
Role of Reductive versus Oxidative Stress in Tumor Progression and Anticancer Drug Resistance




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
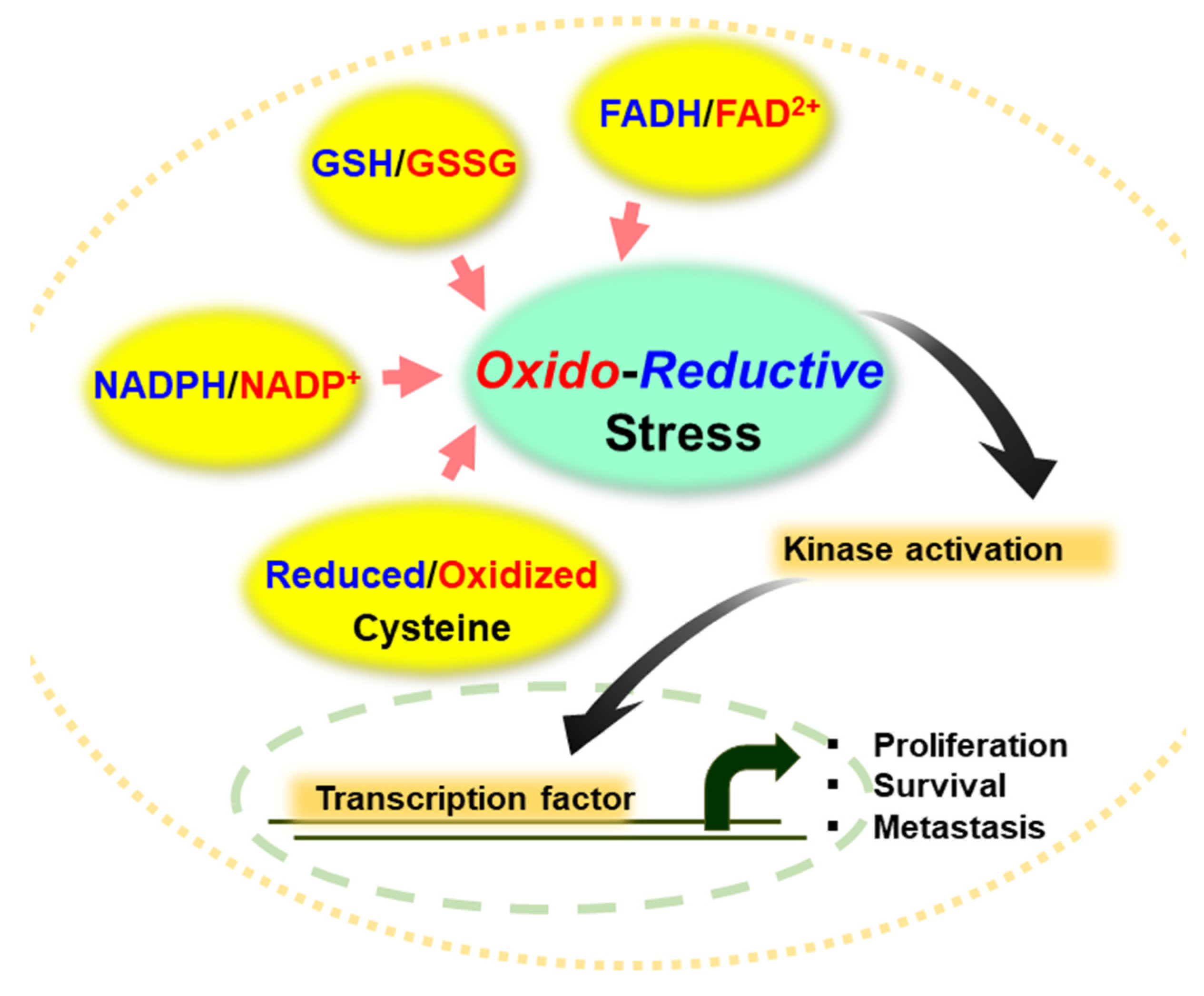
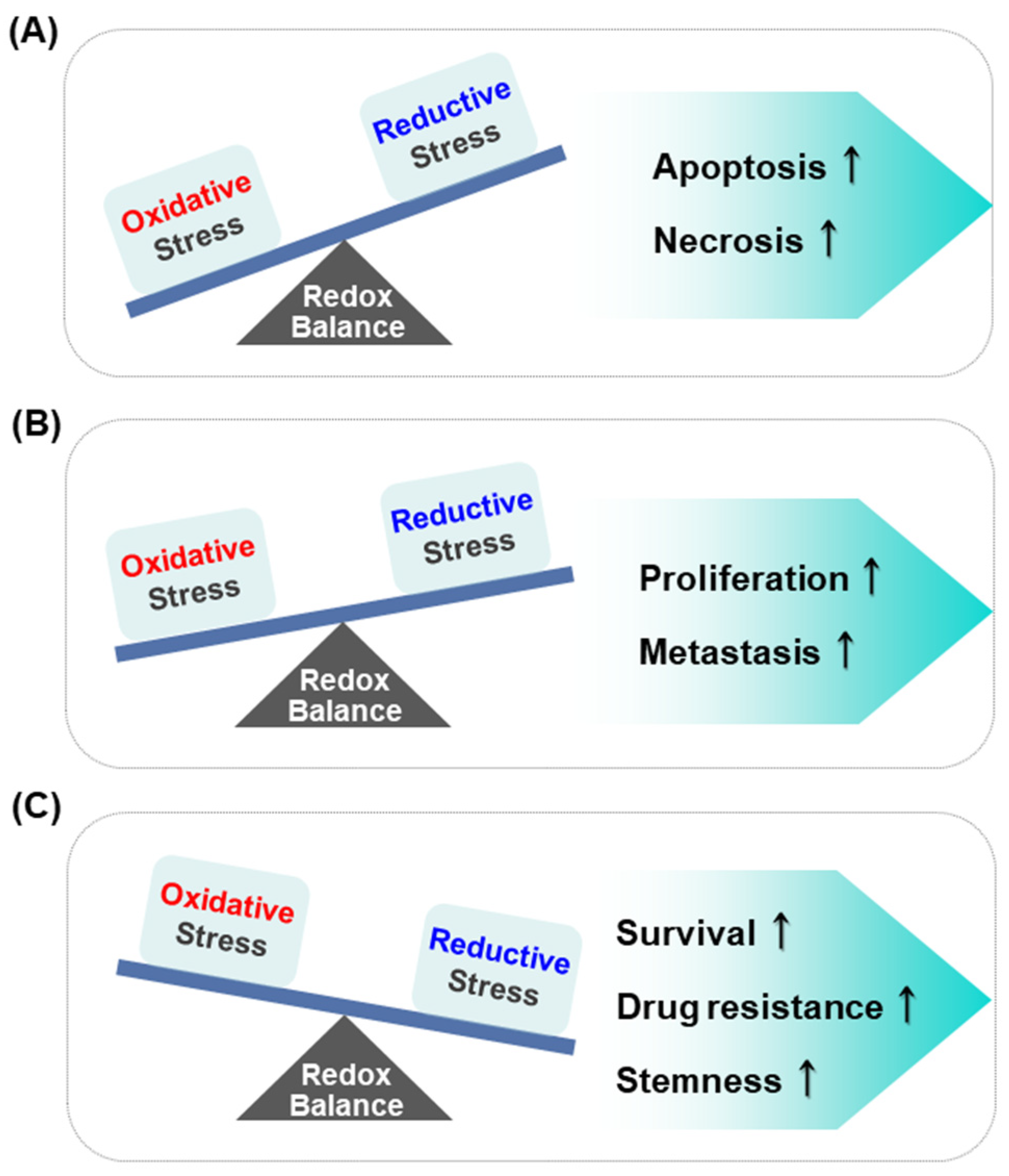
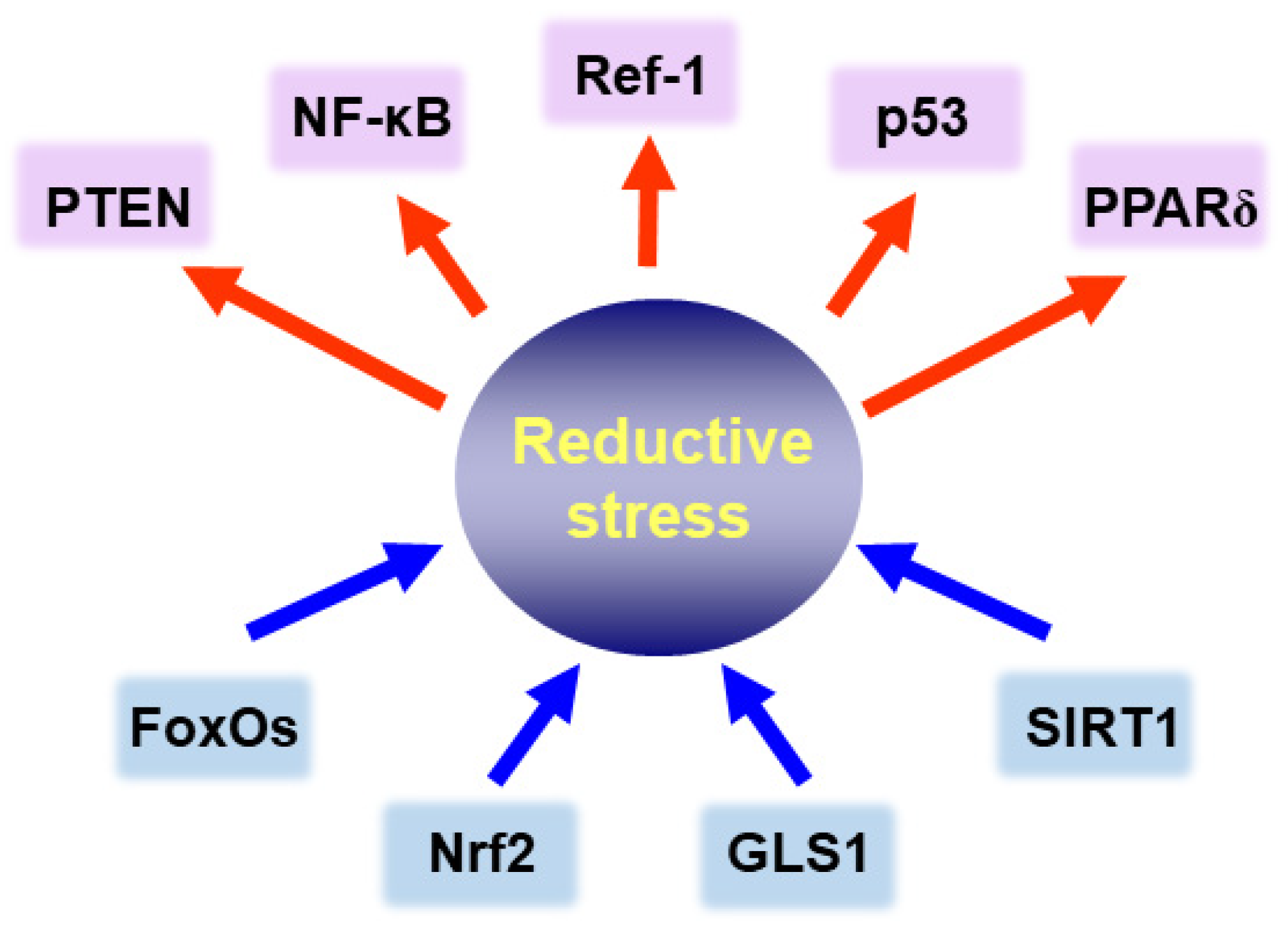
2. Reductive Stress as a Predisposing Factor for Tumorigenesis
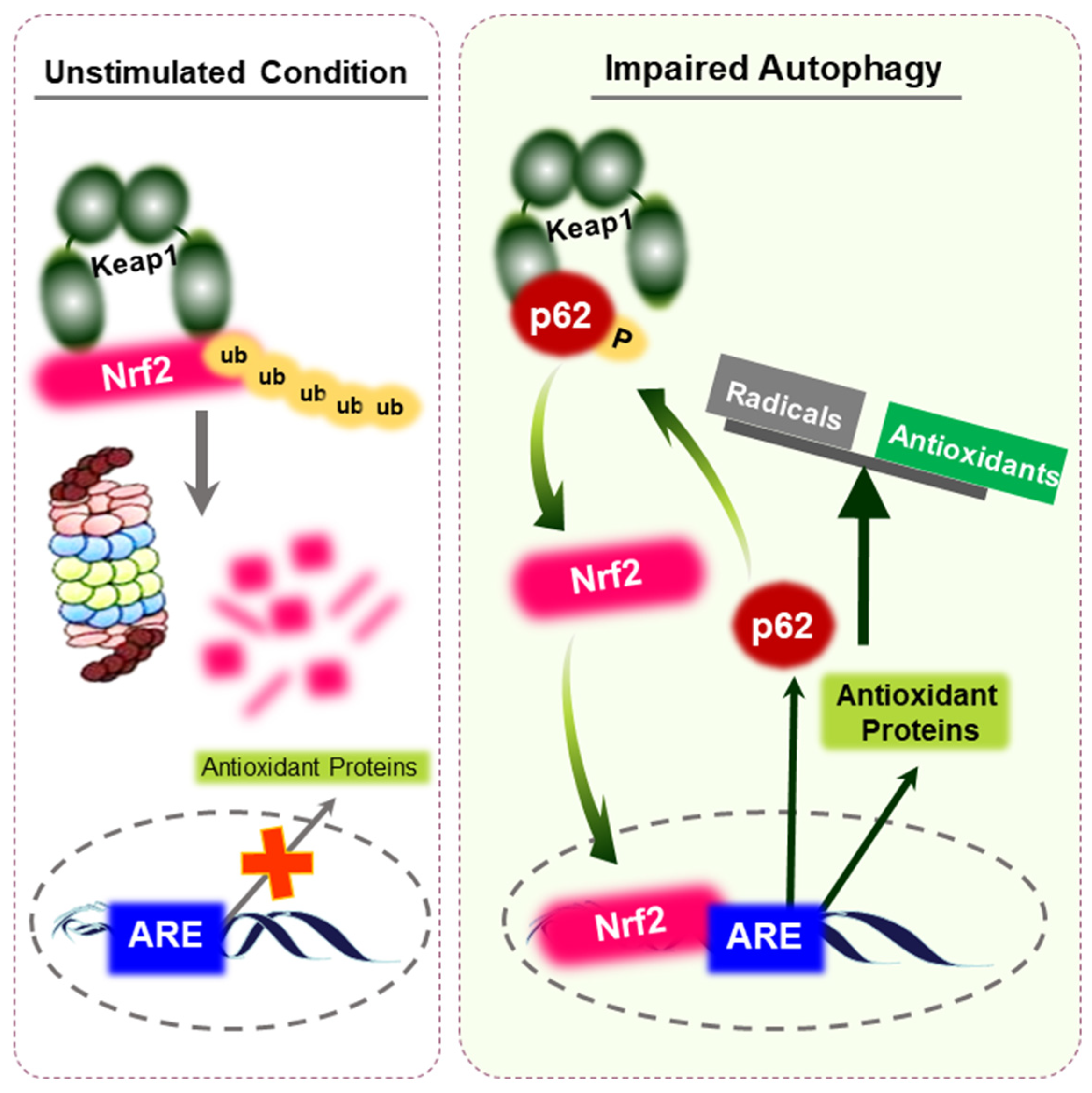
3. Nrf2 as a Master Regulator of Cellular Redox Balance
4. Role of Antioxidant Molecules in Reductive Stress Responsible for Cancer Progression and Drug Resistance
4.1. Glutathione (GSH)
4.2. Manganese Superoxide Dismutase (MnSOD)
4.3. Glutathione S-Transferases (GSTs)
4.4. Heme Oxygenase (HO)
5. Role of Metabolic Pathways in Linking Reductive Stress to Tumorigenesis
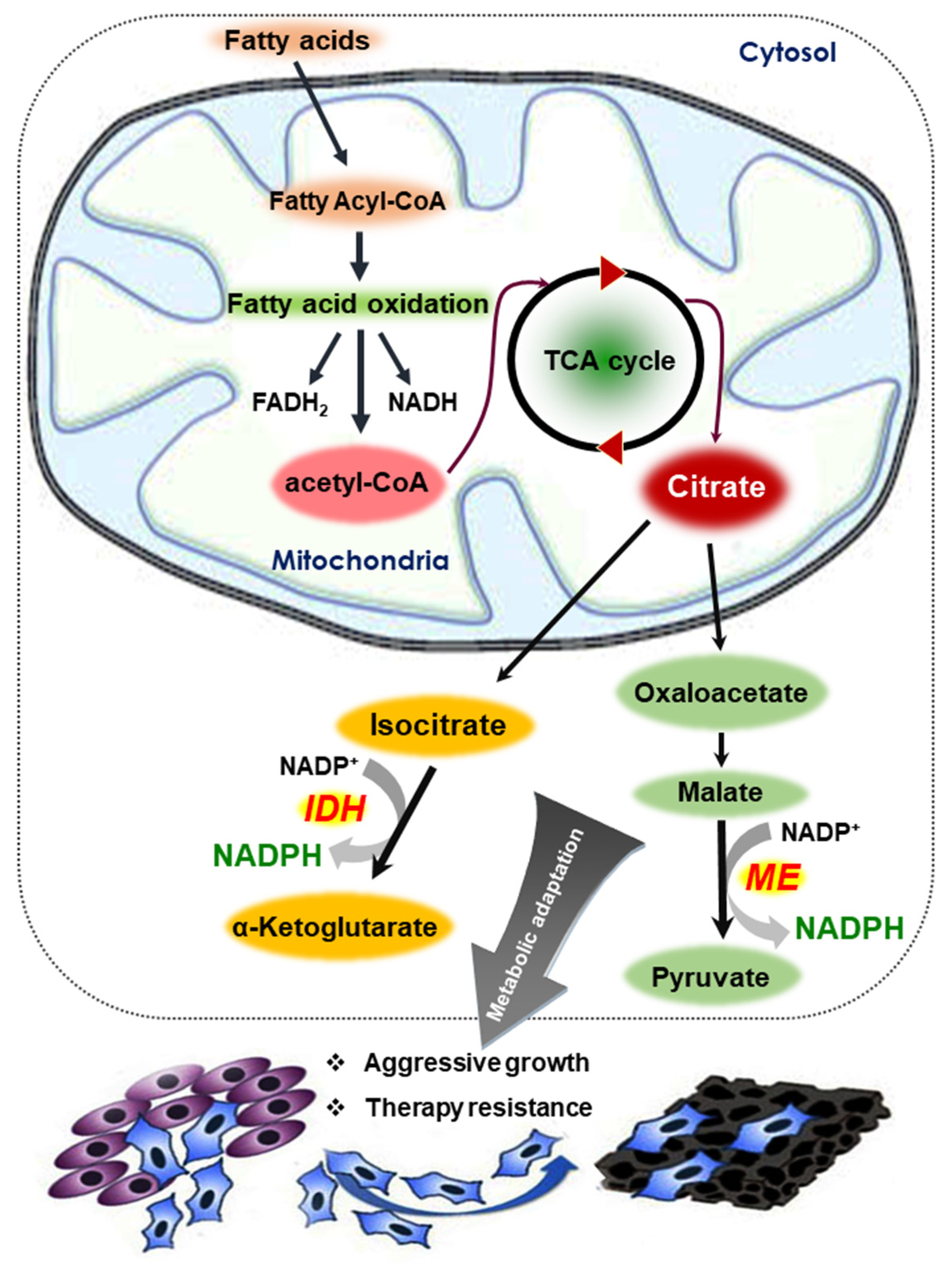
5.1. Fatty Acid Oxidation (FAO)
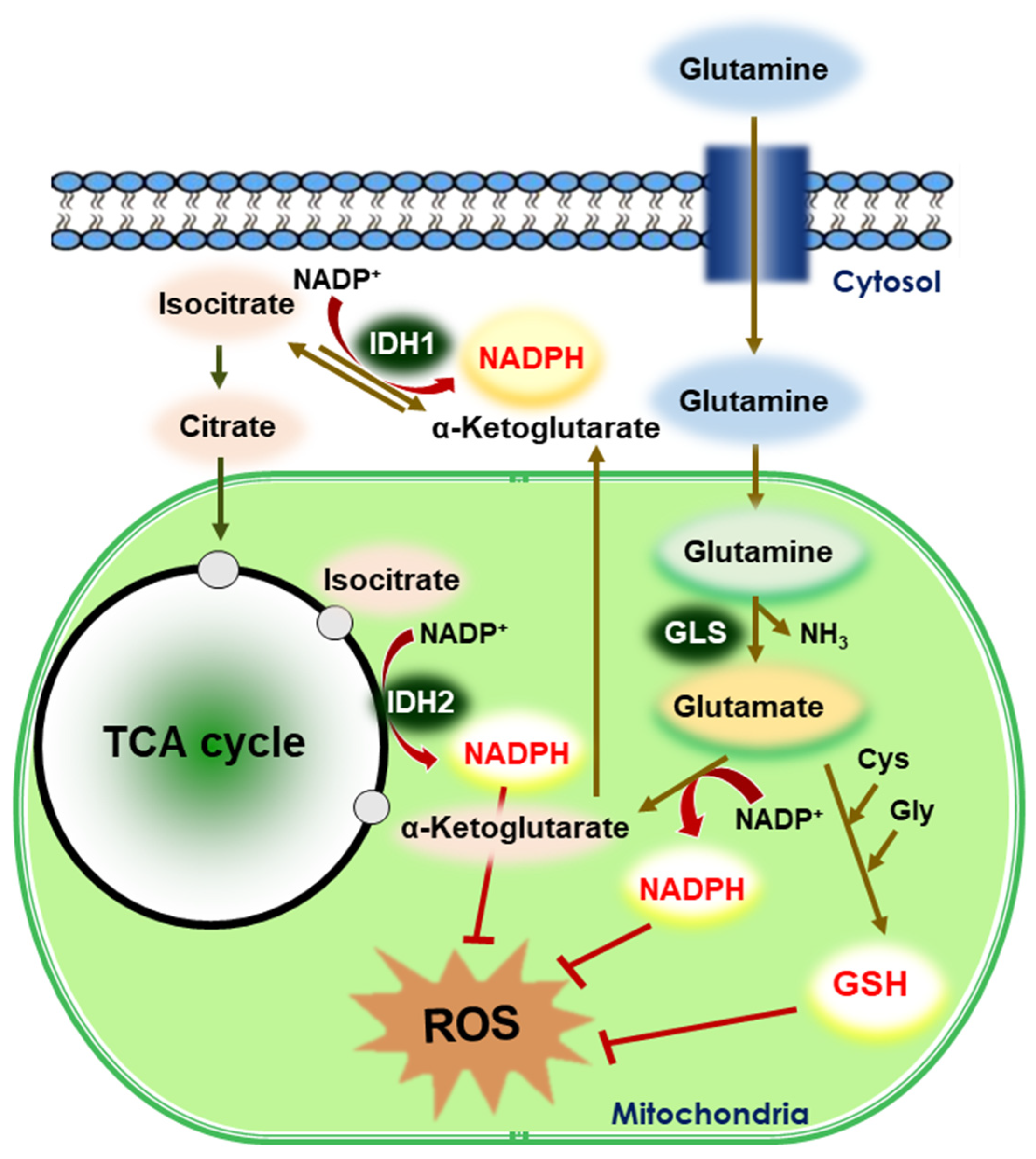
5.2. Glutaminolysis
6. Role of Redox-Regulated Signaling Molecules in Reductive Stress-Associated Tumorigenesis
6.1. Peroxisome Proliferator-Activated Receptor δ (PPAR δ)
6.2. Phosphatidylinositol 3-Kinase (PI3K)/Protein Kinase B (Akt)
6.3. Phosphatase and Tensin Homolog (PTEN)
6.4. Redox Factor-1 (Ref-1)
6.5. Forhead Box O (FoxO)
7. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Imlay, J.A. The molecular mechanisms and physiological consequences of oxidative stress: Lessons from a model bacterium. Nat. Rev. Microbiol. 2013, 11, 443–454. [Google Scholar] [CrossRef] [Green Version]
- Kalyanaraman, B. Teaching the basics of redox biology to medical and graduate students: Oxidants, antioxidants and disease mechanisms. Redox Biol. 2013, 1, 244–257. [Google Scholar] [CrossRef] [Green Version]
- Liochev, S.I. Reactive oxygen species and the free radical theory of aging. Free Radic. Biol. Med. 2013, 60, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Loscalzo, J. Metabolic responses to reductive stress. Antioxid. Redox Signal. 2020, 32, 1330–1347. [Google Scholar] [CrossRef] [Green Version]
- Kehrer, J.P.; Lund, L.G. Cellular reducing equivalents and oxidative stress. Free Radic. Biol. Med. 1994, 17, 65–75. [Google Scholar] [CrossRef]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bae, I.; Fan, S.; Meng, Q.; Rih, J.K.; Kim, H.J.; Kang, H.J.; Xu, J.; Goldberg, I.D.; Jaiswal, A.K.; Rosen, E.M. BRCA1 induces antioxidant gene expression and resistance to oxidative stress. Cancer Res. 2004, 64, 7893–7909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishimoto, T.; Nagano, O.; Yae, T.; Tamada, M.; Motohara, T.; Oshima, H.; Oshima, M.; Ikeda, T.; Asaba, R.; Yagi, H.; et al. CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(-) and thereby promotes tumor growth. Cancer Cell 2011, 19, 387–400. [Google Scholar] [CrossRef] [Green Version]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef]
- Masella, R.; Di Benedetto, R.; Vari, R.; Filesi, C.; Giovannini, C. Novel mechanisms of natural antioxidant compounds in biological systems: Involvement of glutathione and glutathione-related enzymes. J. Nutr. Biochem. 2005, 16, 577–586. [Google Scholar] [CrossRef]
- Liu, Y.; Li, Q.; Zhou, L.; Xie, N.; Nice, E.C.; Zhang, H.; Huang, C.; Lei, Y. Cancer drug resistance: Redox resetting renders a way. Oncotarget 2016, 7, 42740–42761. [Google Scholar] [CrossRef] [Green Version]
- Hayes, J.D.; McMahon, M. The double-edged sword of Nrf2: Subversion of redox homeostasis during the evolution of cancer. Mol. Cell 2006, 21, 732–734. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.D.; Hannink, M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol. Cell. Biol. 2003, 23, 8137–8151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Sporn, M.B.; Liby, K.T. NRF2 and cancer: The good, the bad and the importance of context. Nat. Rev. Cancer 2012, 12, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Ohta, T.; Iijima, K.; Miyamoto, M.; Nakahara, I.; Tanaka, H.; Ohtsuji, M.; Suzuki, T.; Kobayashi, A.; Yokota, J.; Sakiyama, T.; et al. Loss of Keap1 function activates Nrf2 and provides advantages for lung cancer cell growth. Cancer Res. 2008, 68, 1303–1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nioi, P.; Nguyen, T. A mutation of Keap1 found in breast cancer impairs its ability to repress Nrf2 activity. Biochem. Biophys. Res. Commun. 