
HDAC8 Activates AKT through Upregulating PLCB1 and Suppressing DESC1 Expression in MEK1/2 Inhibition-Resistant Cells



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results
3.1. Murine Melanoma B16-BL6 and Human Colorectal HT-29 Cells Develop Resistance to LT in an HDAC8-Dependent Pathway
3.2. LT-Induced Cell Cycle Arrest and Resistance Are Manifested by MEK1/2 Inhibition
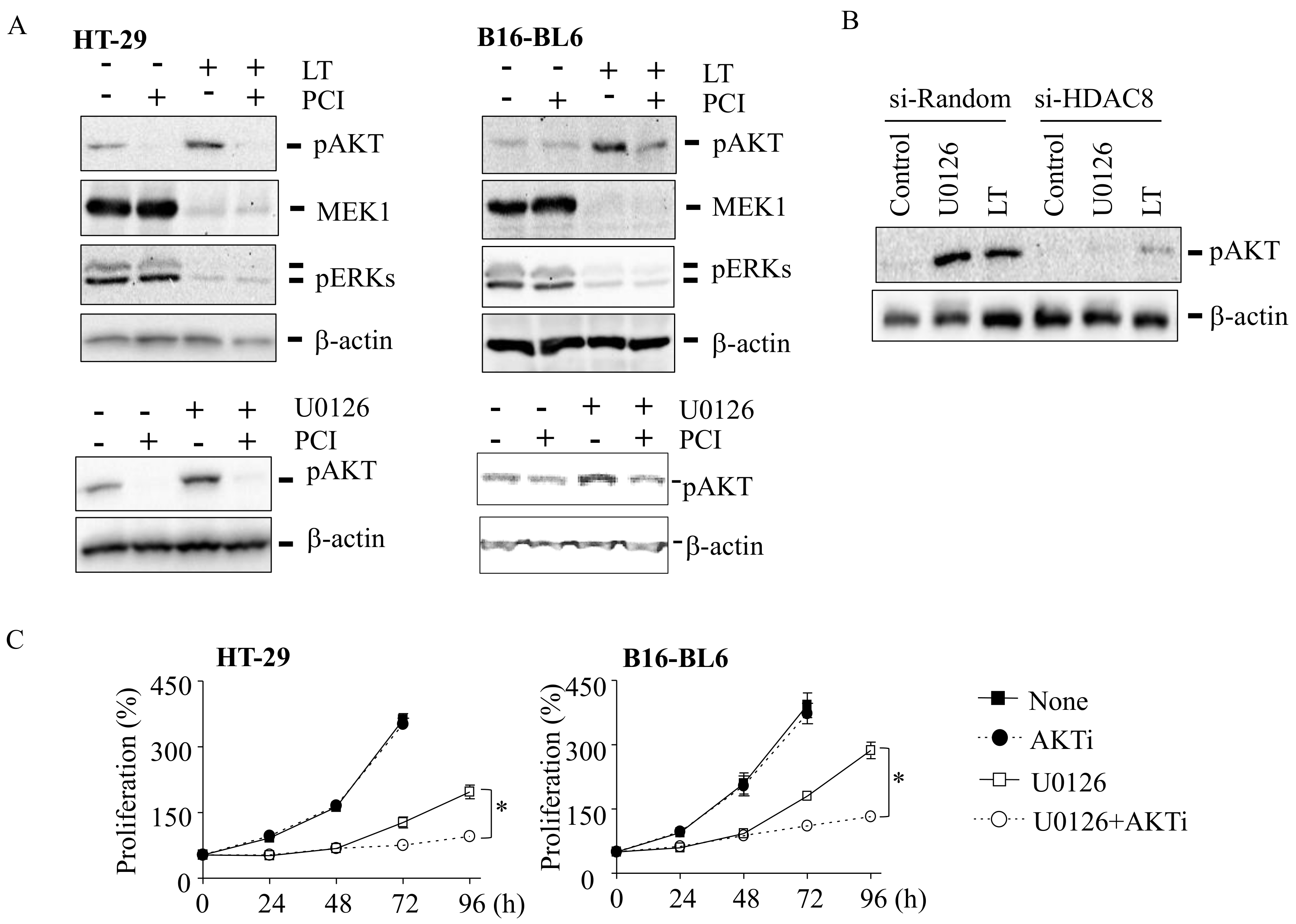
3.3. Activation of AKT Is Mediated by HDAC8 and Required for Cell Proliferation in LT- and U0126-Resistant Cells
3.4. HDAC8 is Involved in Regulating Expression of PLCB1 and DESC1 in MEK1/2 Inhibition-Resistant Cells
3.5. Inhibition of PLCB1 Prevents Resistance to LT and MEK1/2 Inhibition in HT-29 Cells
3.6. DESC1 Prevents Resistance to LT and MEK1/2 Inhibition in HT-29 Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Santarpia, L.; Lippman, S.M.; El-Naggar, A.K. Targeting the mapk-ras-raf signaling pathway in cancer therapy. Expert. Opin. Ther. Targets 2012, 16, 103–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Rauch, J.; Kolch, W. Targeting mapk signaling in cancer: Mechanisms of drug resistance and sensitivity. Int. J. Mol. Sci. 2020, 21, 1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proietti, I.; Skroza, N.; Bernardini, N.; Tolino, E.; Balduzzi, V.; Marchesiello, A.; Michelini, S.; Volpe, S.; Mambrin, A.; Mangino, G.; et al. Mechanisms of acquired braf inhibitor resistance in melanoma: A systematic review. Cancers 2020, 12, 2801. [Google Scholar] [CrossRef] [PubMed]
- Lovly, C.M.; Shaw, A.T. Molecular pathways: Resistance to kinase inhibitors and implications for therapeutic strategies. Clin. Cancer Res. 2014, 20, 2249–2256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noeparast, A.; Giron, P.; Noor, A.; Bahadur Shahi, R.; De Brakeleer, S.; Eggermont, C.; Vandenplas, H.; Boeckx, B.; Lambrechts, D.; De Greve, J.; et al. Craf mutations in lung cancer can be oncogenic and predict sensitivity to combined type ii raf and mek inhibition. Oncogene 2019, 38, 5933–5941. [Google Scholar] [CrossRef] [Green Version]
- Abi-Habib, R.J.; Urieto, J.O.; Liu, S.; Leppla, S.H.; Duesbery, N.S.; Frankel, A.E. Braf status and mitogen-activated protein/extracellular signal-regulated kinase kinase 1/2 activity indicate sensitivity of melanoma cells to anthrax lethal toxin. Mol. Cancer Ther. 2005, 4, 1303–1310. [Google Scholar] [CrossRef] [Green Version]
- Chaudhary, A.; Hilton, M.B.; Seaman, S.; Haines, D.C.; Stevenson, S.; Lemotte, P.K.; Tschantz, W.R.; Zhang, X.M.; Saha, S.; Fleming, T.; et al. Tem8/antxr1 blockade inhibits pathological angiogenesis and potentiates tumoricidal responses against multiple cancer types. Cancer Cell 2012, 21, 212–226. [Google Scholar] [CrossRef] [Green Version]
