
Having an Old Friend for Dinner: The Interplay between Apoptotic Cells and Efferocytes



Abstract
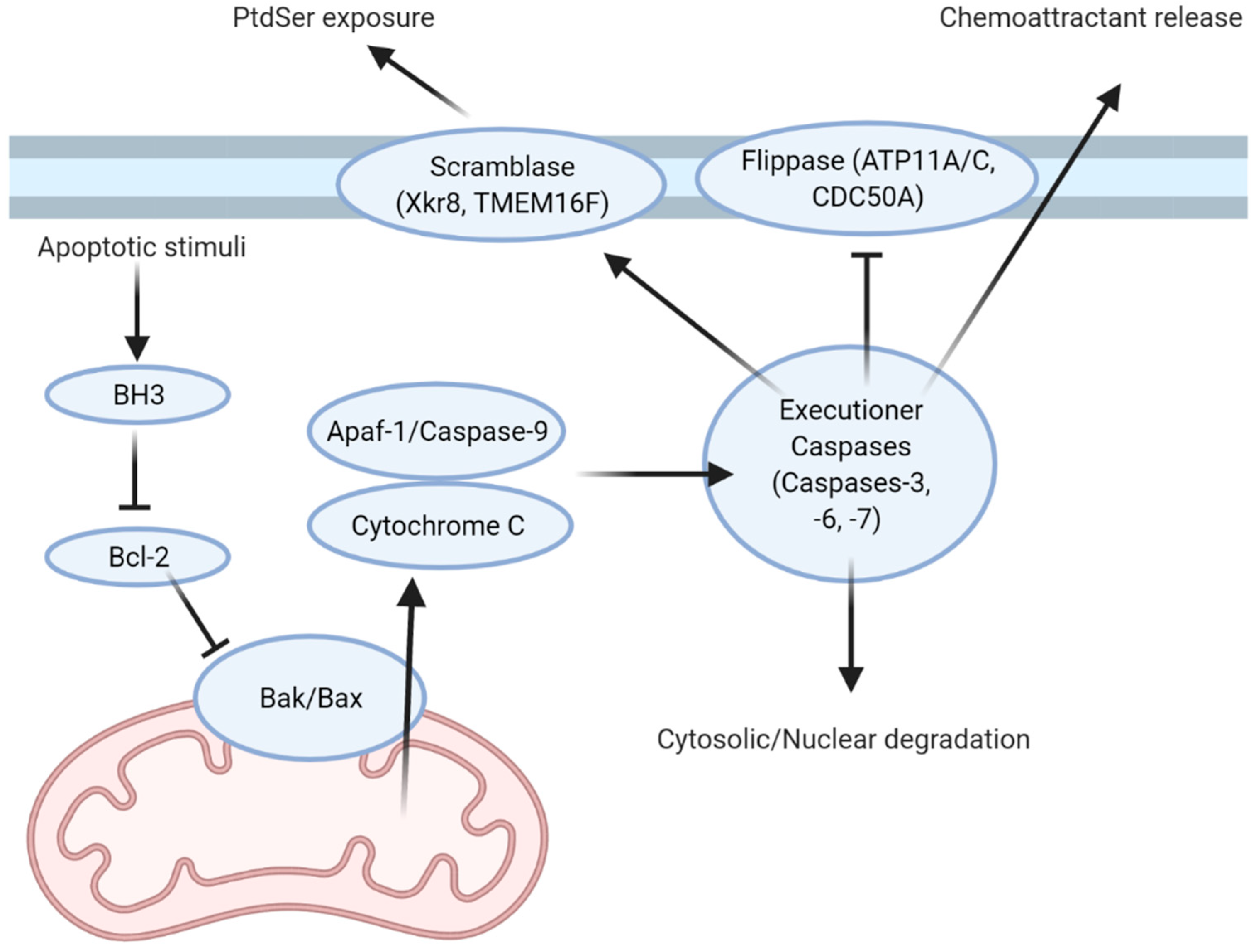
:1. Find Me and Eat Me Signals: Apoptotic Cells Write Their Own Menu
2. Tasting the Apoptotic Cell: Efferocytic Receptors
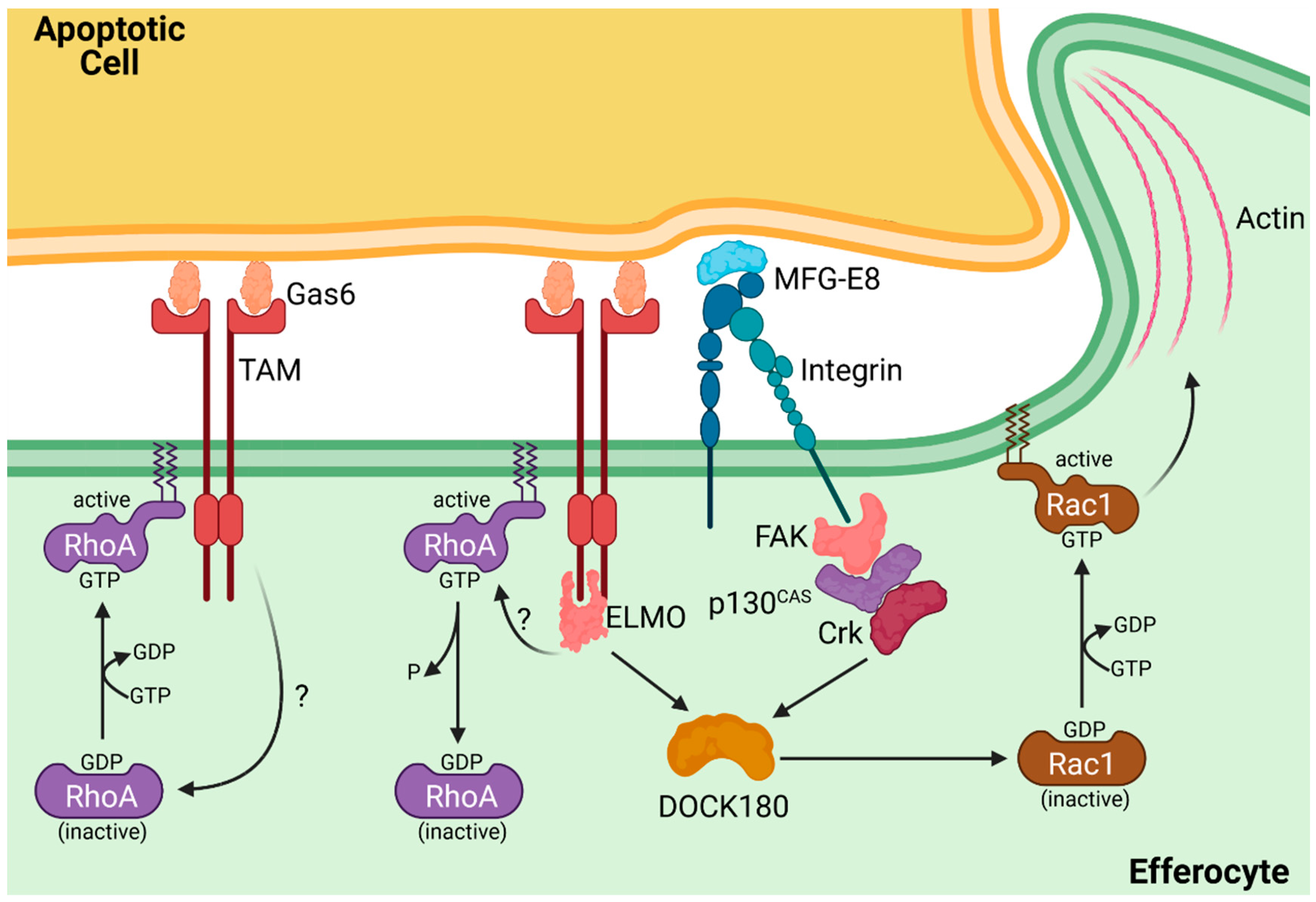
3. Fork and Knife: The TAM Family Takes Center Stage
4. Time to Dine: Engulfment of the Apoptotic Cell
5. Digesting the Apoptotic Meal: Vesicular Trafficking of Apoptotic Cells
6. The Other Menu: Necroptosis, Pyroptosis, and Ferroptosis
7. Spoiling the Meal: Efferocytosis in Disease
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Silva, M.T. Secondary Necrosis: The Natural Outcome of the Complete Apoptotic Program. FEBS Lett. 2010, 584, 4491–4499. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, Y.; Steller, H. Programmed Cell Death in Animal Development and Disease. Cell 2011, 147, 742–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.-Z.; Mapes, J.; Lee, E.-S.; Skeen-Gaar, R.R.; Xue, D. Caspase-Mediated Activation of Caenorhabditis Elegans CED-8 Promotes Apoptosis and Phosphatidylserine Externalization. Nat. Commun. 2013, 4, 2726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mapes, J.; Chen, Y.-Z.; Kim, A.; Mitani, S.; Kang, B.-H.; Xue, D. CED-1, CED-7, and TTR-52 Act in a Pathway to Regulate Exoplasmic Phosphatidylserine Expression on Apoptotic and Phagocytic Cells. Curr. Biol. 2013, 22. [Google Scholar] [CrossRef]
- Hochreiter-hufford, A.; Ravichandran, K.S. Clearing the Dead: Apoptotic Cell Sensing, Recognition, Engulfment, and Digestion. Cold Spring Harb. Perspect. Biol. 2013, 5, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Chung, S.; Gumienny, T.L.; Hengartner, M.O.; Driscoll, M. A Common Set of Engulfment Genes Mediates Removal of Both Apoptotic and Necrotic Cell Corpses in C. Elegans. Nat. Cell Biol. 2000, 2, 931–937. [Google Scholar] [CrossRef] [PubMed]