2007, 362, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Konstantinopoulos, P.A.; Spentzos, D.; Fountzilas, E.; Francoeur, N.; Sanisetty, S.; Grammatikos, A.P.; Hecht, J.L.; Cannistra, S.A. Keap1 mutations and Nrf2 pathway activation in epithelial ovarian cancer. Cancer Res. 2011, 71, 5081–5089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solis, L.M.; Behrens, C.; Dong, W.; Suraokar, M.; Ozburn, N.C.; Moran, C.A.; Corvalan, A.H.; Biswal, S.; Swisher, S.G.; Bekele, B.N.; et al. Nrf2 and Keap1 abnormalities in non-small cell lung carcinoma and association with clinicopathologic features. Clin. Cancer Res. 2010, 16, 3743–3753. [Google Scholar] [CrossRef] [Green Version]
- Kimmelman, A.C.; White, E. Autophagy and tumor metabolism. Cell Metab. 2017, 25, 1037–1043. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Yepes, J.; Burns, M.; Anandhan, A.; Khalimonchuk, O.; del Razo, L.M.; Quintanilla-Vega, B.; Pappa, A.; Panayiotidis, M.I.; Franco, R. Oxidative stress, redox signaling, and autophagy: Cell death versus survival. Antioxid. Redox Signal. 2014, 21, 66–85. [Google Scholar] [CrossRef] [Green Version]
- Bjorkoy, G.; Lamark, T.; Johansen, T. p62/SQSTM1: A missing link between protein aggregates and the autophagy machinery. Autophagy 2006, 2, 138–139. [Google Scholar] [CrossRef] [Green Version]
- Dodson, M.; Redmann, M.; Rajasekaran, N.S.; Darley-Usmar, V.; Zhang, J. KEAP1-NRF2 signalling and autophagy in protection against oxidative and reductive proteotoxicity. Biochem. J. 2015, 469, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Dialynas, G.; Shrestha, O.K.; Ponce, J.M.; Zwerger, M.; Thiemann, D.A.; Young, G.H.; Moore, S.A.; Yu, L.; Lammerding, J.; Wallrath, L.L. Myopathic lamin mutations cause reductive stress and activate the nrf2/keap-1 pathway. PLoS Genet. 2015, 11, e1005231. [Google Scholar] [CrossRef] [Green Version]
- Galavotti, S.; Bartesaghi, S.; Faccenda, D.; Shaked-Rabi, M.; Sanzone, S.; McEvoy, A.; Dinsdale, D.; Condorelli, F.; Brandner, S.; Campanella, M.; et al. The autophagy-associated factors DRAM1 and p62 regulate cell migration and invasion in glioblastoma stem cells. Oncogene 2013, 32, 699–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, D.; Suzuki, T.; Mitsuishi, Y.; Miki, Y.; Suzuki, S.; Sugawara, S.; Watanabe, M.; Sakurada, A.; Endo, C.; Uruno, A.; et al. Accumulation of p62/SQSTM1 is associated with poor prognosis in patients with lung adenocarcinoma. Cancer Sci. 2012, 103, 760–766. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.Z.; Yuan, Z.Y.; Li, M.; Xi, S.Y.; Fu, J.; He, J. Accumulation of p62 is associated with poor prognosis in patients with triple-negative breast cancer. Onco Targets Ther. 2013, 6, 883–888. [Google Scholar] [CrossRef] [Green Version]
- Inami, Y.; Waguri, S.; Sakamoto, A.; Kouno, T.; Nakada, K.; Hino, O.; Watanabe, S.; Ando, J.; Iwadate, M.; Yamamoto, M.; et al. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J. Cell Biol. 2011, 193, 275–284. [Google Scholar] [CrossRef] [Green Version]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef]
- Fojo, T.; Bates, S. Strategies for reversing drug resistance. Oncogene 2003, 22, 7512–7523. [Google Scholar] [CrossRef] [Green Version]
- De Larco, J.E.; Park, C.A.; Dronava, H.; Furcht, L.T. Paradoxical roles for antioxidants in tumor prevention and eradication. Cancer Biol. Ther. 2010, 9, 362–370. [Google Scholar] [CrossRef] [Green Version]
- Meister, A. Glutathione deficiency produced by inhibition of its synthesis, and its reversal; applications in research and therapy. Pharmacol. Ther. 1991, 51, 155–194. [Google Scholar] [CrossRef]
- Ortega, A.L.; Mena, S.; Estrela, J.M. Glutathione in cancer cell death. Cancers 2011, 3, 1285–1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeda, H.; Hori, S.; Ohizumi, H.; Segawa, T.; Kakehi, Y.; Ogawa, O.; Kakizuka, A. Effective treatment of advanced solid tumors by the combination of arsenic trioxide and L-buthionine-sulfoximine. Cell Death Differ. 2004, 11, 737–746. [Google Scholar] [CrossRef] [Green Version]
