
A Propagated Skeleton Approach to High Throughput Screening of Neurite Outgrowth for In Vitro Parkinson’s Disease Modelling

 , , , and
, , , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. SH-SY5Y Cell Culture
2.2. SH-SY5Y Differentiation and Time Course of Experiments
2.3. In Vitro Assay of Primary Mesencephalig Dopaminergic Neurons MDN
2.4. Immunocytochemistry
2.5. Manual Ground Truth Measurement
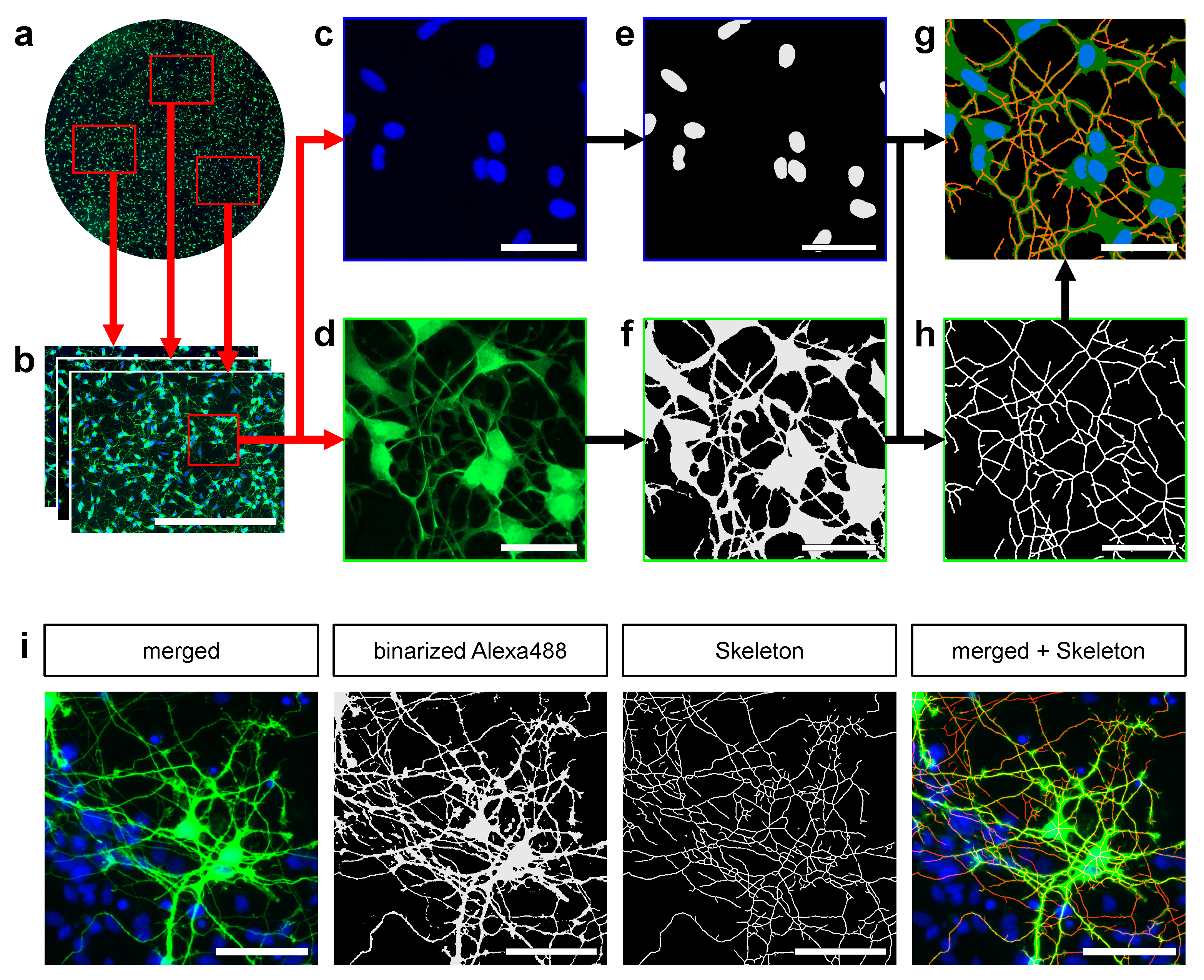
2.6. Automated Image Processing
2.7. Skeletonization and Pruning
2.8. Statistical Analysis and Validation
2.9. Implementation and Hardware
3. Results
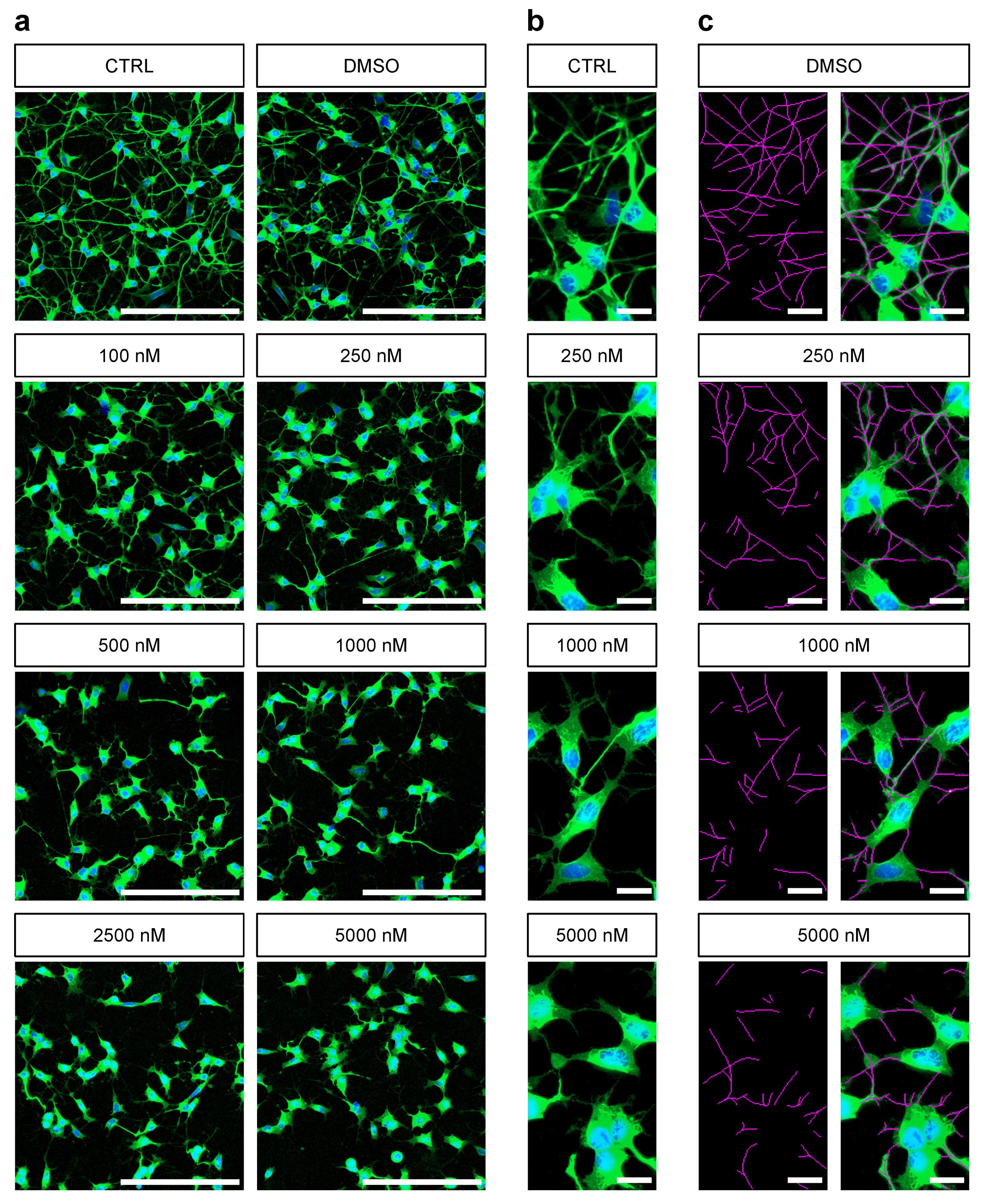
3.1. Manual Analysis of Rotenone-Induced Alterations of Neurite Outgrowth
3.2. Automated Neurite Outgrowth Analysis of Rotenone Treated SH-SY5Y Cells
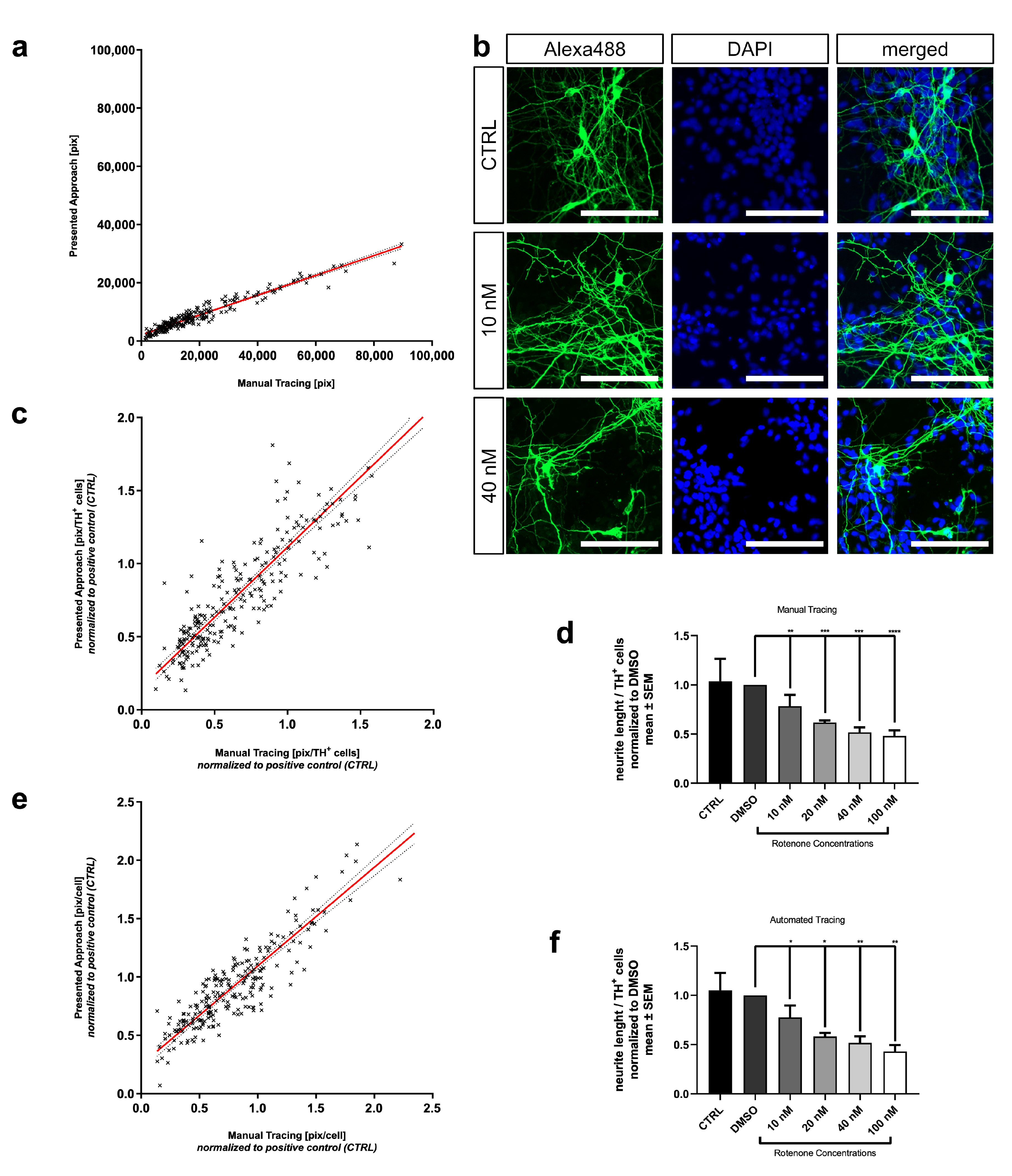
3.3. Comparison of Automated and Manual Analysis Shows High Correlation
3.4. Enhancement with In Vitro Detail Parameters from Automated Neuronal Analysis
3.5. Automated Neurite Outgrowth Quantification of Rotenone Treated MDN
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| PD | Parkinson’s Disease |
| MDN | primary mesencephalic dopaminergic neurons |
| DMSO | dimethylsulfoxide |
| PBS | phosphate buffered saline |
| PBT 1 | PBS with triton |
| PBSA | PBT 1 with bovine serum albumin |
| DAPI | 4,6-diamidino-2-phenylindole |
| SEM | standard error of the mean |
| ANOVA | analysis of variance |
| aSYN | -Synuclein |
| iPSC | induced pluripotent stem cells |
References
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Winklhofer, K.F.; Haass, C. Mitochondrial dysfunction in Parkinson’s disease. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2010, 1802, 29–44. [Google Scholar] [CrossRef]
- Bendor, J.T.; Logan, T.P.; Edwards, R.H. The Function of α-Synuclein. Neuron 2013, 79, 1044–1066. [Google Scholar] [CrossRef] [Green Version]
- Goedert, M.; Spillantini, M.G.; Tredici, K.D.; Braak, H. 100 years of Lewy pathology. Nat. Rev. Neurol. 2013, 9, 13–24. [Google Scholar] [CrossRef]