- Tajbakhsh, A.; Kovanen, P.T.; Rezaee, M.; Banach, M.; Adel, S. Regulation of Efferocytosis by Caspase-Dependent Apoptotic Cell Death in Atherosclerosis. Int. J. Biochem. Cell Biol. 2020, 120, 105684. [Google Scholar] [CrossRef] [PubMed]
- Bournazou, I.; Pound, J.D.; Duffin, R.; Bournazos, S.; Melville, L.A.; Brown, S.B.; Rossi, A.G.; Gregory, C.D. Apoptotic Human Cells Inhibit Migration of Granulocytes via Release of Lactoferrin. J. Clin. Investig. 2009, 119, 20–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuchs, Y.; Steller, H. Live to Die Another Way: Modes of Programmed Cell Death and the Signals Emanating from Dying Cells. Nat. Rev. Mol. Cell Biol. 2015, 16, 329–344. [Google Scholar] [CrossRef]
- Hengartner, M.; Ellis, R.E.; Horvitz, H.R. Caenorhabditis Elegans Gene Ced-9 Protects Cells from Programmed Cell Death. Nature 1992, 356, 494–499. [Google Scholar] [CrossRef] [PubMed]
- Segawa, K.; Nagata, S. An Apoptotic ‘Eat Me’ Signal: Phosphatidylserine Exposure. Trends Cell Biol. 2015, 25, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Fadok, V.A.; Voelker, R.; Campbell, P.A.; Bratton, D.L.; Henson, P.M.; Cohen, J.J. Exposure of Phosphatidylserine on the Surface of Apoptotic Lymphocytes Triggers Specific Recognition and Removal by Macrophages. J. Immunol. 1992, 148, 2207–2216. [Google Scholar] [PubMed]
- Peter, C.; Wesselborg, S.; Herrmann, M.; Lauber, K. Dangerous Attraction: Phagocyte Recruitment and Danger Signals of Apoptotic and Necrotic Cells. Apoptosis 2010, 15, 1007–1028. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Kerr, J.F.R.; Wyllie, A.H.; Currie, A.R. Apoptosis: A Basic Biological Phenomenon with Wide-Ranging Implications in Tissue Kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, S.J.; Reutelingsperger, C.P.; McGahon, A.J.; Rader, J.A.; van Schie, R.C.; LaFace, D.M.; Green, D.R. Early