
Regional Differences in Heat Shock Protein 25 Expression in Brain and Spinal Cord Astrocytes of Wild-Type and SOD1 G93A Mice

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Breeding and Maintenance of (C57BL/6 × SJL) F1 Hybrid WT and SOD1 G93A Mice
2.2. Genotyping
2.3. Primary Murine Cortical and Spinal Cord Mixed Glial Cultures
2.4. Treatment of Primary Mixed Glial Cultures
2.5. Protein Extraction and Quantification
2.6. Antibodies
2.7. Immunoblotting
2.8. Immunolabelling
2.9. Epifluorescence Microscopy
2.10. Confocal Microscopy
2.11. Flow Cytometry
2.12. Statistics
3. Results
3.1. Characterisation of Cortical and Spinal Cord Mixed Glial Cultures
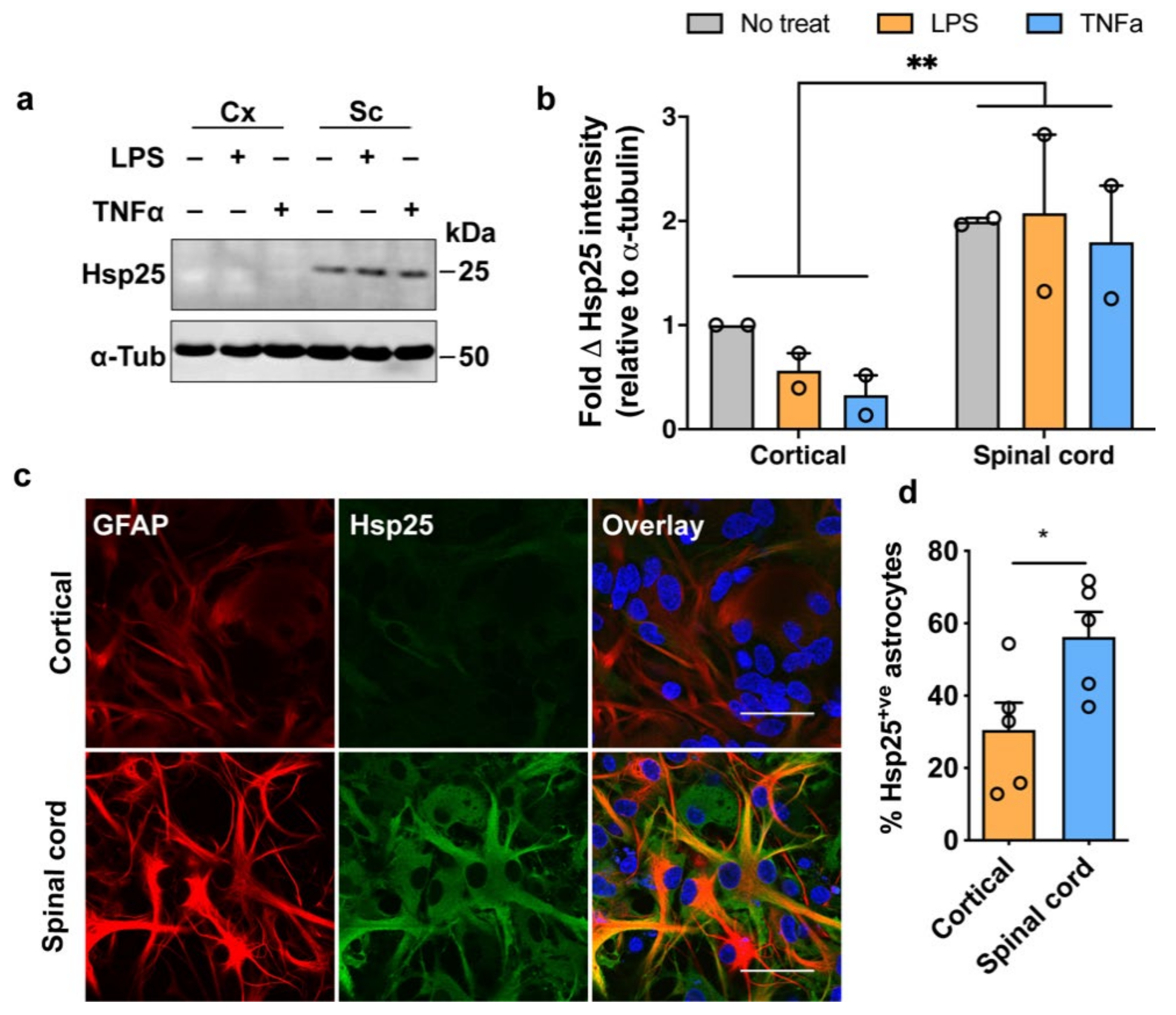
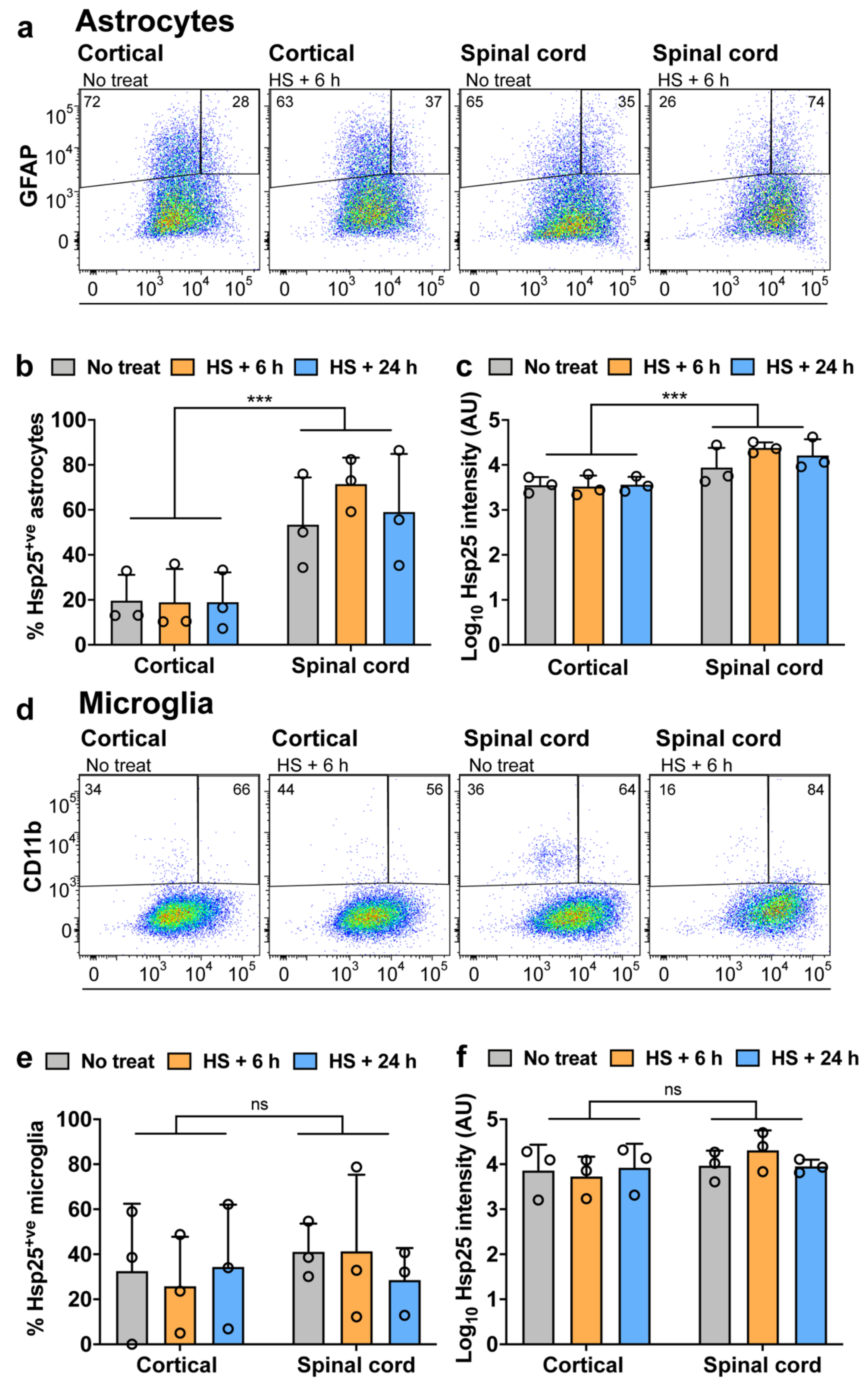
3.2. Expression of Hsp25 in Cortical and Spinal Cord Mixed Glial Cultures
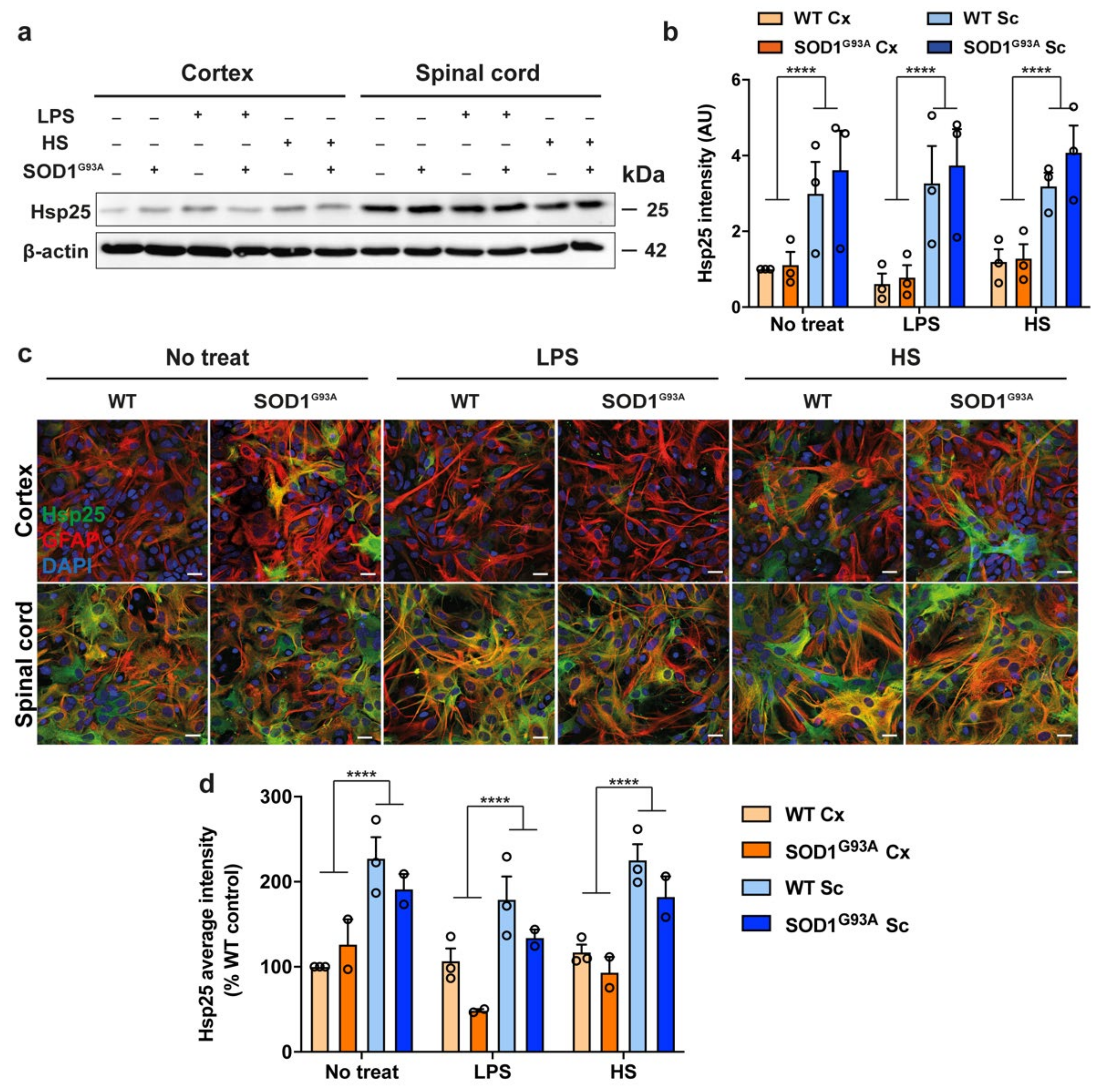
3.3. Hsp25 Expression in Spinal Cord and Cortical Glial Cultures Expressing SOD1 G93A
3.4. Expression of Hsp25 in the Spinal Cord and Brain of Adult Wild-Type and SOD1 G93A Mice
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Allen, N.J.; Lyons, D.A. Glia as architects of central nervous system formation and function. Science 2018, 362, 181–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Haas, A.H.; Boddeke, H.W.G.M.; Biber, K. Region-specific expression of immunoregulatory proteins on microglia in the healthy CNS. Glia 2008, 56, 888–894. [Google Scholar] [CrossRef]
- Farmer, W.T.; Abrahamsson, T.; Chierzi, S.; Lui, C.; Zaelzer, C.; Jones, E.V.; Bally, B.P.; Chen, G.G.; Théroux, J.-F.; Peng, J.; et al. Neurons diversify astrocytes in the adult brain through sonic hedgehog signaling. Science 2016, 351, 849–854. [Google Scholar] [CrossRef]
- Grabert, K.; Michoel, T.; Karavolos, M.H.; Clohisey, S.; Kenneth Baillie, J.; Stevens, M.P.; Freeman, T.C.; Summers, K.M.; McColl, B.W. Microglial brain region—Dependent diversity and selective regional sensitivities to aging. Nat. Neurosci. 2016, 19, 504–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben Haim, L.; Rowitch, D.H. Functional diversity of astrocytes in neural circuit regulation. Nat. Rev. Neurosci. 2017, 18, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Clarke, B.E.; Taha, D.M.; Tyzack, G.E.; Patani, R. Regionally encoded functional heterogeneity of astrocytes in health and disease: A perspective. Glia 2021, 69, 20–27. [Google Scholar] [CrossRef]