- Ganjam, G.K.; Bolte, K.; Matschke, L.A.; Neitemeier, S.; Dolga, A.M.; Höllerhage, M.; Höglinger, G.U.; Adamczyk, A.; Decher, N.; Oertel, W.H.; et al. Mitochondrial damage by α-synuclein causes cell death in human dopaminergic neurons. Cell Death Dis. 2019, 10, 865. [Google Scholar] [CrossRef] [PubMed]
- Lautenschläger, J.; Wagner-Valladolid, S.; Stephens, A.D.; Fernández-Villegas, A.; Hockings, C.; Mishra, A.; Manton, J.D.; Fantham, M.J.; Lu, M.; Rees, E.J.; et al. Intramitochondrial proteostasis is directly coupled to α-synuclein and amyloid β 1-42 pathologies. J. Biol. Chem. 2020. [Google Scholar] [CrossRef]
- Fellner, L.; Irschick, R.; Schanda, K.; Reindl, M.; Klimaschewski, L.; Poewe, W.; Wenning, G.K.; Stefanova, N. Toll-like receptor 4 is required for α-synuclein dependent activation of microglia and astroglia. Glia 2013, 61, 349–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doorn, K.J.; Moors, T.; Drukarch, B.; Berg, W.D.v.d.; Lucassen, P.J.; Dam, A.M.v. Microglial phenotypes and toll-like receptor 2 in the substantia nigra and hippocampus of incidental Lewy body disease cases and Parkinson’s disease patients. Acta Neuropathol. Commun. 2014, 2, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duffy, M.F.; Collier, T.J.; Patterson, J.R.; Kemp, C.J.; Luk, K.C.; Tansey, M.G.; Paumier, K.L.; Kanaan, N.M.; Fischer, D.L.; Polinski, N.K.; et al. Correction to: Lewy body-like alpha-synuclein inclusions trigger reactive microgliosis prior to nigral degeneration. J. Neuroinflamm. 2018, 15, 169. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; MÃŒnch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Yun, S.P.; Kam, T.I.; Panicker, N.; Kim, S.; Oh, Y.; Park, J.S.; Kwon, S.H.; Park, Y.J.; Karuppagounder, S.S.; Park, H.; et al. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat. Med. 2018, 24, 931–938. [Google Scholar] [CrossRef]
