
PARP7 and Mono-ADP-Ribosylation Negatively Regulate Estrogen Receptor α Signaling in Human Breast Cancer Cells


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Plasmids
2.3. Cell Culturing
2.4. Real Time qPCR (RT-qPCR)
2.5. Chromatin Immunoprecipitation
2.6. Generation of Anti-PARP7 Antibody
2.7. Western Blotting
2.8. Reporter Gene Assay
2.9. Generation of Knockout Cells
2.10. Cell Proliferation Assay
2.11. Co-Immunoprecipitation
2.12. Mass Spectrometry
2.13. Statistics
3. Results
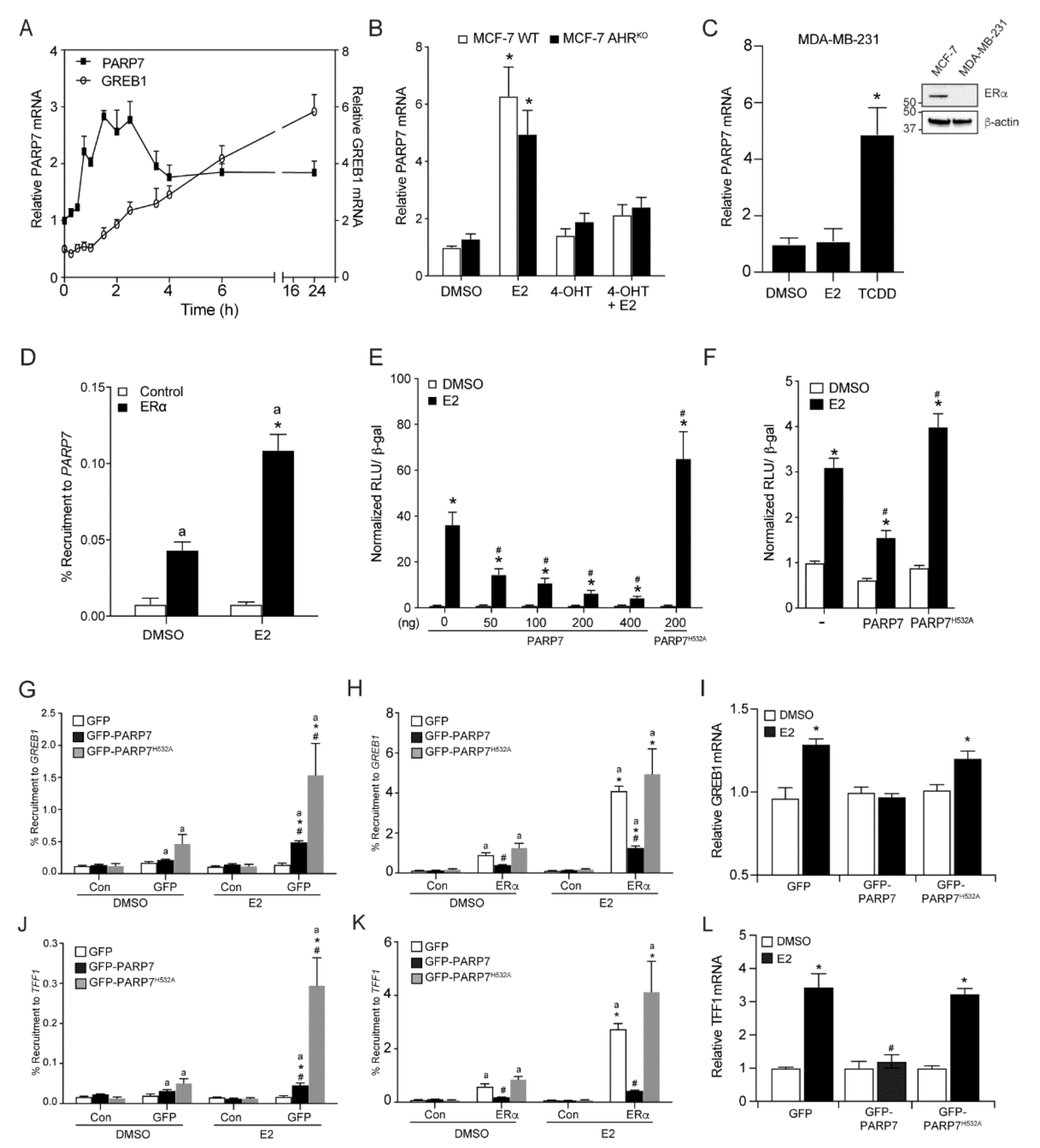
3.1. PARP7 Expression is Induced by ERα
3.2. PARP7 Represses ERα Activity
3.3. The PARP7 Inhibitor, RBN-2397, Increases E2-Dependent GREB1 mRNA Levels and Stabilizes PARP7 and ERα Proteins
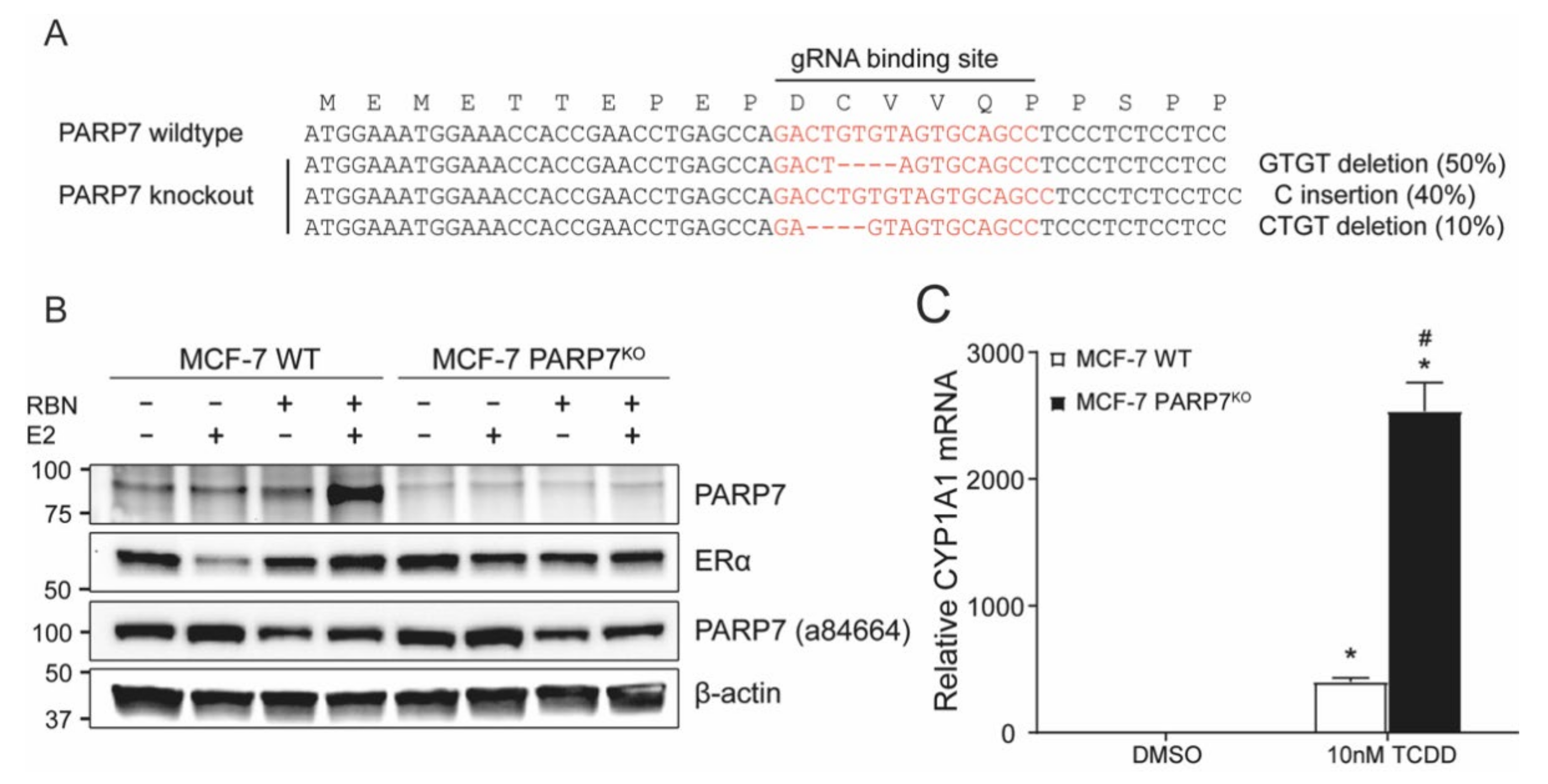
3.4. Generation of CRISPR/Cas9-Mediated PARP7 Knockout MCF-7 Cells
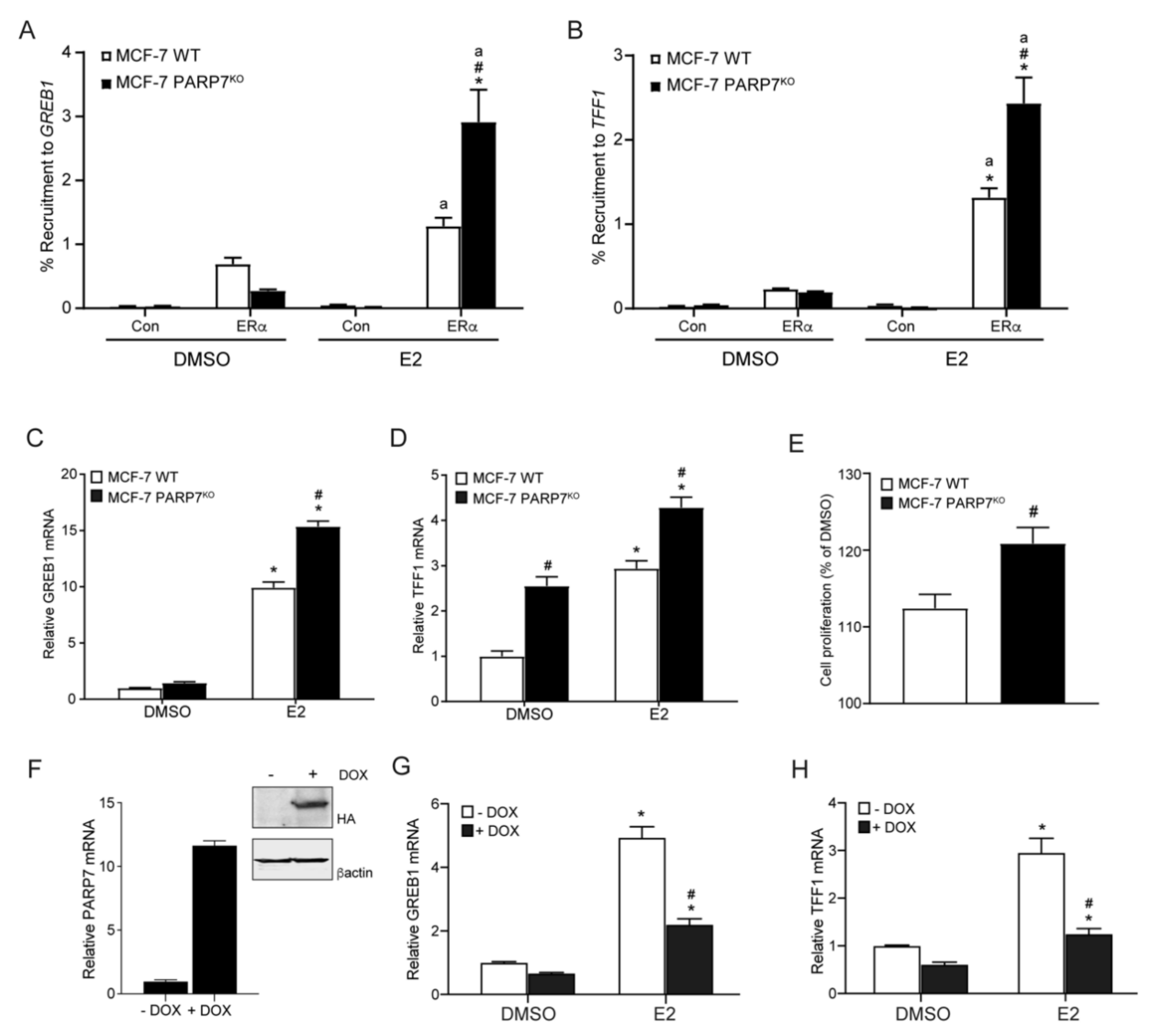
3.5. PARP7 Knockout MCF-7 Cells Display Increased ERα Activity and E2-Induced Proliferation
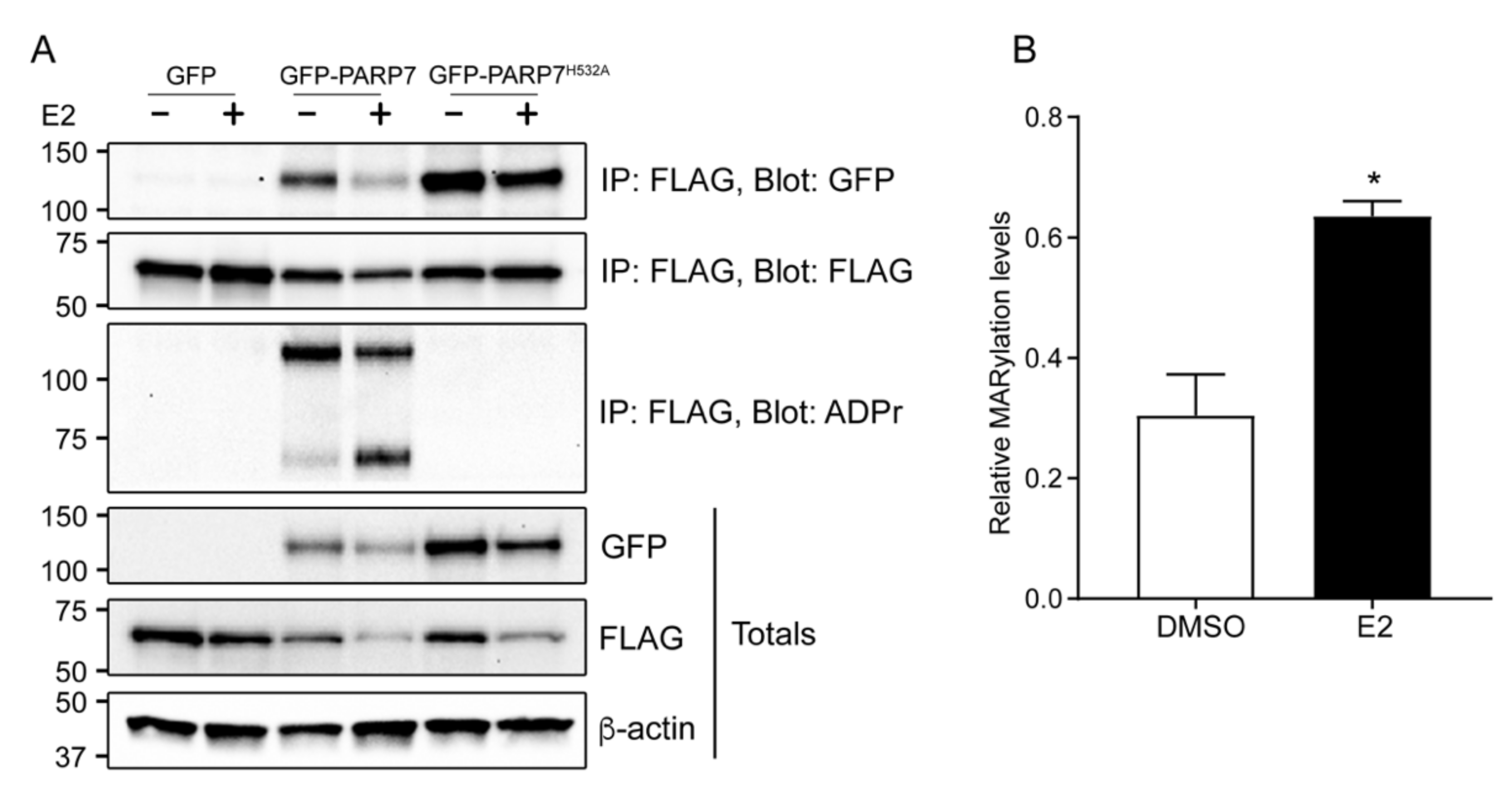
3.6. Overexpressed PARP7 Mono-ADP-ribosylates Overexpressed ERα
