
Th17 Cells in Viral Infections—Friend or Foe?


 and
and
Abstract
1. Introduction
2. Th1/Th2 Paradigm and Discovery of Th17 Cells
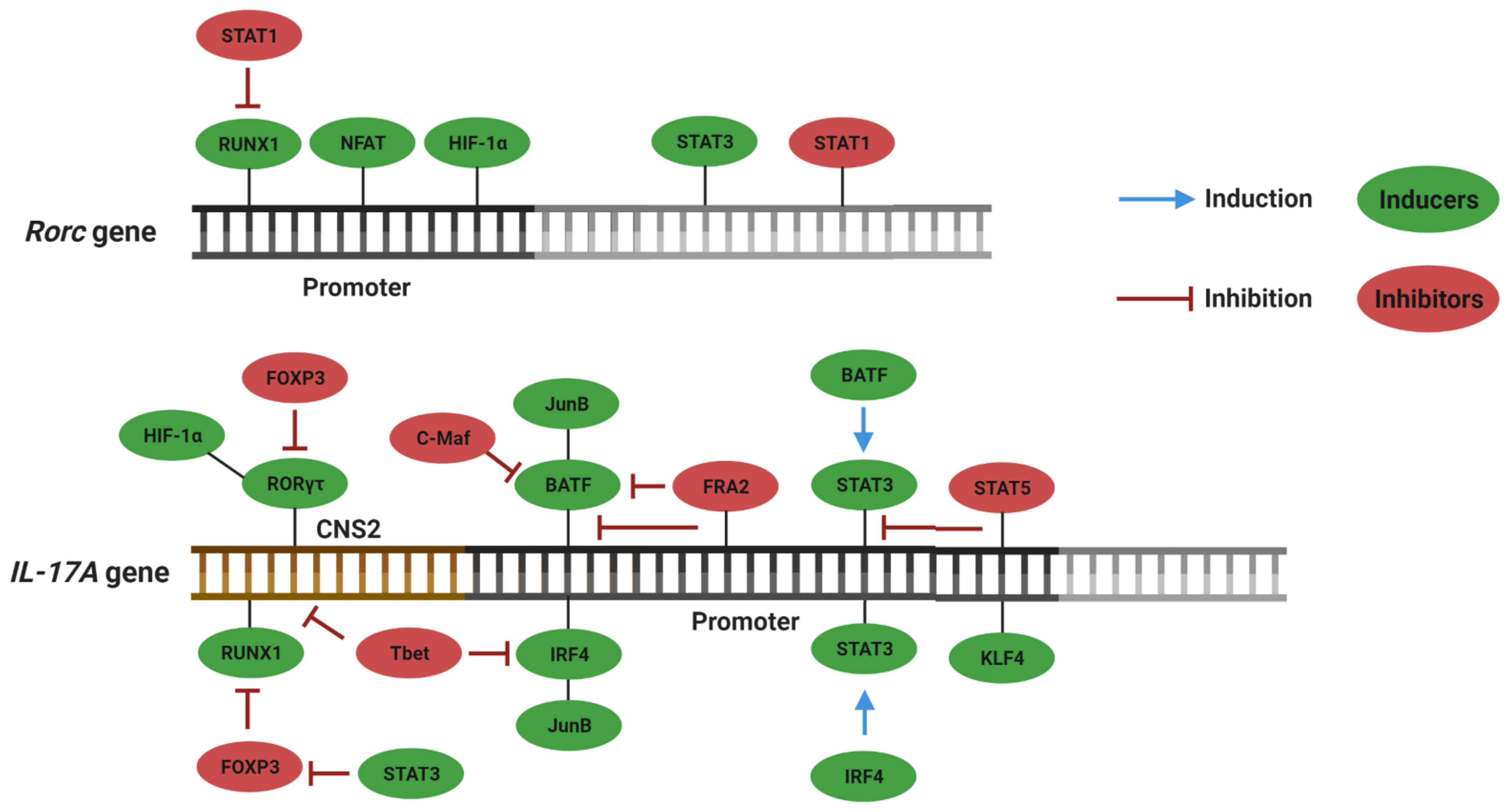
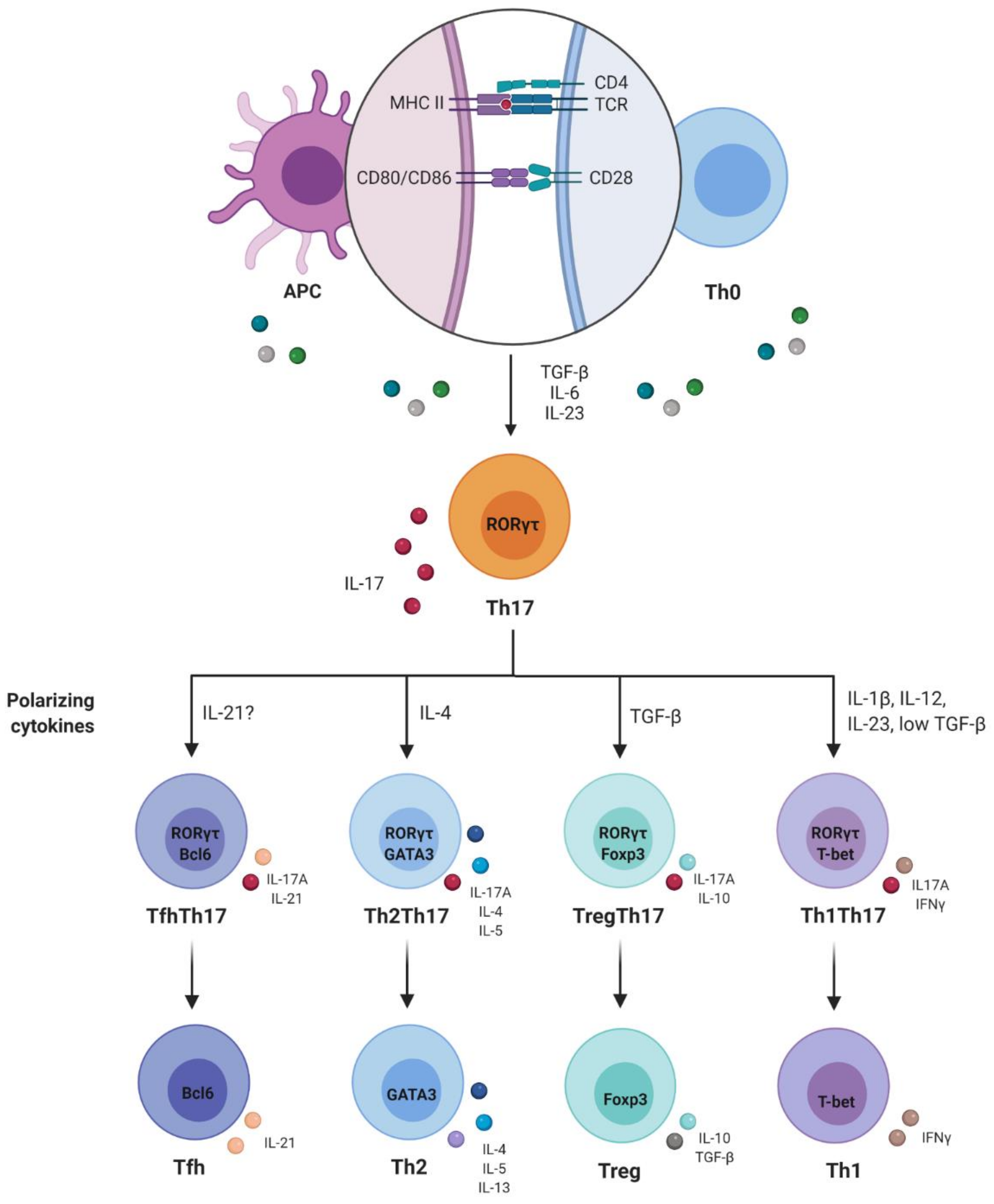
3. Plasticity and Effector Abilities of Th17 Modulated by Inflammatory Cytokines
4. Th17 Cells in Viral Infections
4.1. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2)
4.2. Influenza Virus
4.3. Herpes Simplex Virus (HSV)
4.4. West Nile Virus (WNV) and Adenovirus (Ad)
4.5. Chikungunya (CHIKV), Dengue Virus (DENV) and Zika Virus (ZIKV)
4.6. Viral Myocarditis
4.7. Viral Infection as a Trigger for Multiple Sclerosis
5. Conclusions and Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mosmann, T.R.; Cherwinski, H.; Bond, M.W.; Giedlin, M.A.; Coffman, R.L. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J. Immunol. 1986, 136, 2348–2357. [Google Scholar]
- Coffman, R.L. Origins of the TH1-TH2 model: A personal perspective. Nat. Immunol. 2006, 7, 539–541. [Google Scholar] [CrossRef] [PubMed]
- Coffman, R.L.; Carty, J. A T cell activity that enhances polyclonal IgE production and its inhibition by interferon-gamma. J. Immunol. 1986, 136, 949–954. [Google Scholar] [PubMed]
- Hu-Li, J.; Shevach, E.M.; Mizuguchi, J.; Ohara, J.; Mosmann, T.; Paul, W.E. B cell stimulatory factor I (interleukin 4) is a potent costimulant for normal resting T lymphocytes. J. Exp. Med. 1987, 165, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Cher, D.J.; Mosmann, T.R. Two types of murine helper T cell clone. II. Delayed-type hypersensitivity is mediated by TH1 clones. J. Immunol. 1987, 138, 3688–3694. [Google Scholar]
- Mosmann, T.R.; Coffman, R.L. TH1 and TH2 cells: Different patterns of lymphokine secretion lead to different functional properties. Annu. Rev. Immunol. 1989, 7, 145–173. [Google Scholar] [CrossRef]
- Reiner, S.L.; Locksley, R.M. The regulation of immunity to Leishmania major. Annu. Rev. Immunol. 1995, 13, 151–177. [Google Scholar] [CrossRef]
- Bettelli, E.; Sullivan, B.; Szabo, S.J.; Sobel, R.A.; Glimcher, L.H.; Kuchroo, V.K. Loss of T-bet, but not STAT1, prevents the development of experimental autoimmune encephalomyelitis. J. Exp. Med. 2004, 200, 79–87. [Google Scholar] [CrossRef]
- Krakowski, M.; Owens, T. Interferon-γ confers resistance to experimental allergic encephalomyelitis. Eur. J. Immunol. 1996, 26, 1641–1646. [Google Scholar] [CrossRef]
- Duong, T.T.; Finkelman, F.D.; Singh, B.; Strejan, G.H. Effect of anti-interferon-γ monoclonal antibody treatment on the development of experimental allergic encephalomyelitis in resistant mouse strains. J. Neuroimmunol. 1994, 53, 101–107. [Google Scholar] [CrossRef]
- Oppmann, B.; Lesley, R.; Blom, B.; Timans, J.C.; Xu, Y.; Hunte, B.; Vega, F.; Yu, N.; Wang, J.; Singh, K.; et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity 2000, 13, 715–725. [Google Scholar] [CrossRef]
- Chyuan, I.T.; Lai, J.H. New insights into the IL-12 and IL-23: From a molecular basis to clinical application in immune-mediated inflammation and cancers. Biochem. Pharmacol. 2020, 175, 113928. [Google Scholar] [CrossRef] [PubMed]
- Schnurr, M.; Toy, T.; Shin, A.; Wagner, M.; Cebon, J.; Maraskovsky, E. Extracellular nucleotide signaling by P2 receptors inhibits IL-12 and enhances IL-23 expression in human dendritic cells: A novel role for the cAMP pathway. Blood 2005, 105, 1582–1589. [Google Scholar] [CrossRef]
- Sheibanie, A.F.; Tadmori, I.; Jing, H.; Vassiliou, E.; Ganea, D. Prostaglandin E2 induces IL-23 production in bone marrow-derived dendritic cells. FASEB J. 2004, 18, 1318–1320. [Google Scholar] [CrossRef]
- Aggarwal, S.; Ghilardi, N.; Xie, M.H.; De Sauvage, F.J.; Gurney, A.L. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J. Biol. Chem. 2003, 278, 1910–1914. [Google Scholar] [CrossRef]
- Cua, D.J.; Sherlock, J.; Chen, Y.; Murphy, C.A.; Joyce, B.; Seymour, B.; Lucian, L.; To, W.; Kwan, S.; Churakova, T.; et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature 2003, 421, 744–748. [Google Scholar] [CrossRef]
- Park, H.; Li, Z.; Yang, X.O.; Chang, S.H.; Nurieva, R.; Wang, Y.-H.; Wang, Y.; Hood, L.; Zhu, Z.; Tian, Q.; et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat. Immunol. 2005, 6, 1133–1141. [Google Scholar] [CrossRef]
- Harrington, L.E.; Hatton, R.D.; Mangan, P.R.; Turner, H.; Murphy, T.L.; Murphy, K.M.; Weaver, C.T. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat. Immunol. 2005, 6, 1123–1132. [Google Scholar] [CrossRef]
- Korn, T.; Bettelli, E.; Oukka, M.; Kuchroo, V.K. IL-17 and Th17 cells. Annu. Rev. Immunol. 2009, 27, 485–517. [Google Scholar] [CrossRef] [PubMed]
- Gaffen, S.L.; Jain, R.; Garg, A.V.; Cua, D.J. IL-23-IL-17 immune axis: Discovery, Mechanistic Understanding, and Clinical Testing. Nat. Rev. Immunol. 2014, 14, 585–600. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.I.; McKenzie, B.S.; Zhou, L.; Tadokoro, C.E.; Lepelley, A.; Lafaille, J.J.; Cua, D.J.; Littman, D.R. The Orphan Nuclear Receptor RORγt Directs the Differentiation Program of Proinflammatory IL-17+ T Helper Cells. Cell 2006, 126, 1121–1133. [Google Scholar] [CrossRef]
- Unutmas, D. RORC2: The master of human Th17 cell programming. Eur. J. Immunol. 2009, 39, 1452–1455. [Google Scholar] [CrossRef]
- Zhang, Y.; Luo, X.Y.; Wu, D.H.; Xu, Y. ROR nuclear receptors: Structures, related diseases, and drug discovery. Acta Pharmacol. Sin. 2015, 36, 71–87. [Google Scholar] [CrossRef] [PubMed]