- Hinkle, J.T.; Dawson, V.L.; Dawson, T.M. The A1 astrocyte paradigm: New avenues for pharmacological intervention in neurodegeneration. Mov. Disord. 2019, 34, 959–969. [Google Scholar] [CrossRef]
- Tönges, L.; Frank, T.; Tatenhorst, L.; Saal, K.A.; Koch, J.C.; Szego, E.M.; Bähr, M.; Weishaupt, J.H.; Lingor, P. Inhibition of rho kinase enhances survival of dopaminergic neurons and attenuates axonal loss in a mouse model of Parkinson’s disease. Brain 2012, 135, 3355–3370. [Google Scholar] [CrossRef]
- Koch, J.C.; Saal, K.A.; Tönges, L.; Tatenhorst, L.; Roser, A.E.; Lingor, P. ROCK inhibition in models of neurodegeneration and its potential for clinical translation. Pharmacol. Ther. 2018. [Google Scholar] [CrossRef]
- Ostendorf, F.; Metzdorf, J.; Gold, R.; Haghikia, A.; Tönges, L. Propionic Acid and Fasudil as Treatment Against Rotenone Toxicity in an In Vitro Model of Parkinson’s Disease. Molecules 2020, 25, 2502. [Google Scholar] [CrossRef]
- Qian, H.; Kang, X.; Hu, J.; Zhang, D.; Liang, Z.; Meng, F.; Zhang, X.; Xue, Y.; Maimon, R.; Dowdy, S.F.; et al. Reversing a model of Parkinson’s disease with in situ converted nigral neurons. Nature 2020, 582, 550–556. [Google Scholar] [CrossRef]
- Falkenburger, B.H.; Saridaki, T.; Dinter, E. Cellular models for Parkinson’s disease. J. Neurochem. 2016, 139, 121–130. [Google Scholar] [CrossRef]
- Lázaro, D.F.; Pavlou, M.A.S.; Outeiro, T.F. Cellular models as tools for the study of the role of alpha-synuclein in Parkinson’s disease. Exp. Neurol. 2017, 298, 162–171. [Google Scholar] [CrossRef]
- Kovalevich, J.; Langford, D. Neuronal Cell Culture, Methods and Protocols. Methods Mol. Biol. 2013, 1078, 9–21. [Google Scholar] [CrossRef] [Green Version]
- Forster, J.I.; Köglsberger, S.; Trefois, C.; Boyd, O.; Baumuratov, A.S.; Buck, L.; Balling, R.; Antony, P.M.A. Characterization of Differentiated SH-SY5Y as Neuronal Screening Model Reveals Increased Oxidative Vulnerability. J. Biomol. Screen. 2015, 21, 496–509. [Google Scholar] [CrossRef] [Green Version]
