
Resistance of Hypoxic Cells to Ionizing Radiation Is Mediated in Part via Hypoxia-Induced Quiescence


 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. Cell Lines and Cell Culture Conditions
2.2. Antibodies and Reagents
2.3. Radiation Treatment
2.4. Colony-Forming Assay
2.5. Western Blots
2.6. Flow Cytometry Analysis and Fluorescence-Activated Cell Sorting (FACS)
2.7. Immunofluorescence
2.8. Immunohistochemistry
2.9. Imaging and Image Analysis
2.9.1. Nuclear Foci
2.9.2. Cross-Sections of Multicellular Spheroids
2.10. Live Cell Imaging
2.10.1. Estimation of Cell Cycle Time with FUCCI (Sensors)
2.10.2. Estimation of Quiescence Based on CDK2-Activity Reporter
2.11. Live-Imaging of Multicellular Spheroids
2.12. Analysis of 3D-Imaging of Spheroids
2.13. Statistical Analysis
3. Results
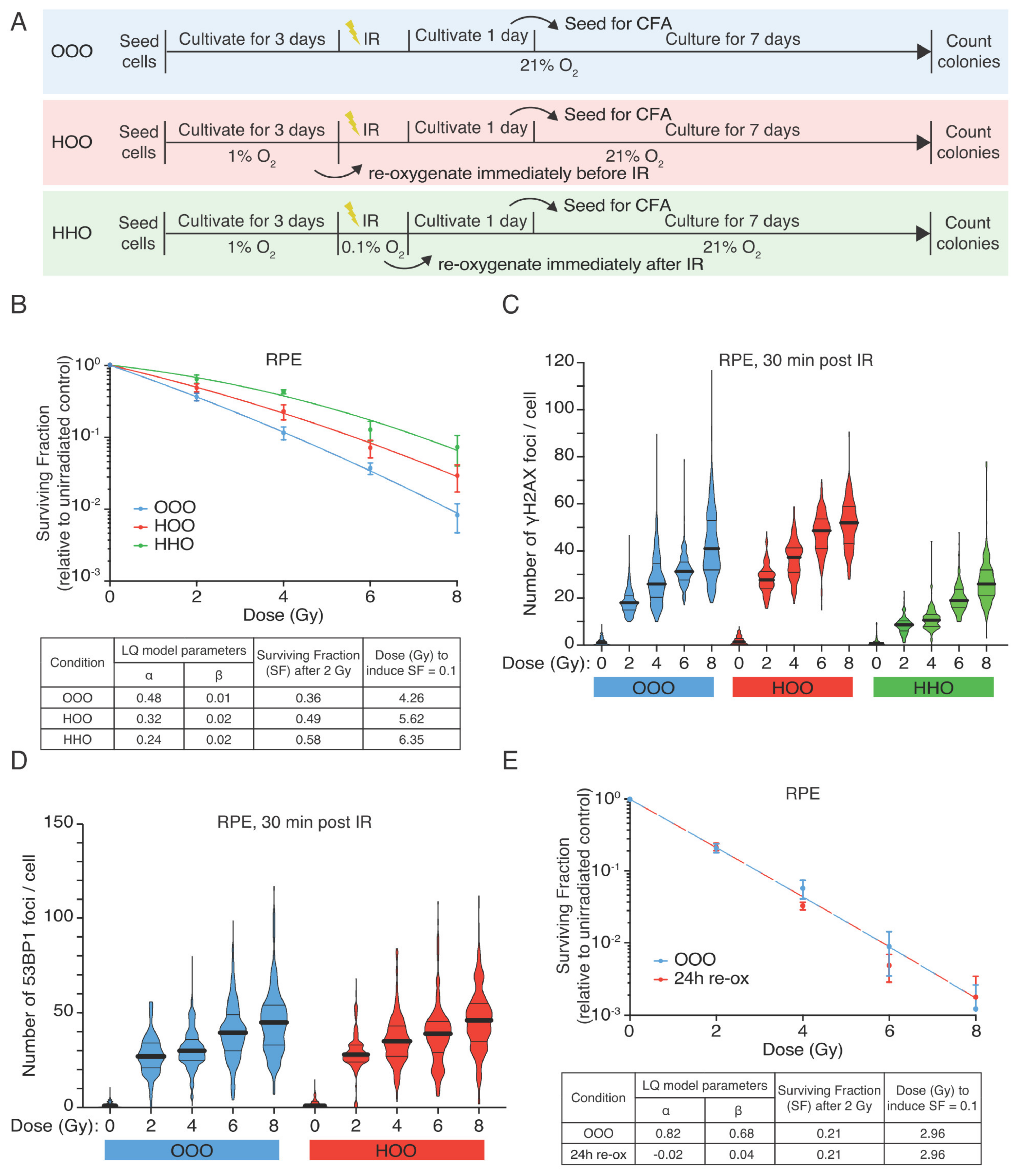
3.1. Hypoxic RPE Cells Retain a Radioresistant Phenotype upon Reoxygenation
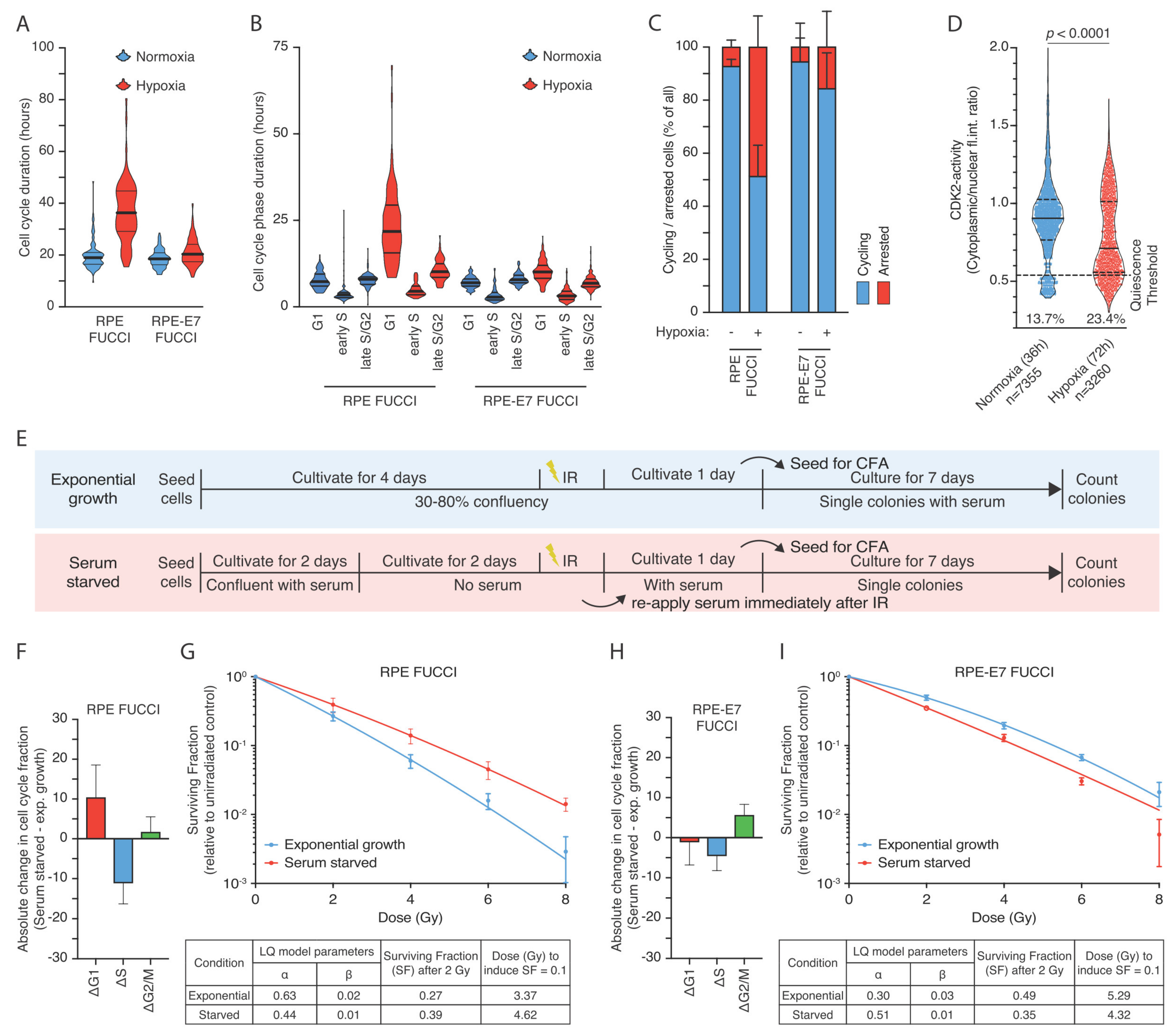
3.2. The Hypoxia-Induced G1-Arrest Causes Continued Radioresistance after Reoxygenation
3.3. Human Papilloma Oncoprotein E7 Prevents the Hypoxia-Induced G1-Arrest and Abolishes the Radioresistance after Reoxygenation
3.4. Hypoxia-Induced Quiescence Determines Radioresistance upon Reoxygenation
3.5. Hypoxia-Induced G1-Arrest Determines Radioresistance upon Reoxygenation in Tumor Cell Lines Depending on HPV Status
3.6. Hypoxia-Induced Cell Cycle Arrest in Multicellular Spheroids
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Harris, A.L. Hypoxia—A key regulatory factor in tumour growth. Nat. Rev. Cancer 2002, 2, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.R.; Hay, M.P. Targeting hypoxia in cancer therapy. Nat. Rev. Cancer 2011, 11, 393–410. [Google Scholar] [CrossRef]
- Lalonde, E.; Ishkanian, A.S.; Sykes, J.; Fraser, M.; Ross-Adams, H.; Erho, N.; Dunning, M.J.; Halim, S.; Lamb, A.D.; Moon, N.C.; et al. Tumour genomic and microenvironmental heterogeneity for integrated prediction of 5-year biochemical recurrence of prostate cancer: A retrospective cohort study. Lancet Oncol. 2014, 15, 1521–1532. [Google Scholar] [CrossRef]
- Linge, A.; Lohaus, F.; Löck, S.; Nowak, A.; Gudziol, V.; Valentini, C.; Von Neubeck, C.; Jütz, M.; Tinhofer, I.; Budach, V.; et al. HPV status, cancer stem cell marker expression, hypoxia gene signatures and tumour volume identify good prognosis subgroups in patients with HNSCC after primary radiochemotherapy: A multicentre retrospective study of the German Cancer Consortium Radiation Oncology Group (DKTK-ROG). Radiother. Oncol. 2016, 121, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Nordsmark, M.; Overgaard, J. Tumor hypoxia is independent of hemoglobin and prognostic for loco-regional tumor control after primary radiotherapy in advanced head and neck cancer. Acta Oncol. 2004, 43, 396–403. [Google Scholar] [CrossRef] [Green Version]