- Peters, D.E.; Hoover, B.; Cloud, L.G.; Liu, S.; Molinolo, A.A.; Leppla, S.H.; Bugge, T.H. Comparative toxicity and efficacy of engineered anthrax lethal toxin variants with broad anti-tumor activities. Toxicol. Appl. Pharmacol. 2014, 279, 220–229. [Google Scholar] [CrossRef] [Green Version]
- Chopra, A.P.; Boone, S.A.; Liang, X.; Duesbery, N.S. Anthrax lethal factor proteolysis and inactivation of mapk kinase. J. Biol. Chem. 2003, 278, 9402–9406. [Google Scholar] [CrossRef] [Green Version]
- Duesbery, N.S.; Webb, C.P.; Leppla, S.H.; Gordon, V.M.; Klimpel, K.R.; Copeland, T.D.; Ahn, N.G.; Oskarsson, M.K.; Fukasawa, K.; Paull, K.D.; et al. Proteolytic inactivation of map-kinase-kinase by anthrax lethal factor. Science 1998, 280, 734–737. [Google Scholar] [CrossRef]
- Popov, S.G.; Villasmil, R.; Bernardi, J.; Grene, E.; Cardwell, J.; Wu, A.; Alibek, D.; Bailey, C.; Alibek, K. Lethal toxin of bacillus anthracis causes apoptosis of macrophages. Biochem. Biophys. Res. Commun. 2002, 293, 349–355. [Google Scholar] [CrossRef]
- Kassam, A.; Der, S.D.; Mogridge, J. Differentiation of human monocytic cell lines confers susceptibility to bacillus anthracis lethal toxin. Cell Microbiol. 2005, 7, 281–292. [Google Scholar] [CrossRef]
- Ha, S.D.; Han, C.Y.; Reid, C.; Kim, S.O. Hdac8-mediated epigenetic reprogramming plays a key role in resistance to anthrax lethal toxin-induced pyroptosis in macrophages. J. Immunol. 2014, 193, 1333–1343. [Google Scholar] [CrossRef] [Green Version]
- Ha, S.D.; Cho, W.; Kim, S.O. Hdac8 prevents anthrax lethal toxin-induced cell cycle arrest through silencing pten in human monocytic thp-1 cells. Toxins 2017, 9, 162. [Google Scholar] [CrossRef] [Green Version]
- Deuker, M.M.; Marsh Durban, V.; Phillips, W.A.; McMahon, M. Pi3’-kinase inhibition forestalls the onset of mek1/2 inhibitor resistance in braf-mutated melanoma. Cancer Discov. 2015, 5, 143–153. [Google Scholar] [CrossRef] [Green Version]
- Schrauwen, S.; Depreeuw, J.; Coenegrachts, L.; Hermans, E.; Lambrechts, D.; Amant, F. Dual blockade of pi3k/akt/mtor (nvp-bez235) and ras/raf/mek (azd6244) pathways synergistically inhibit growth of primary endometrioid endometrial carcinoma cultures, whereas nvp-bez235 reduces tumor growth in the corresponding xenograft models. Gynecol. Oncol. 2015, 138, 165–173. [Google Scholar] [CrossRef] [Green Version]
- Villanueva, J.; Vultur, A.; Lee, J.T.; Somasundaram, R.; Fukunaga-Kalabis, M.; Cipolla, A.K.; Wubbenhorst, B.; Xu, X.; Gimotty, P.A.; Kee, D.; et al. Acquired resistance to braf inhibitors mediated by a raf kinase switch in melanoma can be overcome by cotargeting mek and igf-1r/pi3k. Cancer Cell 2010, 18, 683–695. [Google Scholar] [CrossRef] [Green Version]