- Sherer, T.B.; Betarbet, R.; Testa, C.M.; Seo, B.B.; Richardson, J.R.; Kim, J.H.; Miller, G.W.; Yagi, T.; Matsuno-Yagi, A.; Greenamyre, J.T. Mechanism of Toxicity in Rotenone Models of Parkinson’s Disease. J. Neurosci. 2003, 23, 10756–10764. [Google Scholar] [CrossRef]
- Yuan, Y.; Yan, W.; Sun, J.; Huang, J.; Mu, Z.; Chen, N. The molecular mechanism of rotenone-induced α-synuclein aggregation: Emphasizing the role of the calcium/GSK3β pathway. Toxicol. Lett. 2015, 233, 163–171. [Google Scholar] [CrossRef]
- Cochran, J.N.; Hall, A.M.; Roberson, E.D. The dendritic hypothesis for Alzheimer’s disease pathophysiology. Brain Res. Bull. 2014, 103, 18–28. [Google Scholar] [CrossRef] [Green Version]
- Kweon, J.H.; Kim, S.; Lee, S.B. The cellular basis of dendrite pathology in neurodegenerative diseases. BMB Rep. 2017, 50, 5–11. [Google Scholar] [CrossRef] [Green Version]
- Menon, S.; Gupton, S.L. Chapter Three Building Blocks of Functioning Brain: Cytoskeletal Dynamics in Neuronal Development. Int. Rev. Cell Mol. Biol. 2016, 322, 183–245. [Google Scholar] [CrossRef] [Green Version]
- Meijering, E.; Jacob, M.; Sarria, J.; Steiner, P.; Hirling, H.; Unser, M. Design and validation of a tool for neurite tracing and analysis in fluorescence microscopy images. Cytometry Part A 2004, 58A, 167–176. [Google Scholar] [CrossRef] [Green Version]
- Anderl, J.L.; Redpath, S.; Ball, A.J. A Neuronal and Astrocyte Co-Culture Assay for High Content Analysis of Neurotoxicity. J. Vis. Exp. 2009. [Google Scholar] [CrossRef]
- Schmuck, M.R.; Temme, T.; Dach, K.; de Boer, D.; Barenys, M.; Bendt, F.; Mosig, A.; Fritsche, E. Omnisphero: A high-content image analysis (HCA) approach for phenotypic developmental neurotoxicity (DNT) screenings of organoid neurosphere cultures. Arch. Toxicol. 2017, 91, 2017–2028. [Google Scholar] [CrossRef]
- Monzel, A.S.; Hemmer, K.; Kaoma, T.; Smits, L.M.; Bolognin, S.; Lucarelli, P.; Rosety, I.; Zagare, A.; Antony, P.; Nickels, S.L.; et al. Machine learning-assisted neurotoxicity prediction in human midbrain organoids. Park. Relat. Disord. 2020, 75, 105–109. [Google Scholar] [CrossRef]