- Clement, A.M.; Nguyen, M.D.; Roberts, E.A.; Garcia, M.L.; Boillée, S.; Rule, M.; McMahon, A.P.; Doucette, W.; Siwek, D.; Ferrante, R.J.; et al. Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science 2003, 302, 113–117. [Google Scholar] [CrossRef] [Green Version]
- Boillée, S.; Yamanaka, K.; Lobsiger, C.S.; Copeland, N.G.; Jenkins, N.A.; Kassiotis, G.; Kollias, G.; Cleveland, D.W. Onset and progression in inherited ALS determined by motor neurons and microglia. Science 2006, 312, 1389–1392. [Google Scholar] [CrossRef] [Green Version]
- Yamanaka, K.; Chun, S.J.; Boillee, S.; Fujimori-Tonou, N.; Yamashita, H.; Gutmann, D.H.; Takahashi, R.; Misawa, H.; Cleveland, D.W. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat. Neurosci. 2008, 11, 251–253. [Google Scholar] [CrossRef] [Green Version]
- Ferraiuolo, L.; Higginbottom, A.; Heath, P.R.; Barber, S.; Greenald, D.; Kirby, J.; Shaw, P.J. Dysregulation of astrocyte-motoneuron cross-talk in mutant superoxide dismutase 1-related amyotrophic lateral sclerosis. Brain 2011, 134, 2627–2641. [Google Scholar] [CrossRef] [Green Version]
- Kelley, K.W.; Ben Haim, L.; Schirmer, L.; Tyzack, G.E.; Tolman, M.; Miller, J.G.; Tsai, H.-H.; Chang, S.M.; Molofsky, A.V.; Yang, Y.; et al. Kir4.1-Dependent Astrocyte-Fast Motor Neuron Interactions Are Required for Peak Strength. Neuron 2018, 98, 306–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haidet-Phillips, A.M.; Hester, M.E.; Miranda, C.J.; Meyer, K.; Braun, L.; Frakes, A.; Song, S.; Likhite, S.; Murtha, M.J.; Foust, K.D.; et al. Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat. Biotechnol. 2011, 29, 824–828. [Google Scholar] [CrossRef] [Green Version]
