1. Introduction
The incidence of melanoma is increasing annually at a high rate. Although melanoma represents only 5% percent of skin cancers, it is an aggressive neoplasm and it can rapidly become life-threatening once it has metastasized. Malignant melanoma results in high mortality rates with the higher potential of dissemination among the malignancies. It is responsible for most skin cancer-associated deaths with over 200,000 diagnosed melanoma cases each year worldwide, leading to 55,000 deaths [
1]. It preferentially metastasizes to distant lymph nodes and to organs such as lungs (18–36%), liver (14–20%) and brain (12–20%) [
2]. Notably, pulmonary metastatic melanoma is often asymptomatic. Therefore, early diagnosis and better prevention are important. While environmental factors, lifestyle and phenotypic and genetic susceptibility contribute to the initiation of melanoma, etiology is complex, multifactorial and heterogeneous. For instance, UV radiation is one of the most considerable environmental risk factors, displaying a mutagenic role in melanoma pathogenesis [
2]. Moreover, phenotypic risk factors, including light pigmentation, tendency to sunburn, inability to tan, misshaped lesions with several protrusions and a big number of melanocytic nevi play a major role in the metastatic dissemination of melanoma [
3,
4].
In malignant melanoma, the role of cyclins and especially cyclin D1 and D3 are well described. It is reported that abnormal cyclin D3 expression is correlated with poor clinical outcome, as well as high rate of metastatic melanoma lesions in superficial melanoma [
5]. Cyclin D1 is responsible for promoting cell cycle progression through the G
1-S phase by encoding the regulatory subunit of a holoenzyme, which phosphorylates and inactivates the retinoblastoma protein. Overexpression of cyclin D1 is mainly related to human tumorigenesis and metastasis progression, eventually affecting the development of several human cancer types, such as parathyroid adenoma, breast cancer, colon cancer, prostate cancer, lymphoma and melanoma [
6]. It is worth noticing that NRAS or BRAF mutation are frequent and associated with melanoma subtypes together with cyclin D1 and other factors [
7]. Despite the advances in the treatment of malignant melanoma with BRAF/MEK targeted agents and immunotherapy, a poor prognosis for advanced disease is noted and therefore cyclin-dependent kinase inhibition either as single agent or in combination with established treatment are under clinical trials [
8].
Transcription factor Snail is reported to potentially induce epithelial-to-mesenchymal transition (EMT) endowing cells with increased migratory and invasive properties [
9]. During melanoma progression, cancer cell proliferation is disrupted because of Snail–mediated downregulation of E-cadherin [
10]. Moreover, the inhibition of Snail-triggered EMT resulted in melanoma tumor suppression [
11].
Melanoma metastasis is characterized by an invasion of the tumor cells to the nearest tissues and a formation of secondary sites of tumor growth after colonization of distant tissues. A crucial step for the initiation of melanoma metastasis is the degradation of key components of the basement membrane and macromolecules of the extracellular matrix (ECM) by matrix metalloproteinases (MMPs). Cancer cells have been reported to express high levels of MMP-14 (known also as MT1-MMP) especially in the cell protrusions and extracellular vesicles (EVs) [
12,
13]. Cancer cell-derived EVs are increasingly being recognized as genuine invasive structures as they contribute to many aspects of invasion. A recent report indicates a role of the actin cytoskeleton in the mechanisms underlying EV biogenesis or release. Indeed, Els Beghein et al. have demonstrated a role of the cortactin in EV release [
12]. A contribution of this protein in endosomal trafficking was shown to be a crucial step in EV biogenesis. EVs are preferentially released at invadopodia, the latter being actin rich invasive cell protrusions where cortactin plays a regulatory role. Accordingly, EVs are enriched with invadopodial proteins such as the MMP-14 and exert gelatinolytic activity [
12]. Thus, cortactin plays key roles in EV release by regulating endosomal trafficking or invadopodia formation and function.
Hyaluronan (HA), a linear glycosaminoglycan, with extremely complex functions, plays crucial roles in the development and progression of malignant tumors [
14]. The contribution of HA to tumorigenesis relies on its metabolism, depending on the hyaluronan synthases (HASs) by the mean of the upregulation of HAS2 and HA overexpression as well as by hyaluronidases (HYALs) that degrade HA to various molecular sizes. It has been demonstrated that UDP-glucose 6-dehydrogenase regulates HA production and promotes breast cancer progression [
15]. Moreover, binding of HA to its cellular receptors, mainly CD44 and RHAMM, leads to the activation of several tumorigenic signaling pathways and regulation of cellular functions. It is worth noticing that decreased HA amount is related with elevating content of degrading enzyme HYAL2, and the decreased expression of HAS1 and HAS2 in invasive melanoma [
16,
17].
Heparanase as the only mammalian endoglycosidase, which is able to degrade heparan sulfate side chains of proteoglycans (PGs), plays a crucial role in the remodeling of ECM, and is implicated in the EMT cellular invasion, related to angiogenesis, inflammation and metastasis. Heparanase expression is elevated in several types of tumors and this high expression pattern is mostly related to aggressive type of disease, poor prognosis, as well as increased tumor metastasis [
18,
19,
20].