- Capone, A.; Volpe, E. Transcriptional Regulators of T Helper 17 Cell Differentiation in Health and Autoimmune Diseases. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.O.; Pappu, B.; Nurieva, R.; Akimzhanov, A.; Soon, H.; Chung, Y.; Ma, L.; Shah, B.; Panopoulos, A.D.; Watowich, S.S.; et al. TH17 lineage differentiation is programmed by orphan nuclear receptors RORα and RORγ. Immunity 2008, 28, 29–39. [Google Scholar] [CrossRef]
- Quintana, F.J.; Basso, A.S.; Iglesias, A.H.; Korn, T.; Farez, M.F.; Bettelli, E.; Caccamo, M.; Oukka, M.; Weiner, H.L. Control of Treg and TH17 cell differentiation by the aryl hydrocarbon receptor. Nature 2008, 453, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Durant, L.; Watford, W.T.; Ramos, H.L.; Laurence, A.; Vahedi, G.; Takahashi, H.; Sun, H.; Kanno, Y.; Powrie, F.; Shea, J.J.O. Diverse Targets of the Transcription Factor STAT3 Contribute to T Cell Pathogenicity and Homeostasis. Immunity 2010, 32, 605–615. [Google Scholar] [CrossRef]
- Ciofani, M.; Madar, A.; Galan, C.; Sellars, M.; Mace, K.; Pauli, F.; Agarwal, A.; Huang, W.; Parkhurst, C.N.; Newberry, K.M.; et al. A validated regulatory network for Th17 cell specification. Cell 2012, 151, 289–303. [Google Scholar] [CrossRef]
- Gökmen, M.R.; Dong, R.; Kanhere, A.; Powell, N.; Perucha, E. Genome-wide regulatory analysis reveals T-bet controls Th17 lineage differentiation through direct suppression of IRF41. J. Immunol. 2013, 191, 1–16. [Google Scholar] [CrossRef]
- Lazarevic, V.; Chen, X.; Shim, J.-H.; Hwang, E.-S.; Jang, E.; Bolm, A.N.; Oukka, M.; Kuchroo, V.K.; Glimcher, L.H. T-bet represses TH 17 differentiation by preventing Runx1-mediated activation of the RORγt gene. Physiol. Behav. 2011, 12, 96–104. [Google Scholar] [CrossRef]
- Zhou, L.; Lopes, J.E.; Chong, M.M.W.; Ivanov, I.I.; Min, R.; Gabriel, D.; Shen, Y.; Du, J.; Rubtsov, Y.P.; Rudensky, A.Y.; et al. TGF-β-induced Foxp3 inhibits Th17 cell differentiation by antagonizing RORγt function. Nature 2008, 453, 236–240. [Google Scholar] [CrossRef]
- Zhang, F.; Meng, G.; Strober, W. Interactions among the transcription factors Runx1, RORγt and Foxp3 regulate the differentiation of interleukin 17-producing T cells. Nat. Immunol. 2008, 9, 1297–1306. [Google Scholar] [CrossRef]
- Tripathi, S.K.; Lahesmaa, R. Transcriptional and epigenetic regulation of T-helper lineage specification. Immunol. Rev. 2014, 261, 62–83. [Google Scholar] [CrossRef]
- Annunziato, F.; Cosmi, L.; Santarlasci, V.; Maggi, L.; Liotta, F.; Mazzinghi, B.; Parente, E.; Filì, L.; Ferri, S.; Frosali, F.; et al. Phenotypic and functional features of human Th17 cells. J. Exp. Med. 2007, 204, 1849–1861. [Google Scholar] [CrossRef]
- Acosta-Rodriguez, E.V.; Rivino, L.; Geginat, J.; Jarrossay, D.; Gattorno, M.; Lanzavecchia, A.; Sallusto, F.; Napolitani, G. Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat. Immunol. 2007, 8, 639–646. [Google Scholar] [CrossRef]
- Singh, S.P.; Zhang, H.H.; Foley, J.F.; Hedrick, M.N.; Farber, J.M. Human T Cells That Are Able to Produce IL-17 Express the Chemokine Receptor CCR6. J. Immunol. 2008, 180, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Kunkel, E.J.; Campbell, D.J.; Butcher, E.C. Chemokines in lymphocyte trafficking and intestinal immunity. Microcirculation 2003, 10, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Lowes, M.A.; Suárez-Fariñas, M.; Krueger, J.G. Immunology of psoriasis. Annu. Rev. Immunol. 2014, 32, 227–255. [Google Scholar] [CrossRef] [PubMed]
- Reboldi, A.; Coisne, C.; Baumjohann, D.; Benvenuto, F.; Bottinelli, D.; Lira, S.; Uccelli, A.; Lanzavecchia, A.; Engelhardt, B.; Sallusto, F. C-C chemokine receptor 6-regulated entry of TH-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat. Immunol. 2009, 10, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Cosmi, L.; De Palma, R.; Santarlasci, V.; Maggi, L.; Capone, M.; Frosali, F.; Rodolico, G.; Querci, V.; Abbate, G.; Angeli, R.; et al. Human interleukin 17-producing cells originate from a CD161 +CD4+ T cell precursor. J. Exp. Med. 2008, 205, 1903–1916. [Google Scholar] [CrossRef]
- Zielinski, C.E.; Mele, F.; Aschenbrenner, D.; Jarrossay, D.; Ronchi, F.; Gattorno, M.; Monticelli, S.; Lanzavecchia, A.; Sallusto, F. Pathogen-induced human TH17 cells produce IFN-γ or IL-10 and are regulated by IL-1β. Nature 2012, 484, 514–518. [Google Scholar] [CrossRef] [PubMed]
- Okada, S.; Markle, J.G.; Deenick, E.K.; Mele, F.; Averbuch, D.; Lagos, M.; Alzahrani, M.; Muhsen, S.A.-; Halwani, R.; Ma, C.S.; et al. Impairment of immunity to Candida and Mycobacterium in humans with bi-allelic RORC mutations. Science 2015, 349, 606–613. [Google Scholar] [CrossRef]
- Duhen, T.; Campbell, D.J. IL-1β promotes the differentiation of polyfunctional human CCR6+ CXCR3+ Th1/17 cells that are specific for pathogenic and commensal microbes. J. Immunol. 2014, 193, 120–129. [Google Scholar] [CrossRef]
- Wacleche, V.S.; Landay, A.; Routy, J.P.; Ancuta, P. The Th17 lineage: From barrier surfaces homeostasis to autoimmunity, cancer, and HIV-1 pathogenesis. Viruses 2017, 9, 303. [Google Scholar] [CrossRef]