Redistribution of Plasma Membrane Phosphatidylserine Is a General Feature of Apoptosis Regardless of the Initiating Stimulus: Inhibition by Overexpression of Bcl-2 and Abl. J. Exp. Med. 1995, 182, 1545–1556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gude, D.R.; Alvarez, S.E.; Paugh, S.W.; Mitra, P.; Yu, J.; Griffiths, R.; Barbour, S.E.; Milstien, S.; Spiegel, S. Apoptosis Induces Expression of Sphingosine Kinase 1 to Release Sphingosine-1-Phosphate as a “Come-and- Get-Me” Signal. FASEB J. 2011, 22, 2629–2638. [Google Scholar] [CrossRef] [Green Version]
- Peter, C.; Waibel, M.; Radu, C.G.; Yang, L.V.; Witte, O.N.; Schulze-osthoff, K.; Wesselborg, S.; Lauber, K. Migration to Apoptotic “Find-Me” Signals Is Mediated via the Phagocyte Receptor G2A. J. Biol. Chem. 2008, 283, 5296–5305. [Google Scholar] [CrossRef] [Green Version]
- Truman, L.A.; Ford, C.A.; Pasikowska, M.; Pound, J.D.; Wilkinson, S.J.; Dumitriu, I.E.; Melville, L.; Melrose, L.A.; Ogden, C.A.; Nibbs, R.; et al. CX3CL1 / Fractalkine Is Released from Apoptotic Lymphocytes to Stimulate Macrophage Chemotaxis. Blood 2008, 112, 5026–5036. [Google Scholar] [CrossRef] [Green Version]
- Hundhausen, C.; Misztela, D.; Berkhout, T.A.; Broadway, N.; Saftig, P.; Reiss, K.; Hartmann, D.; Fahrenholz, F.; Postina, R.; Matthews, V.; et al. The Disintegrin-like Metalloproteinase ADAM10 Is Involved in Constitutive Cleavage of CX3CL1 (Fractalkine) and Regulates CX3CL1-Mediated Cell-Cell Adhesion. Blood 2003, 102, 1186–1195. [Google Scholar] [CrossRef] [Green Version]
- Garton, K.J.; Gough, P.J.; Blobel, C.P.; Murphy, G.; David, R.; Dempsey, P.J.; Raines, E.W. TACE (ADAM17) Mediates the Cleavage and Shedding of Fractalkine (CX3CL1). J. Biol. Chem. 2001, 276, 37993–38001. [Google Scholar] [CrossRef]