3.7. Identification of Mono-ADP-ribosylated Peptides in Bacterial Expressed and Purified ERα
3.8. The Hinge Region of ERα is Required for its Mono-ADP-ribosylation by PARP7
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hassa, P.O.; Hottiger, M.O. The diverse biological roles of mammalian PARPS, a small but powerful family of poly-ADP-ribose polymerases. Front. Biosci. 2008, 13, 3046–3082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hottiger, M.O.; Hassa, P.O.; Luscher, B.; Schuler, H.; Koch-Nolte, F. Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends Biochem. Sci. 2010, 35, 208–219. [Google Scholar] [CrossRef]
- Otto, H.; Reche, P.A.; Bazan, F.; Dittmar, K.; Haag, F.; Koch-Nolte, F. In silico characterization of the family of PARP-like poly(ADP-ribosyl)transferases (pARTs). BMC Genom. 2005, 6, 139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleine, H.; Poreba, E.; Lesniewicz, K.; Hassa, P.O.; Hottiger, M.O.; Litchfield, D.W.; Shilton, B.H.; Luscher, B. Substrate-assisted catalysis by PARP10 limits its activity to mono-ADP-ribosylation. Mol. Cell 2008, 32, 57–69. [Google Scholar] [CrossRef]
- Corda, D.; Di Girolamo, M. Functional aspects of protein mono-ADP-ribosylation. EMBO J. 2003, 22, 1953–1958. [Google Scholar] [CrossRef] [Green Version]
- Welsby, I.; Hutin, D.; Leo, O. Complex roles of members of the ADP-ribosyl transferase super family in immune defences: Looking beyond PARP1. Biochem. Pharmacol. 2012, 84, 11–20. [Google Scholar] [CrossRef]
- Feijs, K.L.; Verheugd, P.; Luscher, B. Expanding functions of intracellular resident mono-ADP-ribosylation in cell physiology. FEBS J. 2013, 280, 3519–3529. [Google Scholar] [CrossRef] [PubMed]
- Butepage, M.; Eckei, L.; Verheugd, P.; Luscher, B. Intracellular Mono-ADP-Ribosylation in Signaling and Disease. Cells 2015, 4, 569–595. [Google Scholar] [CrossRef] [Green Version]
- Rosenthal, F.; Feijs, K.L.; Frugier, E.; Bonalli, M.; Forst, A.H.; Imhof, R.; Winkler, H.C.; Fischer, D.; Caflisch, A.; Hassa, P.O.; et al. Macrodomain-containing proteins are new mono-ADP-ribosylhydrolases. Nat. Struct. Mol. Biol. 2013, 20, 502–507. [Google Scholar] [CrossRef]
