
Considerations to Model Heart Disease in Women with Preeclampsia and Cardiovascular Disease

 , , and
, , and 
Abstract
:1. Introduction
2. Preeclampsia Models
2.1. In Vivo Models
2.1.1. Animal Trophoblast Invasion Model
2.1.2. Utero-Placental Ischemia Model
2.1.3. Anti-Angiogenic Response Model
2.1.4. Immune Models
2.2. In Vitro Models
2.2.1. Models of Trophoblast Cells
2.2.2. Placental Explants
2.2.3. Microfluidics Models
2.2.4. In Vitro Models of Endothelial Dysfunction in Preeclampsia
2.3. Additional Considerations of Current In Vivo and In Vitro Models
3. Cardiovascular Models to Mimic Ischemic-Reperfusion Injury
3.1. In Vivo Models of Ischaemic Heart Disease
3.1.1. Small Mammals Models
3.1.2. Large Non-Human Mammals Models
3.2. Ex Vivo Models of Ischemic Heart Disease
3.3. In Vitro Models of Ischaemic Heart Disease
3.3.1. Cardiomyocytes Cell Culture (2D Culture)
3.3.2. Three-Dimensional (3D) Cultures
3.4. Additional Considerations of Current In Vitro and In Vivo Models
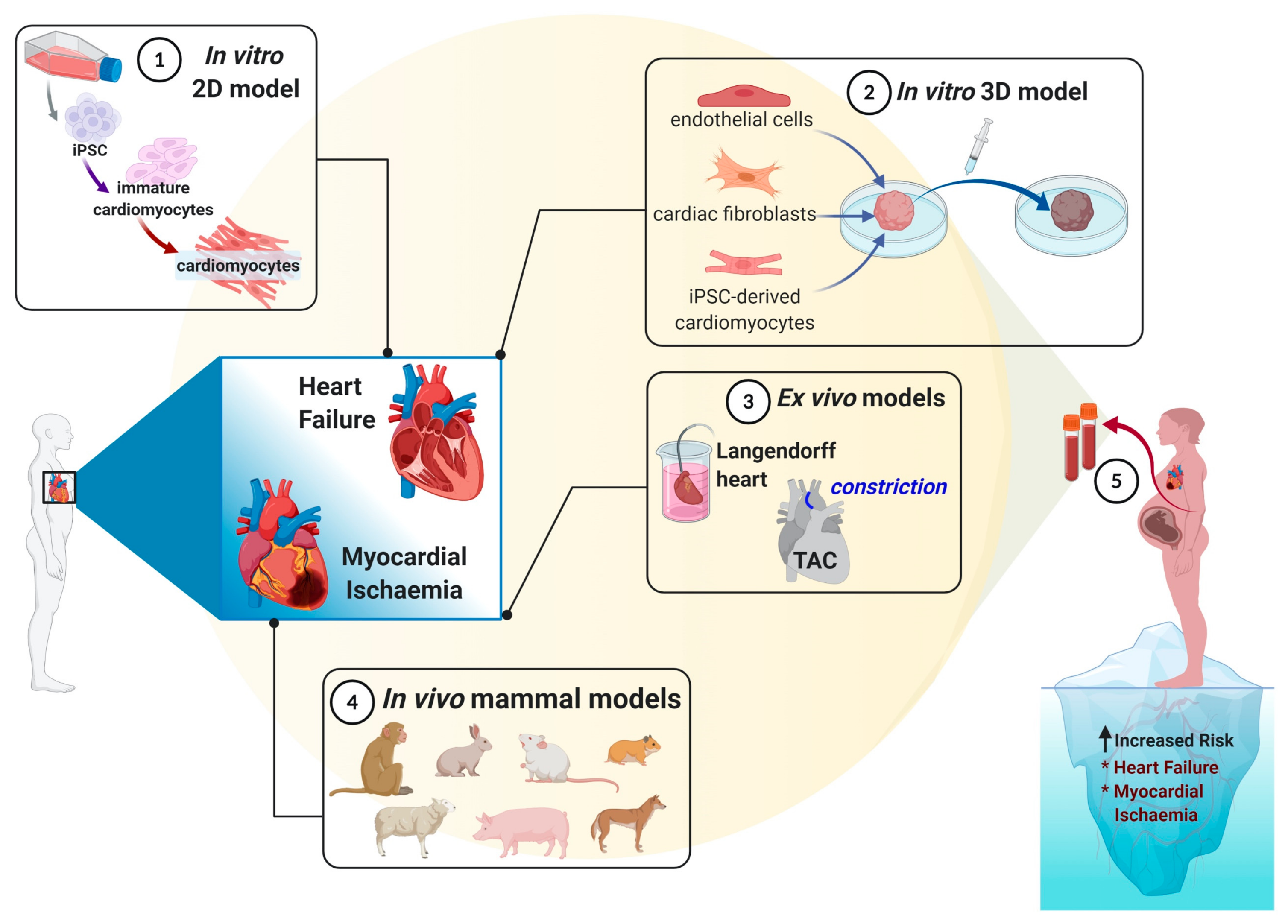
4. Cardiovascular Models to Mimic Heart Failure
4.1. In Vivo Models
4.2. In Vitro Models
4.3. Additional Considerations of Current In Vivo and In Vitro Models
5. Discussion
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bellamy, L.; Casas, J.P.; Hingorani, A.D.; Williams, D.J. Pre-eclampsia and risk of cardiovascular disease and cancer in later life: Systematic review and meta-analysis. BMJ 2007, 335, 974. [Google Scholar] [CrossRef] [Green Version]
- Marshall, S.A.; Hannan, N.J.; Jelinic, M.; Nguyen, T.P.H.; Girling, J.E.; Parry, L.J. Animal models of preeclampsia: Translational failings and why. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 314, R499–R508. [Google Scholar] [CrossRef]
- Pennington, K.A.; Schlitt, J.M.; Jackson, D.L.; Schulz, L.C.; Schust, D.J. Preeclampsia: Multiple approaches for a multifactorial disease. Dmm Dis. Models Mech. 2012, 5, 9–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNally, R.; Alqudah, A.; Obradovic, D.; McClements, L. Elucidating the Pathogenesis of Pre-eclampsia Using In Vitro Models of Spiral Uterine Artery Remodelling. Curr. Hypertens. Rep. 2017, 19, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burton, G.J.; Redman, C.W.; Roberts, J.M.; Moffett, A. Pre-eclampsia: Pathophysiology and clinical implications. BMJ 2019, 366, l2381. [Google Scholar] [CrossRef] [Green Version]
- Leavey, K.; Benton, S.J.; Grynspan, D.; Kingdom, J.C.; Bainbridge, S.A.; Cox, B.J. Unsupervised Placental Gene Expression Profiling Identifies Clinically Relevant Subclasses of Human Preeclampsia. Hypertension 2016, 68, 137–147. [Google Scholar] [CrossRef] [Green Version]
- Hansen, T.; Saleh, S.; Figtree, G.A.; Gentile, C. The Role of Redox Signalling in Cardiovascular Regeneration. In Oxidative Stress in Heart Diseases; Chakraborti, S., Dhalla, N.S., Ganguly, N.K., Dikshit, M., Eds.; Springer: Singapore, 2019; pp. 19–37. [Google Scholar] [CrossRef]
- Lindsey, M.L.; Bolli, R.; Canty, J.M., Jr.; Du, X.J.; Frangogiannis, N.G.; Frantz, S.; Gourdie, R.G.; Holmes, J.W.; Jones, S.P.; Kloner, R.A.; et al. Guidelines for experimental models of myocardial ischemia and infarction. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H812–H838. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J.; Yellon, D.M. Myocardial ischemia-reperfusion injury: A neglected therapeutic target. J. Clin. Investig. 2013, 123, 92–100. [Google Scholar] [CrossRef]
- Sebastião, M.J.; Serra, M.; Pereira, R.; Palacios, I.; Gomes-Alves, P.; Alves, P.M. Human cardiac progenitor cell activation and regeneration mechanisms: Exploring a novel myocardial ischemia/reperfusion in vitro model. Stem Cell Res. Ther. 2019, 10, 77. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, J.P.; Kraus, S.; Mitchell, S.; Perel, P.; Piñeiro, D.; Chioncel, O.; Colque, R.; de Boer, R.; Gomez-Mesa, J.E.; Grancelli, H. World Heart Federation Roadmap for Heart Failure. Glob. Heart 2019, 14, 197. [Google Scholar] [CrossRef]
- Mann, D.L.; Bristow, M.R. Mechanisms and models in heart failure: The biomechanical model and beyond. Circulation 2005, 111, 2837–2849. [Google Scholar] [CrossRef] [Green Version]
- McMurray, J.J.; Pfeffer, M.A. steady increase, age-adjusted rates of admission for heart failure seem to have reached a plateau, or even decreased. Lancet 2005, 365, 1877–1889. [Google Scholar] [CrossRef]
- Ambrosy, A.P.; Fonarow, G.C.; Butler, J.; Chioncel, O.; Greene, S.J.; Vaduganathan, M.; Nodari, S.; Lam, C.S.; Sato, N.; Shah, A.N. The global health and economic burden of hospitalizations for heart failure: Lessons learned from hospitalized heart failure registries. J. Am. Coll. Cardiol. 2014, 63, 1123–1133. [Google Scholar] [CrossRef]
- Ziaeian, B.; Fonarow, G.C. Epidemiology and aetiology of heart failure. Nat. Rev. Cardiol. 2016, 13, 368–378. [Google Scholar] [CrossRef] [Green Version]
- Bui, A.L.; Horwich, T.B.; Fonarow, G.C. Epidemiology and risk profile of heart failure. Nat. Rev. Cardiol. 2011, 8, 30. [Google Scholar] [CrossRef] [Green Version]