Melanoma metastasis correlates with the degradation of macromolecules of the ECM. Among them, PGs are critically implicated in several pathophysiological processes, such as cancer [
14]. Small leucin-rich PGs (SLRPs) are secreted in the ECM and are able to interact with matrix effectors, such as cytokines, growth factors and cell surface proteins [
21]. These interactions endow SLRPs with important cell functional properties, such as regulation of migration, invasion, angiogenesis and metastasis. Lumican is a Class II SLRP, with high molecular heterogeneity due to its glycosylation which is tissue-dependent [
22,
23]. Structurally, its core protein consists of three major domains: a negatively charged N-terminal domain, containing tyrosine sulfates and cysteine residues, a central part, which is characterized by nine leucine rich repeats, and a C- terminal domain of 66 amino acids [
22,
23].
According to the tissue, lumican structure and expression differ qualitatively and quantitatively, respectively. For instance, lumican mRNA exhibits very low expression in liver and sclera, and is almost undetectable in brain and sternum [
23]. It has been shown that lumican delays melanoma growth in mice and plays regulatory roles in functional properties and invadopodia formation in breast cancer cells [
24].
Biologically, lumican is able to regulate various physiological processes, like collagen fibrillo-genesis, cell proliferation, adhesion, migration and invasion [
21,
25,
26]. Moreover, the anticancer effect of lumican was reported in numerous publications [
13,
24,
25,
26,
27,
28,
29,
30,
31,
32,
33,
34,
35]. The former studies [
28,
29] utilized as a melanoma model, B16F1 cells transfected with the human lumican cDNA injected subcutaneously in wild-type C57BL/6J mice. Lumican was shown to inhibit the development of the melanoma primary tumor as well as lung metastatic nodules development [
28,
29]. More recently, another mouse melanoma model was investigated in which melanoma B16F1 cells were transfected with human Snail cDNA and then injected subcutaneously in wild-type and Lum
−/− C57BL/6J mice. In this model, endogenous lumican was demonstrated to inhibit the primary melanoma tumor development, while Snail overexpression was shown to induce EMT and the metastatic potential of melanoma cells [
11,
13]. The potential anti-metastatic role of lumican in melanoma by inhibiting the membrane type matrix metalloproteinase MMP-14 activity and melanoma cell migration in vitro was also reported [
13,
33,
34].
Collectively, it has been previously reported that lumican, among other functions, effectively regulates cell functional properties, expression of ECM effectors, EMT, invadopodia markers, morphology of invading breast cancer cells, as well as MMP-14 and cell migration in melanoma cells [
14,
24,
33,
34]. Notably, lumican was shown to be a strong endogenous inhibitor of tumor growth [
34]. Therefore, the aim of the present study was to investigate the development of metastatic melanoma nodules in the lung following intravenous injection of Snail-transfected B16F1 cells in lumican deficient and wild type C57BL/6J mice as well as to analyze the effect of lumican on Mock and Snail overexpressing B16F1 cells in vivo and in vitro focusing on invadopodia formation and the associated signaling pathways.
2. Materials and Methods
2.1. Extracellular Matrix PROTEINS
Recombinant human lumican (57 kDa) was purchased from R&D Systems (2846-LU-050, R&D Systems, Minneapolis, MN, USA). Type I collagen was extracted from rat tail tendon, as previously described [
30]. Recombinant human (rh) MMP-14 (catalytic domain, aa 89-265, EC 3.4.24.-) was purchased from Merck Millipore
®® (Nottingham, UK).
2.2. Animal Care
Heterozygous Lum+/− mice (C57BL/6J) were kindly provided by Pr. Shukti Chakravarti. After multiple breeding, homozygous Lum−/− mice were obtained. The distinction between homozygous Lum−/− mice and their wild-type (Lum+/+) littermates was performed by PCR genotyping. Two pairs of primers, one pair internally from E2 (forward primer, 5′-CCTGAGGAATAACCAAATCGACC-3′ and reverse primer, 5′-AGGCAGCTTGCTCATCTGAATTGA-3′), the second pair from the pGKneo cassette (forward primer 5′-CATTCGACCACCAAGCGAAAC-3′ and reverse primer 5′-AGCTCTTCAGCAATATCACGGG-3′) were used to amplify a 380-bp and a 300-bp product from the wild type and mutant allele, respectively. All mice procedures conformed to the ethical rules of the University of Reims Champagne-Ardenne ethical committee and were approved by the URCA Animal facility platform (URCAnim, Reims EU0362), and the CNRS. This study was performed respecting the “The French Animal Welfare Act” and “The French Board for Animal Experiments”. Experiments were conducted under approval of the French “Ministère de l’Enseignement Supérieur et de la Recherche” (ethics committee n°C2EA-56) in compliance with the “Directive 2010/63/UE”, Protocol number: APAFIS#17470-2018110911091242V2.
2.3. Cell Culture and Transfection
B16F1 melanoma cells were transfected with the plasmids containing human SNAIL. The pcDNA 3.1-human SNAIL construct was obtained from Prof. Muh-Hwa Yang (Taipei Veterans General Hospital, Taiwan). B16F1 melanoma cells were grown up to 85% confluence in 1 g/L glucose DMEM Biosera LTD (Courtaboeuf CEDEX, France) in presence of 0.1% penicillin/streptomycin and 10% FCS at 37 °C with 5% CO2. The Amaxa Nucleofector X Unit (Lonza, Basel, Switzerland) was used for the transfection of cells with 5 μg DNA/106 cells, according to the manufacturer’s instructions. Cells were cultured in medium with additional 200 μg/mL of Geneticin/G418 (Gibco/LifeTechnologies, Waltham, MA, USA). After two weeks of cell culture and refreshing selection media every two days, the colonies, which were well-separated, were selected. The culture of clones was scaled-up and SNAIL expression was verified by RT-PCR and Western blot analysis. Mock-B16F1 cells, transfected with an empty plasmid, cultured with media supplemented with G418 (200 μg/mL), were used as controls.