- Jankevicius, G.; Hassler, M.; Golia, B.; Rybin, V.; Zacharias, M.; Timinszky, G.; Ladurner, A.G. A family of macrodomain proteins reverses cellular mono-ADP-ribosylation. Nat. Struct. Mol. Biol. 2013, 20, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Fehr, A.R.; Singh, S.A.; Kerr, C.M.; Mukai, S.; Higashi, H.; Aikawa, M. The impact of PARPs and ADP-ribosylation on inflammation and host-pathogen interactions. Genes Dev. 2020, 34, 341–359. [Google Scholar] [CrossRef] [PubMed]
- Matthews, J. AHR toxicity and signaling: Role of TIPARP and ADP-ribosylation. Curr. Opin. Toxicol. 2017, 2, 50–57. [Google Scholar] [CrossRef]
- Gomez, A.; Bindesboll, C.; Satheesh, S.V.; Grimaldi, G.; Hutin, D.; MacPherson, L.; Ahmed, S.; Tamblyn, L.; Cho, T.; Nebb, H.I.; et al. Characterization of TCDD-inducible poly-ADP-ribose polymerase (TIPARP/ARTD14) catalytic activity. Biochem. J. 2018, 475, 3827–3846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozaki, T.; Komano, J.; Kanbayashi, D.; Takahama, M.; Misawa, T.; Satoh, T.; Takeuchi, O.; Kawai, T.; Shimizu, S.; Matsuura, Y.; et al. Mitochondrial