- Black, S.C. In vivo models of myocardial ischemia and reperfusion injury: Application to drug discovery and evaluation. J. Pharmacol. Toxicol. Methods 2000, 43, 153–167. [Google Scholar] [CrossRef]
- Dixon, J.A.; Spinale, F.G. Large animal models of heart failure: A critical link in the translation of basic science to clinical practice. Circ. Heart Fail. 2009, 2, 262–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garbern, J.C.; Mummery, C.L.; Lee, R.T. Model systems for cardiovascular regenerative biology. Cold Spring Harb. Perspect. Med. 2013, 3, a014019. [Google Scholar] [CrossRef] [Green Version]
- Janssen, P.M.; Elnakish, M.T. Modeling heart failure in animal models for novel drug discovery and development. Expert Opin. Drug Discov. 2019, 14, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Verdouw, P.D.; van den Doel, M.A.; de Zeeuw, S.; Duncker, D.J. Animal models in the study of myocardial ischaemia and ischaemic syndromes. Cardiovasc. Res. 1998, 39, 121–135. [Google Scholar] [CrossRef]
- Aryan, L.; Medzikovic, L.; Umar, S.; Eghbali, M. Pregnancy-associated cardiac dysfunction and the regulatory role of microRNAs. Biol. Sex Differ. 2020, 11, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suvakov, S.; Bonner, E.; Nikolic, V.; Jerotic, D.; Simic, T.P.; Garovic, V.D.; Lopez-Campos, G.; McClements, L. Overlapping pathogenic signalling pathways and biomarkers in preeclampsia and cardiovascular disease. Pregnancy Hypertens. 2020, 20, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.G.; Kho, C.; Hajjar, R.J.; Ishikawa, K. Experimental models of cardiac physiology and pathology. Heart Fail. Rev. 2019, 24, 601–615. [Google Scholar] [CrossRef] [PubMed]
- Novakovic, G.V.; Eschenhagen, T.; Mummery, C. Myocardial tissue engineering: In Vitro models. Cold Spring Harb. Perspect. Med. 2014, 4, a014076. [Google Scholar] [CrossRef] [Green Version]
- Davis, R.P.; van den Berg, C.W.; Casini, S.; Braam, S.R.; Mummery, C.L. Pluripotent stem cell models of cardiac disease and their implication for drug discovery and development. Trends Mol. Med. 2011, 17, 475–484. [Google Scholar] [CrossRef]
- Pijnenborg, R.; Vercruysse, L.; Pijnenborg, R.; Brosens, I.; Romero, R. Animal models of deep trophoblast invasion. In Placental Bed Disorders; Brosens, I., Pijnenborg, R., Romero, R., Eds.; Cambridge University Press: Cambridge, UK, 2010; pp. 127–139. [Google Scholar]
- Martinez-Fierro, M.L.; Hernndez-Delgadillo, G.P.; Flores-Morales, V.; Cardenas-Vargas, E.; Mercado-Reyes, M.; Rodriguez-Sanchez, I.P.; Delgado-Enciso, I.; Galvn-Tejada, C.E.; Galvn-Tejada, J.I.; Celaya-Padilla, J.M.; et al. Current model systems for the study of preeclampsia. Exp. Biol. Med. 2018, 243, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Carter, A.M. Animal Models of Human Placentation—A Review. Placenta 2007, 28. [Google Scholar] [CrossRef]
- Verlohren, S.; Geusens, N.; Morton, J.; Verhaegen, I.; Hering, L.; Herse, F.; Dudenhausen, J.W.; Muller, D.N.; Luft, F.C.; Cartwright, J.E.; et al. Inhibition of Trophoblast-Induced Spiral Artery Remodeling Reduces Placental Perfusion in Rat Pregnancy. Hypertension 2010, 56, 304–310. [Google Scholar] [CrossRef] [Green Version]
- Aardema, M.W.; Oosterhof, H.; Timmer, A.; van Rooy, I.; Aarnoudse, J.G. Uterine artery Doppler flow and uteroplacental vascular pathology in normal pregnancies and pregnancies complicated by pre-eclampsia and small for gestational age fetuses. Placenta 2001, 22, 405–411. [Google Scholar] [CrossRef]
- Cavanagh, D.; Rao, P.S.; Tung, K.S.; Gaston, L. Eclamptogenic toxemia: The development of an experimental model in the subhuman primate. Am. J. Obstet. Gynecol. 1974, 120, 183–196. [Google Scholar] [CrossRef]
- Myers, R.E.; Fujikura, T. Placental changes after experimental abruptio placentae and fetal vessel ligation of rhesus monkey placenta. Am. J. Obstet. Gynecol. 1968, 100, 946–951. [Google Scholar] [CrossRef]
- Haynes, D.M. Experimental abruptio placentae in the rabbit. Am. J. Obstet. Gynecol. 1963, 85, 626–645. [Google Scholar] [CrossRef]
- Howard, B.K.; Goodson, J.H. Experimental placental abruption. Obstet. Gynecol. 1953, 2, 442–446. [Google Scholar] [CrossRef]
- Granger, J.P.; LaMarca, B.B.; Cockrell, K.; Sedeek, M.; Balzi, C.; Chandler, D.; Bennett, W. Reduced uterine perfusion pressure (RUPP) model for studying cardiovascular-renal dysfunction in response to placental ischemia. Methods Mol. Med. 2006, 122, 383–392. [Google Scholar] [CrossRef]
- Eder, D.J.; McDonald, M.T. A Role for Brain Angiotensin II in Experimental Pregnancy-Induced Hypertension in Laboratory Rats. Clin. Exp. Hypertens. Part B Hypertens. Pregnancy 1987, 6, 431–451. [Google Scholar] [CrossRef]
- Todd, N.; McNally, R.; Alqudah, A.; Jerotic, D.; Suvakov, S.; Obradovic, D.; Hoch, D.; Hombrebueno, J.R.; Campos, G.L.; Watson, C.J.; et al. Role of A Novel Angiogenesis FKBPL-CD44 Pathway in Preeclampsia Risk Stratification and Mesenchymal Stem Cell Treatment. J. Clin. Endocrinol. Metab. 2020, 106, 26–41. [Google Scholar] [CrossRef]