- Sandhya, T.; Mishra, K.P. Cytotoxic response of breast cancer cell lines, MCF 7 and T 47 D to triphala and its modification by antioxidants. Cancer Lett. 2006, 238, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Kasherman, Y.; Sturup, S.; Gibson, D. Is glutathione the major cellular target of cisplatin? A study of the interactions of cisplatin with cancer cell extracts. J. Med. Chem. 2009, 52, 4319–4328. [Google Scholar] [CrossRef]
- Lan, D.; Wang, L.; He, R.; Ma, J.; Bin, Y.; Chi, X.; Chen, G.; Cai, Z. Exogenous glutathione contributes to cisplatin resistance in lung cancer A549 cells. Am. J. Transl. Res. 2018, 10, 1295–1309. [Google Scholar]
- Wartenberg, M.; Ling, F.C.; Schallenberg, M.; Baumer, A.T.; Petrat, K.; Hescheler, J.; Sauer, H. Down-regulation of intrinsic P-glycoprotein expression in multicellular prostate tumor spheroids by reactive oxygen species. J. Biol. Chem. 2001, 276, 17420–17428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Stokes, W.; Chater, E.; Roy, R.; de Bruin, E.; Hu, Y.; Liu, Z.; Smit, E.F.; Heynen, G.J.; Downward, J.; et al. Decreased glutathione biosynthesis contributes to EGFR T790M-driven erlotinib resistance in non-small cell lung cancer. Cell Discov. 2016, 2, 16031. [Google Scholar] [CrossRef] [Green Version]
- Fu, A.; Ma, S.; Wei, N.; Tan, B.X.; Tan, E.Y.; Luo, K.Q. High expression of MnSOD promotes survival of circulating breast cancer cells and increases their resistance to doxorubicin. Oncotarget 2016, 7, 50239–50257. [Google Scholar] [CrossRef]
- Hart, P.C.; Mao, M.; de Abreu, A.L.; Ansenberger-Fricano, K.; Ekoue, D.N.; Ganini, D.; Kajdacsy-Balla, A.; Diamond, A.M.; Minshall, R.D.; Consolaro, M.E.; et al. MnSOD upregulation sustains the Warburg effect via mitochondrial ROS and AMPK-dependent signalling in cancer. Nat. Commun. 2015, 6, 6053. [Google Scholar] [CrossRef] [Green Version]
- Lai, Y.; Yu, X.; Lin, X.; He, S. Inhibition of mTOR sensitizes breast cancer stem cells to radiation-induced repression of self-renewal through the regulation of MnSOD and Akt. Int. J. Mol. Med. 2016, 37, 369–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.Y.; Saeidi, S.; Kim, E.H.; Kim, D.H.; Na, H.K.; Keum, J.S.; Surh, Y.J. Heregulin-β1 activates NF-E2-related factor 2 and induces manganese superoxide dismutase expression in human breast cancer cells via Akt and extracellular signal-regulated protein kinase signaling pathways. J. Cancer Prev. 2021, in press. [Google Scholar]
- Townsend, D.M.; Tew, K.D. The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene 2003, 22, 7369–7375. [Google Scholar] [CrossRef] [Green Version]
- Cairns, J.; Wright, C.; Cattan, A.R.; Hall, A.G.; Cantwell, B.J.; Harris, A.L.; Horne, C.H. Immunohistochemical demonstration of glutathione S-transferases in primary human breast carcinomas. J. Pathol. 1992, 166, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Di Ilio, C.; Aceto, A.; Bucciarelli, T.; Angelucci, S.; Felaco, M.; Grilli, A.; Zezza, A.; Tenaglia, R.; Federici, G. Glutathione transferase isoenzymes in normal and neoplastic human kidney tissue. Carcinogenesis 1991, 12, 1471–1475. [Google Scholar] [CrossRef]
- Mantovani, G.; Maccio, A.; Massa, E.; Mulas, C.; Mudu, M.C.; Massidda, S.; Massa, D.; Murgia, V.; Ferreli, L.; Succu, G.; et al. Phase II study of induction chemotherapy followed by concomitant chemoradiotherapy in advanced head and neck cancer: Clinical outcome, toxicity and organ/function preservation. Int. J. Oncol. 2000, 16, 1227–1233. [Google Scholar] [CrossRef]
- Kartalou, M.; Essigmann, J.M. Mechanisms of resistance to cisplatin. Mutat. Res. 2001, 478, 23–43. [Google Scholar] [CrossRef]
- Cullen, K.J.; Newkirk, K.A.; Schumaker, L.M.; Aldosari, N.; Rone, J.D.; Haddad, B.R. Glutathione S-transferase π amplification is associated with cisplatin resistance in head and neck squamous cell carcinoma cell lines and primary tumors. Cancer Res. 2003, 63, 8097–8102. [Google Scholar]