- Mazzoni, A.; Maggi, L.; Liotta, F.; Cosmi, L.; Annunziato, F. Biological and clinical significance of T helper 17 cell plasticity. Immunology 2019, 158, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Cosmi, L.; Maggi, L.; Santarlasci, V.; Liotta, F.; Annunziato, F. T helper cells plasticity in inflammation. Cytom. Part. A 2014, 85, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Kamali, A.N.; Noorbakhsh, S.M.; Hamedifar, H.; Jadidi-Niaragh, F.; Yazdani, R.; Bautista, J.M.; Azizi, G. A role for Th1-like Th17 cells in the pathogenesis of inflammatory and autoimmune disorders. Mol. Immunol. 2019, 105, 107–115. [Google Scholar] [CrossRef]
- Wacleche, V.S.; Goulet, J.P.; Gosselin, A.; Monteiro, P.; Soudeyns, H.; Fromentin, R.; Jenabian, M.A.; Vartanian, S.; Deeks, S.G.; Chomont, N.; et al. New insights into the heterogeneity of Th17 subsets contributing to HIV-1 persistence during antiretroviral therapy. Retrovirology 2016, 13, 1–25. [Google Scholar] [CrossRef]
- Sallusto, F. Heterogeneity of Human CD4+ T Cells Against Microbes. Annu. Rev. Immunol. 2016, 34, 317–334. [Google Scholar] [CrossRef]
- Hueber, W.; Sands, B.E.; Lewitzky, S.; Vandemeulebroecke, M.; Reinisch, W.; Higgins, P.D.R.; Wehkamp, J.; Feagan, B.G.; Yao, M.D.; Karczewski, M.; et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: Unexpected results of a randomised, double-blind placebo-controlled trial. Gut 2012, 61, 1693–1700. [Google Scholar] [CrossRef]
- McGeachy, M.J.; Bak-Jensen, K.S.; Chen, Y.; Tato, C.M.; Blumenschein, W.; McClanahan, T.; Cua, D.J. TGF-β and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain TH-17 cell-mediated pathology. Nat. Immunol. 2007, 8, 1390–1397. [Google Scholar] [CrossRef]
- Ghoreschi, K.; Laurence, A.; Yang, X.-P.; Tato, C.M.; McGeachy, M.J.; Konkel, J.; Ramos, H.L.; Wei, L.; Davidson, T.; Bouladoux, N.; et al. Generation of pathogenic TH17 cells in the absence of TGF-b signaling. Nature 2010, 467, 967–971. [Google Scholar] [CrossRef]
- Lee, Y.; Awasthi, A.; Yosef, N.; Quintana, F.J.; Xiao, S.; Peters, A.; Wu, C.; Kleinewietfeld, M.; Kunder, S.; Hafler, D.; et al. Induction and molecular signature of pathogenic TH 17 cells. Nat. Immunol. 2012, 13, 991–999. [Google Scholar] [CrossRef]
- Wu, X.; Tian, J.; Wang, S. Insight into non-pathogenic Th17 cells in autoimmune diseases. Front. Immunol. 2018, 9, 1–8. [Google Scholar] [CrossRef]
- Rivino, L.; Messi, M.; Jarrossay, D.; Lanzavecchia, A.; Sallusto, F.; Geginat, J. Chemokine receptor expression identifies pre-T helper (Th)1, pre-Th2, and nonpolarized cells among human CD4+ central memory T cells. J. Exp. Med. 2004, 200, 725–735. [Google Scholar] [CrossRef] [PubMed]
- Abromson-Leeman, S.; Bronson, R.T.; Dorf, M.E. Encephalitogenic T cells that stably express both T-bet and RORγt consistently produce IFNγ but have a spectrum of IL-17 profiles. J. Neuroimmunol. 2009, 215, 10–24. [Google Scholar] [CrossRef]
- Boniface, K.; Blumenschein, W.M.; Brovont-Porth, K.; McGeachy, M.J.; Basham, B.; Desai, B.; Pierce, R.; McClanahan, T.K.; Sadekova, S.; de Waal Malefyt, R. Human Th17 Cells Comprise Heterogeneous Subsets Including IFN-γ–Producing Cells with Distinct Properties from the Th1 Lineage. J. Immunol. 2010, 185, 679–687. [Google Scholar] [CrossRef]
- Cosmi, L.; Maggi, L.; Santarlasci, V.; Capone, M.; Cardilicchia, E.; Frosali, F.; Querci, V.; Angeli, R.; Matucci, A.; Fambrini, M.; et al. Identification of a novel subset of human circulating memory CD4+ T cells that produce both IL-17A and IL-4. J. Allergy Clin. Immunol. 2010, 125, 222–230.e4. [Google Scholar] [CrossRef]
- Hirota, K.; Turner, J.; Villa, M.; Duarte, J.H.; Demengeot, J. TH 17 cell plasticity in Peyer’s patches is responsible for induction of T cell-dependent IgA responses. Nat. Immunol. 2013, 14, 372–379. [Google Scholar] [CrossRef]
- Hetta, H.F. Role of T Follicular Helper (Tfh) Cells Plasticity in Autoimmune Thyroiditis among Hepatitis C Virus Infection. Gastroenterol. Hepatol. Open Access 2017, 6. [Google Scholar] [CrossRef]
- Bunte, K.; Beikler, T. Th17 cells and the IL-23/IL-17 axis in the pathogenesis of periodontitis and immune-mediated inflammatory diseases. Int. J. Mol. Sci. 2019, 20, 3394. [Google Scholar] [CrossRef]
- Stritesky, G.L.; Yeh, N.; Kaplan, M.H. IL-23 Promotes Maintenance but Not Commitment to the Th17 Lineage. J. Immunol. 2008, 181, 5948–5955. [Google Scholar] [CrossRef]
- Koenen, H.J.P.M.; Smeets, R.L.; Vink, P.M.; Van Rijssen, E.; Boots, A.M.H.; Joosten, I. Human CD25highFoxp3pos regulatory T cells differentiate into IL-17 producing cells. Blood 2008, 112, 2340–2352. [Google Scholar] [CrossRef]
- Valmori, D.; Raffin, C.; Raimbaud, I.; Ayyoub, M. Human RORγt+ TH17 cells preferentially differentiate from naive FOXP3+Treg in the presence of lineagespecific polarizing factors. Proc. Natl. Acad. Sci. USA 2010, 107, 19402–19407. [Google Scholar] [CrossRef]
- Hoechst, B.; Gamrekelashvili, J.; Manns, M.P.; Greten, T.F.; Korangy, F. Plasticity of human Th17 cells and iTregs is orchestrated by different subsets of myeloid cells. Blood 2011, 117, 6532–6541. [Google Scholar] [CrossRef]
- Wang, X.; Ma, C.; Wu, J.; Zhu, J. Roles of T helper 17 cells and interleukin-17 in neuroautoimmune diseases with emphasis on multiple sclerosis and Guillain-Barré syndrome as well as their animal models. J. Neurosci. Res. 2013, 91, 871–881. [Google Scholar] [CrossRef]