damage elicits a TCDD-inducible poly(ADP-ribose) polymerase-mediated antiviral response. Proc. Natl. Acad. Sci. USA 2017, 114, 2681–2686. [Google Scholar] [CrossRef] [Green Version]
- Aravind, L. The WWE domain: A common interaction module in protein ubiquitination and ADP ribosylation. Trends Biochem. Sci. 2001, 26, 273–275. [Google Scholar] [CrossRef]
- Ma, Q.; Baldwin, K.T.; Renzelli, A.J.; McDaniel, A.; Dong, L. TCDD-inducible poly(ADP-ribose) polymerase: A novel response to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Biochem. Biophys. Res. Commun. 2001, 289, 499–506. [Google Scholar] [CrossRef] [PubMed]
- MacPherson, L.; Tamblyn, L.; Rajendra, S.; Bralha, F.; McPherson, J.P.; Matthews, J. 2,3,7,8-tetrachlorodibenzo-p-dioxin poly(ADP-ribose) polymerase (TiPARP, ARTD14) is a mono-ADP-ribosyltransferase and repressor of aryl hydrocarbon receptor transactivation. Nucleic Acids Res. 2013, 41, 1604–1621. [Google Scholar] [CrossRef] [PubMed]
- Bindesboll, C.; Tan, S.; Bott, D.; Cho, T.; Tamblyn, L.; MacPherson, L.; Gronning-Wang, L.; Nebb, H.I.; Matthews, J. TCDD-inducible poly-ADP-ribose polymerase (TIPARP/PARP7) mono-ADP-ribosylates and co-activates liver X receptors. Biochem. J. 2016, 473, 899–910. [Google Scholar] [CrossRef]
- Zhang, L.; Cao, J.; Dong, L.Y.; Lin, H.N. TiPARP forms nuclear condensates to degrade HIF-1 alpha and suppress tumorigenesis. Proc. Natl. Acad. Sci. USA 2020, 117, 13447–13456. [Google Scholar] [CrossRef]
- Yamada, T.; Horimoto, H.; Kameyama, T.; Hayakawa, S.; Yamato, H.; Dazai, M.; Takada, A.; Kida, H.; Bott, D.; Zhou, A.C.; et al. Constitutive aryl hydrocarbon receptor signaling constrains type I interferon-mediated antiviral innate defense. Nat. Immunol. 2016, 17, 687–694. [Google Scholar] [CrossRef] [Green Version]
- Vasbinder, M.M.; Gozgit, J.M.; Abo, R.P.; Kunii, K.; Kuplast-Barr, K.G.; Gui, B.; Lu, A.Z.; Swinger, K.K.; Wigle, T.J.; Blackwell, D.J.; et al. RBN-2397: A first-in-class PARP7 inhibitor targeting a newly discovered cancer vulnerability in stress-signaling pathways. In Proceedings of the AACR Annual Meeting, Philadelphia, PA, USA. 27–28 April 2020 and 22–24 June 2020.