- Maynard, S.E.; Min, J.-Y.; Merchan, J.; Lim, K.-H.; Li, J.; Mondal, S.; Libermann, T.A.; Morgan, J.P.; Sellke, F.W.; Stillman, I.E.; et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J. Clin. Investig. 2003, 111, 649–658. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, F.P.; Kingdom, J.C.; Kenny, L.C.; Walsh, S.K. Animal models of preeclampsia; Uses and limitations. Placenta 2011, 32, 413–419. [Google Scholar] [CrossRef]
- Bergmann, A.; Ahmad, S.; Cudmore, M.; Gruber, A.D.; Wittschen, P.; Lindenmaier, W.; Christofori, G.; Gross, V.; Gonzalves, A.C.d.C.; Gröne, H.-J.; et al. Reduction of circulating soluble Flt-1 alleviates preeclampsia-like symptoms in a mouse model. J. Cell Mol. Med. 2010, 14, 1857–1867. [Google Scholar] [CrossRef] [Green Version]
- Venkatesha, S.; Toporsian, M.; Lam, C.; Hanai, J.I.; Mammoto, T.; Kim, Y.M.; Bdolah, Y.; Lim, K.H.; Yuan, H.T.; Libermann, T.A.; et al. Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat. Med. 2006, 12, 642–649. [Google Scholar] [CrossRef]
- Benyo, D.F.; Smarason, A.; Redman, C.W.; Sims, C.; Conrad, K.P. Expression of inflammatory cytokines in placentas from women with preeclampsia. J. Clin. Endocrinol. Metab. 2001, 86, 2505–2512. [Google Scholar] [CrossRef]
- Kupferminc, M.J.; Peaceman, A.M.; Wigton, T.R.; Rehnberg, K.A.; Socol, M.L. Tumor necrosis factor-alpha is elevated in plasma and amniotic fluid of patients with severe preeclampsia. Am. J. Obstet. Gynecol. 1994, 170, 1752–1757, discussion 1757–1759. [Google Scholar] [CrossRef]
- Vince, G.S.; Starkey, P.M.; Austgulen, R.; Kwiatkowski, D.; Redman, C.W. Interleukin-6, tumour necrosis factor and soluble tumour necrosis factor receptors in women with pre-eclampsia. Br. J. Obstet. Gynaecol. 1995, 102, 20–25. [Google Scholar] [CrossRef]
- Faas, M.M.; Schuiling, G.A.; Baller, J.F.; Visscher, C.A.; Bakker, W.W. A new animal model for human preeclampsia: Ultra-low-dose endotoxin infusion in pregnant rats. Am. J. Obstet. Gynecol. 1994, 171, 158–164. [Google Scholar] [CrossRef]
- Aneman, I.; Pienaar, D.; Suvakov, S.; Simic, T.P.; Garovic, V.D.; McClements, L. Mechanisms of Key Innate Immune Cells in Early- and Late-Onset Preeclampsia. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Lai, Z.; Kalkunte, S.; Sharma, S. A critical role of interleukin-10 in modulating hypoxia-induced preeclampsia-like disease in mice. Hypertension 2011, 57, 505–514. [Google Scholar] [CrossRef] [Green Version]
- Redman, C.W.; Sargent, I.L. Immunology of pre-eclampsia. Am. J. Reprod. Immunol. 2010, 63, 534–543. [Google Scholar] [CrossRef]
- Wenzel, K.; Rajakumar, A.; Haase, H.; Geusens, N.; Hubner, N.; Schulz, H.; Brewer, J.; Roberts, L.; Hubel, C.A.; Herse, F.; et al. Angiotensin II Type 1 Receptor Antibodies and Increased Angiotensin II Sensitivity in Pregnant Rats. Hypertension 2011, 58, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Johansen, M.; Redman, C.W.; Wilkins, T.; Sargent, I.L. Trophoblast deportation in human pregnancy—Its relevance for pre-eclampsia. Placenta 1999, 20, 531–539. [Google Scholar] [CrossRef]
- Abbas, Y.; Turco, M.Y.; Burton, G.J.; Moffett, A. Investigation of human trophoblast invasion in vitro. Hum. Reprod. Update 2020. [Google Scholar] [CrossRef]
- Abou-Kheir, W.; Barrak, J.; Hadadeh, O.; Daoud, G. HTR-8/SVneo cell line contains a mixed population of cells. Placenta 2017, 50, 1–7. [Google Scholar] [CrossRef]
- Li, Z.; Kurosawa, O.; Iwata, H. Establishment of human trophoblast stem cells from human induced pluripotent stem cell-derived cystic cells under micromesh culture. Stem. Cell Res. Ther. 2019, 10, 245. [Google Scholar] [CrossRef]
- Apps, R.; Murphy, S.P.; Fernando, R.; Gardner, L.; Ahad, T.; Moffett, A. Human leucocyte antigen (HLA) expression of primary trophoblast cells and placental cell lines, determined using single antigen beads to characterize allotype specificities of anti-HLA antibodies. Immunology 2009, 127, 26–39. [Google Scholar] [CrossRef]
- Apps, R.; Sharkey, A.; Gardner, L.; Male, V.; Trotter, M.; Miller, N.; North, R.; Founds, S.; Moffett, A. Genome-wide expression profile of first trimester villous and extravillous human trophoblast cells. Placenta 2011, 32, 33–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mi, S.; Lee, X.; Li, X.-P.; Veldman, G.M.; Finnerty, H.; Racie, L.; LaVallie, E.; Tang, X.-Y.; Edouard, P.; Howes, S.; et al. Syncytin is a captive retroviral envelope protein involved in human placental morphogenesis. Nature 2000, 403, 785–789. [Google Scholar] [CrossRef]
- Hiden, U.; Wadsack, C.; Prutsch, N.; Gauster, M.; Weiss, U.; Frank, H.-G.; Schmitz, U.; Fast-Hirsch, C.; Hengstschläger, M.; Pötgens, A.; et al. The first trimester human trophoblast cell line ACH-3P: A novel tool to study autocrine/paracrine regulatory loops of human trophoblast subpopulations—TNF-α stimulates MMP15 expression. BMC Dev. Biol. 2007, 7, 137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morales-Prieto, D.M.; Chaiwangyen, W.; Ospina-Prieto, S.; Schneider, U.; Herrmann, J.; Gruhn, B.; Markert, U.R. MicroRNA expression profiles of trophoblastic cells. Placenta 2012, 33, 725–734. [Google Scholar] [CrossRef]