- Luoto, K.R.; Kumareswaran, R.; Bristow, R.G. Tumor hypoxia as a driving force in genetic instability. Genome Integr. 2013, 4, 5. [Google Scholar] [CrossRef] [Green Version]
- Rankin, E.B.; Giaccia, A.J. Hypoxic control of metastasis. Science 2016, 352, 175–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhandari, V.; Hoey, C.; Liu, L.Y.; LaLonde, E.; Ray, J.; Livingstone, J.; Lesurf, R.; Shiah, Y.-J.; Vujcic, T.; Huang, X.; et al. Molecular landmarks of tumor hypoxia across cancer types. Nat. Genet. 2019, 51, 308–318. [Google Scholar] [CrossRef]
- Gordan, J.D.; Simon, M.C. Hypoxia-inducible factors: Central regulators of the tumor phenotype. Curr. Opin. Genet. Dev. 2007, 17, 71–77. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.M. The hypoxic cell: A target for selective cancer therapy—Eighteenth Bruce, F. Cain Memorial Award lecture. Cancer Res. 1999, 59, 5863–5870. [Google Scholar] [PubMed]
- Menegakis, A.; Eicheler, W.; Yaromina, A.; Thames, H.D.; Krause, M.; Baumann, M. Residual DNA double strand breaks in perfused but not in unperfused areas determine different radiosensitivity of tumours. Radiother. Oncol. 2011, 100, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Olive, P.L.; Banáth, J.P. Phosphorylation of histone H2AX as a measure of radiosensitivity. Int. J. Radiat. Oncol. 2004, 58, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Dengler, V.L.; Galbraith, M.D.; Espinosa, J.M. Transcriptional regulation by hypoxia inducible factors. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greer, S.N.; Metcalf, J.L.; Wang, Y.; Ohh, M. The updated biology of hypoxia-inducible factor. EMBO J. 2012, 31, 2448–2460. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Hypoxia-Inducible Factors in Physiology and Medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubbi, M.E.; Semenza, G.L. Regulation of cell proliferation by hypoxia-inducible factors. Am. J. Physiol. Physiol. 2015, 309, 775–782. [Google Scholar] [CrossRef] [Green Version]
- Ortmann, B.; Druker, J.; Rocha, S. Cell cycle progression in response to oxygen levels. Cell. Mol. Life Sci. 2014, 71, 3569–3582. [Google Scholar] [CrossRef] [Green Version]
- Hochegger, H.; Takeda, S.; Hunt, T. Cyclin-dependent kinases and cell-cycle transitions: Does one fit all? Nat. Rev. Mol. Cell Biol. 2008, 9, 910–916. [Google Scholar] [CrossRef]
- Chen, H.-Z.; Tsai, S.-Y.; Leone, G. Emerging roles of E2Fs in cancer: An exit from cell cycle control. Nat. Rev. Cancer 2009, 9, 785–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardner, L.B.; Li, Q.; Park, M.S.; Flanagan, W.M.; Semenza, G.L.; Dang, C.V. Hypoxia Inhibits G1/S Transition through Regulation of p27 Expression. J. Biol. Chem. 2001, 276, 7919–7926. [Google Scholar] [CrossRef] [Green Version]