- Meyer, K.; Ferraiuolo, L.; Miranda, C.J.; Likhite, S.; McElroy, S.; Renusch, S.; Ditsworth, D.; Lagier-Tourenne, C.; Smith, R.A.; Ravits, J.; et al. Direct conversion of patient fibroblasts demonstrates non-cell autonomous toxicity of astrocytes to motor neurons in familial and sporadic ALS. Proc. Natl. Acad. Sci. USA 2014, 111, 829–832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Clarke, B.E.; Gil, R.S.; Yip, J.; Kalmar, B.; Greensmith, L. Regional differences in the inflammatory and heat shock response in glia: Implications for ALS. Cell Stress Chaperones 2019, 24, 857–870. [Google Scholar] [CrossRef] [Green Version]
- Guttenplan, K.A.; Weigel, M.K.; Adler, D.I.; Couthouis, J.; Liddelow, S.A.; Gitler, A.D.; Barres, B.A. Knockout of reactive astrocyte activating factors slows disease progression in an ALS mouse model. Nat. Commun. 2020, 11, 3753. [Google Scholar] [CrossRef] [PubMed]
- Feinstein, D.L.; Galea, E.; Aquino, D.A.; Li, G.C.; Xu, H.; Reis, D.J. Heat shock protein 70 suppresses astroglial-inducible nitric-oxide synthase expression by decreasing NFκB activation. J. Biol. Chem. 1996, 271, 17724–17732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kieran, D.; Kalmar, B.; Dick, J.R.T.; Riddoch-Contreras, J.; Burnstock, G.; Greensmith, L. Treatment with arimoclomol, a coinducer of heat shock proteins, delays disease progression in ALS mice. Nat. Med. 2004, 10, 402–405. [Google Scholar] [CrossRef]
- Kalmar, B.; Novoselov, S.; Gray, A.; Cheetham, M.E.; Margulis, B.; Greensmith, L. Late stage treatment with arimoclomol delays disease progression and prevents protein aggregation in the SOD1 mouse model of ALS: Arimoclomol is effective in late stage SODG93A mice. J. Neurochem. 2008, 107, 339–350. [Google Scholar] [CrossRef]
- Benatar, M.; Wuu, J.; Andersen, P.M.; Atassi, N.; David, W.; Cudkowicz, M.; Schoenfeld, D. Randomized, double-blind, placebo-controlled trial of arimoclomol in rapidly progressive SOD1 ALS. Neurology 2018, 90, e565–e574. [Google Scholar] [CrossRef] [Green Version]
- Carra, S.; Alberti, S.; Arrigo, P.A.; Benesch, J.L.; Benjamin, I.J.; Boelens, W.; Bartelt-Kirbach, B.; Brundel, B.J.J.M.; Buchner, J.; Bukau, B.; et al. The growing world of small heat shock proteins: From structure to functions. Cell Stress Chaperones 2017, 22, 601–611. [Google Scholar] [CrossRef] [PubMed]