2.4. Lung Metastasis Analysis
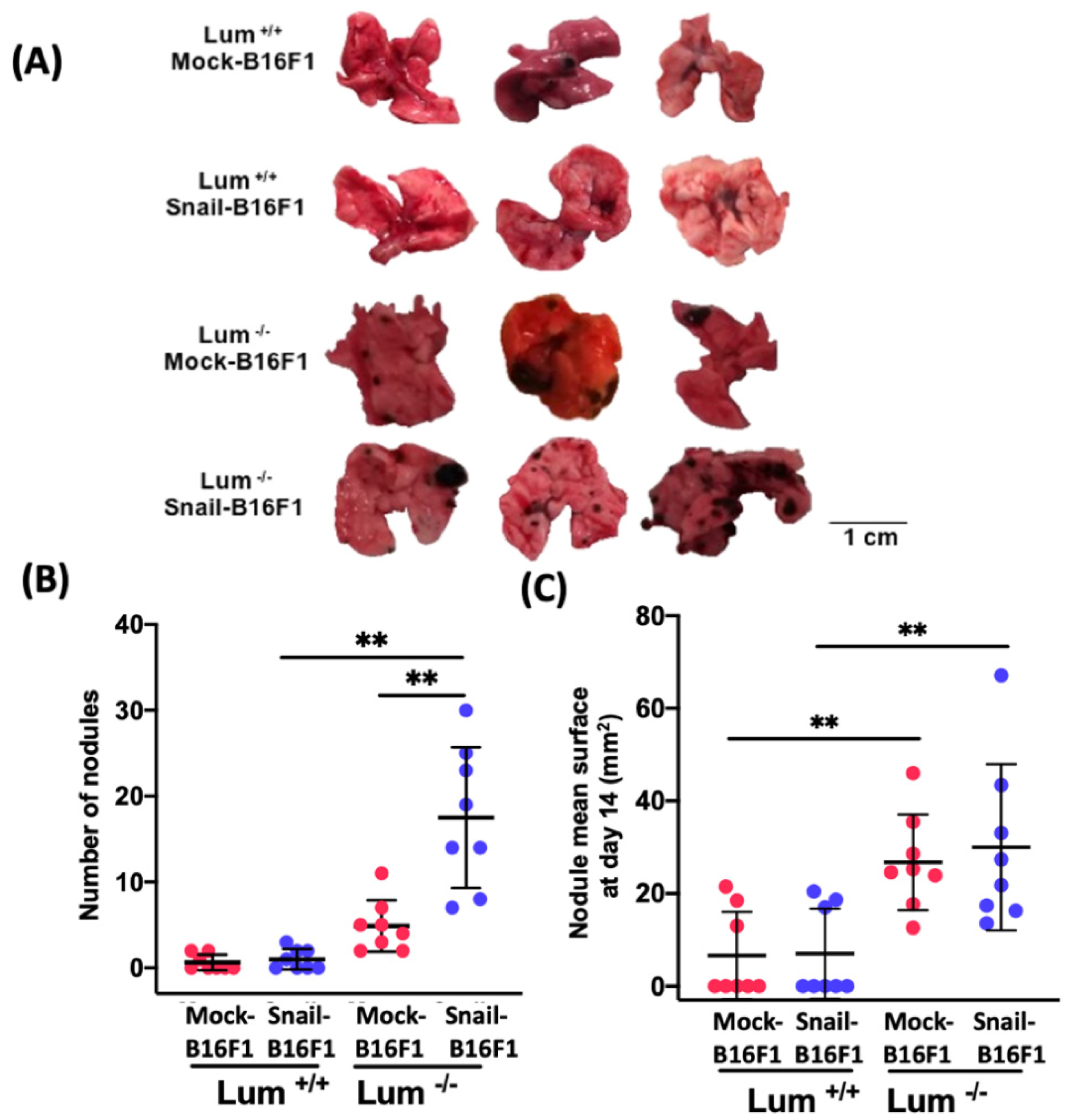
Mock- or Snail-B16F1 cells (n = 2.5 × 105 cells per mouse, 100 μL of cell suspension in DMEM) were injected in the tail vein of a total of 32 mice (Mock-B16F1 cells injected in WT-mice (n = 8), Mock-B16F1 cells injected in lumican deleted mice (n = 8), Snail-B16F1 cells injected in WT-mice (n = 8), Snail-B16F1 cells injected in lumican deleted-mice (n = 8)). At day 14, mice were sacrificed. The lungs were isolated from every animal and were rinsed in PBS. The number of nodules was calculated in lungs. Tissues were paraffin embedded and 5 μm thick sections were performed.
