703. [Google Scholar] [CrossRef]
- Choudhury, A.; Dominguez, M.; Puri, V.; Sharma, D.K.; Narita, K.; Wheatley, C.L.; Marks, D.L.; Pagano, R.E. Rab proteins mediate Golgi transport of caveola-internalized glycosphingolipids and correct lipid trafficking in Niemann-Pick C cells. J. Clin. Investig. 2002, 109, 1541–1550. [Google Scholar] [CrossRef]
- Fröhlich, F.; Petit, C.; Kory, N.; Christiano, R.; Hannibal-Bach, H.-K.; Graham, M.; Liu, X.; Ejsing, C.S.; Farese, R.V.; Walther, T.C. The GARP complex is required for cellular sphingolipid homeostasis. Elife 2015, 4, e08712. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Ogretmen, B. Autophagy paradox and ceramide. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2014, 1841, 783–792. [Google Scholar] [CrossRef] [Green Version]
- Fujioka, Y.; Noda, N.N. Biomolecular condensates in autophagy regulation. Curr. Opin. Cell Biol. 2021, 69, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Dehay, B.; Ramirez, A.; Martinez-Vicente, M.; Perier, C.; Canron, M.-H.; Doudnikoff, E.; Vital, A.; Vila, M.; Klein, C.; Bezard, E. Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration. Proc. Natl. Acad. Sci. USA 2012, 109, 9611–9616. [Google Scholar] [CrossRef] [Green Version]
- Dehay, B.; Martinez-Vicente, M.; Ramirez, A.; Perier, C.; Klein, C.; Vila, M.; Bezard, E. Lysosomal dysfunction in Parkinson disease: ATP13A2 gets into the groove. Autophagy 2012, 8, 1389–1391. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.; Hadeed, A.; Al-Din, A.; Wreikat, A.; Lees, A. Kufor Rakeb disease: Autosomal recessive, levodopa-responsive parkinsonism with pyramidal degeneration, supranuclear gaze palsy, and dementia. Mov. Disord. 