- Alderson, T.R.; Roche, J.; Gastall, H.Y.; Dias, D.M.; Pritišanac, I.; Ying, J.; Bax, A.; Benesch, J.L.P.; Baldwin, A.J. Local unfolding of the HSP27 monomer regulates chaperone activity. Nat. Commun. 2019, 10, 1068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, K.L.; Rahimtula, M.; Mearow, K.M. Hsp27 and axonal growth in adult sensory neurons in vitro. BMC Neurosci. 2005, 6, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casado, P.; Zuazua-Villar, P.; Prado, M.A.; Valle, E.D.; Iglesias, J.M.; Martínez-Campa, C.; Lazo, P.S.; Ramos, S. Characterization of HSP27 phosphorylation induced by microtubule interfering agents: Implication of p38 signalling pathway. Arch. Biochem. Biophys. 2007, 461, 123–129. [Google Scholar] [CrossRef]
- Williams, K.L.; Mearow, K.M. Phosphorylation status of heat shock protein 27 influences neurite growth in adult dorsal root ganglion sensory neurons in vitro. J. Neurosci. Res. 2011, 89, 1160–1172. [Google Scholar] [CrossRef]
- Garrido, C.; Bruey, J.M.; Fromentin, A.; Hammann, A.; Arrigo, A.P.; Solary, E. HSP27 inhibits cytochrome c-dependent activation of procaspase-9. FASEB J. 1999, 13, 2061–2070. [Google Scholar] [CrossRef]
- Charette, S.J.; Lavoie, J.N.; Lambert, H.; Landry, J. Inhibition of Daxx-mediated apoptosis by heat shock protein 27. Mol. Cell. Biol. 2000, 20, 7602–7612. [Google Scholar] [CrossRef] [Green Version]
- Benn, S.C.; Perrelet, D.; Kato, A.C.; Scholz, J.; Decosterd, I.; Mannion, R.J.; Bakowska, J.C.; Woolf, C.J. Hsp27 upregulation and phosphorylation is required for injured sensory and motor neuron survival. Neuron 2002, 36, 45–56. [Google Scholar] [CrossRef] [Green Version]
- Beere, H.M. ‘The stress of dying’: The role of heat shock proteins in the regulation of apoptosis. J. Cell Sci. 2004, 117, 2641–2651. [Google Scholar] [CrossRef] [Green Version]