- Cheng, L.; Li, Z.; Huang, Y.Z.; Zhang, X.; Dai, X.Y.; Shi, L.; Xi, P.W.; Wei, J.F.; Ding, Q. TCDD-Inducible Poly-ADP-Ribose Polymerase (TIPARP), A Novel Therapeutic Target Of Breast Cancer. Cancer Manag. Res. 2019, 11, 8991–9004. [Google Scholar] [CrossRef] [Green Version]
- Couse, J.F.; Korach, K.S. Estrogen receptor null mice: What have we learned and where will they lead us? Endocr. Rev. 1999, 20, 358–417. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Spezia, M.; Huang, S.; Yuan, C.; Zeng, Z.; Zhang, L.; Ji, X.; Liu, W.; Huang, B.; Luo, W.; et al. Breast cancer development and progression: Risk factors, cancer stem cells, signaling pathways, genomics, and molecular pathogenesis. Genes Dis. 2018, 5, 77–106. [Google Scholar] [CrossRef]
- Heldring, N.; Pike, A.; Andersson, S.; Matthews, J.; Cheng, G.; Hartman, J.; Tujague, M.; Strom, A.; Treuter, E.; Warner, M.; et al. Estrogen receptors: How do they signal and what are their targets. Physiol. Rev. 2007, 87, 905–931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burns, K.A.; Li, Y.; Arao, Y.; Petrovich, R.M.; Korach, K.S. Selective mutations in estrogen receptor alpha D-domain alters nuclear translocation and non-estrogen response element gene regulatory mechanisms. J. Biol. Chem. 2011, 286, 12640–12649. [Google Scholar] [CrossRef] [Green Version]
- Tora, L.; Mullick, A.; Metzger, D.; Ponglikitmongkol, M.; Park, I.; Chambon, P. The cloned human oestrogen receptor contains a mutation which alters its hormone binding properties. EMBO J. 1989, 8, 1981–1986. [Google Scholar] [CrossRef]
- Lo, R.; Burgoon, L.; Macpherson, L.; Ahmed, S.; Matthews, J. Estrogen receptor-dependent regulation of CYP2B6 in human breast cancer cells. Biochim. Biophys. Acta 2010, 1799, 469–479. [Google Scholar] [CrossRef] [Green Version]
- MacPherson, L.; Lo, R.; Ahmed, S.; Pansoy, A.; Matthews, J. Activation function 2 mediates dioxin-induced recruitment of estrogen receptor alpha to CYP1A1 and CYP1B1. Biochem. Biophys. Res. Commun. 2009, 385, 263–268. [Google Scholar] [CrossRef]
- Ahmed, S.; Wang, A.; Celius, T.; Matthews, J. Zinc finger nuclease-mediated knockout of AHR or ARNT in human breast cancer cells abolishes basal and ligand-dependent regulation of CYP1B1 and differentially affects estrogen receptor alpha transactivation. Toxicol. Sci. 2014, 138, 89–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, S.; Bott, D.; Gomez, A.; Tamblyn, L.; Rasheed, A.; Cho, T.; MacPherson, L.; Sugamori, K.S.; Yang, Y.; Grant, D.M.; et al. Loss of the Mono-ADP-ribosyltransferase, Tiparp, Increases Sensitivity to Dioxin-induced Steatohepatitis and Lethality. J. Biol. Chem. 2015, 290, 16824–16840. [Google Scholar] [CrossRef] [Green Version]
- Kamata, T.; Yang, C.S.; Melhuish, T.A.; Frierson, H.F., Jr.; Wotton, D.; Paschal, B.M. Post-Transcriptional Regulation of PARP7 Protein Stability Is Controlled by Androgen Signaling. Cells 2021, 10, 363. [Google Scholar] [CrossRef]
- Lumachi, F.; Brunello, A.; Maruzzo, M.; Basso, U.; Basso, S.M. Treatment of estrogen receptor-positive breast cancer. Curr. Med. Chem. 2013, 20, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Fan, M.; Nakshatri, H.; Nephew, K.P. Inhibiting proteasomal proteolysis sustains estrogen receptor-alpha activation. Mol. Endocrinol. 2004, 18, 2603–2615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wuhrer, M.; Deelder, A.M.; van der Burgt, Y.E. Mass spectrometric glycan rearrangements. Mass Spectrom. Rev. 2011, 30, 664–680. [Google Scholar] [CrossRef] [PubMed]