2015, 20, 1264–1267. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Tan, J.; Chen, T.; Han, H.; Tian, R.; Tan, Y.; Wu, Y.; Cui, J.; Chen, F.; Li, J.; et al. ATP13A2 facilitates HDAC6 recruitment to lysosome to promote autophagosome-lysosome fusion. J. Cell Biol. 2019, 218, 267–284. [Google Scholar] [CrossRef]
- Mizushima, N.; Ohsumi, Y.; Yoshimori, T. Autophagosome formation in mammalian cells. Cell Struct. Funct. 2002, 27, 421–429. [Google Scholar] [CrossRef] [Green Version]
- Du, G.; Liu, X.; Chen, X.; Song, M.; Yan, Y.; Jiao, R.; Wang, C.-C. Drosophila histone deacetylase 6 protects dopaminergic neurons against {alpha}-synuclein toxicity by promoting inclusion formation. Mol. Biol. Cell 2010, 21, 2128–2137. [Google Scholar] [CrossRef] [Green Version]
- Mielke, M.M.; Maetzler, W.; Haughey, N.J.; Bandaru, V.V.R.; Savica, R.; Deuschle, C.; Gasser, T.; Hauser, A.-K.; Gräber-Sultan, S.; Schleicher, E.; et al. Plasma Ceramide and Glucosylceramide Metabolism Is Altered in Sporadic Parkinson’s Disease and Associated with Cognitive Impairment: A Pilot Study. PLoS ONE 2013, 8, e73094. [Google Scholar]
- Goya, M.E.; Xue, F.; Sampedro-Torres-Quevedo, C.; Arnaouteli, S.; Riquelme-Dominguez, L.; Romanowski, A.; Brydon, J.; Ball, K.L.; Stanley-Wall, N.R.; Doitsidou, M. Probiotic Bacillus subtilis Protects against α-Synuclein Aggregation in C. elegans. Cell Rep. 2020, 30, 367–380. [Google Scholar] [CrossRef]
- Vos, M.; Geens, A.; Böhm, C.; Deaulmerie, L.; Swerts, J.; Rossi, M.; Craessaerts, K.; Leites, E.P.; Seibler, P.; Rakovic, A.; et al. Cardiolipin promotes electron transport between ubiquinone and complex I to rescue PINK1 deficiency. J. Cell Biol. 2017, 216, 695–708. [Google Scholar] [CrossRef]