2.5. Histological and Immunohistochemical Analyses
Histological analyses of paraffin-embedded sections were performed after hematoxylin and eosin (HE) staining. Paraffin-embedded tissue sections from metastatic lungs were analyzed by immunohistochemistry for protein expression of cyclin D1. Immunohistochemistry was performed using the primary antibody of anti-cyclin D1 (see
Table S1). Biotin-labeled secondary antibodies, streptavidin-HRP, and DAB (3,3′Diaminobenzidine) detection system was provided by Abcam (Abcam, Paris, France). Negative controls were performed by omitting the primary antibody and by using control isotypes (Dako, Les Ulis, France) as negative controls. Cyclin-D1-positive cells were numbered in five random fields of the metastatic lung nodules area at 100× magnification. All quantitative analyses were performed using ImageJ software binarization and thresholding tools.
2.6. Scanning Electron Microscopy (SEM)
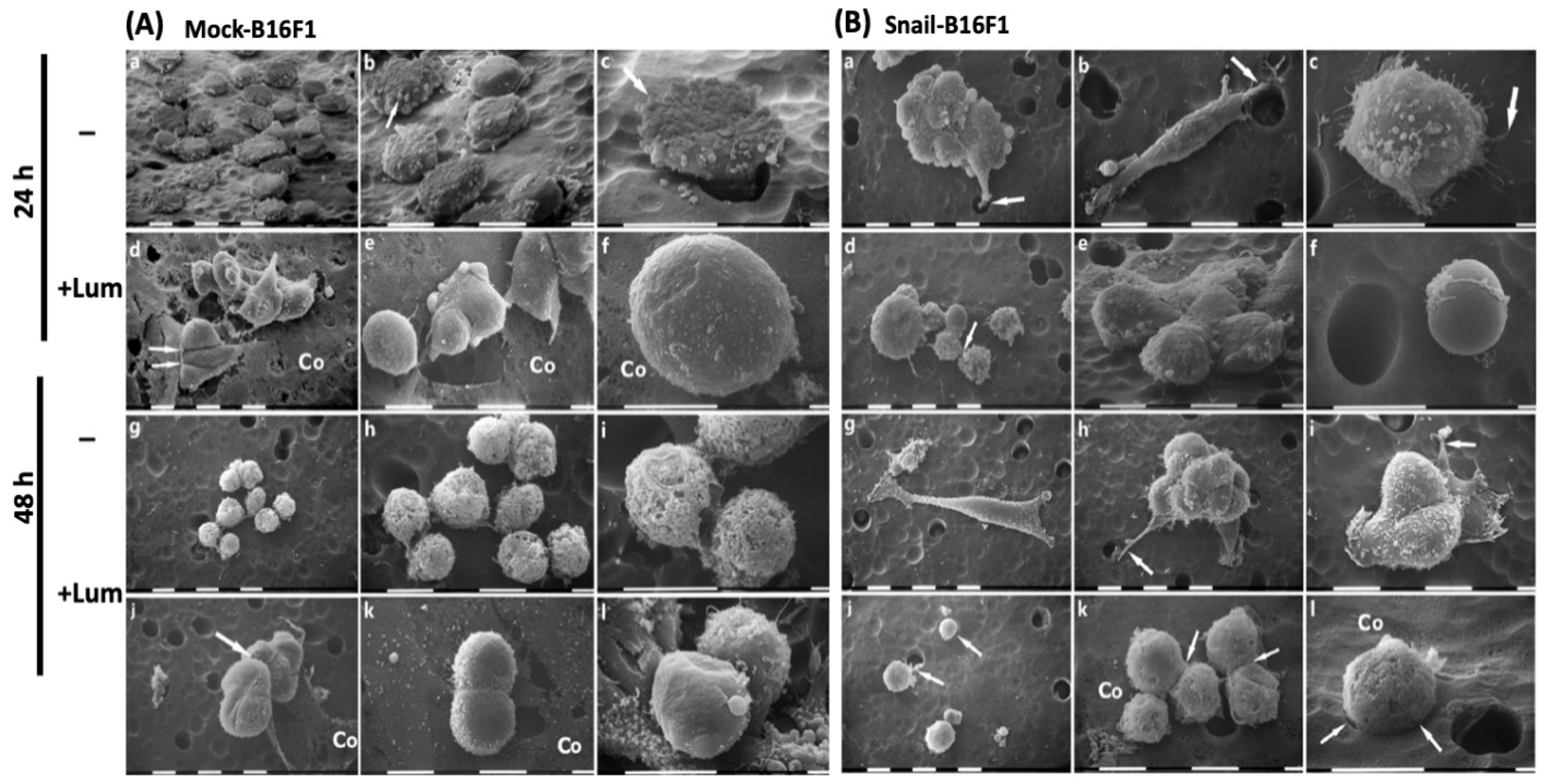
Mock-B16F1 and Snail-B16F1 cells were assessed by using the Boyden-chamber assay. Negative controls were also tested. “Isopore Membrane Filters” (Millipore®®, Milan, Italy) with pore size of 8.0 μm were used for the assays. Twenty-four filters were coated with Type I collagen from rat tendon. For this purpose, collagen was dissolved in 18 mM sterile acetic acid, in order to reach the concentration of 5 μg/100 μL. Initially, 100 μL from the collagen mixture were added in each well of the twenty-four filters trans-well plate, and the plate was incubated overnight at 4 °C. Another portion of 100 μL was added in each well in order to reach a total concentration of 10 μg/100 μL. For every cell line, 2 × 105 cancer cells suspended in 800 μL serum-free medium were seeded on top of the gel in the upper chamber. Conditioned media were added in the lower chamber and were used as chemoattractant.