- Saleh, E.M.; El-Awady, R.A.; Abdel Alim, M.A.; Abdel Wahab, A.H. Altered expression of proliferation-inducing and proliferation-inhibiting genes might contribute to acquired doxorubicin resistance in breast cancer cells. Cell Biochem. Biophys. 2009, 55, 95–105. [Google Scholar] [CrossRef]
- Sun, F.F.; Hu, Y.H.; Xiong, L.P.; Tu, X.Y.; Zhao, J.H.; Chen, S.S.; Song, J.; Ye, X.Q. Enhanced expression of stem cell markers and drug resistance in sphere-forming non-small cell lung cancer cells. Int. J. Clin. Exp. Pathol. 2015, 8, 6287–6300. [Google Scholar]
- Yi, L.; Jenkins, P.M.; Leichert, L.I.; Jakob, U.; Martens, J.R.; Ragsdale, S.W. Heme regulatory motifs in heme oxygenase-2 form a thiol/disulfide redox switch that responds to the cellular redox state. J. Biol. Chem. 2009, 284, 20556–20561. [Google Scholar] [CrossRef] [Green Version]
- Yi, L.; Ragsdale, S.W. Evidence that the heme regulatory motifs in heme oxygenase-2 serve as a thiol/disulfide redox switch regulating heme binding. J. Biol. Chem. 2007, 282, 21056–21067. [Google Scholar] [CrossRef] [Green Version]
- Destefanis, F.; Fiorito, V.; Altruda, F.; Tolosano, E. Investigating the connection between endogenous heme accumulation and COX2 activity in cancer cells. Front. Oncol. 2019, 9, 162. [Google Scholar] [CrossRef]
- Kim, J.J.; Lee, Y.A.; Su, D.; Lee, J.; Park, S.J.; Kim, B.; Jane Lee, J.H.; Liu, X.; Kim, S.S.; Bae, M.A.; et al. A near-infrared probe tracks and treats lung tumor initiating cells by targeting HMOX2. J. Am. Chem. Soc. 2019, 141, 14673–14686. [Google Scholar] [CrossRef]
- Menegon, S.; Columbano, A.; Giordano, S. The dual roles of NRF2 in cancer. Trends Mol. Med. 2016, 22, 578–593. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Butler, E.B.; Tan, M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis. 2013, 4, e532. [Google Scholar] [CrossRef] [Green Version]
- De Santis, M.C.; Porporato, P.E.; Martini, M.; Morandi, A. Signaling pathways regulating redox balance in cancer metabolism. Front. Oncol. 2018, 8, 126. [Google Scholar] [CrossRef]
- Anastasiou, D.; Poulogiannis, G.; Asara, J.M.; Boxer, M.B.; Jiang, J.K.; Shen, M.; Bellinger, G.; Sasaki, A.T.; Locasale, J.W.; Auld, D.S.; et al. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science 2011, 334, 1278–1283. [Google Scholar] [CrossRef] [Green Version]
- Piskounova, E.; Agathocleous, M.; Murphy, M.M.; Hu, Z.; Huddlestun, S.E.; Zhao, Z.; Leitch, A.M.; Johnson, T.M.; DeBerardinis, R.J.; Morrison, S.J. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 2015, 527, 186–191. [Google Scholar] [CrossRef] [Green Version]
- Dang, C.V. Links between metabolism and cancer. Genes Dev. 2012, 26, 877–890. [Google Scholar] [CrossRef] [Green Version]
- Sainz, R.M.; Lombo, F.; Mayo, J.C. Radical decisions in cancer: Redox control of cell growth and death. Cancers 2012, 4, 442–474. [Google Scholar] [CrossRef] [Green Version]
- Carracedo, A.; Cantley, L.C.; Pandolfi, P.P. Cancer metabolism: Fatty acid oxidation in the limelight. Nat. Rev. Cancer 2013, 13, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Schafer, Z.T.; Grassian, A.R.; Song, L.; Jiang, Z.; Gerhart-Hines, Z.; Irie, H.Y.; Gao, S.; Puigserver, P.; Brugge, J.S. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 2009, 461, 109–113. [Google Scholar] [CrossRef] [Green Version]
- Jeon, S.M.; Chandel, N.S.; Hay, N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature 2012, 485, 661–665. [Google Scholar] [CrossRef] [Green Version]
- Bensaad, K.; Favaro, E.; Lewis, C.A.; Peck, B.; Lord, S.; Collins, J.M.; Pinnick, K.E.; Wigfield, S.; Buffa, F.M.; Li, J.L.; et al. Fatty acid uptake and lipid storage induced by HIF-1α contribute to cell growth and survival after hypoxia-reoxygenation. Cell Rep. 2014, 9, 349–365. [Google Scholar] [CrossRef] [Green Version]
- Pike, L.S.; Smift, A.L.; Croteau, N.J.; Ferrick, D.A.; Wu, M. Inhibition of fatty acid oxidation by etomoxir impairs NADPH production and increases reactive oxygen species resulting in ATP depletion and cell death in human glioblastoma cells. Biochim. Biophys. Acta 2011, 1807, 726–734. [Google Scholar] [CrossRef] [Green Version]