- Do, C.B.; Tung, J.Y.; Dorfman, E.; Kiefer, A.K.; Drabant, E.M.; Francke, U.; Mountain, J.L.; Goldman, S.M.; Tanner, C.M.; Langston, J.W.; et al. Web-based genome-wide association study identifies two novel loci and a substantial genetic component for Parkinson’s disease. PLoS Genet. 2011, 7, e1002141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlame, M.; Brody, S.; Hostetler, K. Mitochondrial cardiolipin in diverse eukaryotes. Comparison of biosynthetic reactions and molecular acyl species. Eur. J. Biochem. 1993, 212, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Acehan, D.; Malhotra, A.; Xu, Y.; Ren, M.; Stokes, D.L.; Schlame, M. Cardiolipin Affects the Supramolecular Organization of ATP Synthase in Mitochondria. Biophys. J. 2011, 100, 2184–2192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.-X.; Tsoi, B.; Li, Y.-F.; Kurihara, H.; He, R.-R. Cardiolipin and its different properties in mitophagy and apoptosis. J. Histochem. Cytochem. 2015, 63, 301–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vielhaber, G.; Pfeiffer, S.; Brade, L.; Lindner, B.; Goldmann, T.; Vollmer, E.; Hintze, U.; Wittern, K.; Wepf, R. Localization of ceramide and glucosylceramide in human epidermis by immunogold electron microscopy. J. Investig. Dermatol. 2001, 117, 1126–1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gegg, M.; Schapira, A. The role of glucocerebrosidase in Parkinson disease pathogenesis. FEBS J. 2018, 285, 3591–3603. [Google Scholar] [CrossRef] [Green Version]