- Emmons, M.F.; Faiao-Flores, F.; Sharma, R.; Thapa, R.; Messina, J.L.; Becker, J.C.; Schadendorf, D.; Seto, E.; Sondak, V.K.; Koomen, J.M.; et al. Hdac8 regulates a stress response pathway in melanoma to mediate escape from braf inhibitor therapy. Cancer Res. 2019, 79, 2947–2961. [Google Scholar] [CrossRef] [Green Version]
- Rae, J.M.; Creighton, C.J.; Meck, J.M.; Haddad, B.R.; Johnson, M.D. Mda-mb-435 cells are derived from m14 melanoma cells--a loss for breast cancer, but a boon for melanoma research. Breast Cancer Res. Treat. 2007, 104, 13–19. [Google Scholar] [CrossRef]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the braf gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Corcoran, R.B.; Ebi, H.; Turke, A.B.; Coffee, E.M.; Nishino, M.; Cogdill, A.P.; Brown, R.D.; Della Pelle, P.; Dias-Santagata, D.; Hung, K.E.; et al. Egfr-mediated re-activation of mapk signaling contributes to insensitivity of braf mutant colorectal cancers to raf inhibition with vemurafenib. Cancer Discov. 2012, 2, 227–235. [Google Scholar] [CrossRef] [Green Version]
- Dahlman, K.B.; Xia, J.; Hutchinson, K.; Ng, C.; Hucks, D.; Jia, P.; Atefi, M.; Su, Z.; Branch, S.; Lyle, P.L.; et al. Braf(l597) mutations in melanoma are associated with sensitivity to mek inhibitors. Cancer Discov. 2012, 2, 791–797. [Google Scholar] [CrossRef] [Green Version]
- Sclafani, F.; Gullo, G.; Sheahan, K.; Crown, J. Braf mutations in melanoma and colorectal cancer: A single oncogenic mutation with different tumour phenotypes and clinical implications. Crit. Rev. Oncol. Hematol. 2013, 87, 55–68. [Google Scholar] [CrossRef]
- Vitale, G.; Bernardi, L.; Napolitani, G.; Mock, M.; Montecucco, C. Susceptibility of mitogen-activated protein kinase kinase family members to proteolysis by anthrax lethal factor. Biochem. J. 2000, 352 Pt. 3, 739–745. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Franklin, R.A.; Montalto, G.; Cervello, M.; Libra, M.; Candido, S.; Malaponte, G.; et al. Ras/raf/mek/erk and pi3k/pten/akt/mtor cascade inhibitors: How mutations can result in therapy resistance and how to overcome resistance. Oncotarget 2012, 3, 1068–1111. [Google Scholar] [CrossRef] [Green Version]
- Ha, S.D.; Ng, D.; Pelech, S.L.; Kim, S.O. Critical role of the phosphatidylinositol 3-kinase/akt/glycogen synthase kinase-3 signaling pathway in recovery from anthrax lethal toxin-induced cell cycle arrest and mek cleavage in macrophages. J. Biol. Chem. 2007, 282, 36230–36239. [Google Scholar] [CrossRef] [Green Version]
- Hart, J.R.; Vogt, P.K. Phosphorylation of akt: A mutational analysis. Oncotarget 2011, 2, 467–476. [Google Scholar] [CrossRef] [Green Version]
- Piazzi, M.; Blalock, W.L.; Bavelloni, A.; Faenza, I.; Raffini, M.; Tagliavini, F.; Manzoli, L.; Cocco, L. Pi-plcbeta1b affects akt activation, cyclin e expression, and caspase cleavage, promoting cell survival in pro-b-lymphoblastic cells exposed to oxidative stress. FASEB J. 2015, 29, 1383–1394. [Google Scholar] [CrossRef]