- Rothbauer, M.; Patel, N.; Gondola, H.; Siwetz, M.; Huppertz, B.; Ertl, P. A comparative study of five physiological key parameters between four different human trophoblast-derived cell lines. Sci. Rep. 2017, 7, 5892. [Google Scholar] [CrossRef]
- Miller, R.K.; Genbacev, O.; Turner, M.A.; Aplin, J.D.; Caniggia, I.; Huppertz, B. Human placental explants in culture: Approaches and assessments. Placenta 2005, 26, 439–448. [Google Scholar] [CrossRef]
- Sackmann, E.K.; Fulton, A.L.; Beebe, D.J. The present and future role of microfluidics in biomedical research. Nature 2014, 507, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Romero, R.; Han, Y.M.; Kim, H.C.; Kim, C.J.; Hong, J.S.; Huh, D. Placenta-on-A-chip: A novel platform to study the biology of the human placenta. J. Matern. Fetal Neonatal Med. 2016, 29, 1046–1054. [Google Scholar] [CrossRef] [PubMed]
- Haase, K.; Gillrie, M.R.; Hajal, C.; Kamm, R.D. Pericytes Contribute to Dysfunction in a Human 3D Model of Placental Microvasculature through VEGF-Ang-Tie2 Signaling. Adv. Sci. 2019, 6, 1900878. [Google Scholar] [CrossRef] [Green Version]
- Brownfoot, F.C.; Hastie, R.; Hannan, N.J.; Cannon, P.; Nguyen, T.V.; Tuohey, L.; Cluver, C.; Tong, S.; Kaitu’u-Lino, T.J. Combining metformin and sulfasalazine additively reduces the secretion of antiangiogenic factors from the placenta: Implications for the treatment of preeclampsia. Placenta 2020, 95, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Tian, F.J.; Lin, Y.; Xu, W.M. Oxidative Stress: Placenta Function and Dysfunction. Am. J. Reprod. Immunol. 2016, 76, 258–271. [Google Scholar] [CrossRef] [PubMed]
- De Alwis, N.; Beard, S.; Mangwiro, Y.T.; Binder, N.K.; Kaitu’u-Lino, T.J.; Brownfoot, F.C.; Tong, S.; Hannan, N.J. Pravastatin as the statin of choice for reducing pre-eclampsia-associated endothelial dysfunction. Pregnancy Hypertens. 2020, 20, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Brownfoot, F.C.; Hastie, R.; Hannan, N.J.; Cannon, P.; Tuohey, L.; Parry, L.J.; Senadheera, S.; Illanes, S.E.; Kaitu’u-Lino, T.J.; Tong, S. Metformin as a prevention and treatment for preeclampsia: Effects on soluble fms-like tyrosine kinase 1 and soluble endoglin secretion and endothelial dysfunction. Am. J. Obstet. Gynecol. 2016, 214, 356.e1. [Google Scholar] [CrossRef] [Green Version]
- Brownfoot, F.C.; Hannan, N.J.; Cannon, P.; Nguyen, V.; Hastie, R.; Parry, L.J.; Senadheera, S.; Tuohey, L.; Tong, S.; Kaitu’u-Lino, T.J. Sulfasalazine reduces placental secretion of antiangiogenic factors, up-regulates the secretion of placental growth factor and rescues endothelial dysfunction. EBioMedicine 2019, 41, 636–648. [Google Scholar] [CrossRef] [Green Version]
- Onda, K.; Tong, S.; Beard, S.; Binder, N.; Muto, M.; Senadheera, S.N.; Parry, L.; Dilworth, M.; Renshall, L.; Brownfoot, F.; et al. Proton Pump Inhibitors Decrease Soluble fms-Like Tyrosine Kinase-1 and Soluble Endoglin Secretion, Decrease Hypertension, and Rescue Endothelial Dysfunction. Hypertension 2017, 69, 457–468. [Google Scholar] [CrossRef]
- Beckman, J.A.; Creager, M.A. The nonlipid effects of statins on endothelial function. Trends Cardiovasc. Med. 2006, 16, 156–162. [Google Scholar] [CrossRef]
- Zhou, Q.; Liao, J.K. Statins and cardiovascular diseases: From cholesterol lowering to pleiotropy. Curr. Pharm. Des. 2009, 15, 467–478. [Google Scholar] [CrossRef] [Green Version]
- Xu, B.; Shanmugalingam, R.; Chau, K.; Makris, A.; Hennessy, A. Galectin-1–Related Modulation of Trophoblast Endothelial Interactions by Integrins α1 and β1. Reprod. Sci. 2020, 27, 1097–1109. [Google Scholar] [CrossRef]
- Steegers, E.A.P.A. Pre-eclampsia. Lancet 2010, 376, 631–644. [Google Scholar] [CrossRef]
- Chen, T.; Vunjak-Novakovic, G. Human Tissue-Engineered Model of Myocardial Ischemia–Reperfusion Injury. Tissue Eng. Part A 2019, 25, 711–724. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Vunjak-Novakovic, G. In vitro Models of Ischemia-Reperfusion Injury. Regen. Eng. Transl. Med. 2018, 4, 142–153. [Google Scholar] [CrossRef] [PubMed]
- Lawson, C.S.; Downey, J.M. Preconditioning: State of the art myocardial protection. Cardiovasc. Res. 1993, 27, 542–550. [Google Scholar] [CrossRef]
- Van der Spoel, T.I.G.; Jansen of Lorkeers, S.J.; Agostoni, P.; van Belle, E.; Gyöngyösi, M.; Sluijter, J.P.G.; Cramer, M.J.; Doevendans, P.A.; Chamuleau, S.A.J. Human relevance of pre-clinical studies in stem cell therapy: Systematic review and meta-analysis of large animal models of ischaemic heart disease. Cardiovasc. Res. 2011, 91, 649–658. [Google Scholar] [CrossRef]
- Xu, Z.; McElhanon, K.E.; Beck, E.X.; Weisleder, N. A Murine Model of Myocardial Ischemia-Reperfusion Injury. Methods Mol. Biol. 2018, 1717, 145–153. [Google Scholar] [CrossRef]
- Luther, D.J.; Thodeti, C.K.; Meszaros, J.G. Injury models to study cardiac remodeling in the mouse: Myocardial infarction and ischemia-reperfusion. Methods Mol. Biol. 2013, 1037, 325–342. [Google Scholar]