- Dandana, A.; Ben Khelifa, S.; Chahed, H.; Miled, A.; Ferchichi, S. Gaucher Disease: Clinical, Biological and Therapeutic Aspects. Pathobiology 2016, 83, 13–23. [Google Scholar] [CrossRef]
- Behl, T.; Kaur, G.; Fratila, O.; Buhas, C.; Judea-Pusta, C.T.; Negrut, N.; Bustea, C.; Bungau, S. Cross-talks among GBA mutations, glucocerebrosidase, and α-synuclein in GBA-associated Parkinson’s disease and their targeted therapeutic approaches: A comprehensive review. Transl. Neurodegener. 2021, 10, 4. [Google Scholar] [CrossRef]
- Bandres-Ciga, S.; Diez-Fairen, M.; Kim, J.; Singleton, A. Genetics of Parkinson’s disease: An introspection of its journey towards precision medicine. Neurobiol. Dis. 2020, 137, 104782. [Google Scholar] [CrossRef]
- Robinson, S.W.; Herzyk, P.; Dow, J.A.T.; Leader, D.P. FlyAtlas: Database of gene expression in the tissues of Drosophila melanogaster. Nucleic Acids Res. 2013, 41, D744–D750. [Google Scholar] [CrossRef] [Green Version]
- Cabasso, O.; Paul, S.; Dorot, O.; Maor, G.; Krivoruk, O.; Pasmanik-Chor, M.; Mirzaian, M.; Ferraz, M.; Aerts, J.; Horowitz, M. Drosophila melanogaster Mutated in its GBA1b Ortholog Recapitulates Neuronopathic Gaucher Disease. J. Clin. Med. 2019, 8, 1420. [Google Scholar] [CrossRef] [Green Version]
- Brockmann, K. GBA-Associated Synucleinopathies: Prime Candidates for Alpha-Synuclein Targeting Compounds. Front. Cell Dev. Biol. 2020, 8, 562522. [Google Scholar] [CrossRef] [PubMed]