- Schmuck, M.R.; Keil, K.P.; Sethi, S.; Morgan, R.K.; Lein, P.J. Automated high content image analysis of dendritic arborization in primary mouse hippocampal and rat cortical neurons in culture. J. Neurosci. Methods 2020, 341, 108793. [Google Scholar] [CrossRef]
- Wilson, M.S.; Graham, J.R.; Ball, A.J. Multiparametric High Content Analysis for assessment of neurotoxicity in differentiated neuronal cell lines and human embryonic stem cell-derived neurons. NeuroToxicol. 2014, 42, 33–48. [Google Scholar] [CrossRef]
- Harrill, J.A.; Freudenrich, T.M.; Robinette, B.L.; Mundy, W.R. Comparative sensitivity of human and rat neural cultures to chemical-induced inhibition of neurite outgrowth. Toxicol. Appl. Pharmacol. 2011, 256, 268–280. [Google Scholar] [CrossRef] [PubMed]
- Radio, N.M.; Mundy, W.R. Developmental neurotoxicity testing: Models for assessing chemical effects on neurite outgrowth. NeuroToxicol. 2008, 29, 361–376. [Google Scholar] [CrossRef] [Green Version]
- Eliceiri, K.W.; Berthold, M.R.; Goldberg, I.G.; Ibáñez, L.; Manjunath, B.S.; Martone, M.E.; Murphy, R.F.; Peng, H.; Plant, A.L.; Roysam, B.; et al. Biological imaging software tools. Nat. Methods 2012, 9, 697. [Google Scholar] [CrossRef] [Green Version]
- Durix, B.; Chambon, S.; Leonard, K.; Mari, J.L.; Morin, G. The Propagated Skeleton: A Robust Detail-Preserving Approach. In Discrete Geometry for Computer Imagery; Couprie, M., Cousty, J., Kenmochi, Y., Mustafa, N., Eds.; Springer: Cham, Switzerland, 2019; pp. 343–354. [Google Scholar]
- Durix, B.; Morin, G.; Chambon, S.; Mari, J.L.; Leonard, K. One-step Compact Skeletonization. In Eurographics; Short Papers; The Eurographics Association: Geneve, Switzerland, 2019; pp. 21–24. [Google Scholar]