- Skyschally, A.; van Caster, P.; Iliodromitis, E.K.; Schulz, R.; Kremastinos, D.T.; Heusch, G. Ischemic postconditioning: Experimental models and protocol algorithms. Basic Res. Cardiol. 2009, 104, 469–483. [Google Scholar] [CrossRef]
- Vidavalur, R.; Swarnakar, S.; Thirunavukkarasu, M.; Samuel, S.M.; Maulik, N. Ex vivo and in vivo approaches to study mechanisms of cardioprotection targeting ischemia/reperfusion (i/r) injury: Useful techniques for cardiovascular drug discovery. Curr. Drug Discov. Technol. 2008, 5, 269–278. [Google Scholar] [CrossRef]
- Bohl, S.; Medway, D.J.; Schulz-Menger, J.; Schneider, J.E.; Neubauer, S.; Lygate, C.A. Refined approach for quantification of in vivo ischemia-reperfusion injury in the mouse heart. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H2054–H2058. [Google Scholar] [CrossRef]
- O’Hara, T.; Rudy, Y. Quantitative comparison of cardiac ventricular myocyte electrophysiology and response to drugs in human and nonhuman species. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H1023–H1030. [Google Scholar] [CrossRef] [Green Version]
- Baehr, A.; Klymiuk, N.; Kupatt, C. Evaluating Novel Targets of Ischemia Reperfusion Injury in Pig Models. Int. J. Mol. Sci. 2019, 20, 4749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, R.M.; Mocanu, M.M.; Yellon, D.M. Retrograde heart perfusion: The Langendorff technique of isolated heart perfusion. J. Mol. Cell. Cardiol. 2011, 50, 940–950. [Google Scholar] [CrossRef]
- Sutherland, F.J.; Hearse, D.J. THE ISOLATED BLOOD AND PERFUSION FLUID PERFUSED HEART. Pharmacol. Res. 2000, 41, 613–627. [Google Scholar] [CrossRef]
- Figtree, G.A.; Bubb, K.J.; Tang, O.; Kizana, E.; Gentile, C. Vascularized cardiac spheroids as novel 3D in vitro models to study cardiac fibrosis. Cells Tissues Organs 2017, 204, 191–198. [Google Scholar] [CrossRef]
- Polonchuk, L.; Chabria, M.; Badi, L.; Hoflack, J.-C.; Figtree, G.; Davies, M.J.; Gentile, C. Cardiac spheroids as promising in vitro models to study the human heart microenvironment. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Katare, R.G.; Ando, M.; Kakinuma, Y.; Sato, T. Engineered heart tissue: A novel tool to study the ischemic changes of the heart in vitro. PLoS ONE 2010, 5, e9275. [Google Scholar] [CrossRef]
- Roche, C.D.; Brereton, R.J.; Ashton, A.W.; Jackson, C.; Gentile, C. Current challenges in three-dimensional bioprinting heart tissues for cardiac surgery. Eur. J. Cardio-Thorac. Surg. 2020, 58, 500–510. [Google Scholar] [CrossRef]
- Valdés, G. Preeclampsia and cardiovascular disease: Interconnected paths that enable detection of the subclinical stages of obstetric and cardiovascular diseases. Integr. Blood Press Control 2017, 10, 17–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz, R.J.; Wilson, G.J. Studying ischemic preconditioning in isolated cardiomyocyte models. Cardiovasc. Res. 2006, 70, 286–296. [Google Scholar] [CrossRef] [Green Version]
- Piper, H.; Garcña-Dorado, D.; Ovize, M. A fresh look at reperfusion injury. Cardiovasc. Res. 1998, 38, 291–300. [Google Scholar] [CrossRef] [Green Version]
- Hidalgo, A.; Glass, N.; Ovchinnikov, D.; Yang, S.-K.; Zhang, X.; Mazzone, S.; Chen, C.; Wolvetang, E.; Cooper-White, J. Modelling ischemia-reperfusion injury (IRI) in vitro using metabolically matured induced pluripotent stem cell-derived cardiomyocytes. APL Bioeng. 2018, 2, 026102. [Google Scholar] [CrossRef] [Green Version]
- Kanazawa, H.; Tseliou, E.; Malliaras, K.; Yee, K.; Dawkins, J.F.; De Couto, G.; Smith, R.R.; Kreke, M.; Seinfeld, J.; Middleton, R.C.; et al. Cellular postconditioning: Allogeneic cardiosphere-derived cells reduce infarct size and attenuate microvascular obstruction when administered after reperfusion in pigs with acute myocardial infarction. Circ. Heart Fail. 2015, 8, 322–332. [Google Scholar] [CrossRef] [Green Version]
- Song, H.; Yoon, C.; Kattman, S.J.; Dengler, J.; Massé, S.; Thavaratnam, T.; Gewarges, M.; Nanthakumar, K.; Rubart, M.; Keller, G.M.; et al. Interrogating functional integration between injected pluripotent stem cell-derived cells and surrogate cardiac tissue. Proc. Natl. Acad. Sci. USA 2010, 107, 3329–3334. [Google Scholar] [CrossRef] [Green Version]
- Naito, H.; Melnychenko, I.; Didié, M.; Schneiderbanger, K.; Schubert, P.; Rosenkranz, S.; Eschenhagen, T.; Zimmermann, W.H. Optimizing engineered heart tissue for therapeutic applications as surrogate heart muscle. Circulation 2006, 114, I72–I78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmermann, W.H.; Melnychenko, I.; Wasmeier, G.; Didié, M.; Naito, H.; Nixdorff, U.; Hess, A.; Budinsky, L.; Brune, K.; Michaelis, B.; et al. Engineered heart tissue grafts improve systolic and diastolic function in infarcted rat hearts. Nat. Med. 2006, 12, 452–458. [Google Scholar] [CrossRef]
- Shimizu, T.; Sekine, H.; Yamato, M.; Okano, T. Cell sheet-based myocardial tissue engineering: New hope for damaged heart rescue. Curr. Pharm. Des. 2009, 15, 2807–2814. [Google Scholar] [CrossRef] [PubMed]
