
Placental Glycoredox Dysregulation Associated with Disease Progression in an Animal Model of Superimposed Preeclampsia

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Experimental Design
2.2. Tissue Collection
2.3. Determination of Thiobarbituric Acid Reactive Substances (TBARS)
2.4. Immunohistochemistry for Oxidative Stress Markers, Antioxidant Enzymes and Galectins
2.5. Western Blots
2.6. Glycan Profiling by Lectin-Binding Analysis
2.7. Morphometrical Analyses
2.8. Statistics
3. Results
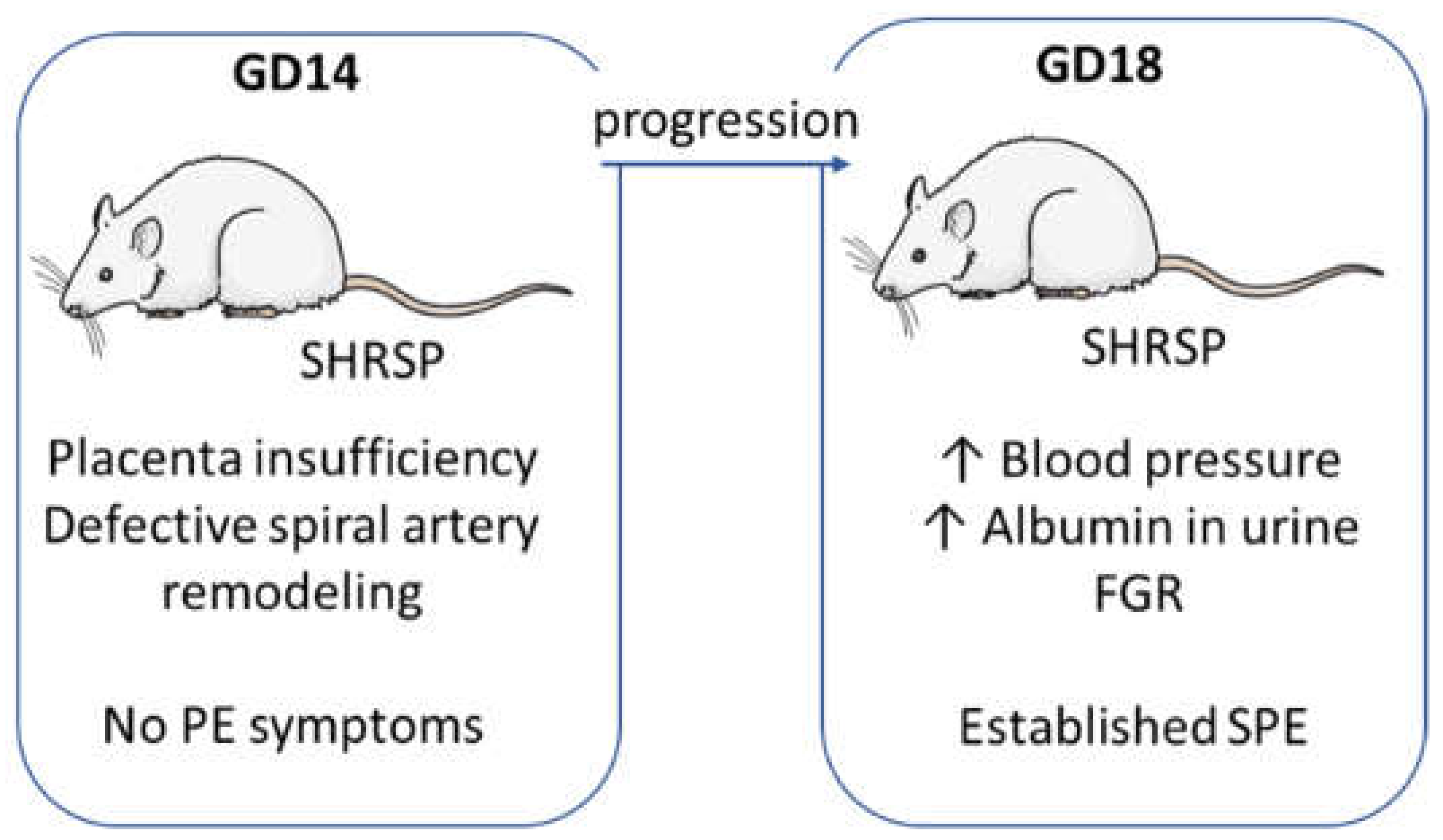
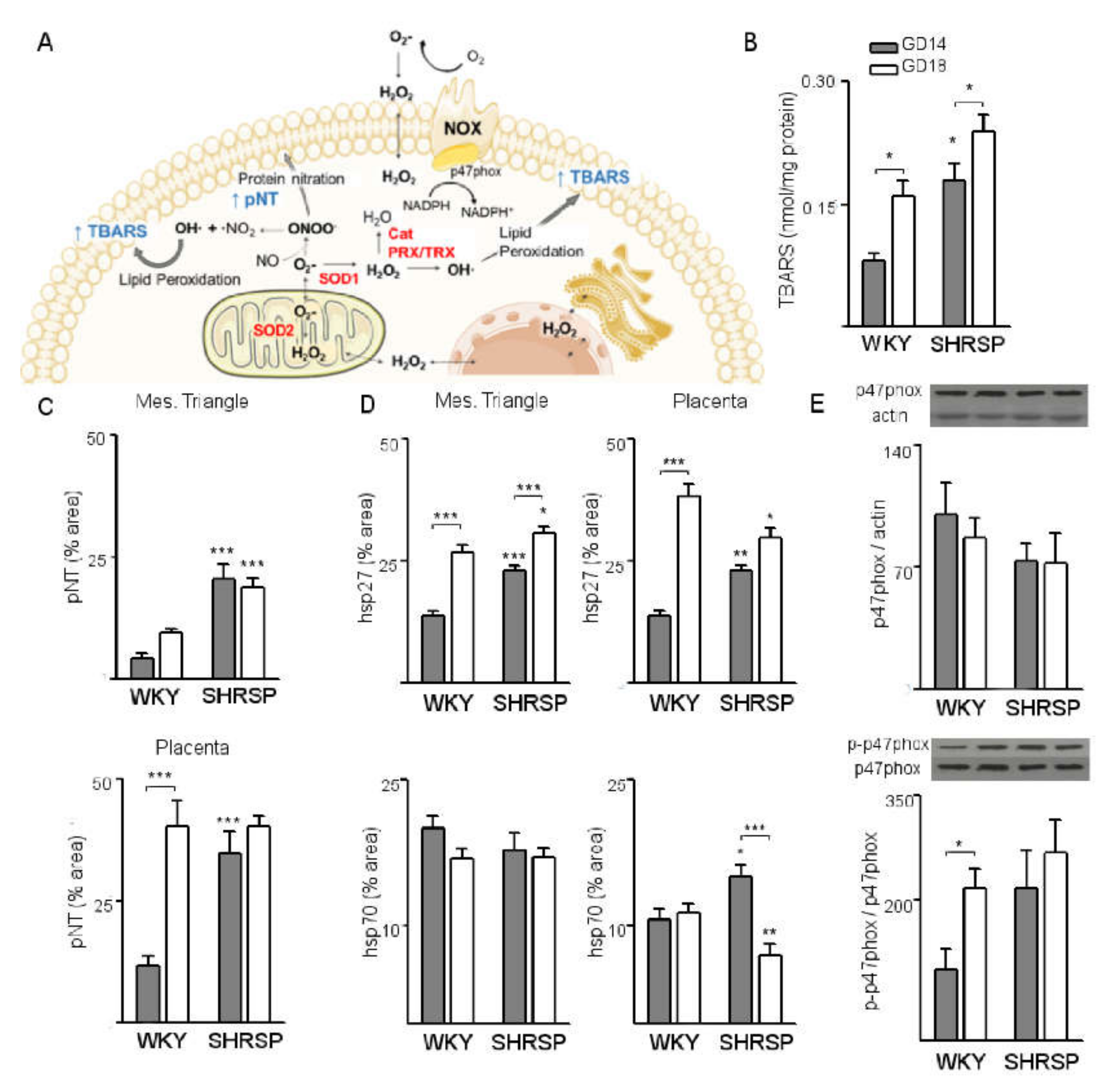
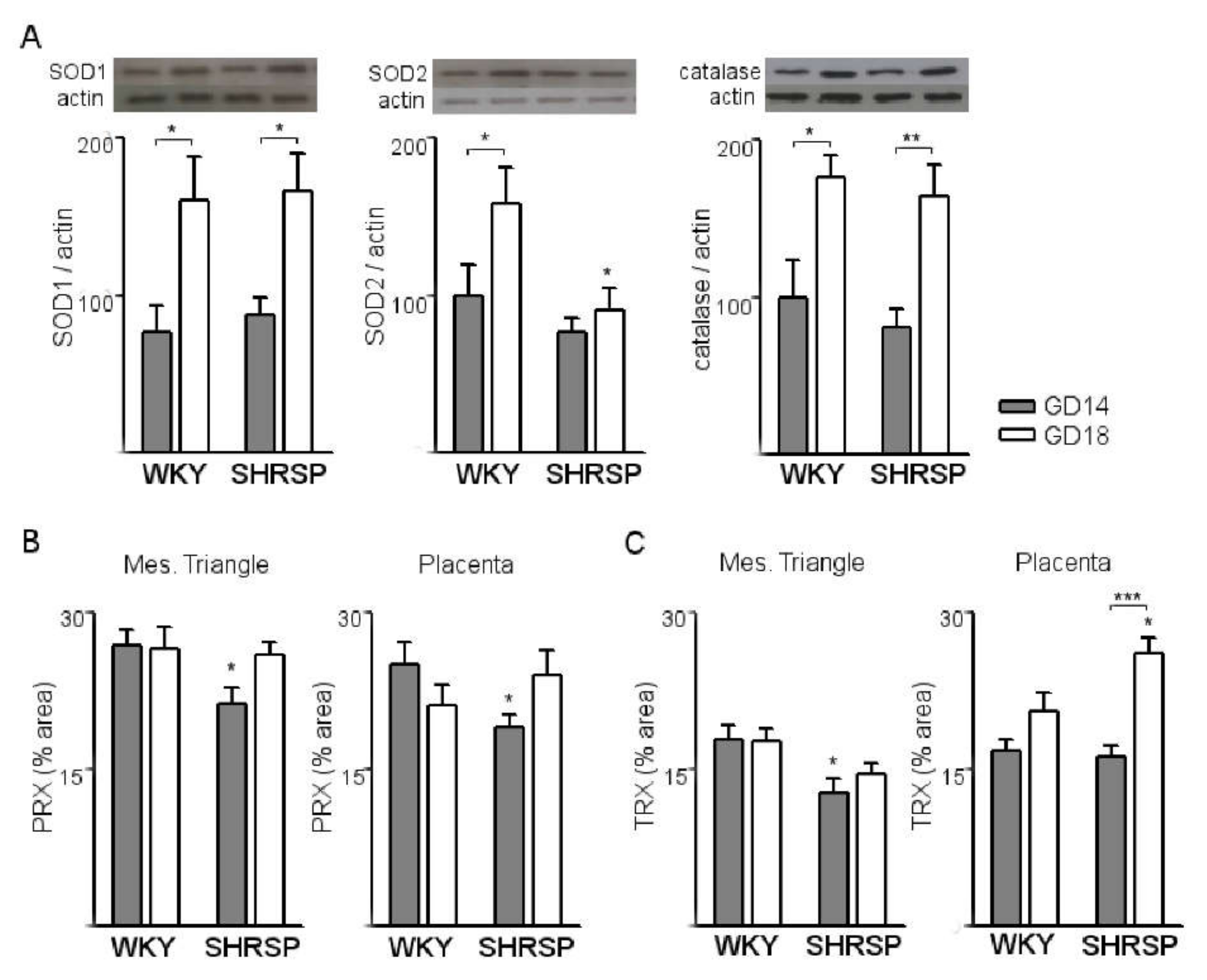
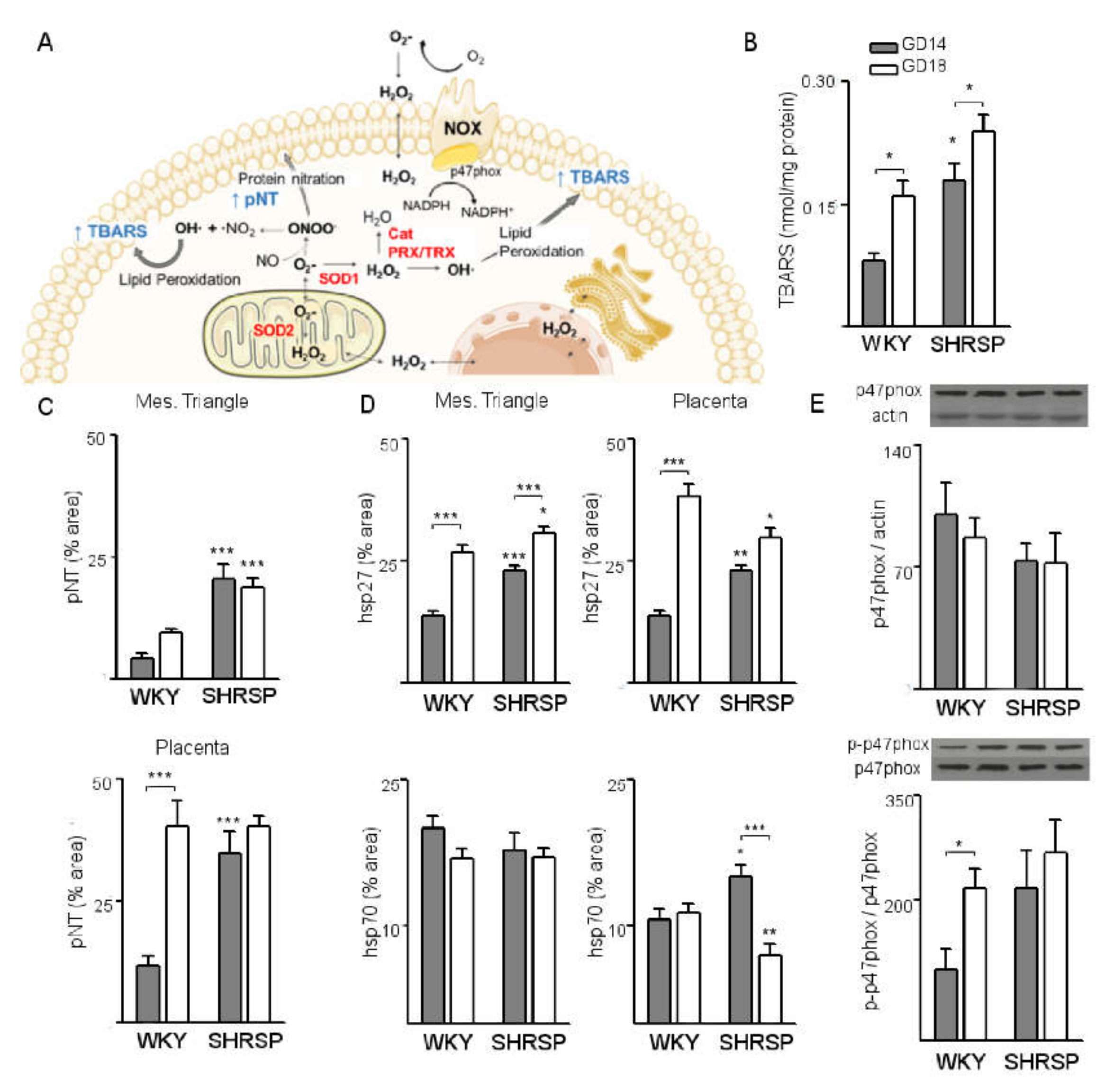
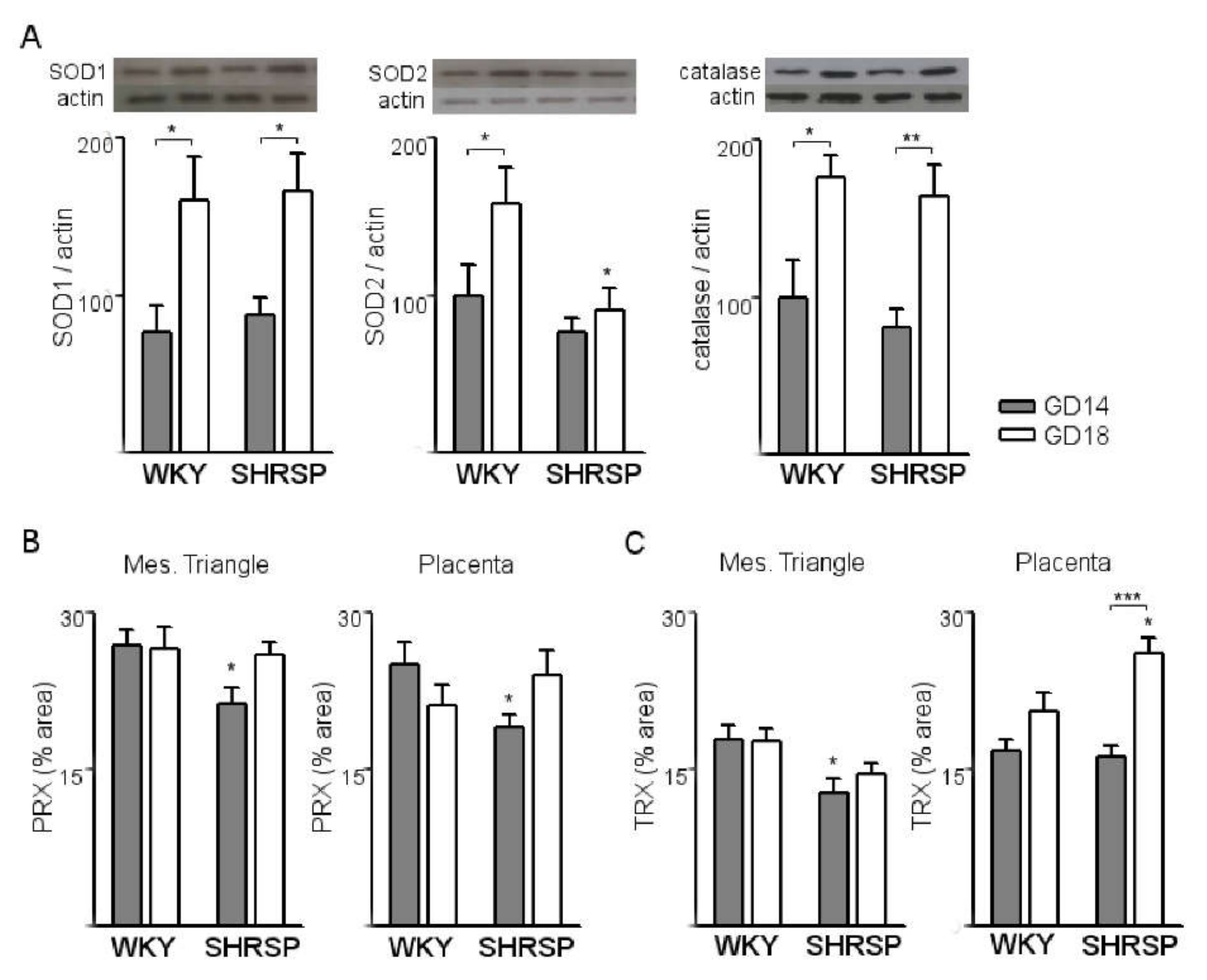
3.1. Onset of SPE in SHRSP Pregnancies Is Marked by an Enhanced Activation of the Placental CSR
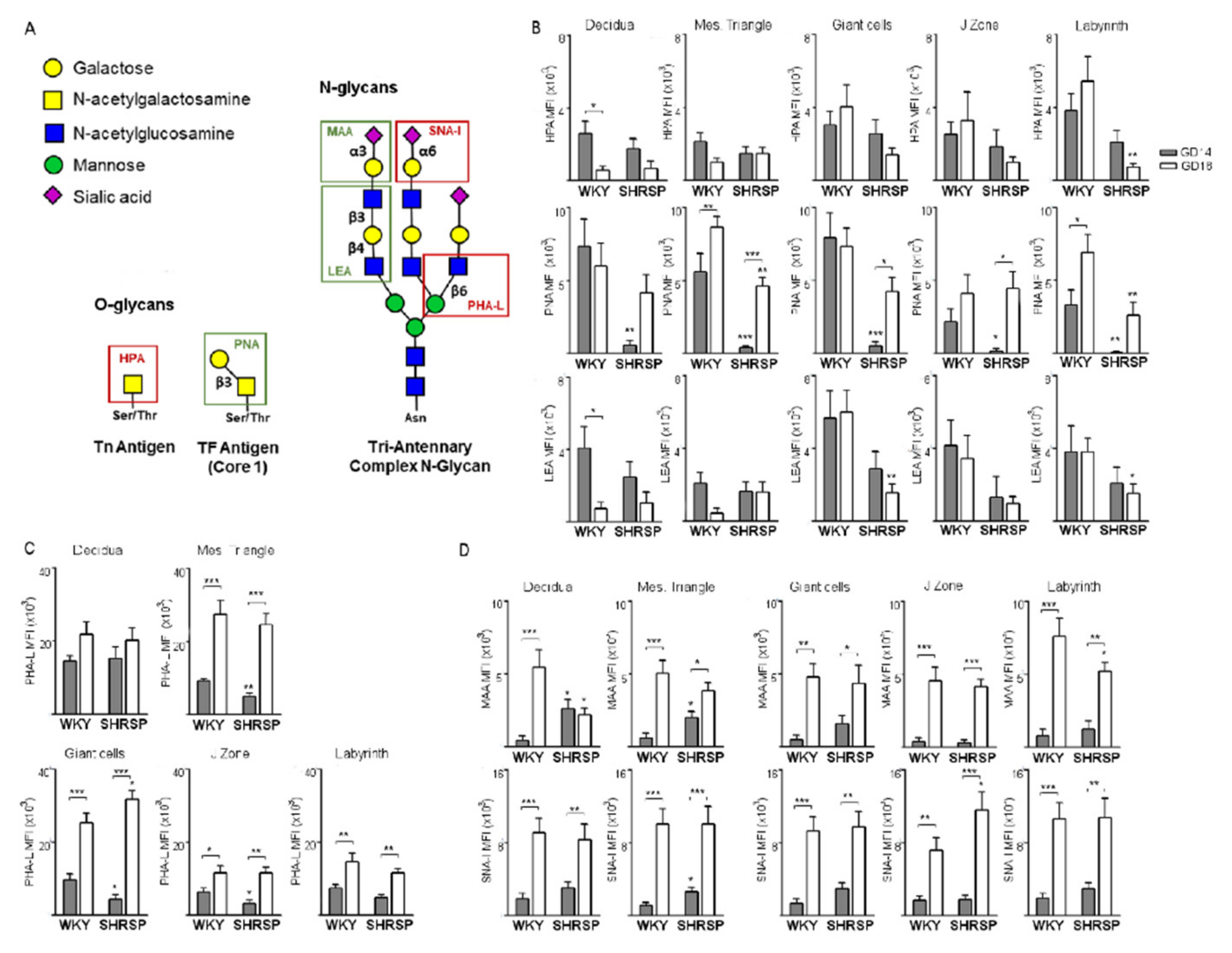
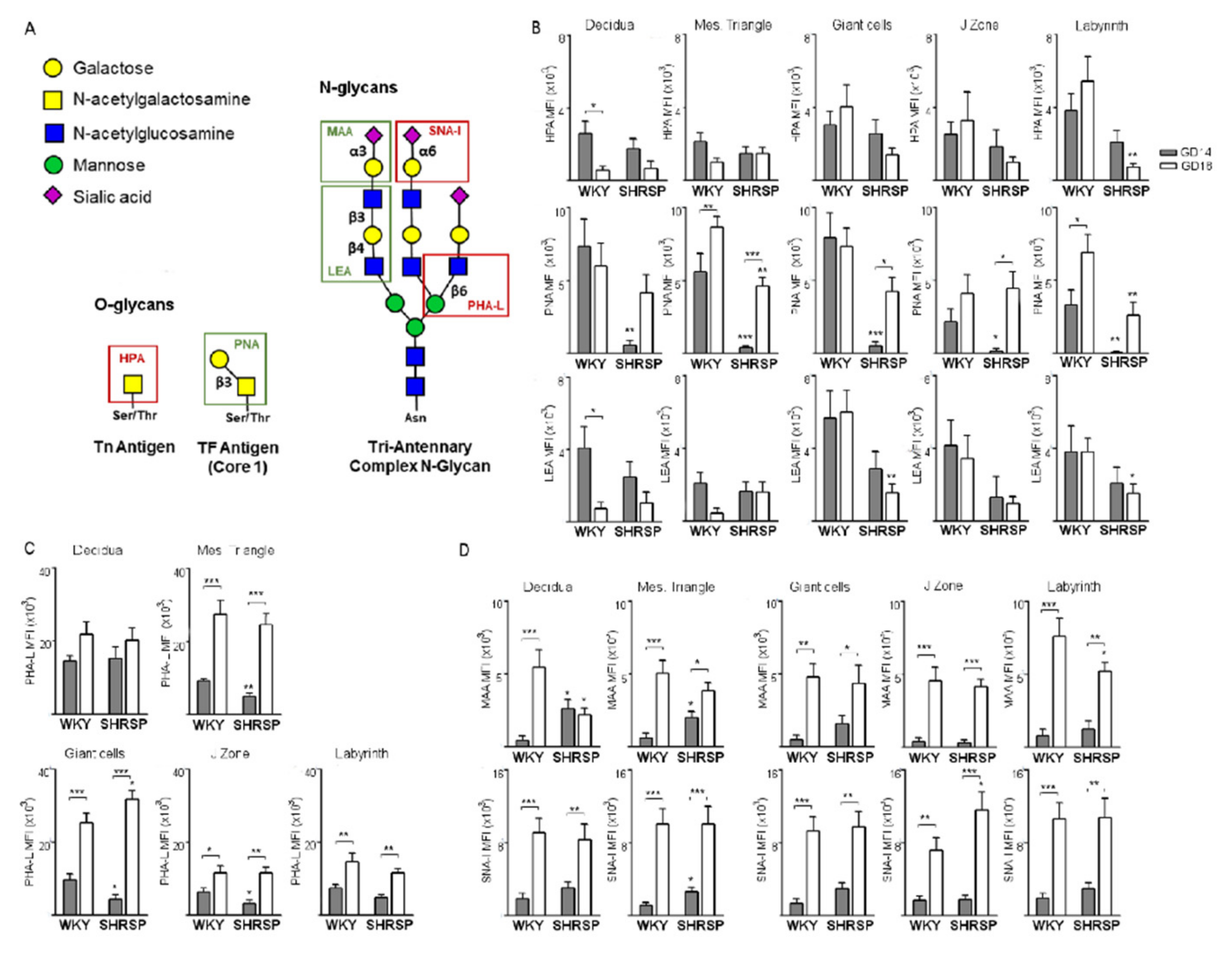
3.2. Marked Changes in Placental Glycosylation Associated with the Onset of SPE in SHRSP Pregnancies
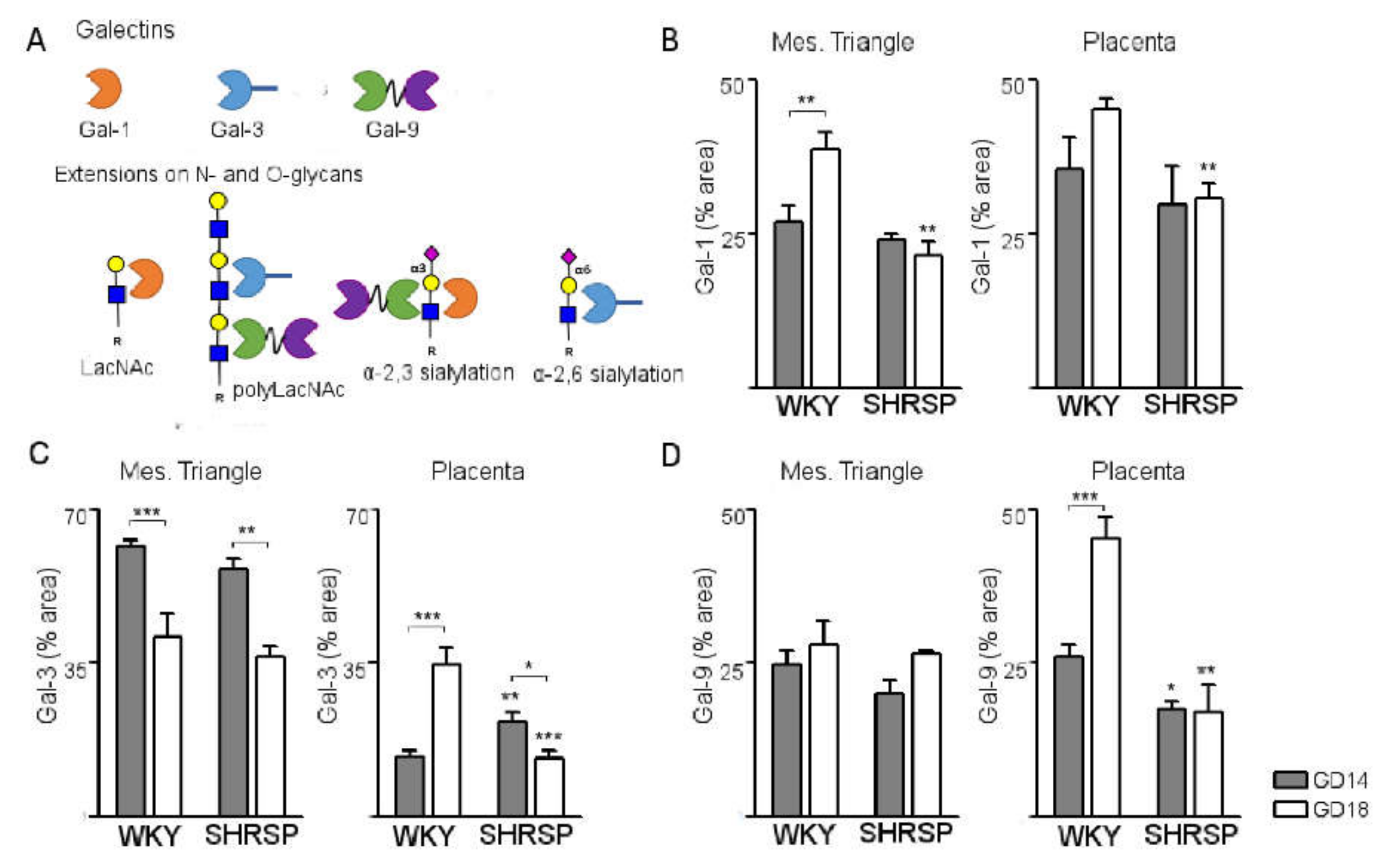
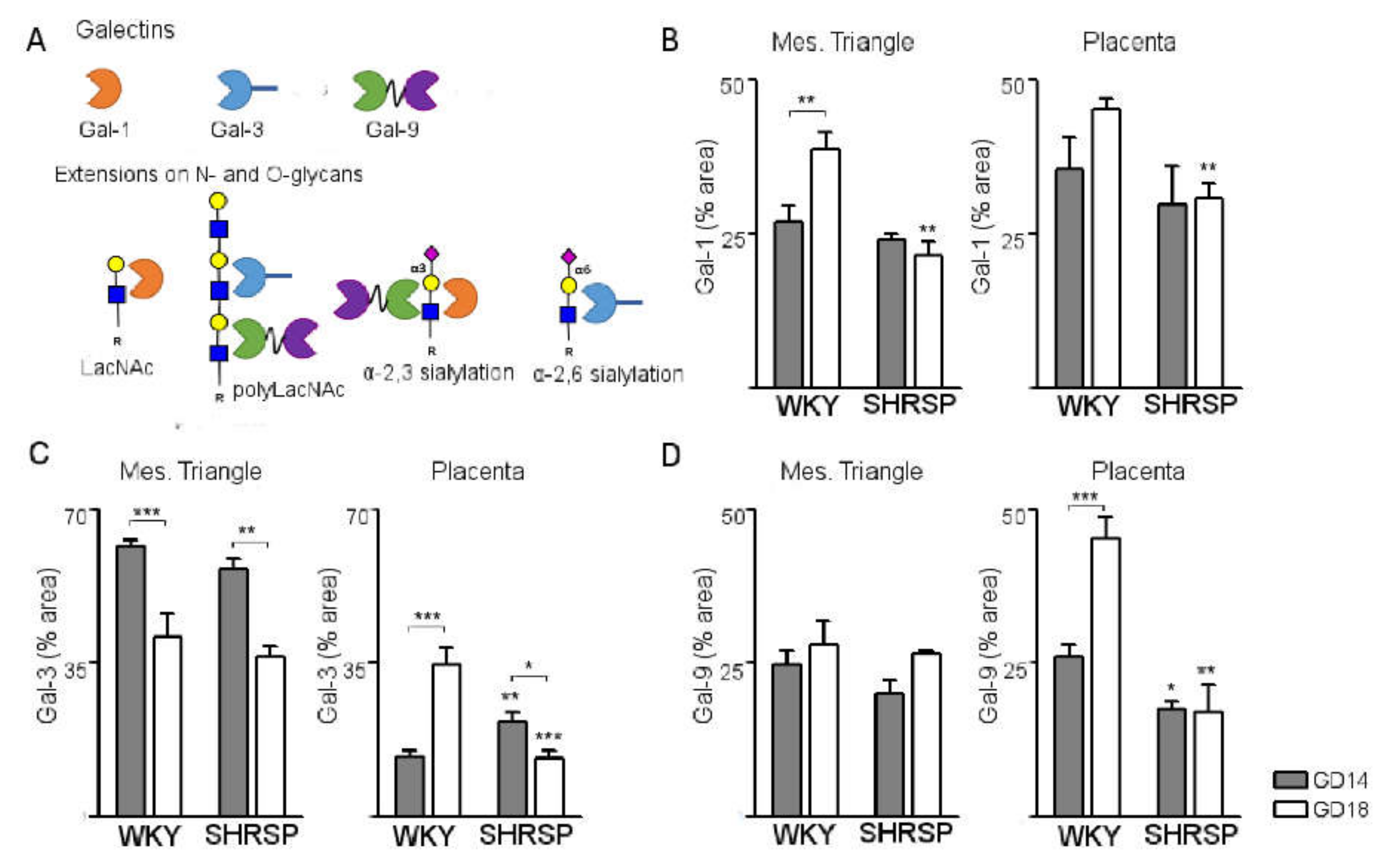
3.3. Development of SPE Induces Changes in the Expression Profile of Stress-Sensitive Galectins in the SHRSP Placenta
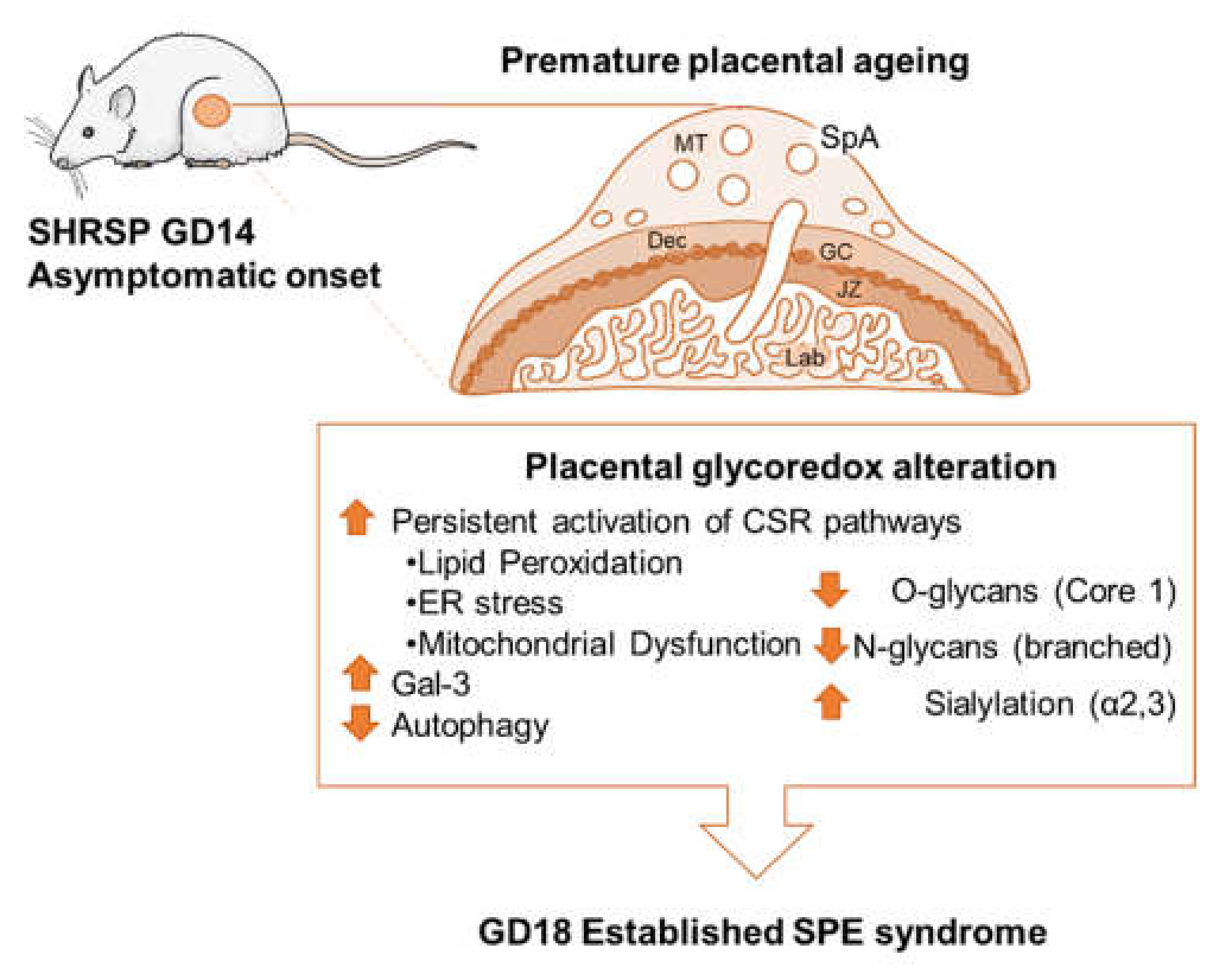
4. Discussion
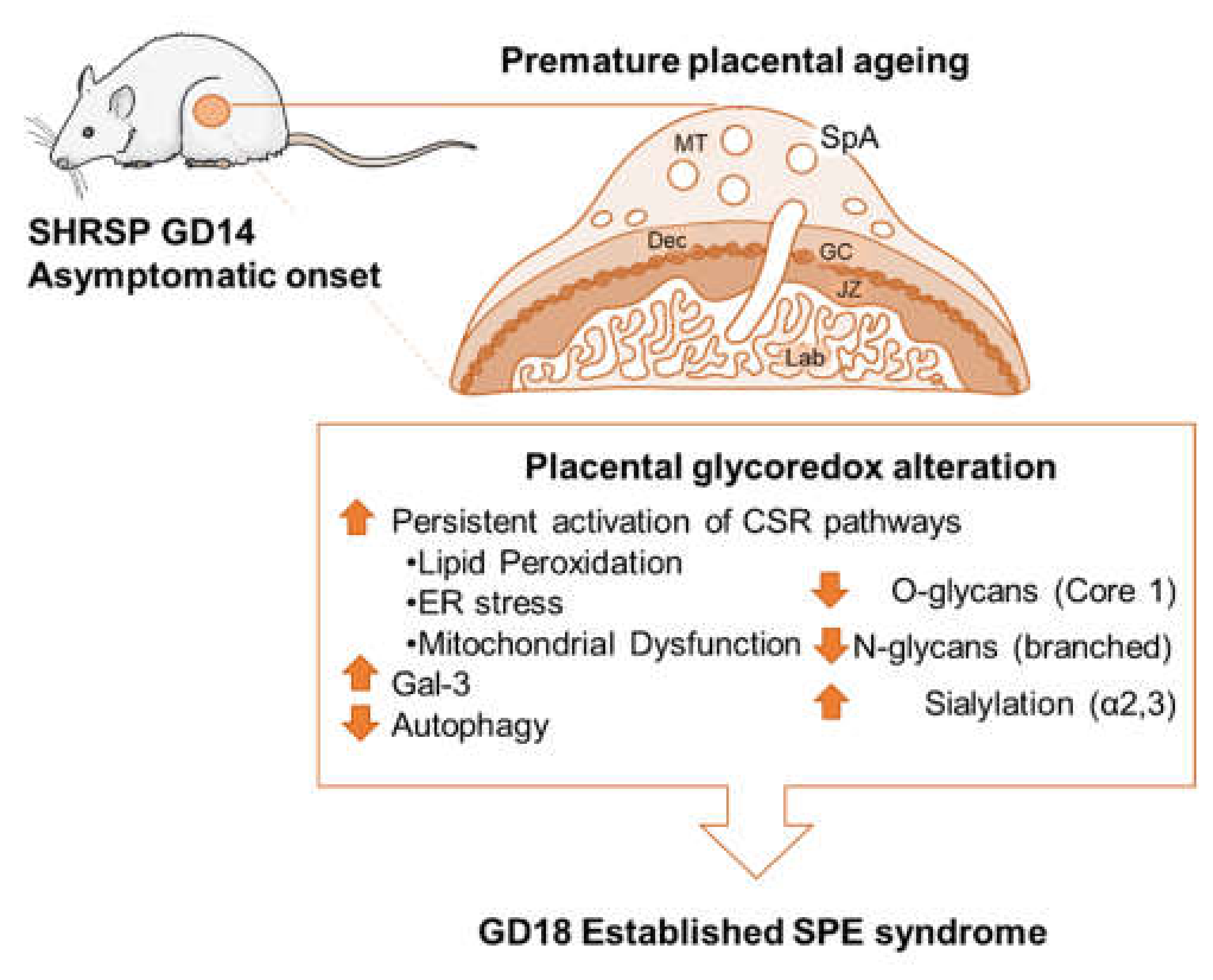
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chappell, L.C.; Enye, S.; Seed, P.; Briley, A.L.; Poston, L.; Shennan, A.H. Adverse Perinatal Outcomes and Risk Factors for Preeclampsia in Women With Chronic Hypertension. Hypertension 2008, 51, 1002–1009. [Google Scholar] [CrossRef] [Green Version]
- Roberts, J.M.; August, P.A.; Bakris, G.; Barton, J.R.; Bernstein, I.M.; Druzin, M.; Gaiser, R.R.; Granger, J.P.; Jeyabalan, A.; Johnson, D.D.; et al. Hypertension in pregnancy. Report of the American College of Obstetricians and Gynecologists’ Task Force on Hypertension in Pregnancy. Obstet. Gynecol. 2013, 122, 1122–1131. [Google Scholar] [CrossRef]
- Bramham, K.; Parnell, B.; Nelson-Piercy, C.; Seed, P.T.; Poston, L.; Chappell, L.C. Chronic hypertension and pregnancy outcomes: Systematic review and meta-analysis. BMJ 2014, 348, g2301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livingston, J.C.; Sibai, B.M. Chronic hypertension in pregnancy. Obstet. Gynecol. Clin. N. Am. 2001, 28, 447–464. [Google Scholar] [CrossRef]
- Roberts, J.M. Pathophysiology of ischemic placental disease. Semin. Perinatol. 2014, 38, 139–145. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, J.S.; Ryan, M.J.; Lamarca, B.B.; Sedeek, M.; Murphy, S.R.; Granger, J.P. Pathophysiology of hypertension during preeclampsia: Linking placental ischemia with endothelial dysfunction. Am. J. Physiol. Circ. Physiol. 2008, 294, H541–H550. [Google Scholar] [CrossRef]