- Carpenter, A.E.; Jones, T.R.; Lamprecht, M.R.; Clarke, C.; Kang, I.H.; Friman, O.; Guertin, D.A.; Chang, J.H.; Lindquist, R.A.; Moffat, J.; et al. CellProfiler: Image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006, 7, R100. [Google Scholar] [CrossRef] [Green Version]
- Pool, M.; Thiemann, J.; Bar-Or, A.; Fournier, A.E. NeuriteTracer: A novel ImageJ plugin for automated quantification of neurite outgrowth. J. Neurosci. Methods 2008, 168, 134–139. [Google Scholar] [CrossRef]
- Ho, S.Y.; Chao, C.Y.; Huang, H.L.; Chiu, T.W.; Charoenkwan, P.; Hwang, E. NeurphologyJ: An automatic neuronal morphology quantification method and its application in pharmacological discovery. BMC Bioinform. 2011, 12, 230. [Google Scholar] [CrossRef] [Green Version]
- Pani, G.; Vos, W.H.; Samari, N.; Saint-Georges, L.; Baatout, S.; Oostveldt, P.; Benotmane, M.A. MorphoNeuroNet: An automated method for dense neurite network analysis. Cytom. Part A 2014, 85, 188–199. [Google Scholar] [CrossRef]
- Phansalkar, N.; More, S.; Sabale, A.; Joshi, M. Adaptive local thresholding for detection of nuclei in diversity stained cytology images. In Proceedings of the IEEE 2011 International Conference on Communications and Signal Processing, Kerala, India, 10–12 February 2011; pp. 218–220. [Google Scholar]
- Meyer, F. Topographic distance and watershed lines. Signal Process. 1994, 38, 113–125. [Google Scholar] [CrossRef]
- Suzuki, S.; be, K. Topological structural analysis of digitized binary images by border following. Comput. Vision Graph. Image Process. 1985, 30, 32–46. [Google Scholar] [CrossRef]
- Saha, P.K.; Borgefors, G.; di Baja, G.S. A survey on skeletonization algorithms and their applications. Pattern Recognit. Lett. 2016, 76, 3–12. [Google Scholar] [CrossRef]
- Kirst, C.; Skriabine, S.; Vieites-Prado, A.; Topilko, T.; Bertin, P.; Gerschenfeld, G.; Verny, F.; Topilko, P.; Michalski, N.; Tessier-Lavigne, M.; et al. Mapping the Fine-Scale Organization and Plasticity of the Brain Vasculature. Cell 2020, 180, 780–795.e25. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Gui, C.P.; Liu, H.K.; Zhang, D.; Mosig, A. An Image Skeletonization-Based Tool for Pollen Tube Morphology Analysis and Phenotyping F. J. Integr. Plant Biol. 2013, 55, 131–141. [Google Scholar] [CrossRef]