For each cell line, three filters were fixed after 24 h and thee additional filters were fixed after 48 h of cell culture (control samples). For each cell line, three different filters were treated with 100 nM of lumican for 24 h and other three for 48 h. All filters with cells seeded on the top were fixed in a Karnovsky’s solution for 20 min and then rinsed three times with 0.1% cacodylate buffer. Afterwards, samples were dehydrated with increasing concentrations of ethanol, and finally dehydrated with hexamethyl disilazane (Sigma-Aldrich Inc., Atlanta, GA, USA) for 15 min. All samples were placed on appropriate stubs, covered with a 5 nm palladium gold film (Emitech 550 sputter-coater, Ashford, UK), to be observed under a SEM (Philips 515, Eindhoven, The Netherlands) operating in secondary-electron mode.
2.7. Laser Scanning Microscopy
For Immunofluorescence/confocal microscopy experiments, cells were seeded on sterile glass coverslips in 24-well plates and grown to 50% confluence before treatment. Snail-B16F1 and Mock-B16F1 cells were seeded at 5 × 10
5 cells/well. Cells were rinsed twice in PBS, fixed using 4% paraformaldehyde (in PBS buffer) pH 7.2 for 15 min at room temperature and washed three times with PBS-Tween buffer. After saturation with 3% BSA for 30 min at room temperature, cells were incubated overnight at 4 °C with the appropriate primary antibody. The references and the dilutions of the primary antibodies are displayed in the
supplementary Table S1. For the detection of actin cytoskeleton, cells were permeabilized with 0.1% Triton X-100 and incubated 1 h at room temperature with Alexa Fluor
®® 488-conjugated phalloidin at 1/200 dilution. Negative controls were also prepared after staining with mouse IgG. Slides were observed under confocal laser scanning microscope (Zeiss LSM 700, Zeiss, Marly le Roi, France).
2.8. Real Time RT-PCR
Total RNA of Mock- and Snail-overexpressing B16F1 cells, was isolated using RNeasy Plus Mini Kit (Qiagen, Courtaboeuf, France) according to the instructions of the Manufacturer. RNA quality was examined and determined on an Agilent 2100 Bioanalyzer (Agilent Technologies, Massy, France). Afterwards, reverse transcription was performed at 50 °C for 30 min using the Maxima First Strand TM cDNA synthesis kit (Thermo Scientific, Villebon and Yvette, France) with 1 μg of total RNA. The Instrument used was the Abi Prism Instrument (Fisher Scientific, Illkirch, France). The primers were purchased from Applied Biosystems (Applied Biosystems Fisher Scientific, Illkirch, France) and are displayed in the
supplementary Table S2. The relative quantifications and finally the results were obtained with the use of the ΔΔCt method. PCR assays were conducted at least three times in triplicates for each sample. Another Instrument used was the Mx3005P Thermocycler (SYBR Green) and the primers were purchased from Abi Taqman, displayed in the
supplementary Table S3. The relative quantifications and finally the results were obtained with the use of ΔΔCt method. PCR assays were conducted at least three times in triplicates for each sample. All primer sequences and sizes of the PCR product of each targeted gene are presented in
supplementary Tables S2 and S3. The specificity of PCR amplification products was assessed by dissociation melting-curve analysis. After the reaction was completed, Ct value was calculated from the amplification plots. The standard curves were generated with serially diluted solutions (1/5–1/3125) of cDNA from B16F1 cells. Each sample was normalized to a housekeeping gene transcript either β-Actin or glyceraldehyde-3-phosphate dehydrogenase (GAPDH).
2.9. Western Immunoblotting
Total cell proteins were prepared from cell monolayers after being washed twice with PBS and detached after scrapping in cell lysis buffer (50 mM Tris-HCl, pH 7.6, 0.5 M NaCl, 0.02% NaN
3, 0.6% NP40, 5 mM EDTA, 1 mM iodoacetamide, 1 mM PMSF, 1 mM Na
3VO
4, Protease Inhibitor Cocktail (Sigma)). The Bradford method was used for the protein concentration. Total cell proteins (30 μg) were mixed with 5× Laemmli buffer (Tris 1.25 M, SDS 10%, sucrose 20%, pH 6.8, bromophenol blue 0.005%) and β-mercapto-ethanol, to produce a final concentration of the latter of 3%. The total volume of the samples is maximum 30 μL, with the precondition that all the loading samples have the same concentration. Samples were denatured for 5 min at 95 °C and were then subjected to electrophoresis in a polyacrylamide gel (concentration range: 7.5 to 15%, containing 0.1% SDS). Proteins were transferred by electroblotting onto Hybond-P PVDF membranes (GE Healthcare, Orsay, France), previously activated for 10s in methanol. The membranes were saturated in TBS-T solution (0.1% Tween 20, 20 mM Tris and 140 mM NaCl, pH 7.6) containing 5% nonfat milk (Bio-Rad) or 5% BSA for 2 h at room temperature. Membranes were incubated overnight at 4 °C with constant gentle shaking with primary antibodies (
Supplementary Table S1). The next day, membranes were washed three times with TBS-T and incubated with a 1:10,000 dilution of the adequate corresponding secondary antibody conjugated to horseradish peroxidase in 1% nonfat milk or 1% BSA, in TBS-T for 1 h at room temperature. After three washes with TBS-T, the bands were revealed by the ECL Prime Chemiluminescence Detection reagent (GE Healthcare, Orsay, France), according to the manufacturer’s instructions. The chemiluminescence signal was captured using a ChemiDocTM MP Imaging (BioRad, Marnes-la-Coquette, France).