- Yan, J.W.; Wang, Y.J.; Peng, W.J.; Tao, J.H.; Wan, Y.N.; Li, B.Z.; Mei, B.; Chen, B.; Yao, H.; Yang, G.J.; et al. Therapeutic potential of interleukin-17 in inflammation and autoimmune diseases. Expert Opin. Ther. Targets 2014, 18, 29–41. [Google Scholar] [CrossRef]
- Wang, C.Q.F.; Akalu, Y.T.; Suarez-farinas, M.; Gonzalez, J.; Mitsui, H.; Lowes, M.A.; Orlow, S.J.; Manga, P.; James, G.; Science, T. IL-17 and TNF synergistically modulate cytokine expression while suppressing melanogenesis: Potential relevance to psoriasis. J. Invest. Dermatol. 2013, 133, 2741–2752. [Google Scholar] [CrossRef]
- Beringer, A.; Thiam, N.; Molle, J.; Bartosch, B.; Miossec, P. Synergistic effect of interleukin-17 and tumour necrosis factor-α on inflammatory response in hepatocytes through interleukin-6-dependent and independent pathways. Clin. Exp. Immunol. 2018, 193, 221–233. [Google Scholar] [CrossRef]
- Hsu, H.C.; Yang, P.A.; Wang, J.; Wu, Q.; Myers, R.; Chen, J.; Yi, J.; Guentert, T.; Tousson, A.; Stanus, A.L.; et al. Interleukin 17-producing T helper cells and interleukin 17 orchestrate autoreactive germinal center development in autoimmune BXD2 mice. Nat. Immunol. 2008, 9, 166–175. [Google Scholar] [CrossRef]
- Shohan, M.; Dehghani, R.; Khodadadi, A.; Dehnavi, S.; Ahmadi, R.; Joudaki, N.; Houshmandfar, S.; Shamshiri, M.; Shojapourian, S.; Bagheri, N. Interleukin-22 and intestinal homeostasis: Protective or destructive? IUBMB Life 2020, 72, 1585–1602. [Google Scholar] [CrossRef]
- Larochette, V.; Miot, C.; Poli, C.; Beaumont, E.; Roingeard, P.; Fickenscher, H.; Jeannin, P.; Delneste, Y. IL-26, a cytokine with roles in extracellular DNA-induced inflammation and microbial defense. Front. Immunol. 2019, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Meller, S.; Di Domizio, J.; Voo, K.S.; Friedrich, H.C.; Ganguly, D.; Conrad, C.; Gregorio, J.; Le Roy, D.; Ladbury, J.E.; Homey, B.; et al. TH17 cells promote microbial killing and innate immune sensing of DNA via interleukin 26. Nat. Immunol. 2015, 16, 970–979. [Google Scholar] [CrossRef] [PubMed]
- Volpe, E.; Touzot, M.; Servant, N.; Marloie-Provost, M.A.; Hupé, P.; Barillot, E.; Soumelis, V. Multiparametric analysis of cytokine-driven human Th17 differentiation reveals a differential regulation of IL-17 and IL-22 production. Blood 2009, 114, 3610–3614. [Google Scholar] [CrossRef]
- Kaabachi, W.; Bouali, E.; Berraïes, A.; Dhifallh, I.B.; Hamdi, B.; Hamzaoui, K.; Hamzaoui, A. Interleukin-26 is overexpressed in Behçet’s disease and enhances Th17 related −cytokines. Immunol. Lett. 2017, 190, 177–184. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Coronavírus Desease (COVID-19) Situation Report; WHO: Geneva, Switzerland, 2020. [Google Scholar]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Xu, Z.; Shi, L.; Wang, Y.; Zhang, J.; Huang, L.; Zhang, C.; Liu, S.; Zhao, P.; Liu, H.; Zhu, L.; et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 2020, 8, 420–422. [Google Scholar] [CrossRef]
- Coomes, E.A.; Haghbayan, H. Interleukin-6 in Covid-19: A systematic review and meta-analysis. Rev. Med. Virol. 2020, 30, 1–9. [Google Scholar] [CrossRef]
- Gil-Etayo, F.J.; Suàrez-Fernández, P.; Cabrera-Marante, O.; Arroyo, D.; Garcinuño, S.; Naranjo, L.; Pleguezuelo, D.E.; Allende, L.M.; Mancebo, E.; Lalueza, A.; et al. T-Helper Cell Subset Response Is a Determining Factor in COVID-19 Progression. Front. Cell. Infect. Microbiol. 2021, 11, 1–11. [Google Scholar] [CrossRef]
- Meckiff, B.J.; Ramírez-Suástegui, C.; Fajardo, V.; Chee, S.J.; Kusnadi, A.; Simon, H.; Eschweiler, S.; Grifoni, A.; Pelosi, E.; Weiskopf, D.; et al. Imbalance of Regulatory and Cytotoxic SARS-CoV-2-Reactive CD4+ T Cells in COVID-19. Cell 2020, 183, 1340–1353.e16. [Google Scholar] [CrossRef]
- Zhao, Y.; Kilian, C.; Turner, J.E.; Bosurgi, L.; Roedl, K.; Bartsch, P.; Gnirck, A.C.; Cortesi, F.; Schultheiß, C.; Hellmig, M.; et al. Clonal expansion and activation of tissue-resident memory-like Th17 cells expressing GM-CSF in the lungs of severe COVID-19 patients. Sci. Immunol. 2021, 6. [Google Scholar] [CrossRef] [PubMed]
- Toor, S.M.; Saleh, R.; Sasidharan Nair, V.; Taha, R.Z.; Elkord, E. T-cell responses and therapies against SARS-CoV-2 infection. Immunology 2021, 162, 30–43. [Google Scholar] [CrossRef]
- De Biasi, S.; Meschiari, M.; Gibellini, L.; Bellinazzi, C.; Borella, R.; Fidanza, L.; Gozzi, L.; Iannone, A.; Lo Tartaro, D.; Mattioli, M.; et al. Marked T cell activation, senescence, exhaustion and skewing towards TH17 in patients with COVID-19 pneumonia. Nat. Commun. 2020, 11, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Tiwari-Heckler, S.; Rauber, C.; Longhi, M.S.; Zörnig, I.; Schnitzler, P.; Jäger, D.; Giese, T.; Merle, U. Dysregulated Host Response in Severe Acute Respiratory Syndrome Coronavirus 2-Induced Critical Illness. Open Forum Infect. Dis. 2021, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Orlov, M.; Wander, P.L.; Morrell, E.D.; Mikacenic, C.; Wurfel, M.M. A Case for Targeting Th17 Cells and IL-17A in SARS-CoV-2 Infections. J. Immunol. 2020, 205, 892–898. [Google Scholar] [CrossRef]
- Aghbash, P.S.; Hemmat, N.; Nahand, J.S.; Shamekh, A.; Memar, M.Y.; Babaei, A.; Baghi, H.B. The role of Th17 cells in viral infections. Int. Immunopharmacol. 2021, 91. [Google Scholar] [CrossRef]
- Hu, B.; Huang, S.; Yin, L. The cytokine storm and COVID-19. J. Med. Virol. 2021, 93, 250–256. [Google Scholar] [CrossRef]