- Abràmoff, M.D.; Magalhães, P.J.; Ram, S.J. Image processing with ImageJ. Biophotonics Int. 2004, 11, 36–42. [Google Scholar]
- Bradski, G. The opencv library. Dr Dobb’s J. Softw. Tools 2000, 25, 120–125. [Google Scholar]
- Merkel, D. Docker: Lightweight linux containers for consistent development and deployment. Linux J. 2014, 2014, 2. [Google Scholar]
- Wu, X.; Liang, Y.; Jing, X.; Lin, D.; Chen, Y.; Zhou, T.; Peng, S.; Zheng, D.; Zeng, Z.; Lei, M.; et al. Rifampicin Prevents SH-SY5Y Cells from Rotenone-Induced Apoptosis via the PI3K/Akt/GSK-3β/CREB Signaling Pathway. Neurochem. Res. 2018, 43, 886–893. [Google Scholar] [CrossRef]
- Sun, C.; Mo, M.; Wang, Y.; Yu, W.; Song, C.; Wang, X.; Chen, S.; Liu, Y. Activation of the immunoproteasome protects SH-SY5Y cells from the toxicity of rotenone. NeuroToxicology 2019, 73, 112–119. [Google Scholar] [CrossRef]
- Mouhape, C.; Costa, G.; Ferreira, M.; Abin-Carriquiry, J.A.; Dajas, F.; Prunell, G. Nicotine-Induced Neuroprotection in Rotenone In Vivo and In Vitro Models of Parkinson’s Disease: Evidences for the Involvement of the Labile Iron Pool Level as the Underlying Mechanism. Neurotox. Res. 2019, 35, 71–82. [Google Scholar] [CrossRef]
- Bai, X.; Latecki, L.J.; Liu, W.Y. Skeleton pruning by contour partitioning with discrete curve evolution. IEEE Trans. Pattern Anal. Mach. Intell. 2007, 29, 449–462. [Google Scholar] [CrossRef]
- Selbach, L.; Kowalski, T.; Gerwert, K.; Buchin, M.; Mosig, A. Shape Decomposition Algorithms for Laser Capture Microdissection. In Proceedings of the 20th International Workshop on Algorithms in Bioinformatics (WABI 2020), Pisa, Italy, 7–9 September 2020; Kingsford, C., Pisanti, N., Eds.; Leibniz International Proceedings in Informatics (LIPIcs). Schloss Dagstuhl–Leibniz-Zentrum für Informatik: Dagstuhl, Germany, 2020; Volume 172, pp. 13:1–13:17. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Degree of Automation | Morphology Measurements | Platform |