- Havasi, A.; Li, Z.; Wang, Z.; Martin, J.L.; Botla, V.; Ruchalski, K.; Schwartz, J.H.; Borkan, S.C. Hsp27 inhibits Bax activation and apoptosis via a phosphatidylinositol 3-kinase-dependent mechanism. J. Biol. Chem. 2008, 283, 12305–12313. [Google Scholar] [CrossRef] [Green Version]
- Park, K.-J.; Gaynor, R.B.; Kwak, Y.T. Heat shock protein 27 association with the IκB kinase complex regulates tumor necrosis factor α-induced NF-κB activation. J. Biol. Chem. 2003, 278, 35272–35278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plumier, J.C.; Hopkins, D.A.; Robertson, H.A.; Currie, R.W. Constitutive expression of the 27-kDa heat shock protein (Hsp27) in sensory and motor neurons of the rat nervous system. J. Comp. Neurol. 1997, 384, 409–428. [Google Scholar] [CrossRef]
- Kalmár, B.; Burnstock, G.; Vrbová, G.; Greensmith, L. The effect of neonatal nerve injury on the expression of heat shock proteins in developing rat motoneurones. J. Neurotrauma 2002, 19, 667–679. [Google Scholar] [CrossRef] [PubMed]
- Treweek, T.M.; Meehan, S.; Ecroyd, H.; Carver, J.A. Small heat-shock proteins: Important players in regulating cellular proteostasis. Cell. Mol. Life Sci. 2015, 72, 429–451. [Google Scholar] [CrossRef] [PubMed]
- Cox, D.; Whiten, D.R.; Brown, J.W.P.; Horrocks, M.H.; Gil, R.S.; Dobson, C.M.; Klenerman, D.; van Oijen, A.M.; Ecroyd, H. The small heat shock protein Hsp27 binds α-synuclein fibrils, preventing elongation and cytotoxicity. J. Biol. Chem. 2018, 293, 4486–4497. [Google Scholar] [CrossRef] [Green Version]
- Yerbury, J.J.; Gower, D.; Vanags, L.; Roberts, K.; Lee, J.A.; Ecroyd, H. The small heat shock proteins αB-crystallin and Hsp27 suppress SOD1 aggregation in vitro. Cell Stress Chaperones 2013, 18, 251–257. [Google Scholar] [CrossRef] [Green Version]
- Cox, D.; Selig, E.; Griffin, M.D.W.; Carver, J.A.; Ecroyd, H. Small Heat-shock Proteins Prevent α-Synuclein Aggregation via Transient Interactions and Their Efficacy Is Affected by the Rate of Aggregation. J. Biol. Chem. 2016, 291, 22618–22629. [Google Scholar] [CrossRef] [Green Version]
- Kojima, K.; Musch, M.W.; Ropeleski, M.J.; Boone, D.L.; Ma, A.; Chang, E.B. Escherichia coli LPS induces heat shock protein 25 in intestinal epithelial cells through MAP kinase activation. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 286, G645–G652. [Google Scholar] [CrossRef] [Green Version]