- Ng, H.Y.; Ko, J.M.; Yu, V.Z.; Ip, J.C.; Dai, W.; Cal, S.; Lung, M.L. Desc1, a novel tumor suppressor, sensitizes cells to apoptosis by downregulating the egfr/akt pathway in esophageal squamous cell carcinoma. Int. J. Cancer 2016, 138, 2940–2951. [Google Scholar] [CrossRef] [Green Version]
- Powis, G.; Seewald, M.J.; Gratas, C.; Melder, D.; Riebow, J.; Modest, E.J. Selective inhibition of phosphatidylinositol phospholipase c by cytotoxic ether lipid analogues. Cancer Res. 1992, 52, 2835–2840. [Google Scholar]
- Viloria, C.G.; Peinado, J.R.; Astudillo, A.; Garcia-Suarez, O.; Gonzalez, M.V.; Suarez, C.; Cal, S. Human desc1 serine protease confers tumorigenic properties to mdck cells and it is upregulated in tumours of different origin. Br. J. Cancer 2007, 97, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Haarberg, H.E.; Smalley, K.S. Resistance to raf inhibition in cancer. Drug Discov. Today Technol. 2014, 11, 27–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, M.; Bode, A.M.; Dong, Z.; Lee, M.H. Akt as a therapeutic target for cancer. Cancer Res. 2019, 79, 1019–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, G.; Wang, J.; Xin, X.; Ke, Z.; Luo, J. Phosphorylation of glycogen synthase kinase-3 beta at serine 9 confers cisplatin resistance in ovarian cancer cells. Int. J. Oncol. 2007, 31, 657–662. [Google Scholar]
- Zhang, Q.; Kuang, H.; Chen, C.; Yan, J.; Do-Umehara, H.C.; Liu, X.Y.; Dada, L.; Ridge, K.M.; Chandel, N.S.; Liu, J. The kinase jnk2 promotes stress-induced mitophagy by targeting the small mitochondrial form of the tumor suppressor arf for degradation. Nat. Immunol. 2015, 16, 458–466. [Google Scholar] [CrossRef] [Green Version]
- Duda, P.; Akula, S.M.; Abrams, S.L.; Steelman, L.S.; Martelli, A.M.; Cocco, L.; Ratti, S.; Candido, S.; Libra, M.; Montalto, G.; et al. Targeting gsk3 and associated signaling pathways involved in cancer. Cells 2020, 9, 1110. [Google Scholar] [CrossRef]
- Gresset, A.; Sondek, J.; Harden, T.K. The phospholipase c isozymes and their regulation. Subcell Biochem. 2012, 58, 61–94. [Google Scholar]
- Piazzi, M.; Blalock, W.L.; Bavelloni, A.; Faenza, I.; D’Angelo, A.; Maraldi, N.M.; Cocco, L. Phosphoinositide-specific phospholipase c beta 1b (pi-plcbeta1b) interactome: Affinity purification-mass spectrometry analysis of pi-plcbeta1b with nuclear protein. Mol. Cell Proteom. 2013, 12, 2220–2235. [Google Scholar] [CrossRef] [Green Version]
- Poli, A.; Faenza, I.; Chiarini, F.; Matteucci, A.; McCubrey, J.A.; Cocco, L. K562 cell proliferation is modulated by plcbeta1 through a pkcalpha-mediated pathway. Cell Cycle 2013, 12, 1713–1721. [Google Scholar] [CrossRef] [Green Version]