- Abul Khair, S.B.; Dhanushkodi, N.R.; Ardah, M.T.; Chen, W.; Yang, Y.; Haque, M.E. Silencing of Glucocerebrosidase Gene in Drosophila Enhances the Aggregation of Parkinson’s Disease Associated α-Synuclein Mutant A53T and Affects Locomotor Activity. Front. Neurosci. 2018, 12, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maor, G.; Cabasso, O.; Krivoruk, O.; Rodriguez, J.; Steller, H.; Segal, D.; Horowitz, M. The contribution of mutant GBA to the development of Parkinson disease in Drosophila. Hum. Mol. Genet. 2016, 25, 2712–2727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Julienne, H.; Buhl, E.; Leslie, D.S.; Hodge, J.J.L. Drosophila PINK1 and parkin loss-of-function mutants display a range of non-motor Parkinson’s disease phenotypes. Neurobiol. Dis. 2017, 104, 15–23. [Google Scholar] [CrossRef]
- Zarranz, J.J.; Alegre, J.; Gómez-Esteban, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atarés, B.; et al. The New Mutation, E46K, of Alpha-Synuclein Causes Parkinson and Lewy Body Dementia. Ann. Neurol. 2004, 55, 164–173. [Google Scholar] [CrossRef]
- Ito, K.; Kawasaki, H.; Suzuki, T.; Takahara, T.; Ishida, N. Effects of Kamikihito and Unkei-to on Sleep Behavior of Wild Type and Parkinson Model in Drosophila. Front. Psychiatry 2017, 8, 132. [Google Scholar] [CrossRef] [Green Version]
- Celardo, I.; Costa, A.C.; Lehmann, S.; Jones, C.; Wood, N.; Mencacci, N.E.; Mallucci, G.R.; Loh, S.H.Y.; Martins, L.M. Mitofusin-mediated ER stress triggers neurodegeneration in pink1/parkin models of Parkinson’s disease. Cell Death Dis. 2016, 7, e2271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valadas, J.S.; Esposito, G.; Vandekerkhove, D.; Miskiewicz, K.; Deaulmerie, L.; Raitano, S.; Seibler, P.; Klein, C.; Verstreken, P. ER Lipid Defects in Neuropeptidergic Neurons Impair Sleep Patterns in Parkinson’s Disease. Neuron 2018, 98, 