2.10. Phosphorylation Analysis
Western Blot experiments concerning the signaling pathways require a specific method of preparation of the cell lysates. For every cell line, 5 × 106 cells were required as a starting point. Cells were rinsed twice with a buffer containing 50 mM HEPES, 126 mM NaCl, 5 mM KCl, 1 mM Na2EDTA. Then, cells were incubated for 5 min at 37 °C to detach in the above buffer. Serum-free basal medium was added and the cell suspensions were centrifuged for 1 min at 400× g in 4 °C. The cell pellets were resuspended in serum-free basal medium and separated in low-binding tubes.
Every tube corresponds to a time point: 0, 5, 10, 15 min. Cell suspensions were prepared both in the absence and presence of lumican (57 kDa). Lumican 100 nM was added at 0 min time point. All samples were placed in a carousel at 37 °C and were pulled according to the time point. All the gathered samples (final volume 100 μL) were placed on ice and then, centrifuged at 400× g at 4 °C. Pellets were resuspended with lysis buffer (2% SDS in 50 mM Tris, pH 7.4, 150 mM NaCl, 5 mM EDTA, 10 mM NaF, 2 mM Na3VO4, 1 mM PMSF, 10 μg/mL leupeptin, 10 μg/mL aprotinin). Samples were centrifuged at 1400× g at 4 °C for 15 min. Supernatants were collected and their protein concentration was measured by Bradford Assay. Proteins were denatured at 95 °C for 5 min before western immunoblotting.
2.11. Invasion Assay
The invasive potential of B16F1 melanoma cells was evaluated as follows: ThinCertTM cell culture inserts (24-well, pore size 8 μm; Greiner, Bio-One, Courtaboeuf, France) were used and 5 × 104 cells/chamber were seeded in absence or presence of 100 nM of lumican when needed. Every cell invasion chamber includes an 8-micron pore size PET membrane coated with 50 μg of Matrigel. (BD, Biosciences, San Jose, CA, USA). Matrigel. was polymerized at 37 °C for 1 h. In the upper chamber, 200 μL medium used was serum-free with 0.5% BSA, while the lower chamber contains 800 μL of medium, containing 5% FBS, as chemoattractant, and 2% BSA. Negative controls, containing 2% BSA, were also prepared. After 48 h, the non-invasive cells were removed with a cotton swab along with cells that invaded through the Matrigel. The invasion of B16F1 cells was determined by counting the number of stained nuclei by crystal violet under Å~20 magnification using a Zeiss Axiovert-25 inverted microscope equipped with a digital camera (Carl Zeiss). Each individual experiment (n = 3) was made of three inserts per condition and three microscopic fields were counted per insert.
2.12. Statistical Analyses
Results were expressed as mean ± SD. Statistical significance between groups was assessed by unpaired Student’s t-test or, when necessary, by one-way analysis of variance. p value < 0.05 was considered statistically significant. Every standard deviation (SD) was obtained from at least three experiments, made in triplicate. Statistical analysis and graphs were made using GraphPad Prism 6 (GraphPad Software, San Diego, CA, USA).
4. Discussion
Previous in vivo study demonstrated that the size of primary melanoma tumors was significantly smaller in the group of wild-type mice (Lum
+/+) injected subcutaneously with Snail-B16F1 cells in comparison with the mice deficient in lumican gene (Lum
−/−) [
13]. Moreover, the stimulatory effect of SNAIL in the MMP-14 activity and the in vitro melanoma cell migration countered by lumican and its derived peptide in the MMP-14 activity [
25,
33,
34] led us to investigate the effect of lumican in vivo.
The present study demonstrated the pro-invasive role of SNAIL and the inhibitory effect in the formation of pulmonary metastasis by endogenous lumican after intra vascular injection of mock and Snail-B16F1 cells in wild type and lumican deficient mice (Lum−/−).
The results show that endogenous lumican inhibits the cell proliferation in lung metastatic nodules of melanoma cells. Progression of cell cycle is regulated by cyclins, cyclin-dependent kinases, and the respective cyclin-dependent kinase inhibitory proteins. Cyclin D1 overexpression is reported to correlate with short cancer patient survival, as well as tumor progression, and can lead to oncogenesis by inducing anchorage-independent growth and angiogenesis through VEGF promotion [
36] In the present report, cyclin D1 was abundantly expressed in the lumican knockout-mice, injected with either Mock-B16F1 or with Snail-B16F1 melanoma cells, while it was less expressed in the wild type lumican group, rendering lumican a powerful metastasis suppressor.
Analysis at SEM demonstrated that lumican treatment reduces the aggressiveness of both Mock-B16F1 and Snail-B16F1 cells by reducing the cytoplasmic protrusions and microvilli which are related to cancer cells’ aggressiveness [
24]. Lumican treatment also favors grouping of cells and cell–cell tight contacts and modifies the method of cell invasion/invagination in Mock-B16F1. It is of interest that in Snail-B16F1 cells, lumican also inhibits the mesenchymal phenotypes which are expression of individual cell invasion through the invadopodia development and a proteolytic process [
24,
37].