- Crotty, S. Raging evolution of a B cell response to a viral infection. Nat. Rev. Immunol. 2018, 18, 79. [Google Scholar] [CrossRef]
- Wang, X.; Chan, C.C.S.; Yang, M.; Deng, J.; Poon, V.K.M.; Leung, V.H.C.; Ko, K.H.; Zhou, J.; Yung Yuen, K.; Zheng, B.J.; et al. A critical role of IL-17 in modulating the B-cell response during H5N1 influenza virus infection. Cell. Mol. Immunol. 2011, 8, 462–468. [Google Scholar] [CrossRef]
- Wang, X.; Ma, K.; Chen, M.; Ko, K.H.; Zheng, B.J.; Lu, L. IL-17A Promotes Pulmonary B-1a Cell Differentiation via Induction of Blimp-1 Expression during Influenza Virus Infection. PLoS Pathog. 2016, 12, 1–19. [Google Scholar] [CrossRef]
- Mckinstry, K.K.; Strutt, T.M.; Buck, A.; Curtis, J.D.; John, P.; Huston, G.; Tighe, M.; Hamada, H.; Sell, S.; Richard, W.; et al. IL-10 deficiency unleashes an influenza-specific Th17 response and enhances survival against high dose challenge. J. Immunol. 2009, 182, 7353–7363. [Google Scholar] [CrossRef]
- Harpur, C.M.; Kato, Y.; Dewi, S.T.; Stankovic, S.; Johnson, D.N.; Bedoui, S.; Whitney, P.G.; Lahoud, M.H.; Caminschi, I.; Heath, W.R.; et al. Classical Type 1 Dendritic Cells Dominate Priming of Th1 Responses to Herpes Simplex Virus Type 1 Skin Infection. J. Immunol. 2019, 202, 653–663. [Google Scholar] [CrossRef]
- Anipindi, V.C.; Bagri, P.; Roth, K.; Dizzell, S.E.; Nguyen, P.V.; Shaler, C.R.; Chu, D.K.; Jiménez-Saiz, R.; Liang, H.; Swift, S.; et al. Estradiol Enhances CD4+ T-Cell Anti-Viral Immunity by Priming Vaginal DCs to Induce Th17 Responses via an IL-1-Dependent Pathway. PLoS Pathog. 2016, 12, 1–27. [Google Scholar] [CrossRef]
- Bagri, P.; Anipindi, V.C.; Nguyen, P.V.; Vitali, D.; Stämpfli, M.R.; Kaushic, C. Novel Role for Interleukin-17 in Enhancing Type 1 Helper T Cell Immunity in the Female Genital Tract following Mucosal Herpes Simplex Virus 2 Vaccination. J. Virol. 2017, 91, 1–18. [Google Scholar] [CrossRef]
- Mei, X.X.; Lei, S.S.; Xu, L.; Wu, S.; Gu, H.P.; Du, Y.; Zhao, T.; Xie, G.Q.; Fan, Y.S.; Pan, X.P.; et al. Herpes simplex virus type I–infected disorders alter the balance between Treg and Th17 cells in recurrent herpes labialis patients. Int. J. Immunopathol. Pharmacol. 2020, 34, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Acharya, D.; Wang, P.; Paul, A.M.; Dai, J.; Gate, D.; Lowery, J.E.; Stokic, D.S.; Leis, A.A.; Flavell, R.A.; Town, T.; et al. Interleukin-17A Promotes CD8+ T Cell Cytotoxicity To Facilitate West Nile Virus Clearance. J. Virol. 2017, 91, 1–22. [Google Scholar] [CrossRef]
- Jie, Z.; Liang, Y.; Hou, L.; Dong, C.; Iwakura, Y.; Soong, L.; Cong, Y.; Sun, J. Intrahepatic Innate Lymphoid Cells Secrete IL-17A and IL-17F That Are Crucial for T Cell Priming in Viral Infection. J. Immunol. 2014, 192, 3289–3300. [Google Scholar] [CrossRef]
- Hoarau, J.-J.; Jaffar Bandjee, M.-C.; Krejbich Trotot, P.; Das, T.; Li-Pat-Yuen, G.; Dassa, B.; Denizot, M.; Guichard, E.; Ribera, A.; Henni, T.; et al. Persistent Chronic Inflammation and Infection by Chikungunya Arthritogenic Alphavirus in Spite of a Robust Host Immune Response. J. Immunol. 2010 2010, 184, 5914–5927. [Google Scholar] [CrossRef]
- Phuklia, W.; Kasisith, J.; Modhiran, N.; Rodpai, E.; Thannagith, M.; Thongsakulprasert, T.; Smith, D.R.; Ubol, S. Osteoclastogenesis induced by CHIKV-infected fibroblast-like synoviocytes: A possible interplay between synoviocytes and monocytes/macrophages in CHIKV-induced arthralgia/arthritis. Virus Res. 2013, 177, 179–188. [Google Scholar] [CrossRef]
- Noret, M.; Herrero, L.; Rulli, N.; Rolph, M.; Smith, P.N.; Li, R.W.; Roques, P.; Gras, G.; Mahalingam, S. Interleukin 6, RANKL, and osteoprotegerin expression by chikungunya virus-infected human osteoblasts. J. Infect. Dis. 2012, 206, 455–457. [Google Scholar] [CrossRef] [PubMed]
- Teo, T.-H.; Lum, F.-M.; Claser, C.; Lulla, V.; Lulla, A.; Merits, A.; Rénia, L.; Ng, L.F.P. A Pathogenic Role for CD4 + T Cells during Chikungunya Virus Infection in Mice. J. Immunol. 2013, 190, 259–269. [Google Scholar] [CrossRef]
- Chow, A.; Her, Z.; Ong, E.K.S.; Chen, J.M.; Dimatatac, F.; Kwek, D.J.C.; Barkham, T.; Yang, H.; Rénia, L.; Leo, Y.S.; et al. Persistent arthralgia induced by Chikungunya virus infection is associated with interleukin-6 and granulocyte macrophage colony-stimulating factor. J. Infect. Dis. 2011, 203, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Ng, L.F.P.; Chow, A.; Sun, Y.J.; Kwek, D.J.C.; Lim, P.L.; Dimatatac, F.; Ng, L.C.; Ooi, E.E.; Chao, K.H.; Her, Z.; et al. IL-1β, IL-6, and RANTES as biomarkers of Chikungunya severity. PLoS ONE 2009, 4, e4261. [Google Scholar] [CrossRef] [PubMed]
- Van Bezooijen, R.L.; Papapoulos, S.E.; Löwik, C.W.G.M. Effect of interleukin-17 on nitric oxide production and osteoclastic bone resorption: Is there dependency on nuclear factor-κB and receptor activator of nuclear factor κB (RANK)/RANK ligand signaling? Bone 2001, 28, 378–386. [Google Scholar] [CrossRef]
- Chabaud, M.; Garnero, P.; Dayer, J.M.; Guerne, P.A.; Fossiez, F.; Miossec, P. Contribution of interleukin 17 to synovium matrix destruction in rheumatoid arthritis. Cytokine 2000, 12, 1092–1099. [Google Scholar] [CrossRef]
- Robert, M.; Miossec, P. IL-17 in rheumatoid arthritis and precision medicine: From synovitis expression to circulating bioactive levels. Front. Med. 2019, 6, 1–10. [Google Scholar] [CrossRef]