1155–1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sliter, D.A.; Martinez, J.; Hao, L.; Chen, X.; Sun, N.; Fischer, T.D.; Burman, J.L.; Li, Y.; Zhang, Z.; Narendra, D.P.; et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature 2018, 561, 258–262. [Google Scholar] [CrossRef]
- Martin, M.; Hiroyasu, A.; Guzman, R.M.; Roberts, S.A.; Goodman, A.G. Analysis of Drosophila STING Reveals an Evolutionarily Conserved Antimicrobial Function. Cell Rep. 2018, 23, 3537–3550.e6. [Google Scholar] [CrossRef]
- Lee, J.J.; Andreazza, S.; Whitworth, A.J. The STING pathway does not contribute to behavioural or mitochondrial phenotypes in Drosophila Pink1/parkin or mtDNA mutator models. Sci. Rep. 2020, 10, 2693. [Google Scholar] [CrossRef] [PubMed]
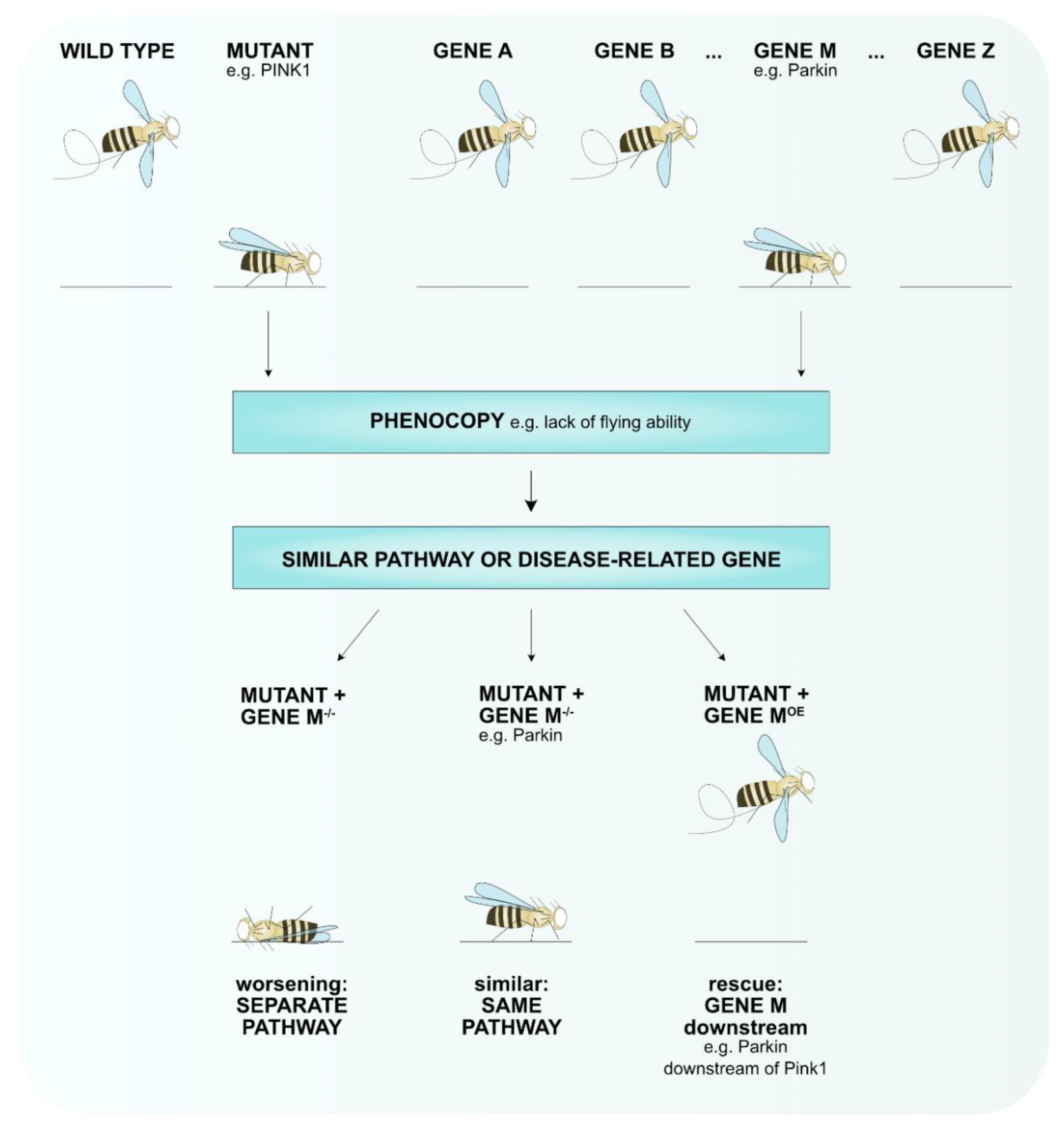
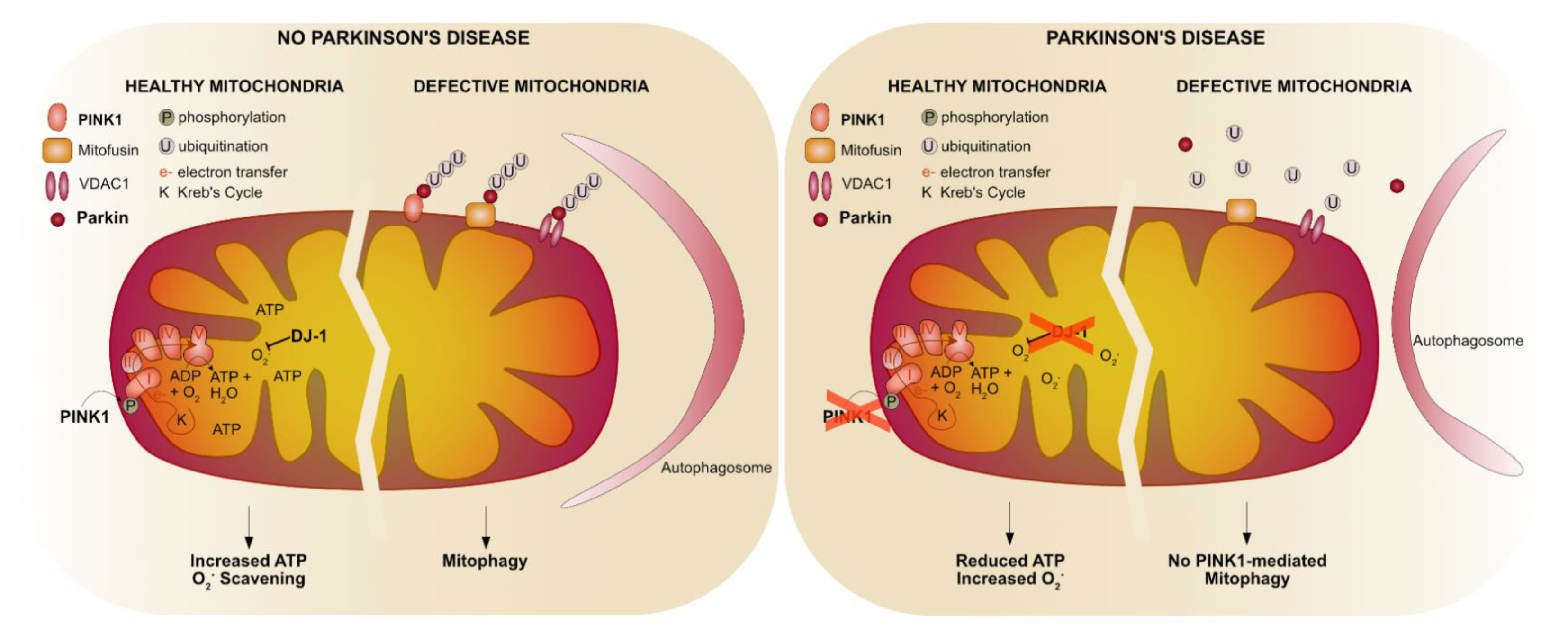
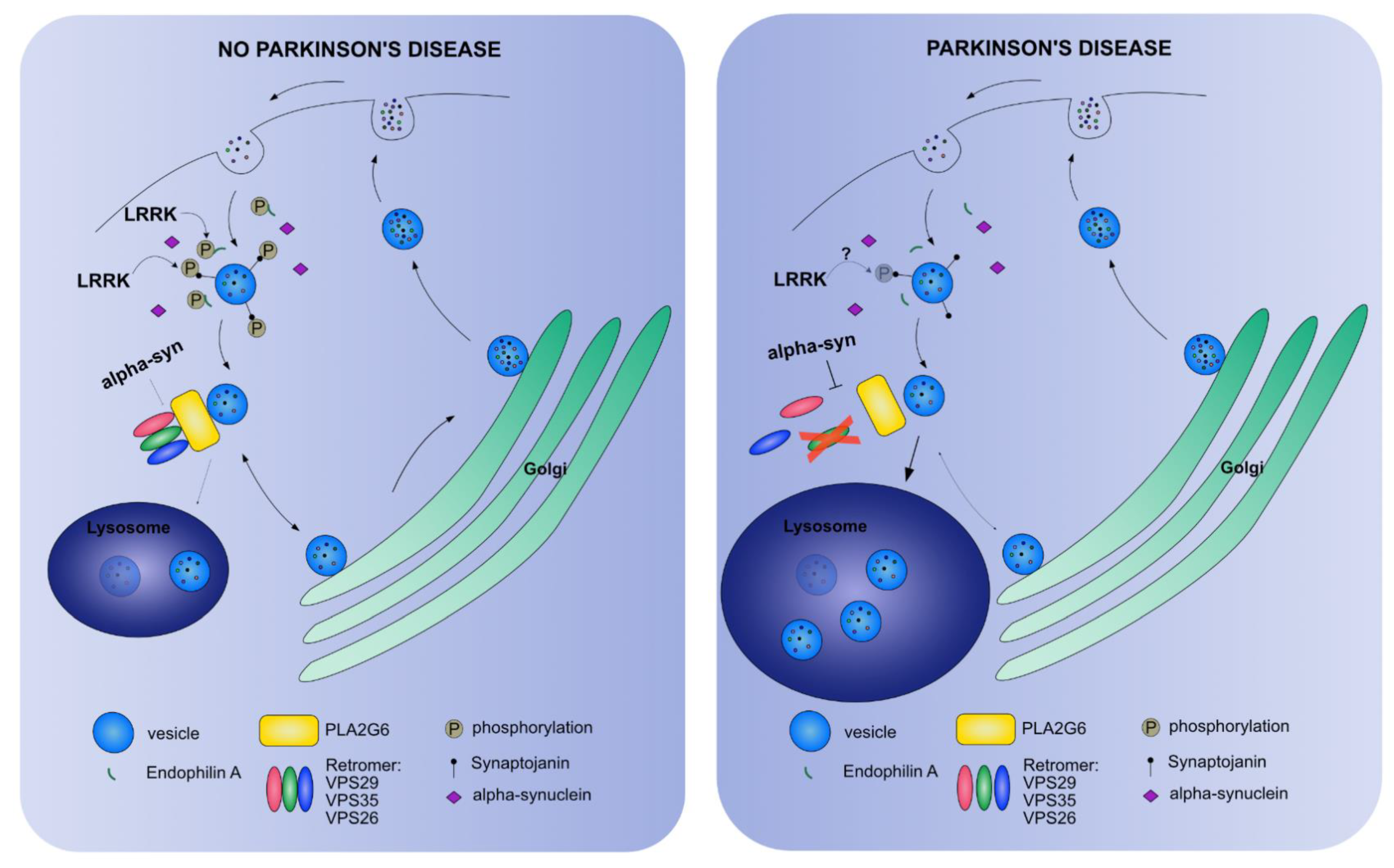

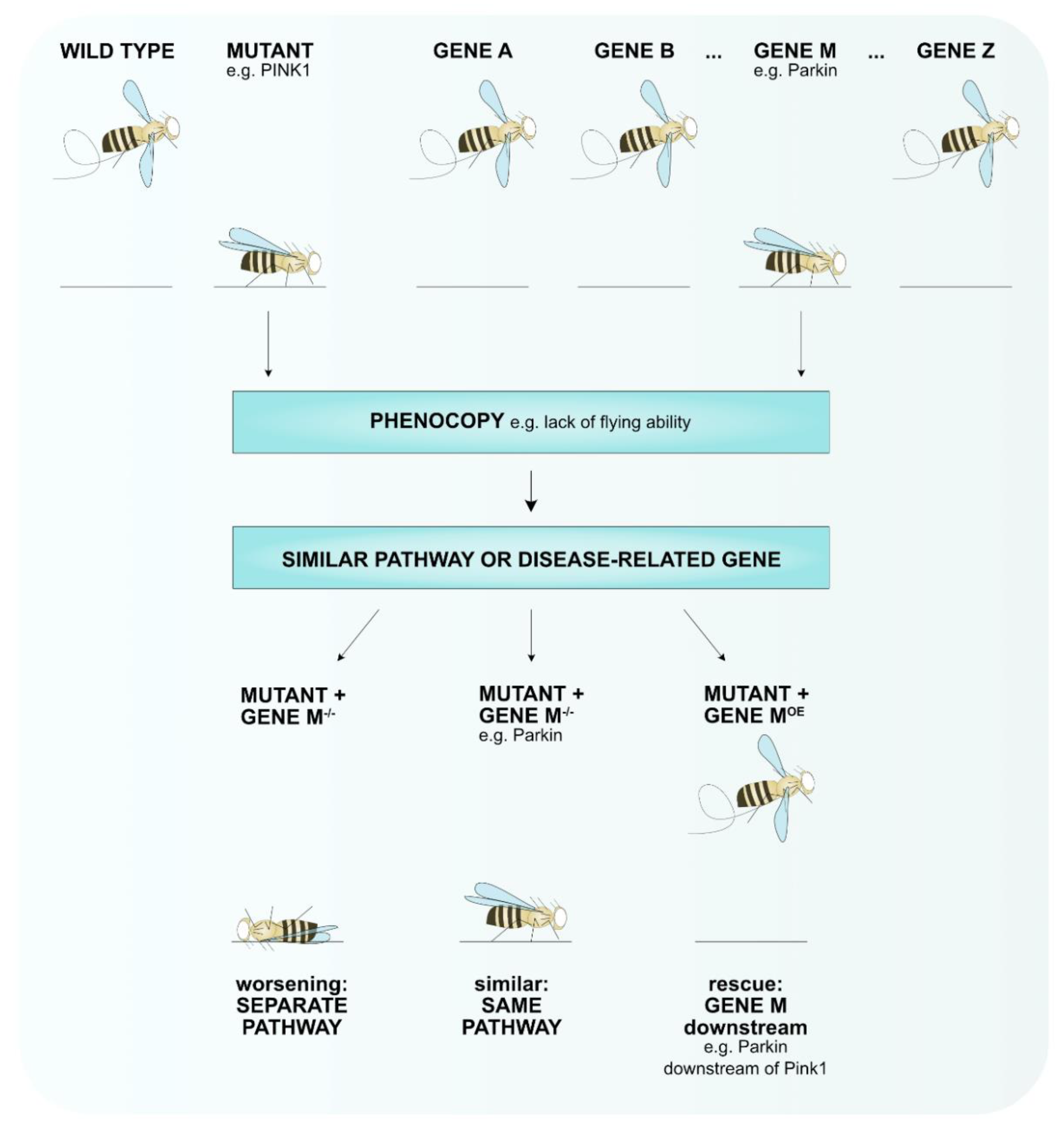{kind=link}
{kind=link}
{kind=link}
| Feature | Limitation | Advantage |
|---|---|---|
| Life cycle of ~10 days | Too short to study late-life stage signs | A lot of flies in a short amount of time |
| Behavior | Not all aspects can be analyzed | Locomotion, sleep, circadian rhythm, can be analyzed |
| Brain | Neuronal circuitry is not evolutionarily conserved | Complex neuronal circuitry (including dopaminergic neurons) |
| UAS-gal4 system | Off-target effects Overexpression not controlled: too much protein, and thus less physiological condition | Overexpression of human disease genes Knockdown of genes to mimic a loss of function |
| Genome | Only 4 chromosomes versus 23 in human | 75% of the disease-causing genes have a fly ortholog |