- Ohtake, F.; Baba, A.; Takada, I.; Okada, M.; Iwasaki, K.; Miki, H.; Takahashi, S.; Kouzmenko, A.; Nohara, K.; Chiba, T.; et al. Dioxin receptor is a ligand-dependent E3 ubiquitin ligase. Nature 2007, 446, 562–566. [Google Scholar] [CrossRef] [PubMed]
- Buch-Larsen, S.C.; Hendriks, I.A.; Lodge, J.M.; Rykaer, M.; Furtwangler, B.; Shishkova, E.; Westphall, M.S.; Coon, J.J.; Nielsen, M.L. Mapping Physiological ADP-Ribosylation Using Activated Ion Electron Transfer Dissociation. Cell Rep. 2020, 32, 108176. [Google Scholar] [CrossRef] [PubMed]
- Larsen, S.C.; Hendriks, I.A.; Lyon, D.; Jensen, L.J.; Nielsen, M.L. Systems-wide Analysis of Serine ADP-Ribosylation Reveals Widespread Occurrence and Site-Specific Overlap with Phosphorylation. Cell Rep. 2018, 24, 2493–2505.e4. [Google Scholar] [CrossRef] [Green Version]
- Han, W.D.; Zhao, Y.L.; Meng, Y.G.; Zang, L.; Wu, Z.Q.; Li, Q.; Si, Y.L.; Huang, K.; Ba, J.M.; Morinaga, H.; et al. Estrogenically regulated LRP16 interacts with estrogen receptor alpha and enhances the receptor’s transcriptional activity. Endocr. Relat. Cancer 2007, 14, 741–753. [Google Scholar] [CrossRef] [PubMed]
- Agnew, T.; Munnur, D.; Crawford, K.; Palazzo, L.; Mikoc, A.; Ahel, I. MacroD1 Is a Promiscuous ADP-Ribosyl Hydrolase Localized to Mitochondria. Front. Microbiol. 2018, 9, 20. [Google Scholar] [CrossRef] [Green Version]
- Bolton, E.C.; So, A.Y.; Chaivorapol, C.; Haqq, C.M.; Li, H.; Yamamoto, K.R. Cell- and gene-specific regulation of primary target genes by the androgen receptor. Genes Dev. 2007, 21, 2005–2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massie, C.E.; Lynch, A.; Ramos-Montoya, A.; Boren, J.; Stark, R.; Fazli, L.; Warren, A.; Scott, H.; Madhu, B.; Sharma, N.; et al. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J. 2011, 30, 2719–2733. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Zhao, Y.L.; Wu, Z.Q.; Si, Y.L.; Meng, Y.G.; Fu, X.B.; Mu, Y.M.; Han, W.D. The single-macro domain protein LRP16 is an essential cofactor of androgen receptor. Endocr. Relat. Cancer 2009, 16, 139–153. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rasmussen, M.; Tan, S.; Somisetty, V.S.; Hutin, D.; Olafsen, N.E.; Moen, A.; Anonsen, J.H.; Grant, D.M.; Matthews, J. PARP7 and Mono-ADP-Ribosylation Negatively Regulate Estrogen Receptor α Signaling in Human Breast Cancer Cells. Cells 2021, 10, 623. https://doi.org/10.3390/cells10030623
Rasmussen M, Tan S, Somisetty VS, Hutin D, Olafsen NE, Moen A, Anonsen JH, Grant DM, Matthews J. PARP7 and Mono-ADP-Ribosylation Negatively Regulate Estrogen Receptor α Signaling in Human Breast Cancer Cells. Cells. 2021; 10(3):623. https://doi.org/10.3390/cells10030623
Chicago/Turabian StyleRasmussen, Marit, Susanna Tan, Venkata S. Somisetty, David Hutin, Ninni Elise Olafsen, Anders Moen, Jan H. Anonsen, Denis M. Grant, and Jason Matthews. 2021. "PARP7 and Mono-ADP-Ribosylation Negatively Regulate Estrogen Receptor α Signaling in Human Breast Cancer Cells" Cells 10, no. 3: 623. https://doi.org/10.3390/cells10030623
APA StyleRasmussen, M., Tan, S., Somisetty, V. S., Hutin, D., Olafsen, N. E., Moen, A., Anonsen, J. H., Grant, D. M., & Matthews, J. (2021). PARP7 and Mono-ADP-Ribosylation Negatively Regulate Estrogen Receptor α Signaling in Human Breast Cancer Cells. Cells, 10(3), 623. https://doi.org/10.3390/cells10030623