- Gentile, C. Filling the Gaps between the In Vivo and In Vitro Microenvironment: Engineering of Spheroids for Stem Cell Technology. Curr. Stem Cell Res. Ther. 2016, 11, 652–665. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.S.; Park, C.-Y.; Kim, J.-H.; Joo, H.J.; Choi, S.-C.; Choi, J.-H.; Lim, I.R.; Park, J.H.; Hong, S.J.; Lim, D.-S. Cardioprotective effects of genetically engineered cardiac stem cells by spheroid formation on ischemic cardiomyocytes. Mol. Med. 2020, 26, 15. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhang, L.; Li, Y.; Chen, L.; Wang, X.; Guo, W.; Zhang, X.; Qin, G.; He, S.-h.; Zimmerman, A.; et al. Exosomes/microvesicles from induced pluripotent stem cells deliver cardioprotective miRNAs and prevent cardiomyocyte apoptosis in the ischemic myocardium. Int. J. Cardiol. 2015, 192, 61–69. [Google Scholar] [CrossRef] [Green Version]
- Sebastião, M.J.; Gomes-Alves, P.; Reis, I.; Sanchez, B.; Palacios, I.; Serra, M.; Alves, P.M. Bioreactor-based 3D human myocardial ischemia/reperfusion in vitro model: A novel tool to unveil key paracrine factors upon acute myocardial infarction. Transl. Res. 2020, 215, 57–74. [Google Scholar] [CrossRef]
- Shahul, S.; Rhee, J.; Hacker, M.R.; Gulati, G.; Mitchell, J.D.; Hess, P.; Mahmood, F.; Arany, Z.; Rana, S.; Talmor, D. Subclinical left ventricular dysfunction in preeclamptic women with preserved left ventricular ejection fraction: A 2D speckle-tracking imaging study. Circ. Cardiovasc. Imaging 2012, 5, 734–739. [Google Scholar] [CrossRef] [Green Version]
- Houser, S.R.; Margulies, K.B.; Murphy, A.M.; Spinale, F.G.; Francis, G.S.; Prabhu, S.D.; Rockman, H.A.; Kass, D.A.; Molkentin, J.D.; Sussman, M.A. Animal models of heart failure: A scientific statement from the American Heart Association. Circ. Res. 2012, 111, 131–150. [Google Scholar] [CrossRef] [Green Version]
- Patten, R.D.; Hall-Porter, M.R. Small animal models of heart failure: Development of novel therapies, past and present. Circ. Heart Fail. 2009, 2, 138–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rockman, H.A.; Ross, R.S.; Harris, A.N.; Knowlton, K.U.; Steinhelper, M.E.; Field, L.J.; Ross, J.; Chien, K.R. Segregation of atrial-specific and inducible expression of an atrial natriuretic factor transgene in an in vivo murine model of cardiac hypertrophy. Proc. Natl. Acad. Sci. USA 1991, 88, 8277–8281. [Google Scholar] [CrossRef] [Green Version]
- Richards, D.A.; Aronovitz, M.J.; Calamaras, T.D.; Tam, K.; Martin, G.L.; Liu, P.; Bowditch, H.K.; Zhang, P.; Huggins, G.S.; Blanton, R.M. Distinct phenotypes Induced by three Degrees of transverse Aortic Constriction in Mice. Sci. Rep. 2019, 9, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irion, C.I.; John-Williams, K.; Chahdi, A.; Yousefi, K.; Fernandez, Y.R.; Hatzistergos, K.E.; Hare, J.M.; Webster, K.; Shehadeh, L.A. Osteopontin Regulates Adult Cardiomyocyte Division in a Mouse Model of Pressure Overload Induced Heart Failure. Circ. Res. 2019, 125, A123. [Google Scholar] [CrossRef]
- Veeraveedu, P.T.; Sanada, S.; Okuda, K.; Fu, H.Y.; Matsuzaki, T.; Araki, R.; Yamato, M.; Yasuda, K.; Sakata, Y.; Yoshimoto, T. Ablation of IL-33 gene exacerbate myocardial remodeling in mice with heart failure induced by mechanical stress. Biochem. Pharmacol. 2017, 138, 73–80. [Google Scholar] [CrossRef]
- Weinberg, E.O.; Schoen, F.J.; George, D.; Kagaya, Y.; Douglas, P.S.; Litwin, S.E.; Schunkert, H.; Benedict, C.R.; Lorell, B.H. Angiotensin-converting enzyme inhibition prolongs survival and modifies the transition to heart failure in rats with pressure overload hypertrophy due to ascending aortic stenosis. Circulation 1994, 90, 1410–1422. [Google Scholar] [CrossRef] [Green Version]
- Pfeffer, M.A.; Pfeffer, J.M.; Fishbein, M.C.; Fletcher, P.J.; Spadaro, J.; Kloner, R.A.; Braunwald, E. Myocardial infarct size and ventricular function in rats. Circ. Res. 1979, 44, 503–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spannbauer, A.; Traxler, D.; Zlabinger, K.; Gugerell, A.; Winkler, J.; Mester-Tonczar, J.; Lukovic, D.; Müller, C.; Riesenhuber, M.; Pavo, N.; et al. Large Animal Models of Heart Failure With Reduced Ejection Fraction (HFrEF). Front. Cardiovasc. Med. 2019, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinbane, J.S.; Wood, M.A.; Jensen, D.N.; Ellenbogen, K.A.; Fitzpatrick, A.P.; Scheinman, M.M. Tachycardia-induced cardiomyopathy: A review of animal models and clinical studies. J. Am. Coll. Cardiol. 1997, 29, 709–715. [Google Scholar] [CrossRef] [Green Version]
- Dandamudi, G.; Rampurwala, A.Y.; Mahenthiran, J.; Miller, J.M.; Das, M.K. Persistent left ventricular dilatation in tachycardia-induced cardiomyopathy patients after appropriate treatment and normalization of ejection fraction. Heart Rhythm 2008, 5, 1111–1114. [Google Scholar] [CrossRef] [PubMed]
- McMahon, W.S.; Mukherjee, R.; Gillette, P.C.; Crawford, F.A.; Spinale, F.G. Right and left ventricular geometry and myocyte contractile processes with dilated cardiomyopathy: Myocyte growth and β-adrenergic responsiveness. Cardiovasc. Res. 1996, 31, 314–323. [Google Scholar]