- Samudio, I.; Harmancey, R.; Fiegl, M.; Kantarjian, H.; Konopleva, M.; Korchin, B.; Kaluarachchi, K.; Bornmann, W.; Duvvuri, S.; Taegtmeyer, H.; et al. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J. Clin. Investig. 2010, 120, 142–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andela, V.B.; Altuwaijri, S.; Wood, J.; Rosier, R.N. Inhibition of beta-oxidative respiration is a therapeutic window associated with the cancer chemo-preventive activity of PPARγ agonists. FEBS Lett. 2005, 579, 1765–1769. [Google Scholar] [CrossRef] [Green Version]
- Coloff, J.L.; Brugge, J.S. Coping with the metabolic stress of leaving home. Cell Res. 2016, 26, 757–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molenaar, R.J.; Botman, D.; Smits, M.A.; Hira, V.V.; van Lith, S.A.; Stap, J.; Henneman, P.; Khurshed, M.; Lenting, K.; Mul, A.N.; et al. Radioprotection of IDH1-mutated cancer cells by the IDH1-mutant inhibitor AGI-5198. Cancer Res. 2015, 75, 4790–4802. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Sun, B.; Shi, W.; Zuo, H.; Cui, D.; Ni, L.; Chen, J. Decreasing GSH and increasing ROS in chemosensitivity gliomas with IDH1 mutation. Tumour Biol. 2015, 36, 655–662. [Google Scholar] [CrossRef]
- Gelman, S.J.; Naser, F.; Mahieu, N.G.; McKenzie, L.D.; Dunn, G.P.; Chheda, M.G.; Patti, G.J. Consumption of NADPH for 2-HG synthesis increases pentose phosphate pathway flux and sensitizes cells to oxidative stress. Cell Rep. 2018, 22, 512–522. [Google Scholar] [CrossRef] [Green Version]
- Calvert, A.E.; Chalastanis, A.; Wu, Y.; Hurley, L.A.; Kouri, F.M.; Bi, Y.; Kachman, M.; May, J.L.; Bartom, E.; Hua, Y.; et al. Cancer-associated IDH1 promotes growth and resistance to targeted therapies in the absence of mutation. Cell Rep. 2017, 19, 1858–1873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Perez, M.V.; Sanchez-Jimenez, F.; Alonso, F.J.; Segura, J.A.; Marquez, J.; Medina, M.A. Glutamine, glucose and other fuels for cancer. Curr. Pharm. Des. 2014, 20, 2557–2579. [Google Scholar] [CrossRef] [PubMed]
- Daye, D.; Wellen, K.E. Metabolic reprogramming in cancer: Unraveling the role of glutamine in tumorigenesis. Semin. Cell Dev. Biol. 2012, 23, 362–369. [Google Scholar] [CrossRef]
- Hensley, C.T.; Wasti, A.T.; DeBerardinis, R.J. Glutamine and cancer: Cell biology, physiology, and clinical opportunities. J. Clin. Investig. 2013, 123, 3678–3684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izaki, S.; Goto, H.; Yokota, S. Increased chemosensitivity and elevated reactive oxygen species are mediated by glutathione reduction in glutamine deprived neuroblastoma cells. J. Cancer Res. Clin. Oncol. 2008, 134, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Goto, M.; Miwa, H.; Shikami, M.; Tsunekawa-Imai, N.; Suganuma, K.; Mizuno, S.; Takahashi, M.; Mizutani, M.; Hanamura, I.; Nitta, M. Importance of glutamine metabolism in leukemia cells by energy production through TCA cycle and by redox homeostasis. Cancer Investig. 2014, 32, 241–247. [Google Scholar] [CrossRef]
- Jiang, L.; Shestov, A.A.; Swain, P.; Yang, C.; Parker, S.J.; Wang, Q.A.; Terada, L.S.; Adams, N.D.; McCabe, M.T.; Pietrak, B.; et al. Reductive carboxylation supports redox homeostasis during anchorage-independent growth. Nature 2016, 532, 255–258. [Google Scholar] [CrossRef]
- Tennant, D.A.; Duran, R.V.; Gottlieb, E. Targeting metabolic transformation for cancer therapy. Nat. Rev. Cancer 2010, 10, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Le, A.; Hancock, C.; Lane, A.N.; Dang, C.V.; Fan, T.W.; Phang, J.M. Reprogramming of proline and glutamine metabolism contributes to the proliferative and metabolic responses regulated by oncogenic transcription factor c-MYC. Proc. Natl. Acad. Sci. USA 2012, 109, 8983–8988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Csibi, A.; Fendt, S.M.; Li, C.; Poulogiannis, G.; Choo, A.Y.; Chapski, D.J.; Jeong, S.M.; Dempsey, J.M.; Parkhitko, A.; Morrison, T.; et al. The mTORC1 pathway stimulates glutamine metabolism and cell proliferation by repressing SIRT4. Cell 2013, 153, 840–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, K.; Sasayama, T.; Irino, Y.; Takata, K.; Nagashima, H.; Satoh, N.; Kyotani, K.; Mizowaki, T.; Imahori, T.; Ejima, Y.; et al. Compensatory glutamine metabolism promotes glioblastoma resistance to mTOR inhibitor treatment. J. Clin. Investig. 2015, 125, 1591–1602. [Google Scholar] [CrossRef]