- Koshiji, M.; Kageyama, Y.; Pete, E.A.; Horikawa, I.; Barrett, J.C.; Huang, L.E. HIF-1α induces cell cycle arrest by functionally counteracting Myc. EMBO J. 2004, 23, 1949–1956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubbi, M.E.; Luo, W.; Baek, J.H.; Semenza, G.L. MCM Proteins Are Negative Regulators of Hypoxia-Inducible Factor 1. Mol. Cell 2011, 42, 700–712. [Google Scholar] [CrossRef] [Green Version]
- Krenning, L.; Berg, J.V.D.; Medema, R.H. Life or Death after a Break: What Determines the Choice? Mol. Cell 2019, 76, 346–358. [Google Scholar] [CrossRef] [PubMed]
- Shaltiel, A.I.; Krenning, L.; Bruinsma, W.; Medema, R.H. The same, only different—DNA damage checkpoints and their reversal throughout the cell cycle. J. Cell Sci. 2015, 128, 607–620. [Google Scholar] [CrossRef] [Green Version]
- Sinclair, W.K.; Morton, R.A. X-ray Sensitivity during the Cell Generation Cycle of Cultured Chinese Hamster Cells. Radiat. Res. 1966, 29, 450. [Google Scholar] [CrossRef]
- Terasima, T.; Tolmach, L.J. X-ray Sensitivity and DNA Synthesis in Synchronous Populations of HeLa Cells. Science 1963, 140, 490–492. [Google Scholar] [CrossRef] [PubMed]
- Barazas, M.; Gasparini, A.; Huang, Y.; Küçükosmanoğlu, A.; Annunziato, S.; Bouwman, P.; Sol, W.; Kersbergen, A.; Proost, N.; De Korte-Grimmerink, R.; et al. Radiosensitivity Is an Acquired Vulnerability of PARPi-Resistant BRCA1-Deficient Tumors. Cancer Res. 2019, 79, 452–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otani, K.; Naito, Y.; Sakaguchi, Y.; Seo, Y.; Takahashi, Y.; Kikuta, J.; Ogawa, K.; Ishii, M. Cell-cycle-controlled radiation therapy was effective for treating a murine malignant melanoma cell line in vitro and in vivo. Sci. Rep. 2016, 6, 30689. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.-H.; Altschuler, S.J.; Wu, L.F. Patterns of Early p21 Dynamics Determine Proliferation-Senescence Cell Fate after Chemotherapy. Cell 2019, 178, 361–373. [Google Scholar] [CrossRef] [PubMed]
- Ryl, T.; Kuchen, E.E.; Bell, E.; Shao, C.; Flórez, A.F.; Mönke, G.; Gogolin, S.; Friedrich, M.; Lamprecht, F.; Westermann, F.; et al. Cell-Cycle Position of Single MYC-Driven Cancer Cells Dictates Their Susceptibility to a Chemotherapeutic Drug. Cell Syst. 2017, 5, 237–250. [Google Scholar] [CrossRef] [Green Version]