- Harada, K.; Ohira, S.; Isse, K.; Ozaki, S.; Zen, Y.; Sato, Y.; Nakanuma, Y. Lipopolysaccharide activates nuclear factor kappaB through toll-like receptors and related molecules in cultured biliary epithelial cells. Lab. Invest. 2003, 83, 1657–1667. [Google Scholar] [CrossRef] [Green Version]
- Elliott, J.L. Cytokine upregulation in a murine model of familial amyotrophic lateral sclerosis. Brain Res. Mol. Brain Res. 2001, 95, 172–178. [Google Scholar] [CrossRef]
- Hensley, K.; Fedynyshyn, J.; Ferrell, S.; Floyd, R.A.; Gordon, B.; Grammas, P.; Hamdheydari, L.; Mhatre, M.; Mou, S.; Pye, Q.N.; et al. Message and protein-level elevation of tumor necrosis factor α (TNFα) and TNFα-modulating cytokines in spinal cords of the G93A-SOD1 mouse model for amyotrophic lateral sclerosis. Neurobiol. Dis. 2003, 14, 74–80. [Google Scholar] [CrossRef]
- Roberts, K.; Zeineddine, R.; Corcoran, L.; Li, W.; Campbell, I.L.; Yerbury, J.J. Extracellular aggregated Cu/Zn superoxide dismutase activates microglia to give a cytotoxic phenotype. Glia 2013, 61, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Duan, W.; Liu, Y.; Liu, Y.; Liu, C.; Li, Y.; Wen, D.; Li, Z.; Li, C. IGF1 affects macrophage invasion and activation and TNF-α production in the sciatic nerves of female SOD1G93A mice. Neurosci. Lett. 2018, 668, 1–6. [Google Scholar] [CrossRef]
- Satoh, J.I.; Kim, S.U. Differential expression of heat shock protein HSP27 in human neurons and glial cells in culture. J. Neurosci. Res. 1995, 41, 805–818. [Google Scholar] [CrossRef] [PubMed]
- Bruijn, L.I.; Becher, M.W.; Lee, M.K.; Anderson, K.L.; Jenkins, N.A.; Copeland, N.G.; Sisodia, S.S.; Rothstein, J.D.; Borchelt, D.R.; Price, D.L.; et al. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron 1997, 18, 327–338. [Google Scholar] [CrossRef] [Green Version]
- Hall, E.D.; Oostveen, J.A.; Gurney, M.E. Relationship of microglial and astrocytic activation to disease onset and progression in a transgenic model of familial ALS. Glia 1998, 23, 249–256. [Google Scholar] [CrossRef]