- Jeon, Y.J.; Khelifa, S.; Ratnikov, B.; Scott, D.A.; Feng, Y.; Parisi, F.; Ruller, C.; Lau, E.; Kim, H.; Brill, L.M.; et al. Regulation of glutamine carrier proteins by RNF5 determines breast cancer response to ER stress-inducing chemotherapies. Cancer Cell 2015, 27, 354–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2011, 481, 380–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullen, A.R.; Wheaton, W.W.; Jin, E.S.; Chen, P.H.; Sullivan, L.B.; Cheng, T.; Yang, Y.; Linehan, W.M.; Chandel, N.S.; DeBerardinis, R.J. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 2011, 481, 385–388. [Google Scholar] [CrossRef] [Green Version]
- Wise, D.R.; Ward, P.S.; Shay, J.E.; Cross, J.R.; Gruber, J.J.; Sachdeva, U.M.; Platt, J.M.; DeMatteo, R.G.; Simon, M.C.; Thompson, C.B. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of α-ketoglutarate to citrate to support cell growth and viability. Proc. Natl. Acad. Sci. USA 2011, 108, 19611–19616. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, M.; Numazawa, S.; Yoshida, T. Redox regulation of the transcriptional repressor Bach1. Free Radic. Biol. Med. 2005, 38, 1344–1352. [Google Scholar] [CrossRef]
- Liu, H.; Colavitti, R.; Rovira, I.I.; Finkel, T. Redox-dependent transcriptional regulation. Circ. Res. 2005, 97, 967–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaikhali, J.; Noren, L.; de Dios Barajas-Lopez, J.; Srivastava, V.; Konig, J.; Sauer, U.H.; Wingsle, G.; Dietz, K.J.; Strand, A. Redox-mediated mechanisms regulate DNA binding activity of the G-group of basic region leucine zipper (bZIP) transcription factors in Arabidopsis. J. Biol. Chem. 2012, 287, 27510–27525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearen, M.A.; Muscat, G.E. Orphan nuclear receptors and the regulation of nutrient metabolism: Understanding obesity. Physiology 2012, 27, 156–166. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.X.; Lee, C.H.; Tiep, S.; Yu, R.T.; Ham, J.; Kang, H.; Evans, R.M. Peroxisome-proliferator-activated receptor δ activates fat metabolism to prevent obesity. Cell 2003, 113, 159–170. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Wang, H.; Guo, Y.; Ning, W.; Katkuri, S.; Wahli, W.; Desvergne, B.; Dey, S.K.; DuBois, R.N. Crosstalk between peroxisome proliferator-activated δ and VEGF stimulates cancer progression. Proc. Natl. Acad. Sci. USA 2006, 103, 19069–19074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.J.; Choi, Y.K.; Park, S.Y.; Jang, S.Y.; Lee, J.Y.; Ham, H.J.; Kim, B.G.; Jeon, H.J.; Kim, J.H.; Kim, J.G.; et al. PPARδ reprograms glutamine metabolism in sorafenib-resistant HCC. Mol. Cancer Res. 2017, 15, 1230–1242. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wang, G.; Shi, Y.; Sun, L.; Gorczynski, R.; Li, Y.J.; Xu, Z.; Spaner, D.E. PPAR-δ promotes survival of breast cancer cells in harsh metabolic conditions. Oncogenesis 2016, 5, e232. [Google Scholar] [CrossRef] [Green Version]
- Spitz, D.R.; Sim, J.E.; Ridnour, L.A.; Galoforo, S.S.; Lee, Y.J. Glucose deprivation-induced oxidative stress in human tumor cells. A fundamental defect in metabolism? Ann. N. Y. Acad. Sci. 2000, 899, 349–362. [Google Scholar] [CrossRef]
- Kim, T.; Yang, Q. Peroxisome-proliferator-activated receptors regulate redox signaling in the cardiovascular system. World J. Cardiol. 2013, 5, 164–174. [Google Scholar] [CrossRef]
- Fruman, D.A.; Rommel, C. PI3K and cancer: Lessons, challenges and opportunities. Nat. Rev. Drug Discov. 2014, 13, 140–156. [Google Scholar] [CrossRef] [Green Version]