- Godet, I.; Shin, Y.J.; Ju, J.A.; Ye, I.C.; Wang, G.; Gilkes, D.M. Fate-mapping post-hypoxic tumor cells reveals a ROS-resistant phenotype that promotes metastasis. Nat. Commun. 2019, 10, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fluegen, G.; Avivar-Valderas, A.; Wang, Y.; Padgen, M.R.; Williams, J.K.; Nobre, A.R.; Calvo, V.; Cheung, J.F.; Bravo-Cordero, J.J.; Entenberg, D.; et al. Phenotypic heterogeneity of disseminated tumour cells is preset by primary tumour hypoxic microenvironments. Nat. Cell Biol. 2017, 19, 120–132. [Google Scholar] [CrossRef] [Green Version]
- Zielke, N.; Edgar, B.A. FUCCI sensors: Powerful new tools for analysis of cell proliferation. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 469–487. [Google Scholar] [CrossRef]
- Krenning, L.; Feringa, F.M.; Shaltiel, I.A.; Berg, J.V.D.; Medema, R.H. Transient Activation of p53 in G2 Phase Is Sufficient to Induce Senescence. Mol. Cell 2014, 55, 59–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feringa, F.M.; Krenning, L.; Koch, A.; Berg, J.V.D.; Broek, B.V.D.; Jalink, K.; Medema, R.H. Hypersensitivity to DNA damage in antephase as a safeguard for genome stability. Nat. Commun. 2016, 7, 12618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weigert, M.; Schmidt, U.; Boothe, T.; Müller, A.; Dibrov, A.; Jain, A.; Wilhelm, B.; Schmidt, D.; Broaddus, C.; Culley, S.; et al. Content-aware image restoration: Pushing the limits of fluorescence microscopy. Nat. Methods 2018, 15, 1090–1097. [Google Scholar] [CrossRef] [Green Version]
- Haase, R.; Royer, L.A.; Steinbach, P.; Schmidt, D.; Dibrov, A.; Schmidt, U.; Weigert, M.; Maghelli, N.; Tomancak, P.; Jug, F.; et al. CLIJ: GPU-accelerated image processing for everyone. Nat. Methods 2019, 17, 5–6. [Google Scholar] [CrossRef] [PubMed]
- Gookin, S.; Mingyu, C.; Phadke, H.; Chung, M.; Moser, J.; Miller, I.; Carter, D.; Spencer, S.L. A map of protein dynamics during cell-cycle progression and cell-cycle exit. PLoS Biol. 2017, 15, e2003268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLaughlin-Drubin, M.E.; Münger, K. The human papillomavirus E7 oncoprotein. Virology 2009, 384, 335–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spencer, S.L.; Cappell, S.D.; Tsai, F.-C.; Overton, K.W.; Wang, C.L.; Meyer, T. The Proliferation-Quiescence Decision Is Controlled by a Bifurcation in CDK2 Activity at Mitotic Exit. Cell 2013, 155, 369–383. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Adler, A.S.; Segal, E.; Chang, H.Y. A Transcriptional Program Mediating Entry into Cellular Quiescence. PLoS Genet. 2007, 3, 91. [Google Scholar] [CrossRef] [PubMed]
- Broceño, C.; Wilkie, S.; Mittnacht, S. RB activation defect in tumor cell lines. Proc. Natl. Acad. Sci. USA 2002, 99, 14200–14205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, J.M.; Wilson, W.R. Exploiting tumour hypoxia in cancer treatment. Nat. Rev. Cancer 2004, 4, 437–447. [Google Scholar] [CrossRef]