- Levine, J.B.; Kong, J.; Nadler, M.; Xu, Z. Astrocytes interact intimately with degenerating motor neurons in mouse amyotrophic lateral sclerosis (ALS). Glia 1999, 28, 215–224. [Google Scholar] [CrossRef]
- Ince, P.G.; Lowe, J.; Shaw, P.J. Amyotrophic lateral sclerosis: Current issues in classification, pathogenesis and molecular pathology. Neuropathol. Appl. Neurobiol. 1998, 24, 104–117. [Google Scholar] [CrossRef]
- Barbeito, L.H.; Pehar, M.; Cassina, P.; Vargas, M.R.; Peluffo, H.; Viera, L.; Estévez, A.G.; Beckman, J.S. A role for astrocytes in motor neuron loss in amyotrophic lateral sclerosis. Brain Res. Brain Res. Rev. 2004, 47, 263–274. [Google Scholar] [CrossRef]
- Morimoto, R.I. Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev. 2008, 22, 1427–1438. [Google Scholar] [CrossRef] [Green Version]
- Ruegsegger, C.; Saxena, S. Proteostasis impairment in ALS. Brain Res. 2016, 1648, 571–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahheydari, H.; Ragagnin, A.; Walker, A.K.; Toth, R.P.; Vidal, M.; Jagaraj, C.J.; Perri, E.R.; Konopka, A.; Sultana, J.M.; Atkin, J.D. Protein Quality Control and the Amyotrophic Lateral Sclerosis/Frontotemporal Dementia Continuum. Front. Mol. Neurosci. 2017, 10, 119. [Google Scholar] [CrossRef] [PubMed]
- San Gil, R.; Ooi, L.; Yerbury, J.J.; Ecroyd, H. The heat shock response in neurons and astroglia and its role in neurodegenerative diseases. Mol. Neurodegener. 2017, 12, 65. [Google Scholar] [CrossRef]
- Gorter, R.P.; Nutma, E.; Jahrei, M.-C.; de Jonge, J.C.; Quinlan, R.A.; van der Valk, P.; van Noort, J.M.; Baron, W.; Amor, S. Heat shock proteins are differentially expressed in brain and spinal cord: Implications for multiple sclerosis. Clin. Exp. Immunol. 2018, 194, 137–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calabrese, V.; Bates, T.E.; Stella, A.M. NO synthase and NO-dependent signal pathways in brain aging and neurodegenerative disorders: The role of oxidant/antioxidant balance. Neurochem. Res. 2000, 25, 1315–1341. [Google Scholar] [CrossRef] [PubMed]