- Spinale, F.G.; Tomita, M.; Zellner, J.L.; Cook, J.C.; Crawford, F.A.; Zile, M.R. Collagen remodeling and changes in LV function during development and recovery from supraventricular tachycardia. Am. J. Physiol. Heart Circ. Physiol. 1991, 261, H308–H318. [Google Scholar] [CrossRef]
- Watkins, S.J.; Borthwick, G.M.; Arthur, H.M. The H9C2 cell line and primary neonatal cardiomyocyte cells show similar hypertrophic responses in vitro. Vitr. Cell. Dev. Biol. Anim. 2011, 47, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Carlson, C.; Koonce, C.; Aoyama, N.; Einhorn, S.; Fiene, S.; Thompson, A.; Swanson, B.; Anson, B.; Kattman, S. Phenotypic Screening with Human iPS Cell–Derived Cardiomyocytes: HTS-Compatible Assays for Interrogating Cardiac Hypertrophy. J. Biomol. Screen. 2013, 18, 1203–1211. [Google Scholar] [CrossRef] [Green Version]
- Hirt, M.N.; Sörensen, N.A.; Bartholdt, L.M.; Boeddinghaus, J.; Schaaf, S.; Eder, A.; Vollert, I.; Stöhr, A.; Schulze, T.; Witten, A. Increased afterload induces pathological cardiac hypertrophy: A new in vitro model. Basic Res. Cardiol. 2012, 107, 307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouten, C.; Dankers, P.; Driessen-Mol, A.; Pedron, S.; Brizard, A.; Baaijens, F. Substrates for cardiovascular tissue engineering. Adv. Drug Deliv. Rev. 2011, 63, 221–241. [Google Scholar] [CrossRef]
- Wang, Y.; Hill, J.A. Electrophysiological remodeling in heart failure. J. Mol. Cell. Cardiol. 2010, 48, 619–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarbrough, W.M.; Spinale, F.G. Large animal models of congestive heart failure: A critical step in translating basic observations into clinical applications. J. Nucl. Cardiol. 2003, 10, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Chang, Y.H.; Xiong, Q.; Zhang, P.; Zhang, L.; Somasundaram, P.; Lepley, M.; Swingen, C.; Su, L.; Wendel, J.S.; et al. Cardiac repair in a porcine model of acute myocardial infarction with human induced pluripotent stem cell-derived cardiovascular cells. Cell Stem Cell 2014, 15, 750–761. [Google Scholar] [CrossRef] [Green Version]
- Nomura, Y.; John, R.M.; Janssen, A.B.; Davey, C.; Finik, J.; Buthmann, J.; Glover, V.; Lambertini, L. Neurodevelopmental consequences in offspring of mothers with preeclampsia during pregnancy: Underlying biological mechanism via imprinting genes. Arch. Gynecol. Obstet. 2017, 295, 1319–1329. [Google Scholar] [CrossRef] [PubMed]
- Carty, D.M.; Delles, C.; Dominiczak, A.F. Novel biomarkers for predicting preeclampsia. Trends Cardiovasc. Med. 2008, 18, 186–194. [Google Scholar] [CrossRef] [Green Version]
- Eastabrook, G.; Aksoy, T.; Bedell, S.; Penava, D.; de Vrijer, B. Preeclampsia biomarkers: An assessment of maternal cardiometabolic health. Pregnancy Hypertens. 2018, 13, 204–213. [Google Scholar] [CrossRef]
- Melchiorre, K.; Sutherland, G.R.; Liberati, M.; Thilaganathan, B. Preeclampsia Is Associated With Persistent Postpartum Cardiovascular Impairment. Hypertension 2011, 58, 709–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
| Model | Typical Features | Advantages | Disadvantages | References |
|---|---|---|---|---|
| Large Nonhuman mammals (dogs, sheep, pigs or nonhuman primates) | -They capture the process of hypoxia-reoxygenation but does not fully model the clinical setting. - Interactions between various cell types. | -Pigs are the closest analogues to humans, followed by sheeps and dogs (comparable heart size and heart rate to humans). | - Difficult and expensive to work with. - Ethical considerations. | [17,19,76,77,78] |
| Small mammals (rodents, mice, rabbits) | -They capture the process of hypoxia-reoxygenation but does not fully model the clinical setting. -Interactions between various cell types. | - Physiologically relevant. -Cheaper compared to big animals. -Easier to genetically manipulate compared to larger animals. -Effective to evaluate therapeutic approaches to regenerate the heart after injury. | -Effectiveness and safety for humans remain to be determined. -Rodent hearts have a much higher intrinsic beating rate, higher cardiac basal metabolism and different electrophysiology compared with the human heart. - Ethical considerations. | [76,79,80] |
| Model | Typical Features | Advantages | Disadvantages | References |
|---|---|---|---|---|
| 2D Cultures (monocellular and multicellular cell layers) | -High control of various confounding factors (temperature, pH, CO2). -Widely used to study pathways of IRI and test the candidate therapeutic options. | Monocellular cultures -Testing of the electromechanical properties of individual cardiomyocytes (cardiac physiology). -Individual cardiomyocytes can be controlled by numerous factors such as stress, strain, stiffness. -Effective technique for expanding cell lines Multicellular cultures -Can examine cardiomyocytes culture electrically using microelectrode arrays. -Optimal control over environmental parameters. | Monocellular cultures -Isolated cardiomyocytes can behave differently and show different responses to drugs from cells that are cultured with other cells. -Limited maturity. Multicellular cultures -No cell to cell interaction in 3D and static conditions. - Response to drugs, toxins or signalling modifiers may be misleading. | [8,25,76] |