- Redman, C.; Sargent, I.; Staff, A. IFPA Senior Award Lecture: Making sense of pre-eclampsia—Two placental causes of preeclampsia? Placenta 2014, 35, S20–S25. [Google Scholar] [CrossRef]
- Corrêa, R.R.M.; Gilio, D.B.; Cavellani, C.L.; Paschoini, M.C.; Oliveira, F.A.; Peres, L.C.; Reis, M.A.; Teixeira, V.P.A.; Castro, E.C.C. Placental morphometrical and histopathology changes in the different clinical presentations of Hypertensive Syndromes in Pregnancy. Arch. Gynecol. Obstet. 2008, 277, 201–206. [Google Scholar] [CrossRef]
- Gratacós, E.; Casals, E.; Deulofeu, R.; Gómez, O.; Cararach, V.; Alonso, P.L.; Fortuny, A. Serum and Placental Lipid Peroxides in Chronic Hypertension During Pregnancy with and without Superimposed Preeclampsia. Hypertens. Pregnancy 1999, 18, 139–146. [Google Scholar] [CrossRef]
- Vashukova, E.S.; Glotov, A.S.; Fedotov, P.V.; Efimova, O.A.; Pakin, V.S.; Mozgovaya, E.V.; Pendina, A.A.; Tikhonov, A.V.; Koltsova, A.S.; Baranov, V.S. Placental microRNA expression in pregnancies complicated by superimposed pre-eclampsia on chronic hypertension. Mol. Med. Rep. 2016, 14, 22–32. [Google Scholar] [CrossRef]
- Barrientos, G.; Pussetto, M.; Rose, M.; Staff, A.; Blois, S.; Toblli, J. Defective trophoblast invasion underlies fetal growth restriction and preeclampsia-like symptoms in the stroke-prone spontaneously hypertensive rat. Mol. Hum. Reprod. 2017, 23, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Gillis, E.E.; Williams, J.M.; Garrett, M.R.; Mooney, J.N.; Sasser, J.M. The Dahl salt-sensitive rat is a spontaneous model of superimposed preeclampsia. Am. J. Physiol. Integr. Comp. Physiol. 2015, 309, R62–R70. [Google Scholar] [CrossRef]
- Maeda, K.J.; Showmaker, K.C.; Johnson, A.C.; Garrett, M.R.; Sasser, J.M. Spontaneous superimposed preeclampsia: Chronology and expression unveiled by temporal transcriptomic analysis. Physiol. Genom. 2019, 51, 342–355. [Google Scholar] [CrossRef] [PubMed]
- Tenório, M.B.; Ferreira, R.C.; Moura, F.A.; Bueno, N.B.; De Oliveira, A.C.M.; Goulart, M.O.F. Cross-Talk between Oxidative Stress and Inflammation in Preeclampsia. Oxidative Med. Cell. Longev. 2019, 2019, 1–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takamiya, R.; Ohtsubo, K.; Taniguchi, N. Glycoredox: New Paradigm for Glycosylation and Redox Signaling Research. In Glycoscience: Biology and Medicine; Taniguchi, N., Endo, T., Hart, G.W., Seeberger, P.H., Wong, C.-H., Eds.; Springer: Tokyo, Japan, 2015; pp. 1275–1281. [Google Scholar]