- Rockwell, S.; Dobrucki, I.T.; Kim, E.Y.; Marrison, S.T.; Vu, V.T. Hypoxia and Radiation Therapy: Past History, Ongoing Research, and Future Promise. Curr. Mol. Med. 2009, 9, 442–458. [Google Scholar] [CrossRef] [Green Version]
- Goda, N.; Ryan, H.E.; Khadivi, B.; McNulty, W.; Rickert, R.C.; Johnson, R.S. Hypoxia-Inducible Factor 1α Is Essential for Cell Cycle Arrest during Hypoxia. Mol. Cell. Biol. 2003, 23, 359–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordan, J.D.; Thompson, C.B.; Simon, M.C. HIF and c-Myc: Sibling Rivals for Control of Cancer Cell Metabolism and Proliferation. Cancer Cell 2007, 12, 108–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonissi, F.; Lattanzio, L.; Astesana, V.; Cavicchioli, F.; Ghiglia, A.; Monteverde, M.; Vivenza, D.; Gianello, L.; Russi, E.; Merlano, M.; et al. Reoxygenation Reverses Hypoxia-related Radioresistance in Head and Neck Cancer Cell Lines. Anticancer. Res. 2016, 36, 2211–2215. [Google Scholar] [PubMed]
- Hoppe-Seyler, K.; Bossler, F.; Lohrey, C.; Bulkescher, J.; Rösl, F.; Jansen, L.; Mayer, A.; Vaupel, P.; Dürst, M.; Hoppe-Seyler, F. Induction of dormancy in hypoxic human papillomavirus-positive cancer cells. Proc. Natl. Acad. Sci. USA 2017, 114, 990–998. [Google Scholar] [CrossRef] [Green Version]
- Bindra, R.S.; Schaffer, P.J.; Meng, A.; Woo, J.; Maseide, K.; Roth, M.E.; Lizardi, P.; Hedley, D.W.; Bristow, R.G.; Glazer, P.M. Down-Regulation of Rad51 and Decreased Homologous Recombination in Hypoxic Cancer Cells. Mol. Cell. Biol. 2004, 24, 8504–8518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, N.; Koritzinsky, M.; Zhao, H.; Bindra, R.; Glazer, P.M.; Powell, S.; Belmaaza, A.; Wouters, B.; Bristow, R.G. Chronic Hypoxia Decreases Synthesis of Homologous Recombination Proteins to Offset Chemoresistance and Radioresistance. Cancer Res. 2008, 68, 605–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Chu, A.; Turker, M.S.; Glazer, P.M. Hypoxia-Induced Epigenetic Regulation and Silencing of the BRCA1 Promoter. Mol. Cell. Biol. 2011, 31, 3339–3350. [Google Scholar] [CrossRef] [Green Version]
- Lee, P.; Chandel, N.S.; Simon, M.C. Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nat. Rev. Mol. Cell Biol. 2020, 21, 268–283. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, A.N.; Chauhan, A.; Khanna, S.; Rai, Y.; Singh, S.; Soni, R.; Kalra, N.; Dwarakanath, B.S. Transient elevation of glycolysis confers radio-resistance by facilitating DNA repair in cells. BMC Cancer 2015, 15, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Shimura, T.; Noma, N.; Sano, Y.; Ochiai, Y.; Oikawa, T.; Fukumoto, M.; Kunugita, N. AKT-mediated enhanced aerobic glycolysis causes acquired radioresistance by human tumor cells. Radiother. Oncol. 2014, 112, 302–307. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Wei, F.; Wu, Y.; He, Y.; Shi, L.; Xiong, F.; Gong, Z.; Guo, C.; Li, X.; Deng, H.; et al. Role of metabolism in cancer cell radioresistance and radiosensitization methods. J. Exp. Clin. Cancer Res. 2018, 37, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Intlekofer, A.M.; Wang, B.; Liu, H.; Shah, H.; Carmona-Fontaine, C.; Rustenburg, A.S.; Salah, S.; Gunner, M.R.; Chodera, J.D.; Cross, J.R.; et al. L-2-Hydroxyglutarate production arises from noncanonical enzyme function at acidic pH. Nat. Chem. Biol. 2017, 13, 494–500. [Google Scholar] [CrossRef]
- Nadtochiy, S.M.; Schafer, X.; Fu, D.; Nehrke, K.; Munger, J.; Brookes, P.S. Acidic pH Is a Metabolic Switch for 2-Hydroxyglutarate Generation and Signaling. J. Biol. Chem. 2016, 291, 20188–20197. [Google Scholar] [CrossRef] [Green Version]
- Fu, S.; Li, Z.; Xiao, L.; Hu, W.; Zhang, L.; Xie, B.; Zhou, Q.; He, J.; Qiu, Y.; Wen, M.; et al. Glutamine Synthetase Promotes Radiation Resistance via Facilitating Nucleotide Metabolism and Subsequent DNA Damage Repair. Cell Rep. 2019, 28, 1136–1143.e4. [Google Scholar] [CrossRef] [Green Version]
- Sun, R.C.; Denko, N.C. Hypoxic Regulation of Glutamine Metabolism through HIF1 and SIAH2 Supports Lipid Synthesis that Is Necessary for Tumor Growth. Cell Metab. 2014, 19, 285–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Floberg, J.M.; Schwarz, J.K. Manipulation of Glucose and Hydroperoxide Metabolism to Improve Radiation Response. Semin. Radiat. Oncol. 2019, 29, 33–41. [Google Scholar] [CrossRef]
- Rashmi, R.; Huang, X.; Floberg, J.M.; Elhammali, A.; McCormick, M.L.; Patti, G.J.; Spitz, U.R.; Schwarz, J.K. Radioresistant Cervical Cancers Are Sensitive to Inhibition of Glycolysis and Redox Metabolism. Cancer Res. 2018, 78, 1392–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Palma, M.; Biziato, D.; Petrova, T.V. Microenvironmental regulation of tumour angiogenesis. Nat. Rev. Cancer. 2017, 17, 457–474. [Google Scholar] [CrossRef] [PubMed]
- Stapor, P.; Wang, X.; Goveia, J.; Moens, S.; Carmeliet, P. Angiogenesis revisited—Role and therapeutic potential of targeting endothelial metabolism. J. Cell Sci. 2014, 127, 4331–4341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, Y.; Yashiro, M.; Fuyuhiro, Y.; Kashiwagi, S.; Matsuoka, J.; Hirakawa, T.; Noda, S.; Aomatsu, N.; Hasegawa, T.; Matsuzaki, T.; et al. Effects of acute and chronic hypoxia on the radiosensitivity of gastric and esophageal cancer cells. Anticancer. Res. 2011, 31, 3369–3375. [Google Scholar] [PubMed]