- Chabaud, M.; Lubberts, E.; Joosten, L.; Van Den Berg, W.; Miossec, P. IL-17 derived from juxta-articular bone and synovium contributes to joint degradation in rheumatoid arthritis. Arthritis Res. 2001, 3, 168–177. [Google Scholar] [CrossRef]
- Neumann, E.; Lefèvre, S.; Zimmermann, B.; Gay, S.; Müller-Ladner, U. Rheumatoid arthritis progression mediated by activated synovial fibroblasts. Trends Mol. Med. 2010, 16, 458–468. [Google Scholar] [CrossRef]
- Jain, A.; Pandey, N.; Garg, R.K.; Kumar, R. IL-17 level in patients with dengue virus infection & its association with severity of illness. J. Clin. Immunol. 2013, 33, 613–618. [Google Scholar] [CrossRef]
- Malavige, G.N.; Huang, L.C.; Salimi, M.; Gomes, L.; Jayaratne, S.D.; Ogg, G.S. Cellular and Cytokine Correlates of Severe Dengue Infection. PLoS ONE 2012, 7, e50387. [Google Scholar] [CrossRef]
- Guabiraba, R.; Besnard, A.G.; Marques, R.E.; Maillet, I.; Fagundes, C.T.; Conceição, T.M.; Rust, N.M.; Charreau, S.; Paris, I.; Lecron, J.C.; et al. IL-22 modulates IL-17A production and controls inflammation and tissue damage in experimental dengue infection. Eur. J. Immunol. 2013, 43, 1529–1544. [Google Scholar] [CrossRef]
- Guabiraba, R.; Ryffel, B. Dengue virus infection: Current concepts in immune mechanisms and lessons from murine models. Immunology 2014, 141, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Vargas, L.A.; Hernández-Flores, K.G.; Thomas-Dupont, P.; Izaguirre-Hernández, I.Y.; Sánchez-Marce, E.E.; Remes-Ruiz, R.; Fonseca-Coronado, S.; Hernández-Romano, P.A.; Flores-Collins, M.E.; Vivanco-Cid, H. Characterization of the IL-17 and CD4+ Th17 Cells in the Clinical Course of Dengue Virus Infections. Viruses 2020, 12, 1435. [Google Scholar] [CrossRef] [PubMed]
- Tappe, D.; Pérez-Girón, J.V.; Zammarchi, L.; Rissland, J.; Ferreira, D.F.; Jaenisch, T.; Gómez-Medina, S.; Günther, S.; Bartoloni, A.; Muñoz-Fontela, C.; et al. Cytokine kinetics of Zika virus-infected patients from acute to reconvalescent phase. Med. Microbiol. Immunol. 2016, 205, 269–273. [Google Scholar] [CrossRef]
- Fares-Gusmao, R.; Rocha, B.C.; Sippert, E.; Lanteri, M.C.; Áñez, G.; Rios, M. Differential Pattern of Soluble Immune Markers in Asymptomatic Dengue, West Nile and Zika Virus Infections. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef]
- Naveca, F.G.; Pontes, G.S.; Chang, A.Y.H.; da Silva, G.A.V.; do Nascimento, V.A.; Monteiro, D.C.d.S.; da Silva, M.S.; Abdalla, L.F.; Santos, J.H.A.; de Almeida, T.A.P.; et al. Analysis of the immunological biomarker profile during acute zika virus infection reveals the overexpression of CXCL10, a chemokine linked to neuronal damage. Mem. Inst. Oswaldo Cruz 2018, 113, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, R.S.S.; De Sousa, J.R.; Araujo, M.T.F.; Martins Filho, A.J.; De Alcantara, B.N.; Araujo, F.M.C.; Queiroz, M.G.L.; Cruz, A.C.R.; Vasconcelos, B.H.B.; Chiang, J.O.; et al. In situ immune response and mechanisms of cell damage in central nervous system of fatal cases microcephaly by Zika virus. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef]
- Whitton, J.L.; Feuer, R. Myocarditis, microbes and autoimmunity. Autoimmunity 2004, 37, 375–386. [Google Scholar] [CrossRef]
- Yajima, T.; Knowlton, K.U. Viral myocarditis from the perspective of the virus. Circulation 2009, 119, 2615–2624. [Google Scholar] [CrossRef]
- Martinez, N.E.; Sato, F.; Kawai, E.; Omura, S.; Chervenak, R.P.; Tsunoda, I. Regulatory T cells and Th17 cells in viral infections: Implications for multiple sclerosis and myocarditis. Future Virol. 2012, 7, 593–608. [Google Scholar] [CrossRef]
- Mason, J.W.; O’Connell, J.B.; Herskowitz, A.; Rose, N.R.; McManus, B.M.; Billingham, M.E.; Moon, T.E. A Clinical Trial of Immunosuppressive Therapy for Myocarditis. N. Engl. J. Med. 1995, 333, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Cao, A.L.; Yu, M.; Lin, Q.W.; Yu, X.; Zhang, J.H.; Wang, M.; Guo, H.P.; Liao, Y.H. Th17 cells facilitate the humoral immune response in patients with acute viral myocarditis. J. Clin. Immunol. 2010, 30, 226–234. [Google Scholar] [CrossRef]
- Yuan, J.; Yu, M.; Lin, Q.-W.; Cao, A.-L.; Yu, X.; Dong, J.-H.; Wang, J.-P.; Zhang, J.-H.; Wang, M.; Guo, H.-P.; et al. Th17 Cells Contribute to Viral Replication in Coxsackievirus B3-Induced Acute Viral Myocarditis. J. Immunol. 2010, 185, 4004–4010. [Google Scholar] [CrossRef]
- Yuan, J.; Yu, M.; Lin, Q.W.; Cao, A.L.; Yu, X.; Dong, J.H.; Wang, J.P.; Zhang, J.H.; Wang, M.; Guo, H.P.; et al. Neutralization of IL-17 inhibits the production of anti-ANT autoantibodies in CVB3-induced acute viral myocarditis. Int. Immunopharmacol. 2010, 10, 272–276. [Google Scholar] [CrossRef]
- Wei, B.; Deng, Y.; Huang, Y.; Gao, X.; Wu, W. IL-10-producing B cells attenuate cardiac inflammation by regulating Th1 and Th17 cells in acute viral myocarditis induced by coxsackie virus B3. Life Sci. 2019, 235, 116838. [Google Scholar] [CrossRef]
- Chen, R.; Cao, Y.; Tian, Y.; Gu, Y.; Lu, H.; Zhang, S.; Xu, H.; Su, Z. PGE2 ameliorated viral myocarditis development and promoted IL-10-producing regulatory B cell expansion via MAPKs/AKT-AP1 axis or AhR signaling. Cell. Immunol. 2020, 347, 104025. [Google Scholar] [CrossRef]
- Huang, Y.; Li, Y.; Wei, B.; Wu, W.; Gao, X. CD80 Regulates Th17 Cell Differentiation in Coxsackie Virus B3-Induced Acute Myocarditis. Inflammation 2018, 41, 232–239. [Google Scholar] [CrossRef]