| Human Gene | Drosophila Gene | Disease-Causing OE | LOF | KD | DA Neuron Loss | Motor Deficits | Non-Motor Symptoms | Mito Dysfunction | Endo-Lysosomal Pathway | Lipid Homeostasis | Key Findings in Drosophila |
|---|---|---|---|---|---|---|---|---|---|---|---|
| SNCA (AD) | / | x | / | / | + | + | + | + | + | + | - link to retromer and sphingolipids |
| Parkin (AR) | parkin | x | x | +/− | + | + | + | - functions in the same pathway with Pink1-circadian rhythm - age-dependent mitophagy | |||
| PINK1 (AR) | pink1 | x | x | +/− | + | + | + | + | - functions in the same pathway with Parkin - circadian rhythm - age-dependent mitophagy - complex I dysfunction - lipid alterations | ||
| DJ-1 (AR) | dj-1α dj-1β | x | x | + | + | ||||||
| LRRK2 (AD) | Lrrk | x | x | x | + | + | + | - link to Rab proteins-link to autophagy | |||
| VPS35 (AD) | Vps35 | x | x | x | + | + | + | + | - recycling of sphingolipids | ||
| Risk genes | |||||||||||
| GBA | Gba1a Gba1b | x | x | x | + | + | + | + |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vos, M.; Klein, C. The Importance of Drosophila melanogaster Research to UnCover Cellular Pathways Underlying Parkinson’s Disease. Cells 2021, 10, 579. https://doi.org/10.3390/cells10030579
Vos M, Klein C. The Importance of Drosophila melanogaster Research to UnCover Cellular Pathways Underlying Parkinson’s Disease. Cells. 2021; 10(3):579. https://doi.org/10.3390/cells10030579
Chicago/Turabian StyleVos, Melissa, and Christine Klein. 2021. "The Importance of Drosophila melanogaster Research to UnCover Cellular Pathways Underlying Parkinson’s Disease" Cells 10, no. 3: 579. https://doi.org/10.3390/cells10030579