- Löck, S.; Perrin, R.; Seidlitz, A.; Bandurska-Luque, A.; Zschaeck, S.; Zöphel, K.; Krause, M.; Steinbach, J.; Kotzerke, J.; Zips, D.; et al. Residual tumour hypoxia in head-and-neck cancer patients undergoing primary radiochemotherapy, final results of a prospective trial on repeat FMISO-PET imaging. Radiother. Oncol. 2017, 124, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Petersen, C.; Zips, D.; Krause, M.; Schöne, K.; Eicheler, W.; Hoinkis, C.; Thames, H.D.; Baumann, M. Repopulation of FaDu human squamous cell carcinoma during fractionated radiotherapy correlates with reoxygenation. Int. J. Radiat. Oncol. Biol. Phys. 2001, 51, 483–493. [Google Scholar] [CrossRef]
- Lenos, K.J.; Miedema, D.M.; Lodestijn, S.C.; Nijman, L.E.; Bosch, T.V.D.; Ros, X.R.; Lourenço, F.C.; Lecca, M.C.; Van Der Heijden, M.; Van Neerven, S.M.; et al. Stem cell functionality is microenvironmentally defined during tumour expansion and therapy response in colon cancer. Nat. Cell Biol. 2018, 20, 1193–1202. [Google Scholar] [CrossRef]
- Crokart, N.; Jordan, B.F.; Baudelet, C.; Ansiaux, R.; Sonveaux, P.; Gregoire, V.; Beghein, N.; Dewever, J.; Bouzin, C.; Feron, O.; et al. Early reoxygenation in tumors after irradiation: Determining factors and consequences for radiotherapy regimens using daily multiple fractions. Int. J. Radiat. Oncol. 2005, 63, 901–910. [Google Scholar] [CrossRef]
- Willett, C.G.; Kozin, S.V.; Duda, D.G.; Di Tomaso, E.; Kozak, K.R.; Boucher, Y.; Jain, R.K. Combined Vascular Endothelial Growth Factor–Targeted Therapy and Radiotherapy for Rectal Cancer: Theory and Clinical Practice. Semin. Oncol. 2006, 33, 35–40. [Google Scholar] [CrossRef] [Green Version]
- Janssens, G.O.; Rademakers, S.E.; Terhaard, C.H.; Doornaert, P.A.; Bijl, H.P.; Ende, P.V.D.; Chin, A.; Marres, H.A.; De Bree, R.; Van Der Kogel, A.J.; et al. Accelerated Radiotherapy with Carbogen and Nicotinamide for Laryngeal Cancer: Results of a Phase III Randomized Trial. J. Clin. Oncol. 2012, 30, 1777–1783. [Google Scholar] [CrossRef] [PubMed]
- Janssens, G.O.; Rademakers, S.E.; Terhaard, C.H.; Doornaert, P.A.; Bijl, H.P.; Ende, P.V.D.; Chin, A.; Takes, R.P.; De Bree, R.; Hoogsteen, I.J.; et al. Improved Recurrence-Free Survival with ARCON for Anemic Patients with Laryngeal Cancer. Clin. Cancer Res. 2014, 20, 1345–1354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Overgaard, J. Hypoxic Radiosensitization: Adored and Ignored. J. Clin. Oncol. 2007, 25, 4066–4074. [Google Scholar] [CrossRef] [Green Version]
- Overgaard, J. Hypoxic modification of radiotherapy in squamous cell carcinoma of the head and neck—A systematic review and meta-analysis. Radiother. Oncol. 2011, 100, 22–32. [Google Scholar] [CrossRef]