- Jones, C.J.P.; Aplin, J.D. Glycosylation at the fetomaternal interface: Does the glycocode play a critical role in implantation? Glycoconj. J. 2008, 26, 359–366. [Google Scholar] [CrossRef]
- Sgambati, E.; Biagiotti, R.; Marini, M.; Brizzi, E. Lectin histochemistry in the human placenta of pregnancies complicated by intrauterine growth retardation based on absent or reversed diastolic flow. Placenta 2002, 23, 503–515. [Google Scholar] [CrossRef]
- Marini, M.; Bonaccini, L.; Thyrion, G.D.Z.; Vichi, D.; Parretti, E.; Sgambati, E. Distribution of sugar residues in human placentas from pregnancies complicated by hypertensive disorders. Acta Histochem. 2011, 113, 815–825. [Google Scholar] [CrossRef]
- Tannetta, D.; Masliukaite, I.; Vatish, M.; Redman, C.; Sargent, I. Update of syncytiotrophoblast derived extracellular vesicles in normal pregnancy and preeclampsia. J. Reprod. Immunol. 2017, 119, 98–106. [Google Scholar] [CrossRef] [Green Version]
- Blois, S.M.; Dveksler, G.; Vasta, G.R.; Freitag, N.; Blanchard, V.; Barrientos, G. Pregnancy Galectinology: Insights Into a Complex Network of Glycan Binding Proteins. Front. Immunol. 2019, 10, 1166. [Google Scholar] [CrossRef]
- Yang, R.-Y.; Liu, F.-T. Galectins in cell growth and apoptosis. Cell. Mol. Life Sci. 2003, 60, 267–276. [Google Scholar] [CrossRef]
- Seyrek, K.; Richter, M.; Lavrik, I.N. Decoding the sweet regulation of apoptosis: The role of glycosylation and galectins in apoptotic signaling pathways. Cell Death Differ. 2019, 26, 981–993. [Google Scholar] [CrossRef] [PubMed]
- Balogh, A.; Pozsgay, J.; Matkó, J.; Dong, Z.; Kim, C.J.; Várkonyi, T.; Sammar, M.; Rigó, J.; Meiri, H.; Romero, R.; et al. Placental protein 13 (PP13/galectin-13) undergoes lipid raft-associated subcellular redistribution in the syncytiotrophoblast in preterm preeclampsia and HELLP syndrome. Am. J. Obstet. Gynecol. 2011, 205, 156.e1–156.e14. [Google Scholar] [CrossRef] [Green Version]
- Steichen, A.L.; Simonson, T.J.; Salmon, S.L.; Metzger, D.W.; Mishra, B.B.; Sharma, J. Alarmin Function of Galectin-9 in Murine Respiratory Tularemia. PLoS ONE 2015, 10, e0123573. [Google Scholar] [CrossRef] [PubMed]
- Yip, P.K.; Carrillo-Jimenez, A.; King, P.; Vilalta, A.; Nomura, K.; Chau, C.C.; Egerton, A.M.S.; Liu, Z.-H.; Shetty, A.J.; Tremoleda, J.L.; et al. Galectin-3 released in response to traumatic brain injury acts as an alarmin orchestrating brain immune response and promoting neurodegeneration. Sci. Rep. 2017, 7, srep41689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russo, A.J.; Vasudevan, S.O.; Méndez-Huergo, S.P.; Kumari, P.; Menoret, A.; Duduskar, S.; Wang, C.; Sáez, J.M.P.; Fettis, M.M.; Li, C.; et al. Intracellular immune sensing promotes inflammation via gasdermin D–driven release of a lectin alarmin. Nat. Immunol. 2021, 22, 154–165. [Google Scholar] [CrossRef]