- Bajramović, J.J.; Bsibsi, M.; Geutskens, S.B.; Hassankhan, R.; Verhulst, K.C.; Stege, G.J.; de Groot, C.J.; van Noort, J.M. Differential expression of stress proteins in human adult astrocytes in response to cytokines. J. Neuroimmunol. 2000, 106, 14–22. [Google Scholar] [CrossRef]
- Krueger-Naug, A.M.R.; Hopkins, D.A.; Armstrong, J.N.; Plumier, J.-C.L.; William Currie, R. Hyperthermic induction of the 27-kDa heat shock protein (Hsp27) in neuroglia and neurons of the rat central nervous system. J. Comp. Neurol. 2000, 428, 495–510. [Google Scholar] [CrossRef]
- Bechtold, D.A.; Brown, I.R. Induction of Hsp27 and Hsp32 stress proteins and vimentin in glial cells of the rat hippocampus following hyperthermia. Neurochem. Res. 2003, 28, 1163–1173. [Google Scholar] [CrossRef]
- Acarin, L.; Paris, J.; González, B.; Castellano, B. Glial expression of small heat shock proteins following an excitotoxic lesion in the immature rat brain. Glia 2002, 38, 1–14. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
San Gil, R.; Clarke, B.E.; Ecroyd, H.; Kalmar, B.; Greensmith, L. Regional Differences in Heat Shock Protein 25 Expression in Brain and Spinal Cord Astrocytes of Wild-Type and SOD1 G93A Mice. Cells 2021, 10, 1257. https://doi.org/10.3390/cells10051257
San Gil R, Clarke BE, Ecroyd H, Kalmar B, Greensmith L. Regional Differences in Heat Shock Protein 25 Expression in Brain and Spinal Cord Astrocytes of Wild-Type and SOD1 G93A Mice. Cells. 2021; 10(5):1257. https://doi.org/10.3390/cells10051257
Chicago/Turabian StyleSan Gil, Rebecca, Benjamin E. Clarke, Heath Ecroyd, Bernadett Kalmar, and Linda Greensmith. 2021. "Regional Differences in Heat Shock Protein 25 Expression in Brain and Spinal Cord Astrocytes of Wild-Type and SOD1 G93A Mice" Cells 10, no. 5: 1257. https://doi.org/10.3390/cells10051257
APA StyleSan Gil, R., Clarke, B. E., Ecroyd, H., Kalmar, B., & Greensmith, L. (2021). Regional Differences in Heat Shock Protein 25 Expression in Brain and Spinal Cord Astrocytes of Wild-Type and SOD1 G93A Mice. Cells, 10(5), 1257. https://doi.org/10.3390/cells10051257