- Nakaso, K.; Yano, H.; Fukuhara, Y.; Takeshima, T.; Wada-Isoe, K.; Nakashima, K. PI3K is a key molecule in the Nrf2-mediated regulation of antioxidative proteins by hemin in human neuroblastoma cells. FEBS Lett. 2003, 546, 181–184. [Google Scholar] [CrossRef] [Green Version]
- Gorrini, C.; Gang, B.P.; Bassi, C.; Wakeham, A.; Baniasadi, S.P.; Hao, Z.; Li, W.Y.; Cescon, D.W.; Li, Y.T.; Molyneux, S.; et al. Estrogen controls the survival of BRCA1-deficient cells via a PI3K-NRF2-regulated pathway. Proc. Natl. Acad. Sci. USA 2014, 111, 4472–4477. [Google Scholar] [CrossRef] [Green Version]
- Nogueira, V.; Park, Y.; Chen, C.C.; Xu, P.Z.; Chen, M.L.; Tonic, I.; Unterman, T.; Hay, N. Akt determines replicative senescence and oxidative or oncogenic premature senescence and sensitizes cells to oxidative apoptosis. Cancer Cell 2008, 14, 458–470. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.H.; Suh, J.; Surh, Y.J.; Na, H.K. Regulation of the tumor suppressor PTEN by natural anticancer compounds. Ann. N. Y. Acad. Sci. 2017, 1401, 136–149. [Google Scholar] [CrossRef]
- Ali, I.U.; Schriml, L.M.; Dean, M. Mutational spectra of PTEN/MMAC1 gene: A tumor suppressor with lipid phosphatase activity. J. Natl. Cancer Inst. 1999, 91, 1922–1932. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.H.; Lindsay, Y.; Safrany, S.T.; Lorenzo, O.; Villa, F.; Toth, R.; Clague, M.J.; Downes, C.P.; Leslie, N.R. Differential redox regulation within the PTP superfamily. Cell Signal. 2007, 19, 1521–1530. [Google Scholar] [CrossRef]
- Leslie, N.R.; Bennett, D.; Lindsay, Y.E.; Stewart, H.; Gray, A.; Downes, C.P. Redox regulation of PI 3-kinase signalling via inactivation of PTEN. EMBO J. 2003, 22, 5501–5510. [Google Scholar] [CrossRef]
- Cho, S.H.; Lee, C.H.; Ahn, Y.; Kim, H.; Kim, H.; Ahn, C.Y.; Yang, K.S.; Lee, S.R. Redox regulation of PTEN and protein tyrosine phosphatases in H2O2 mediated cell signaling. FEBS Lett. 2004, 560, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Song, Y.B.; Kim, T.Y.; Kim, I.; Han, S.J.; Ahn, Y.; Cho, S.H.; Choi, C.Y.; Chay, K.O.; Yang, S.Y.; et al. Redox regulation of the tumor suppressor PTEN by glutathione. FEBS Lett. 2010, 584, 3550–3556. [Google Scholar] [CrossRef]
- Cao, J.; Schulte, J.; Knight, A.; Leslie, N.R.; Zagozdzon, A.; Bronson, R.; Manevich, Y.; Beeson, C.; Neumann, C.A. Prdx1 inhibits tumorigenesis via regulating PTEN/AKT activity. EMBO J. 2009, 28, 1505–1517. [Google Scholar] [CrossRef] [Green Version]
- Schwertassek, U.; Haque, A.; Krishnan, N.; Greiner, R.; Weingarten, L.; Dick, T.P.; Tonks, N.K. Reactivation of oxidized PTP1B and PTEN by thioredoxin 1. FEBS J. 2014, 281, 3545–3558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Chay, K.O.; Kim, I.; Song, Y.B.; Kim, T.Y.; Han, S.J.; Ahn, Y.; Cho, S.H.; Hoe, K.L.; Ahn, B.W.; et al. Redox regulation of the tumor suppressor PTEN by glutaredoxin 5 and Ycp4. Biochem. Biophys. Res. Commun. 2011, 407, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Clerkin, J.S.; Naughton, R.; Quiney, C.; Cotter, T.G. Mechanisms of ROS modulated cell survival during carcinogenesis. Cancer Lett. 2008, 266, 30–36. [Google Scholar] [CrossRef]
- Bonneau, D.; Longy, M. Mutations of the human PTEN gene. Hum. Mutat. 2000, 16, 109–122. [Google Scholar] [CrossRef]
- Tell, G.; Quadrifoglio, F.; Tiribelli, C.; Kelley, M.R. The many functions of APE1/Ref-1: Not only a DNA repair enzyme. Antioxid. Redox Signal. 2009, 11, 601–620. [Google Scholar] [CrossRef] [Green Version]
- Seemann, S.; Hainaut, P. Roles of thioredoxin reductase 1 and APE/Ref-1 in the control of basal p53 stability and activity. Oncogene 2005, 24, 3853–3863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xanthoudakis, S.; Curran, T. Identification and characterization of Ref-1, a nuclear protein that facilitates AP-1 DNA-binding activity. EMBO J. 1992, 11, 653–665. [Google Scholar] [CrossRef]
- Xanthoudakis, S.; Miao, G.G.; Curran, T. The redox and DNA-repair activities of Ref-1 are encoded by nonoverlapping domains. Proc. Natl. Acad. Sci. USA 1994, 91, 23–27. [Google Scholar] [CrossRef] [Green Version]