- Geusens, N.; Hering, L.; Verlohren, S.; Luyten, C.; Drijkoningen, K.; Taube, M.; Vercruysse, L.; Hanssens, M.; Dechend, R.; Pijnenborg, R. Changes in endovascular trophoblast invasion and spiral artery remodelling at term in a transgenic preeclamptic rat model. Placenta 2010, 31, 320–326. [Google Scholar] [CrossRef]
- Fraga, C.G.; Leibovitz, B.E.; Tappel, A.L. Halogenated compounds as inducers of lipid peroxidation in tissue slices. Free. Radic. Biol. Med. 1987, 3, 119–123. [Google Scholar] [CrossRef]
- Conrad, M.L.; Freitag, N.; Diessler, M.E.; Hernandez, R.; Barrientos, G.; Rose, M.; Casas, L.A.; Barbeito, C.G.; Blois, S.M. Differential Spatiotemporal Patterns of Galectin Expression are a Hallmark of Endotheliochorial Placentation. Am. J. Reprod. Immunol. 2015, 75, 317–325. [Google Scholar] [CrossRef] [Green Version]
- Borowski, S.; Tirado-Gonzalez, I.; Freitag, N.; Garcia, M.G.; Barrientos, G.; Blois, S.M. Altered Glycosylation Contributes to Placental Dysfunction Upon Early Disruption of the NK Cell-DC Dynamics. Front. Immunol. 2020, 11, 1316. [Google Scholar] [CrossRef]
- Jauniaux, E.; Watson, A.L.; Hempstock, J.; Bao, Y.-P.; Skepper, J.N.; Burton, G.J. Onset of Maternal Arterial Blood Flow and Placental Oxidative Stress. Am. J. Pathol. 2000, 157, 2111–2122. [Google Scholar] [CrossRef]
- Buelke-Sam, J.; Holson, J.F.; Nelson, C.J. Blood flow during pregnancy in the rat: II. Dynamics of and litter variability in uterine flow. Teratology 1982, 26, 279–288. [Google Scholar] [CrossRef]
- Hewitt, D.P.; Mark, P.J.; Waddell, B.J. Glucocorticoids Prevent the Normal Increase in Placental Vascular Endothelial Growth Factor Expression and Placental Vascularity during Late Pregnancy in the Rat. Endocrinology 2006, 147, 5568–5574. [Google Scholar] [CrossRef] [Green Version]
- Silva, J.F.; Serakides, R. Intrauterine trophoblast migration: A comparative view of humans and rodents. Cell Adhes. Migr. 2016, 10, 88–110. [Google Scholar] [CrossRef] [Green Version]
- Basu, J.; Bendek, B.; Agamasu, E.; Salafia, C.M.; Mishra, A.; Benfield, N.; Patel, R.; Mikhail, M. Placental Oxidative Status throughout Normal Gestation in Women with Uncomplicated Pregnancies. Obstet. Gynecol. Int. 2015, 2015, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Jones, M.L.; Mark, P.J.; Lewis, J.L.; Mori, T.A.; Keelan, J.A.; Waddell, B.J. Antioxidant Defenses in the Rat Placenta in Late Gestation: Increased Labyrinthine Expression of Superoxide Dismutases, Glutathione Peroxidase 3, and Uncoupling Protein 21. Biol. Reprod. 2010, 83, 254–260. [Google Scholar] [CrossRef] [Green Version]