- De-Pu, Z.; Li-Sha, G.; Guang-Yi, C.; Xiaohong, G.; Chao, X.; Cheng, Z.; Wen-Wu, Z.; Jia, L.; Jia-Feng, L.; Maoping, C.; et al. The cholinergic anti-inflammatory pathway ameliorates acute viral myocarditis in mice by regulating cd4 + t cell differentiation. Virulence 2018, 9, 1364–1376. [Google Scholar] [CrossRef]
- Dendrou, C.A.; Fugger, L.; Friese, M.A. Immunopathology of multiple sclerosis. Nat. Rev. Immunol. 2015, 15, 545–558. [Google Scholar] [CrossRef]
- Ontaneda, D.; Fox, R.J.; Chataway, J. Clinical trials in progressive multiple sclerosis: Lessons learned and future perspectives. Lancet Neurol. 2015, 14, 208–223. [Google Scholar] [CrossRef]
- Geginat, J.; Paroni, M.; Pagani, M.; Galimberti, D.; De Francesco, R.; Scarpini, E.; Abrignani, S. The Enigmatic Role of Viruses in Multiple Sclerosis: Molecular Mimicry or Disturbed Immune Surveillance? Trends Immunol. 2017, 38, 498–512. [Google Scholar] [CrossRef] [PubMed]
- Hafler, D.A. Multiple sclerosis. J. Clin. Investig. 2004, 113, 788–794. [Google Scholar] [CrossRef] [PubMed]
- Tselis, A. Evidence for viral etiology of multiple sclerosis. Semin. Neurol. 2011, 31, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Jarius, S.; Eichhorn, P.; Jacobi, C.; Wildemann, B.; Wick, M.; Voltz, R. The intrathecal, polyspecific antiviral immune response: Specific for MS or a general marker of CNS autoimmunity? J. Neurol. Sci. 2009, 280, 98–100. [Google Scholar] [CrossRef]
- Tarlinton, R.E.; Martynova, E.; Rizvanov, A.A.; Khaiboullina, S.; Verma, S. Role of Viruses in the Pathogenesis of Multiple Sclerosis. Viruses 2020, 12, 643. [Google Scholar] [CrossRef]
- Kyewski, B.; Klein, L. A central role for central tolerance. Annu. Rev. Immunol. 2006, 24, 571–606. [Google Scholar] [CrossRef]
- Wucherpfennig, K.W.; Strominger, J.L. Molecular mimicry in T cell-mediated autoimmunity: Viral peptides activate human T cell clones specific for myelin basic protein. Cell 1995, 80, 695–705. [Google Scholar] [CrossRef]
- Lünemann, J.D.; Jelčić, I.; Roberts, S.; Lutterotti, A.; Tackenberg, B.; Martin, R.; Münz, C. EBNA1-specific T cells from patients with multiple sclerosis cross react with myelin antigens and co-produce IFN-γ and IL-2. J. Exp. Med. 2008, 205, 1763–1773. [Google Scholar] [CrossRef]
- Fletcher, J.M.; Lalor, S.J.; Sweeney, C.M.; Tubridy, N.; Mills, K.H.G. T cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Clin. Exp. Immunol. 2010, 162, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Dal Canto, M.C.; Kim, B.S.; Miller, S.D.; Melvold, R.W. Theiler’s murine encephalomyelitis virus (TMEV)-induced demyelination: A model for human multiple sclerosis. Methods A Companion Methods Enzymol. 1996, 10, 453–461. [Google Scholar] [CrossRef]
- Hou, W.; Kang, H.S.; Kim, B.S. Th17 cells enhance viral persistence and inhibit T cell cytotoxicity in a model of chronic virus infection. J. Exp. Med. 2009, 206, 313–328. [Google Scholar] [CrossRef] [PubMed]
- Balashov, K.E.; Rottman, J.B.; Weiner, H.L.; Hancock, W.W. CCR5+ and CXCR3+ T cells are increased in multiple sclerosis and their ligands MIP-1α and IP-10 are expressed in demyelinating brain lesions. Proc. Natl. Acad. Sci. USA 1999, 96, 6873–6878. [Google Scholar] [CrossRef]
- Noor, S.; Wilson, E.H. Role of C-C chemokine receptor type 7 and its ligands during neuroinflammation. J. Neuroinflammation 2012, 9, 1–9. [Google Scholar] [CrossRef]
- Huppert, J.; Closhen, D.; Croxford, A.; White, R.; Kulig, P.; Pietrowski, E.; Bechmann, I.; Becher, B.; Luhmann, H.J.; Waisman, A.; et al. Cellular mechanisms of IL-17-induced blood-brain barrier disruption. FASEB J. 2010, 24, 1023–1034. [Google Scholar] [CrossRef]
- Minton, K. IL-17A brings new recruits to EAE. Nat. Rev. Immunol. 2020, 20, 137. [Google Scholar] [CrossRef] [PubMed]
- Kapil, P.; Atkinson, R.; Ramakrishna, C.; Cua, D.J.; Bergmann, C.C.; Stohlman, S.A. Interleukin-12 (IL-12), but Not IL-23, Deficiency Ameliorates Viral Encephalitis without Affecting Viral Control. J. Virol. 2009, 83, 5978–5986. [Google Scholar] [CrossRef] [PubMed]
- Kebir, H.; Ifergan, I.; Alvarez, J.I.; Bernard, M.; Poirier, J.; Arbour, N.; Duquette, P.; Prat, A. Preferential recruitment of interferon-γ-expressing TH17 cells in multiple sclerosis. Ann. Neurol. 2009, 66, 390–402. [Google Scholar] [CrossRef] [PubMed]
- Codarri, L.; Gyülvészii, G.; Tosevski, V.; Hesske, L.; Fontana, A.; Magnenat, L.; Suter, T.; Becher, B. RORγ3t drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat. Immunol. 2011, 12, 560–567. [Google Scholar] [CrossRef]
- Noster, R.; Riedel, R.; Mashreghi, M.F.; Radbruch, H.; Harms, L.; Haftmann, C.; Chang, H.D.; Radbruch, A.; Zielinski, C.E. IL-17 and GM-CSF expression are antagonistically regulated by human T helper cells. Sci. Transl. Med. 2014, 6. [Google Scholar] [CrossRef]
- Cervantes-Barragán, L.; Firner, S.; Bechmann, I.; Waisman, A.; Lahl, K.; Sparwasser, T.; Thiel, V.; Ludewig, B. Regulatory T Cells Selectively Preserve Immune Privilege of Self-Antigens during Viral Central Nervous System Infection. J. Immunol. 2012, 188, 3678–3685. [Google Scholar] [CrossRef]