| 3D Cultures (cardiac spheroids, scaffold-based approaches and organ-on-a-chip models) | -Useful to evaluate more physiologically relevant mechanisms for the prevention and treatment of ischemia/reperfusion injury -Rely on isolated cardiomyocytes from animals, immortalised cell lines, or HiPS-CMs | -Prolonged viability and retain contractile properties. - Mimic key aspects of the phenotypical and cellular heterogeneity as well as microenvironmental aspects. -Cardiac tissue engineering uisng hiPS-CMs aims at promoting cardiac cell maturation and developing a more predictive human tissue model of IRI as well as be patient-specific. | -Expensive cultures. -Tissue culture skills optimal for these cultures are required. -Cell phenotype can be dramatically affected by the culture geometry. | [8,88,89,90,91,92] |
| Model | Typical Features | Advantages | Disadvantages | References |
|---|---|---|---|---|
| In Vitro 2D Cell Culture | - Monolayer cell cultures of cardiac cells (either transformed cell lines or iPSC-derived cardiac myocytes) can be co-cultured with other cardiac cells to better recapitulate the in vivo cardiac environment. (commonly employed for genetic studies and drug discovery). - Treatment with endothelin-1 is commonly used as a positive control for cardiac hypertrophy (the largest risk factor for heart failure). | - Culturing cells in 2D is significantly cost-effective when using immortallised cell lines. -Both transformed cells and iPSC-derived cells could be human derived. -Extensive literature using transformed cell lines for drug discovery and cardiotoxic effects. - Cardiac myocytes can be employed for studies of genetic mutations in response to hypertension and cardiac hypertrophy. | -Transformed cells have fundamentally altered genomes. - Two-dimensional culturing lacks the full 3D architecture present in vivo (i.e.,interactions with other cells and the ECM). -They cannot fully recapitulate the human heart pathophysiology. | [23,113,117,119,124] |
| In Vitro 3D Cell Culture | - Often including a biomaterial (i.e., a hydrogel or biocompatible polymer) for optimal stiffness and electrical signals. | - Improved models of the in vivo physiological, morphological, biochemical and genetic profile. - Engineered 3D environments also use structural features not present in 2D to mimic mechanical cues (i.e., increased afterload). | -Increased complexity of experimental design. -Directly inducing heart failure is still a challenge for in vitro models when compared to in vivo counterparts. | [22,115,116,117] |
| Small Animal In Vivo | - Transverse aortic constriction (TAC) surgery (greater pressure in the left ventricle and subsequently cardiac hypertrophy, fibrosis as well as cardiac output dysfunction).-In periods of up to 4–6 weeks, this progresses to clinical heart failure. | -TAC procedure is a well established method (it can be easily replicated with consistent results). - Can use transgenic mice-Low maintenance costs when compared to in vivo models in large animals. | -Translatability of results is challenging in small animals. - Features of the heart are functionally different when compared to the human heart. - Slight variations can result in greater pathological stimuli than intended. - Ethical considerations. | [106,107,109,110,111] |
| Large Animal In Vivo | - A progressive aortic constriction in dogs, sheeps and pigs, is induced in a similar fashion to small animals. - Another method involves tachycardia-induced cardiomyopathy that results in heart failure after several weeks of continuation. | - Increased translatability to human physiology. - Allows live monitoring. | -Research facilities are rarely equipped for significant large animal studies. - Higher costs compared to small animals; - Multidisciplinary teams required for handling. - Ethical considerations. | [18,20,24,106,115,116,125] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu Chung Ming, C.; Sesperez, K.; Ben-Sefer, E.; Arpon, D.; McGrath, K.; McClements, L.; Gentile, C. Considerations to Model Heart Disease in Women with Preeclampsia and Cardiovascular Disease. Cells 2021, 10, 899. https://doi.org/10.3390/cells10040899
Liu Chung Ming C, Sesperez K, Ben-Sefer E, Arpon D, McGrath K, McClements L, Gentile C. Considerations to Model Heart Disease in Women with Preeclampsia and Cardiovascular Disease. Cells. 2021; 10(4):899. https://doi.org/10.3390/cells10040899
Chicago/Turabian StyleLiu Chung Ming, Clara, Kimberly Sesperez, Eitan Ben-Sefer, David Arpon, Kristine McGrath, Lana McClements, and Carmine Gentile. 2021. "Considerations to Model Heart Disease in Women with Preeclampsia and Cardiovascular Disease" Cells 10, no. 4: 899. https://doi.org/10.3390/cells10040899