- Vaka, V.R.; McMaster, K.M.; Cunningham, M.W.; Ibrahim, T.; Hazlewood, R.; Usry, N.; Cornelius, D.C.; Amaral, L.M.; Lamarca, B. Role of Mitochondrial Dysfunction and Reactive Oxygen Species in Mediating Hypertension in the Reduced Uterine Perfusion Pressure Rat Model of Preeclampsia. Hypertension 2018, 72, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Marín, R.; Chiarello, D.I.; Abad, C.; Rojas, D.; Toledo, F.; Sobrevia, L. Oxidative stress and mitochondrial dysfunction in early-onset and late-onset preeclampsia. Biochim. et Biophys. Acta (BBA) Mol. Basis Dis. 2020, 1866, 165961. [Google Scholar] [CrossRef] [PubMed]
- Holland, O.J.; Cuffe, J.S.M.; Nitert, M.D.; Callaway, L.; Cheung, K.A.K.; Radenkovic, F.; Perkins, A.V. Placental mitochondrial adaptations in preeclampsia associated with progression to term delivery. Cell Death Dis. 2018, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Padmini, E.; Lavanya, S.; Uthra, V. Preeclamptic placental stress and over expression of mitochondrial HSP70. Clin. Chem. Lab. Med. 2009, 47, 1073–1080. [Google Scholar] [CrossRef] [PubMed]
- Rubattu, S.; Stanzione, R.; Volpe, M. Mitochondrial Dysfunction Contributes to Hypertensive Target Organ Damage: Lessons from an Animal Model of Human Disease. Oxidative Med. Cell. Longev. 2016, 2016, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Small, H.Y.; Morgan, H.; Beattie, E.; Griffin, S.; Indahl, M.; Delles, C.; Graham, D. Abnormal uterine artery remodelling in the stroke prone spontaneously hypertensive rat. Placenta 2016, 37, 34–44. [Google Scholar] [CrossRef] [Green Version]
- Thaete, L.G.; Khan, S.; Neerhof, M.G. Endothelin Receptor A Antagonism Prevents Damage to Glycogen-Rich Placental Cells Following Uterine Ischemia–Reperfusion in the Rat. Reprod. Sci. 2016, 23, 1518–1525. [Google Scholar] [CrossRef] [Green Version]
- Flood-Nichols, S.K.; Kazanjian, A.A.; Tinnemore, D.; Gafken, P.R.; Ogata, Y.; Napolitano, P.G.; Stallings, J.D.; Ippolito, D.L. Aberrant Glycosylation of Plasma Proteins in Severe Preeclampsia Promotes Monocyte Adhesion. Reprod. Sci. 2013, 21, 204–214. [Google Scholar] [CrossRef] [Green Version]
- Tannetta, D.; Collett, G.; Vatish, M.; Redman, C.; Sargent, I. Syncytiotrophoblast extracellular vesicles—Circulating biopsies reflecting placental health. Placenta 2017, 52, 134–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcos, N.T.; Bennett, E.P.; Gomes, J.; Magalhaes, A.; Gomes, C.; David, L.; Dar, I.; Jeanneau, C.; DeFrees, S.; Krustrup, D.; et al. ST6GalNAc-I controls expression of sialyl-Tn antigen in gastrointestinal tissues. Front. Biosci. 2011, 3, 1443–1455. [Google Scholar] [CrossRef]