- Baran, N.; Konopleva, M. Molecular Pathways: Hypoxia-Activated Prodrugs in Cancer Therapy. Clin. Cancer Res. 2017, 23, 2382–2390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mistry, I.N.; Thomas, M.; Calder, E.D.; Conway, S.J.; Hammond, E.M. Clinical Advances of Hypoxia-Activated Prodrugs in Combination With Radiation Therapy. Int. J. Radiat. Oncol. 2017, 98, 1183–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saggar, J.K.; Tannock, I.F. Abstract A51: Chemotherapy rescues hypoxic tumor cells and induces reoxygenation and repopulation—An effect that is inhibited by the hypoxia-activated pro-drug TH-302. Transl. Ther. Potential Tumor Microenviron. 2015, 21, 2107–2114. [Google Scholar] [CrossRef]
- Allo, G.; Yap, M.L.; Cuartero, J.; Milosevic, M.; Ferguson, S.; Mackay, H.; Kamel-Reid, S.; Weinreb, I.; Ghazarian, D.; Pintilie, M.; et al. HPV-independent Vulvar Squamous Cell Carcinoma is Associated With Significantly Worse Prognosis Compared With HPV-associated Tumors. Int. J. Gynecol. Pathol. 2019, 39, 391–399. [Google Scholar] [CrossRef]
- Lei, J.; Ploner, A.; Lagheden, C.; Eklund, C.; Kleppe, S.N.; Andrae, B.; Elfström, K.M.; Dillner, J.; Sparén, P.; Sundström, K. High-risk human papillomavirus status and prognosis in invasive cervical cancer: A nationwide cohort study. PLoS Med. 2018, 15, e1002666. [Google Scholar] [CrossRef] [PubMed]
- Lohaus, F.; Linge, A.; Tinhofer, I.; Budach, V.; Gkika, E.; Stuschke, M.; Balermpas, P.; Rödel, C.; Avlar, M.; Grosu, A.-L.; et al. HPV16 DNA status is a strong prognosticator of loco-regional control after postoperative radiochemotherapy of locally advanced oropharyngeal carcinoma: Results from a multicentre explorative study of the German Cancer Consortium Radiation Oncology Group (DKTK-ROG). Radiother. Oncol. 2014, 113, 317–323. [Google Scholar] [CrossRef] [Green Version]
- Göttgens, E.L.; Ostheimer, C.; Span, P.N.; Bussink, J.; Hammond, E.M. Hpv, hypoxia and radiation response in head and neck cancer. Br. J. Radiol. 2019, 92, 20180047. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Menegakis, A.; Klompmaker, R.; Vennin, C.; Arbusà, A.; Damen, M.; van den Broek, B.; Zips, D.; van Rheenen, J.; Krenning, L.; Medema, R.H. Resistance of Hypoxic Cells to Ionizing Radiation Is Mediated in Part via Hypoxia-Induced Quiescence. Cells 2021, 10, 610. https://doi.org/10.3390/cells10030610
Menegakis A, Klompmaker R, Vennin C, Arbusà A, Damen M, van den Broek B, Zips D, van Rheenen J, Krenning L, Medema RH. Resistance of Hypoxic Cells to Ionizing Radiation Is Mediated in Part via Hypoxia-Induced Quiescence. Cells. 2021; 10(3):610. https://doi.org/10.3390/cells10030610
Chicago/Turabian StyleMenegakis, Apostolos, Rob Klompmaker, Claire Vennin, Aina Arbusà, Maartje Damen, Bram van den Broek, Daniel Zips, Jacco van Rheenen, Lenno Krenning, and René H. Medema. 2021. "Resistance of Hypoxic Cells to Ionizing Radiation Is Mediated in Part via Hypoxia-Induced Quiescence" Cells 10, no. 3: 610. https://doi.org/10.3390/cells10030610