- Fonovic, U.P.; Jevnikar, Z.; Kos, J. Cathepsin S Generates Soluble CX3CL1 (Fractalkine) in Vascular Smooth Muscle Cells. Biol. Chem. 2013, 394, 1349–1352. [Google Scholar] [CrossRef] [PubMed]
- Saederup, N.; Chan, L.; Lira, S.A.; Charo, I.F. Fractalkine Deficiency Marketdly Reduces Macrophage Accumulation and Atheroslcerotic Lesion Formation in CCR2 −/− Mice. Circulation 2008, 117, 1642–1648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teupser, D.; Pavlides, S.; Tan, M.; Kolbeck, R.; Breslow, J.L. Major Reduction of Atherosclerosis in Fractalkine (CX3CL1) -Deficient Mice Is at the Brachiocephalic Artery, Not the Aortic Root. Proc. Natl. Acad. Sci. USA 2004, 101, 17795–17800. [Google Scholar] [CrossRef] [Green Version]
- Noda, M.; Doi, Y.; Liang, J.; Kawanokuchi, J.; Sonobe, Y.; Takeuchi, H.; Mizuno, T.; Suzumura, A. Fractalkine Attenuates Excito-Neurotoxicity via Microglial Clearance of Damaged Neurons and Antioxidant Enzyme Heme. J. Biol. Chem. 2011, 286, 2308–2319. [Google Scholar] [CrossRef] [Green Version]
- Mizuno, T.; Kawanokuchi, J.; Numata, K.; Suzumura, A. Production and Neuroprotective Functions of Fractalkine in the Central Nervous System. Brain Res. 2009, 979, 65–70. [Google Scholar] [CrossRef]
- Miksa, M.; Amin, D.; Wu, R.; Dong, W.; Ravikumar, T.S.; Wang, P. Fractalkine-Induced MFG-E8 Leads to Enhanced Apoptotic Cell Clearance by Macrophages. Mol. Med. 2007, 13, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Gershov, D.; Ma, X.; Brot, N.; Elkon, K.B. I-PLA2 Activation during Apoptosis Promotes the Exposure of Membrane Lysophosphatidylcholine Leading to Binding by Natural Immunoglobulin M Antibodies and Complement Activation. J. Exp. Med. 2002, 196, 655–665. [Google Scholar] [CrossRef] [Green Version]
- Lauber, K.; Bohn, E.; Xiao, Y.; Blumenthal, S.G.; Lindemann, R.K.; Marini, P.; Wiedig, C.; Zobywalski, A.; Baksh, S.; Xu, Y.; et al. Apoptotic Cells Induce Migration of Phagocytes via Caspase-3-Mediated Release of a Lipid Attraction Signal. Cell 2003, 113, 717–730. [Google Scholar] [CrossRef] [Green Version]