- Fiume, R.; Ramazzotti, G.; Teti, G.; Chiarini, F.; Faenza, I.; Mazzotti, G.; Billi, A.M.; Cocco, L. Involvement of nuclear plcbeta1 in lamin b1 phosphorylation and g2/m cell cycle progression. FASEB J. 2009, 23, 957–966. [Google Scholar] [CrossRef]
- Lee, Y.H.; Kim, S.Y.; Kim, J.R.; Yoh, K.T.; Baek, S.H.; Kim, M.J.; Ryu, S.H.; Suh, P.G.; Kim, J.H. Overexpression of phospholipase cbeta-1 protects nih3t3 cells from oxidative stress-induced cell death. Life Sci. 2000, 67, 827–837. [Google Scholar] [CrossRef]
- Johannessen, C.M.; Johnson, L.A.; Piccioni, F.; Townes, A.; Frederick, D.T.; Donahue, M.K.; Narayan, R.; Flaherty, K.T.; Wargo, J.A.; Root, D.E.; et al. A melanocyte lineage program confers resistance to map kinase pathway inhibition. Nature 2013, 504, 138–142. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zhao, X.; Wang, D.; He, W.; Zhang, S.; Cao, W.; Huang, Y.; Wang, L.; Zhou, S.; Luo, K. Up-regulated expression of phospholipase c, beta1 is associated with tumor cell proliferation and poor prognosis in hepatocellular carcinoma. Onco. Targets Ther. 2016, 9, 1697–1706. [Google Scholar]
- Lin, D.; Fu, Z.; Yang, G.; Gao, D.; Wang, T.; Liu, Z.; Li, G.; Wang, Y. Exportin-5 sumoylation promotes hepatocellular carcinoma progression. Exp. Cell Res. 2020, 395, 112219. [Google Scholar] [CrossRef]
- Zhang, T.; Song, X.; Liao, X.; Wang, X.; Zhu, G.; Yang, C.; Xie, X. Distinct prognostic values of phospholipase c beta family members for non-small cell lung carcinoma. Biomed. Res. Int. 2019, 2019, 1–11. [Google Scholar] [CrossRef]
- Lu, M.L.; Zhang, Y.; Li, J.; Fu, Y.; Li, W.H.; Zhao, G.F.; Li, X.H.; Wei, L.; Liu, G.B.; Huang, H. Microrna-124 inhibits colorectal cancer cell proliferation and suppresses tumor growth by interacting with plcb1 and regulating wnt/beta-catenin signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 121–136. [Google Scholar]
- Wang, Q.; Ye, J.; Fang, D.; Lv, L.; Wu, W.; Shi, D.; Li, Y.; Yang, L.; Bian, X.; Wu, J.; et al. Multi-omic profiling reveals associations between the gut mucosal microbiome, the metabolome, and host DNA methylation associated gene expression in patients with colorectal cancer. BMC Microbiol. 2020, 20, 83. [Google Scholar] [CrossRef]
- Molinari, C.; Medri, L.; Follo, M.Y.; Piazzi, M.; Mariani, G.A.; Calistri, D.; Cocco, L. Pi-plcbeta1 gene copy number alterations in breast cancer. Oncol Rep. 2012, 27, 403–408. [Google Scholar]
- Damm, F.; Lange, K.; Heuser, M.; Oberacker, T.; Morgan, M.; Wagner, K.; Krauter, J.; Schlegelberger, B.; Ganser, A.; Gohring, G. Phosphoinositide phospholipase cbeta1 (pi-plcbeta1) gene in myelodysplastic syndromes and cytogenetically normal acute myeloid leukemia: Not a deletion, but increased pi-plcbeta1 expression is an independent prognostic factor. J. Clin. Oncol 2010, 28, e384–e387, author reply e388-9. [Google Scholar] [CrossRef]