- Sewell, R.; Bäckström, M.; Dalziel, M.; Gschmeissner, S.; Karlsson, H.; Noll, T.; Gätgens, J.; Clausen, H.; Hansson, G.C.; Burchell, J.; et al. The ST6GalNAc-I Sialyltransferase Localizes throughout the Golgi and Is Responsible for the Synthesis of the Tumor-associated Sialyl-Tn O-Glycan in Human Breast Cancer. J. Biol. Chem. 2006, 281, 3586–3594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clement, M.; Rocher, J.; Loirand, G.; Le Pendu, J. Expression of sialyl-Tn epitopes on 1 integrin alters epithelial cell phenotype, proliferation and haptotaxis. J. Cell Sci. 2004, 117, 5059–5069. [Google Scholar] [CrossRef] [Green Version]
- Julien, S.; Adriaenssens, E.; Ottenberg, K.; Furlan, A.J.; Courtand, G.; Vercoutter-Edouart, A.-S.; Hanisch, F.-G.; Delannoy, P.; Le Bourhis, X. ST6GalNAc I expression in MDA-MB-231 breast cancer cells greatly modifies their O-glycosylation pattern and enhances their tumourigenicity. Glycobiology 2005, 16, 54–64. [Google Scholar] [CrossRef] [Green Version]
- Julien, S.; Picco, G.; Sewell, R.; Vercoutter-Edouart, A.-S.; Tarp, M.; Miles, D.; Clausen, H.; Taylor-Papadimitriou, J.; Burchell, J.M. Sialyl-Tn vaccine induces antibody-mediated tumour protection in a relevant murine model. Br. J. Cancer 2009, 100, 1746–1754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozaki, H.; Matsuzaki, H.; Ando, H.; Kaji, H.; Nakanishi, H.; Ikehara, Y.; Narimatsu, H. Enhancement of metastatic ability by ectopic expression of ST6GalNAcI on a gastric cancer cell line in a mouse model. Clin. Exp. Metastasis 2012, 29, 229–238. [Google Scholar] [CrossRef] [Green Version]
- Jeschke, U.; Richter, D.; Hammer, A.; Briese, V.; Friese, K.; Karsten, U. Expression of the Thomsen-Friedenreich antigen and of its putative carrier protein mucin 1 in the human placenta and in trophoblast cells in vitro. Histochem. Cell Biol. 2002, 117, 219–226. [Google Scholar] [CrossRef]
- Scholz, C.; Hermann, C.; Kachler, A.; Kainer, F.; Friese, K.; Makrigiannakis, A.; Jeschke, U. Association of placental inflammation with fetomaternal hemorrhage and loss of placental mucin-1. Arch. Gynecol. Obstet. 2011, 285, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Shalom-Barak, T.; Nicholas, J.M.; Wang, Y.; Zhang, X.; Ong, E.S.; Young, T.H.; Gendler, S.J.; Evans, R.M.; Barak, Y. Peroxisome Proliferator-Activated Receptor γ Controls Muc1 Transcription in Trophoblasts. Mol. Cell. Biol. 2004, 24, 10661–10669. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, F.P.; Drewlo, S.; English, F.A.; Kingdom, J.; Johns, E.J.; Kenny, L.C.; Walsh, S.K. Evidence Implicating Peroxisome Proliferator-Activated Receptor-γ in the Pathogenesis of Preeclampsia. Hypertension 2011, 58, 882–887. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, F.P.; Drewlo, S.; Kingdom, J.; Johns, E.J.; Walsh, S.K.; Kenny, L.C. Peroxisome Proliferator-Activated Receptor-γ as a Potential Therapeutic Target in the Treatment of Preeclampsia. Hypertension 2011, 58, 280–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadam, L.; Kohan-Ghadr, H.R.; Drewlo, S. The balancing act—PPAR-γ’s roles at the maternal-fetal interface. Syst. Biol. Reprod. Med. 2014, 61, 65–71. [Google Scholar] [CrossRef]
- Patnaik, S.K.; Potvin, B.; Carlsson, S.; Sturm, D.; Leffler, H.; Stanley, P. Complex N-glycans are the major ligands for galectin-1, -3, and -8 on Chinese hamster ovary cells. Glycobiology 2005, 16, 305–317. [Google Scholar] [CrossRef]
- Dam, T.K.; Gabius, H.-J.; André, S.; Kaltner, H.; Lensch, A.M.; Brewer, C.F. Galectins Bind to the Multivalent Glycoprotein Asialofetuin with Enhanced Affinities and a Gradient of Decreasing Binding Constants. Biochemistry 2005, 44, 12564–12571. [Google Scholar] [CrossRef]
- Hirabayashi, J.; Hashidate, T.; Arata, Y.; Nishi, N.; Nakamura, T.; Hirashima, M.; Urashima, T.; Oka, T.; Futai, M.; Muller, W.E.; et al. Oligosaccharide specificity of galectins: A search by frontal affinity chromatography. Biochim. Biophys. Acta 2002, 1572, 232–254. [Google Scholar] [CrossRef]
- Tsuboi, S.; Suzuki, Y.; Sutoh, M.; Hatakeyama, S.; Mori, K.; Yamamoto, H.; Koie, T.; Saitoh, H.; Yamaya, K.; Funyu, T.; et al. MUC1 carrying core 2 O-glycans functions as a molecular shield against NK cell attack, promoting bladder tumor metastasis. Int. J. Oncol. 2012, 40, 1831–1838. [Google Scholar] [CrossRef] [Green Version]
- Stowell, S.R.; Arthur, C.M.; Mehta, P.; Slanina, K.A.; Blixt, O.; Leffler, H.; Smith, D.F.; Cummings, R.D. Galectin-1, -2, and -3 Exhibit Differential Recognition of Sialylated Glycans and Blood Group Antigens. J. Biol. Chem. 2008, 283, 10109–10123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiodelli, P.; Urbinati, C.; Paiardi, G.; Monti, E.; Rusnati, M. Sialic acid as a target for the development of novel antiangiogenic strategies. Futur. Med. Chem. 2018, 10, 2835–2854. [Google Scholar] [CrossRef] [PubMed]
- Kitazume, S.; Imamaki, R.; Ogawa, K.; Taniguchi, N. Sweet role of platelet endothelial cell adhesion molecule in understanding angiogenesis. Glycobiology 2014, 24, 1260–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abeln, M.; Albers, I.; Peters-Bernard, U.; Flächsig-Schulz, K.; Kats, E.; Kispert, A.; Tomlinson, S.; Gerardy-Schahn, R.; Münster-Kühnel, A.; Weinhold, B. Sialic acid is a critical fetal defense against maternal complement attack. J. Clin. Investig. 2018, 129, 422–436. [Google Scholar] [CrossRef] [Green Version]