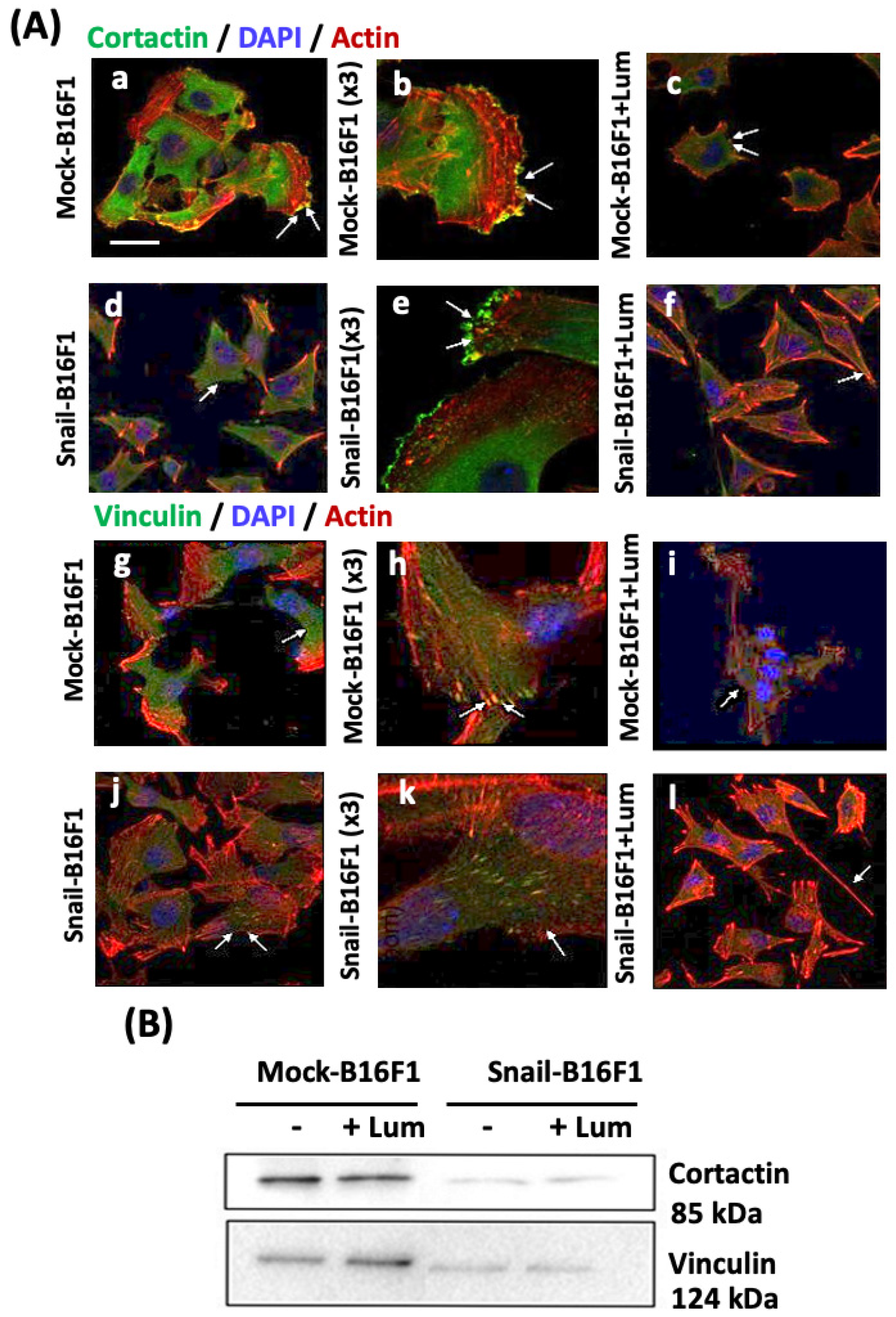
Invadopodia formation was analyzed first by confocal microscopy and later by immunostaining following the distribution and expression of two specific biomarkers. Cortactin is a regulator protein of the formation and structure of invadopodia [
38]. The co-localization with actin is expected, since cortactin is essential for the maintenance of F-actin-enriched invadopodia core structures. The direct relation of SNAIL and the number of formed invadopodia and particularly the expression of cortactin, marker of invadopodia, has been shown. Lumican significantly inhibited the expression of cortactin. Inhibition of cortactin leads to the blocking of Src signaling, which results in the inhibition of invadopodia formation and, in the long term, the inhibition of metastasis, confirming the anti-metastatic action of lumican [
39].
Vinculin overexpression facilitates cell adhesion and recruitment of cytoskeletal proteins at the domains of integrin binding at the sites of focal adhesions [
40]. Inhibition of vinculin leads to the alteration of many cell functions, such as formation of fewer focal adhesions and inhibition of lamellipodia protrusions [
41]. Vinculin is part of the focal adhesion complexes and consists of essential invadopodia markers [
42]. Thus, the role of vinculin in the morphology of the melanoma cells was addressed. Regarding vinculin staining, its expression was significantly decreased by the treatment of lumican at the focal adhesion complexes, accompanied by decreased formation of invadopodia as observed by confocal microscopy. Vinculin increased expression facilitates cell adhesion and recruitment of cytoskeletal proteins at the domains of integrin binding at the sites of focal adhesions. On the other hand, in Snail-B16F1 melanoma cells treated with lumican, vinculin expression is significantly reduced, leading to the formation of fewer focal adhesions and inhibition of lamellipodia protrusions, further suggesting the anti-metastatic effect of lumican.