|---|---|---|---|
| NeuronJ [26] | semi-automatic | neurite length | ImageJ |
| Cell Profiler [37] | semi-automatic | neurite length | Python |
| NeuriteTracer [38] | automatic | neurite length, soma number | ImageJ |
| NeurophologyJ [39] | automatic | neurite length, soma number and size, neurite attachment points, neurite ending points | ImageJ |
| MorphoNeuroNet [40] | automatic | neurite length, soma number and size, nucleus number, neurite attachment points, neurite ending points | ImageJ |
| Omnisphero [28] | automatic | neurite area, neurite length, neurite branching points | Matlab |
| presented approach | automatic | neurite length, soma number and size, nucleus number and size, neurite ending points, neurite branching points | C++, ImageJ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schikora, J.; Kiwatrowski, N.; Förster, N.; Selbach, L.; Ostendorf, F.; Pallapies, F.; Hasse, B.; Metzdorf, J.; Gold, R.; Mosig, A.; et al. A Propagated Skeleton Approach to High Throughput Screening of Neurite Outgrowth for In Vitro Parkinson’s Disease Modelling. Cells 2021, 10, 931. https://doi.org/10.3390/cells10040931
Schikora J, Kiwatrowski N, Förster N, Selbach L, Ostendorf F, Pallapies F, Hasse B, Metzdorf J, Gold R, Mosig A, et al. A Propagated Skeleton Approach to High Throughput Screening of Neurite Outgrowth for In Vitro Parkinson’s Disease Modelling. Cells. 2021; 10(4):931. https://doi.org/10.3390/cells10040931
Chicago/Turabian StyleSchikora, Justus, Nina Kiwatrowski, Nils Förster, Leonie Selbach, Friederike Ostendorf, Frida Pallapies, Britta Hasse, Judith Metzdorf, Ralf Gold, Axel Mosig, and et al. 2021. "A Propagated Skeleton Approach to High Throughput Screening of Neurite Outgrowth for In Vitro Parkinson’s Disease Modelling" Cells 10, no. 4: 931. https://doi.org/10.3390/cells10040931