- Nakashima, A.; Tsuda, S.; Kusabiraki, T.; Aoki, A.; Ushijima, A.; Shima, T.; Cheng, S.-B.; Sharma, S.; Saito, S. Current Understanding of Autophagy in Pregnancy. Int. J. Mol. Sci. 2019, 20, 2342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakashima, A.; Yamanaka-Tatematsu, M.; Fujita, N.; Koizumi, K.; Shima, T.; Yoshida, T.; Nikaido, T.; Okamoto, A.; Yoshimori, T.; Saito, S. Impaired autophagy by soluble endoglin, under physiological hypoxia in early pregnant period, is involved in poor placentation in preeclampsia. Autophagy 2013, 9, 303–316. [Google Scholar] [CrossRef] [Green Version]
- Aoki, A.; Nakashima, A.; Kusabiraki, T.; Ono, Y.; Yoshino, O.; Muto, M.; Kumasawa, K.; Yoshimori, T.; Ikawa, M.; Saito, S. Trophoblast-Specific Conditional Atg7 Knockout Mice Develop Gestational Hypertension. Am. J. Pathol. 2018, 188, 2474–2486. [Google Scholar] [CrossRef] [Green Version]
- Bustos, S.O.; Pereira, G.J.D.S.; Saito, R.D.F.; Gil, C.D.; Zanatta, D.B.; Smaili, S.S.; Chammas, R. Galectin-3 sensitized melanoma cell lines to vemurafenib (PLX4032) induced cell death through prevention of autophagy. Oncotarget 2018, 9, 14567–14579. [Google Scholar] [CrossRef] [Green Version]
- Qin, A.; Zhong, T.; Zou, H.; Wan, X.; Yao, B.; Zheng, X.; Yin, D. Critical role of Tim-3 mediated autophagy in chronic stress induced immunosuppression. Cell Biosci. 2019, 9, 13. [Google Scholar] [CrossRef] [Green Version]
- Sudhakar, J.N.; Lu, H.-H.; Chiang, H.-Y.; Suen, C.-S.; Hwang, M.-J.; Wu, S.-Y.; Shen, C.-N.; Chang, Y.-M.; Li, F.-A.; Liu, F.-T.; et al. Lumenal Galectin-9-Lamp2 interaction regulates lysosome and autophagy to prevent pathogenesis in the intestine and pancreas. Nat. Commun. 2020, 11, 4286. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blois, S.M.; Prince, P.D.; Borowski, S.; Galleano, M.; Barrientos, G. Placental Glycoredox Dysregulation Associated with Disease Progression in an Animal Model of Superimposed Preeclampsia. Cells 2021, 10, 800. https://doi.org/10.3390/cells10040800
Blois SM, Prince PD, Borowski S, Galleano M, Barrientos G. Placental Glycoredox Dysregulation Associated with Disease Progression in an Animal Model of Superimposed Preeclampsia. Cells. 2021; 10(4):800. https://doi.org/10.3390/cells10040800
Chicago/Turabian StyleBlois, Sandra M., Paula D. Prince, Sophia Borowski, Monica Galleano, and Gabriela Barrientos. 2021. "Placental Glycoredox Dysregulation Associated with Disease Progression in an Animal Model of Superimposed Preeclampsia" Cells 10, no. 4: 800. https://doi.org/10.3390/cells10040800