The effect of lumican on invadopodia markers, prompted us to monitor the effect of lumican of hyaluronan synthesis markers, such as synthase HAS2 and HAS2-AS1. Hyaluronan synthesis is induced not only by the cross-talk between stromal and tumor cells, but also by tumor cells themselves promoting their aggressive potential [
43]. Moreover, antisense inhibition of HAS2 in aggressive breast cancer has been reported to inhibit the in vivo formation of tumors [
17]. It is known that the metastatic ability of cancer cells is associated with the degradation of the ECM and increased heparanase expression. Heparanase degrades heparan sulfate chains and it contributes to ECM remodeling. Heparanase is eventually involved in ECM remodeling and EMT [
44,
45]. When heparanase is downregulated, it can inhibit tumor metastasis [
44,
45]. Previous studies from our laboratories showed that MMP-14 activity was inhibited by lumican in melanoma [
33].
Cell invasion is a crucial biological property, and therefore the effect of lumican on melanoma cell invasion properties was analyzed by an in vitro cell invasion assay (
Figure 5C). We investigated whether lumican could prevent invasion through type I collagen coating. The demonstrated inhibitory effect of lumican on melanoma cell invasion may suggest that lumican potentially inhibited cell migration by involvement of FAK phosphorylation in EMT-like melanoma cells.
MMP-14 plays an important role in cell migration, not only by regulating the activity and expression of downstream MMPs [
46], but also by processing and activating migration-associated molecules, such as integrins or CD44. MMP-14 interacts with CD44 and localizes at invadopodia, rendering MMP-14 a crucial regulator of cell migration and invasion. Lumican was shown to be an inhibitor of MMP-14 expression and thus of cell invasion and metastasis.
MMP-14 also activates a variety of intracellular signaling pathways, such as MAPK, FAK, Src and Rac [
47,
48]. The inhibition of melanoma cell migration by lumican was related to the inhibition of the phosphorylation of focal adhesion kinase (FAK) [
28], and as observed, a significant decrease of the ratio pFAK/FAK was also shown in presence of lumican [
28]. In this study, in the presence of lumican, inhibition of pY397FAK phosphorylation was also observed. The ratio pFAK/FAK was decreased by 40% compared with control. Therefore, the decreased FAK phosphorylation and the inhibition of MMP-14 activity induced by lumican in melanoma cells might explain, at least in part, the anti-invasive effect of this SLRP.
Proteolytic degradation of ECM by MMP-14 induces integrin cell–cell and cell–matrix contacts, focal adhesion stability, FAK phosphorylation at Tyr-397, AKT activation, and ERK activation, resulting in the promotion of cell migration [
47]. Our hypothesis is that lumican inhibits the degradation of ECM by inhibiting MMP-14, then influencing integrin clustering, modulating focal adhesion stability and FAK phosphorylation at Tyr-397, and inhibiting cell migration. Furthermore, it was also demonstrated that activation of RTK, like EGFR, leads to a phosphorylation of FAK and promotes cell migration [
48]. Therefore, lumican’s anti-cancer effect in respect to the decrease of EGFR phosphorylation and the downregulation of the downstream signaling, which results in decreased phosphorylation of FAK and inhibition of cell migration, could be considered as a potential mechanism that needs further evaluation.
The inhibition of FAK phosphorylation induced by lumican leads to downregulation of the phosphorylation of p130 Cas and AKT. The inhibition of p130 Cas phosphorylation makes this protein inactive. As a consequence, a down-regulation of the downstream signaling events may explain the inhibition in lamellipodia formation and MMP production resulting in an inhibition of cell migration. Moreover, the inhibition of AKT kinase results in a decrease of its activity and to a reduction of the downstream signaling cascade which may cause inhibition of cell growth. The AKT–mediated phosphorylation of serine-9 in GSK3α/β significantly decreases the availability of the active site for its substrate, such as β-catenin. However, lumican by inhibiting phosphorylation of AKT causes decreased phosphorylation of GSK3α/β at serine-9 resulting in possible increased activity of this enzyme. Degraded proteins cannot be translocated to the nucleus and act as transcription factors for genes connected with cell proliferation and cell migration. This may explain decreased melanoma cell growth and cell motility in the presence of lumican. Further investigation to evaluate the involvement of MAPK pathway related to MMPs as well as cyclin D1 transcription and p21, which is a cyclin dependent kinase inhibitor, will be useful to evaluate the underlying mechanisms of the lumican action and its potential use for future pharmacological disease targeting.
Altogether, the results are summarized in
Figure 7. These results are promising and indicate a potential mechanism of action for lumican in melanoma metastasis inhibition. Functionalized nanoparticles containing or coated with lumican (or lumican-derived peptides able to mimic the anti-tumor effect of lumican) could be inoculated in vivo by intravascular injection in mice melanoma models to investigate whether it could down-regulate the development of lung metastatic nodules of B16F1 melanoma cells. Then, if the effect is demonstrated in animal models in pre-clinical assays, and that no cytotoxic effect is demonstrated, potential clinical assays could be designed.



 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