- Cardoso, A.A.; Jiang, Y.; Luo, M.; Reed, A.M.; Shahda, S.; He, Y.; Maitra, A.; Kelley, M.R.; Fishel, M.L. APE1/Ref-1 regulates STAT3 transcriptional activity and APE1/Ref-1-STAT3 dual-targeting effectively inhibits pancreatic cancer cell survival. PLoS ONE 2012, 7, e47462. [Google Scholar] [CrossRef]
- Fishel, M.L.; Wu, X.; Devlin, C.M.; Logsdon, D.P.; Jiang, Y.; Luo, M.; He, Y.; Yu, Z.; Tong, Y.; Lipking, K.P.; et al. Apurinic/apyrimidinic endonuclease/redox factor-1 (APE1/Ref-1) redox function negatively regulates NRF2. J. Biol. Chem. 2015, 290, 3057–3068. [Google Scholar] [CrossRef] [Green Version]
- Zou, G.M.; Maitra, A. Small-molecule inhibitor of the AP endonuclease 1/REF-1 E3330 inhibits pancreatic cancer cell growth and migration. Mol. Cancer Ther. 2008, 7, 2012–2021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watroba, M.; Maslinska, D.; Maslinski, S. Current overview of functions of FoxO proteins, with special regards to cellular homeostasis, cell response to stress, as well as inflammation and aging. Adv. Med. Sci. 2012, 57, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Sweeney, L.B.; Sturgill, J.F.; Chua, K.F.; Greer, P.L.; Lin, Y.; Tran, H.; Ross, S.E.; Mostoslavsky, R.; Cohen, H.Y.; et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 2004, 303, 2011–2015. [Google Scholar] [CrossRef] [Green Version]
- Imai, S.; Guarente, L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014, 24, 464–471. [Google Scholar] [CrossRef]
- Kalous, K.S.; Wynia-Smith, S.L.; Olp, M.D.; Smith, B.C. Mechanism of Sirt1 NAD+-dependent protein deacetylase inhibition by cysteine S-Nitrosation. J. Biol. Chem. 2016, 291, 25398–25410. [Google Scholar] [CrossRef] [Green Version]
- Matsuzaki, H.; Daitoku, H.; Hatta, M.; Aoyama, H.; Yoshimochi, K.; Fukamizu, A. Acetylation of Foxo1 alters its DNA-binding ability and sensitivity to phosphorylation. Proc. Natl. Acad. Sci. USA 2005, 102, 11278–11283. [Google Scholar] [CrossRef] [Green Version]
- Nakae, J.; Oki, M.; Cao, Y. The FoxO transcription factors and metabolic regulation. FEBS Lett. 2008, 582, 54–67. [Google Scholar] [CrossRef] [Green Version]
- Choi, H.K.; Cho, K.B.; Phuong, N.T.; Han, C.Y.; Han, H.K.; Hien, T.T.; Choi, H.S.; Kang, K.W. SIRT1-mediated FoxO1 deacetylation is essential for multidrug resistance-associated protein 2 expression in tamoxifen-resistant breast cancer cells. Mol. Pharm. 2013, 10, 2517–2527. [Google Scholar] [CrossRef]
- Beatty, A.; Fink, L.S.; Singh, T.; Strigun, A.; Peter, E.; Ferrer, C.M.; Nicolas, E.; Cai, K.Q.; Moran, T.P.; Reginato, M.J.; et al. Metabolite profiling reveals the glutathione biosynthetic pathway as a therapeutic target in triple-negative breast cancer. Mol. Cancer Ther. 2018, 17, 264–275. [Google Scholar] [CrossRef] [Green Version]
- Schafer, M.; Dutsch, S.; auf dem Keller, U.; Navid, F.; Schwarz, A.; Johnson, D.A.; Johnson, J.A.; Werner, S. Nrf2 establishes a glutathione-mediated gradient of UVB cytoprotection in the epidermis. Genes Dev. 2010, 24, 1045–1058. [Google Scholar] [CrossRef] [Green Version]
- Metere, A.; Graves, C.E.; Chirico, M.; Caramujo, M.J.; Pisanu, M.E.; Iorio, E. Metabolomic reprogramming detected by 1H-NMR dpectroscopy in human thyroid cancer tissues. Biology 2020, 9, 112. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chun, K.-S.; Kim, D.-H.; Surh, Y.-J. Role of Reductive versus Oxidative Stress in Tumor Progression and Anticancer Drug Resistance. Cells 2021, 10, 758. https://doi.org/10.3390/cells10040758
Chun K-S, Kim D-H, Surh Y-J. Role of Reductive versus Oxidative Stress in Tumor Progression and Anticancer Drug Resistance. Cells. 2021; 10(4):758. https://doi.org/10.3390/cells10040758
Chicago/Turabian StyleChun, Kyung-Soo, Do-Hee Kim, and Young-Joon Surh. 2021. "Role of Reductive versus Oxidative Stress in Tumor Progression and Anticancer Drug Resistance" Cells 10, no. 4: 758. https://doi.org/10.3390/cells10040758
APA StyleChun, K.-S., Kim, D.-H., & Surh, Y.-J. (2021). Role of Reductive versus Oxidative Stress in Tumor Progression and Anticancer Drug Resistance. Cells, 10(4), 758. https://doi.org/10.3390/cells10040758