- Lang, J.C.; Schuller, D.E. Differential expression of a novel serine protease homologue in squamous cell carcinoma of the head and neck. Br. J. Cancer 2001, 84, 237–243. [Google Scholar] [CrossRef] [Green Version]
- Zinovyeva, M.V.; Monastyrskaya, G.S.; Kopantzev, E.P.; Vinogradova, T.V.; Kostina, M.B.; Sass, A.V.; Filyukova, O.B.; Uspenskaya, N.Y.; Sukhikh, G.T.; Sverdlov, E.D. Identification of some human genes oppositely regulated during esophageal squamous cell carcinoma formation and human embryonic esophagus development. Dis. Esophagus 2010, 23, 260–270. [Google Scholar] [CrossRef] [PubMed]
- Chang, Z.W.; Jia, Y.X.; Zhang, W.J.; Song, L.J.; Gao, M.; Li, M.J.; Zhao, R.H.; Li, J.; Zhong, Y.L.; Sun, Q.Z.; et al. Lncrna-tusc7/mir-224 affected chemotherapy resistance of esophageal squamous cell carcinoma by competitively regulating desc1. J. Exp. Clin. Cancer Res. 2018, 37, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.J.; Seto, E. Collaborative spirit of histone deacetylases in regulating chromatin structure and gene expression. Curr. Opin. Genet. Dev. 2003, 13, 143–153. [Google Scholar] [CrossRef]
- Chakrabarti, A.; Oehme, I.; Witt, O.; Oliveira, G.; Sippl, W.; Romier, C.; Pierce, R.J.; Jung, M. Hdac8: A multifaceted target for therapeutic interventions. Trends Pharmacol. Sci. 2015, 36, 481–492. [Google Scholar] [CrossRef] [Green Version]
- Zhao, P.; Sun, S.; Zhai, Y.; Tian, Q.; Zhou, T.; Li, J. Mir-423-5p inhibits the proliferation and metastasis of glioblastoma cells by targeting phospholipase c beta 1. Int. J. Clin. Exp. Pathol. 2019, 12, 2941–2950. [Google Scholar]
- Zhao, G.; Wang, G.; Bai, H.; Li, T.; Gong, F.; Yang, H.; Wen, J.; Wang, W. Targeted inhibition of hdac8 increases the doxorubicin sensitivity of neuroblastoma cells via up regulation of mir-137. Eur. J. Pharmacol. 2017, 802, 20–26. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ha, S.-D.; Lewin, N.; Li, S.S.C.; Kim, S.-O. HDAC8 Activates AKT through Upregulating PLCB1 and Suppressing DESC1 Expression in MEK1/2 Inhibition-Resistant Cells. Cells 2021, 10, 1101. https://doi.org/10.3390/cells10051101
Ha S-D, Lewin N, Li SSC, Kim S-O. HDAC8 Activates AKT through Upregulating PLCB1 and Suppressing DESC1 Expression in MEK1/2 Inhibition-Resistant Cells. Cells. 2021; 10(5):1101. https://doi.org/10.3390/cells10051101
Chicago/Turabian StyleHa, Soon-Duck, Naomi Lewin, Shawn S. C. Li, and Sung-Ouk Kim. 2021. "HDAC8 Activates AKT through Upregulating PLCB1 and Suppressing DESC1 Expression in MEK1/2 Inhibition-Resistant Cells" Cells 10, no. 5: 1101. https://doi.org/10.3390/cells10051101
APA StyleHa, S.-D., Lewin, N., Li, S. S. C., & Kim, S.-O. (2021). HDAC8 Activates AKT through Upregulating PLCB1 and Suppressing DESC1 Expression in MEK1/2 Inhibition-Resistant Cells. Cells, 10(5), 1101. https://doi.org/10.3390/cells10051101