- Kleinewietfeld, M.; Hafler, D.A. The plasticity of human Treg and Th17 cells and its role in autoimmunity. Semin. Immunol. 2013, 25, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Paroni, M.; Maltese, V.; De Simone, M.; Ranzani, V.; Larghi, P.; Fenoglio, C.; Pietroboni, A.M.; De Riz, M.A.; Crosti, M.C.; Maglie, S.; et al. Recognition of viral and self-antigens by TH1 and TH1/TH17 central memory cells in patients with multiple sclerosis reveals distinct roles in immune surveillance and relapses. J. Allergy Clin. Immunol. 2017, 140, 797–808. [Google Scholar] [CrossRef] [PubMed]
- Matusevicius, D.; Kivisäkk, P.; He, B.; Kostulas, N.; Özenci, V.; Fredrikson, S.; Link, H. Interleukin-17 mRNA expression in blood and CSF mononuclear cells is augmented in multiple sclerosis. Mult. Scler. 1999, 5, 101–104. [Google Scholar] [CrossRef]
- Hedegaard, C.J.; Krakauer, M.; Bendtzen, K.; Lund, H.; Sellebjerg, F.; Nielsen, C.H. T helper cell type 1 (Th1), Th2 and Th17 responses to myelin basic protein and disease activity in multiple sclerosis. Immunology 2008, 125, 161–169. [Google Scholar] [CrossRef]
- Restorick, S.M.; Durant, L.; Kalra, S.; Hassan-Smith, G.; Rathbone, E.; Douglas, M.R.; Curnow, S.J. CCR6+ Th cells in the cerebrospinal fluid of persons with multiple sclerosis are dominated by pathogenic non-classic Th1 cells and GM-CSF-only-secreting Th cells. Brain. Behav. Immun. 2017, 64, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Van Langelaar, J.; Van Der Vuurst De Vries, R.M.; Janssen, M.; Wierenga-Wolf, A.F.; Spilt, I.M.; Siepman, T.A.; Dankers, W.; Verjans, G.M.G.M.; De Vries, H.E.; Lubberts, E.; et al. T helper 17.1 cells associate with multiple sclerosis disease activity: Perspectives for early intervention. Brain 2018, 141, 1334–1349. [Google Scholar] [CrossRef]
- Jadidi-Niaragh, F.; Mirshafiey, A. Th17 Cell, the new player of neuroinflammatory process in multiple sclerosis. Scand. J. Immunol. 2011, 74, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kebir, H.; Kreymborg, K.; Ifergan, I.; Dodelet-Devillers, A.; Cayrol, R.; Bernard, M.; Giuliani, F.; Arbour, N.; Becher, B.; Prat, A. Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat. Med. 2007, 13, 1173–1175. [Google Scholar] [CrossRef]
- Ghoreschi, K.; Laurence, A.; Yang, X.P.; Hirahara, K.; O’Shea, J.J. T helper 17 cell heterogeneity and pathogenicity in autoimmune disease. Trends Immunol. 2011, 32, 395–401. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus | Disease | Organism | Friend or Foe? | Evidence | Ref. |
|---|---|---|---|---|---|
| SARS-CoV2 | COVID-19 | Human | foe | Cytokine storm; polyfunctional Th1 and Th17 cells underrepresented in the repertoire of T cells reactive to SARS-CoV-2; lung tissue-resident memory-like Th17 cells; high frequency of Th17 cells and IL-17 levels in severe cases | [78,81,82,85] |
| Influenza Virus | Flu | Mice | friend | Role in the recruitment of B cells into the lungs; B1 cells differentiation and IgM production | [90,91] |
| HSV-2 | Herpes | Mice | friend | Enhancement of DCs ability to induce a Th1 response | [94,95] |
| HSV-1 | Herpes (RHL) | Human | foe | Increased Th17/Treg ratio and Th17 related cytokines in RHL patients | [96] |
| WNV | West Nile fever | Human | friend | Less permissiveness of viral invasion in the brain; activation of CD8 T cells | [97] |
| Ad | Hepatitis | Mice | foe | Expansion of IL-17A and IL-17F producing T cells in the liver; absence of IL-17F led to better clinical outcome | [98] |
| CHIKV | Chikungunya fever | Mice Human | foe | High levels of Th17 related cytokines in patients and CHIKV-infected cultures; high IL-17 levels involved in the progression to the chronic phase | [101,103] |
| DENV | Dengue fever | Mice Human | foe | High IL-17 levels in circulation and liver; high frequency of Th17 in DHF and DSS patients | [110,111,112,113,114] |
| ZIKV | Zika fever | Human | foe | High levels Th17-related cytokines in concomitant with viremia peaks; Th17 cytokines in the brain of microcephalic babies | [115,116,117,118] |
| Enteroviruses, adenovirus, parvoviruses B19, EBV, HHV-6, CMV, CVB3 | Viral myocarditis | Human Mice | foe | Increased frequencies of Th17, IL-17 mRNA expression and Th17-related cytokines in AVMC patients and mice; induction of anti-ANT autoantibodies | [123,124,125,126,127,129] |
| EBV, measles, rubella, VZV, TMEV | Multiple sclerosis | Human Mice | foe | IL-17 inhibit activity of cytotoxic T cells; viral persistence; high levels of IL-17A in the CSF in MS patients; presence of Th1Th17 cells in brain lesions; migration of inflammatory cells to the brain through BBB disruption | [143,146,148,149,154,155,156,157,158,159,160] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paiva, I.A.; Badolato-Corrêa, J.; Familiar-Macedo, D.; de-Oliveira-Pinto, L.M. Th17 Cells in Viral Infections—Friend or Foe? Cells 2021, 10, 1159. https://doi.org/10.3390/cells10051159
Paiva IA, Badolato-Corrêa J, Familiar-Macedo D, de-Oliveira-Pinto LM. Th17 Cells in Viral Infections—Friend or Foe? Cells. 2021; 10(5):1159. https://doi.org/10.3390/cells10051159
Chicago/Turabian StylePaiva, Iury Amancio, Jéssica Badolato-Corrêa, Débora Familiar-Macedo, and Luzia Maria de-Oliveira-Pinto. 2021. "Th17 Cells in Viral Infections—Friend or Foe?" Cells 10, no. 5: 1159. https://doi.org/10.3390/cells10051159
APA StylePaiva, I. A., Badolato-Corrêa, J., Familiar-Macedo, D., & de-Oliveira-Pinto, L. M. (2021). Th17 Cells in Viral Infections—Friend or Foe? Cells, 10(5), 1159. https://doi.org/10.3390/cells10051159