- Murakami, N.; Yokomizo, T.; Okuno, T.; Shimizu, T. G2A Is a Proton-Sensing G-Protein-Coupled Receptor Antagonized by Lysophosphatidylcholine. J. Biol. Chem. 2004, 279, 42484–42491. [Google Scholar] [CrossRef] [Green Version]
- Le, L.Q.; Kabarowski, J.H.S.; Weng, Z.; Satterthwaite, A.B.; Harvill, E.T.; Jensen, E.R.; Miller, J.F.; Witte, O.N. Mice Lacking the Orphan G Protein-Coupled Receptor G2A Develop a Late-Onset Autoimmune Syndrome. Immunity 2001, 14, 561–571. [Google Scholar] [CrossRef] [Green Version]
- Weigert, A.; Tzieply, N.; Von Knethen, A.; Johann, A.M.; Schmidt, H.; Geisslinger, G.; Brune, B. Tumor Cell Apoptosis Polarizes Macrophages — Role of Sphingosine-1-Phosphate. Mol. Biol. Cell 2007, 18, 3810–3819. [Google Scholar] [CrossRef] [Green Version]
- Weigert, A.; Cremer, S.; Schmidt, M.V.; Von Knethen, A.; Angioni, C.; Geisslinger, G. Cleavage of Sphingosine Kinase 2 by Caspase-1 Provokes Its Release from Apoptotic Cells. Blood 2010, 115, 3531–3540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Toman, R.E.; Goparaju, S.K.; Maceyka, M.; Nava, V.E.; Sankala, H.; Payne, S.G.; Bektas, M.; Ishii, I.; Chun, J.; et al. Sphingosine Kinase Type 2 Is a Putative BH3-Only Protein That Induces Apoptosis. J. Biol. Chem. 2003, 278, 40330–40336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medina, C.B.; Ravichandran, K.S. Do Not Let Death Do Us Part: ‘Find-Me’ Signals in Communication between Dying Cells and the Phagocytes. Cell Death Differ. 2016, 23, 979–989. [Google Scholar] [CrossRef] [Green Version]
- Luo, B.; Gan, W.; Liu, Z.; Jiang, M.; Luo, B.; Gan, W.; Liu, Z.; Shen, Z.; Wang, J.; Shi, R.; et al. Erythropoeitin Signaling in Macrophages Promotes Dying Cell Clearance and Immune Tolerance Article Erythropoeitin Signaling in Macrophages Promotes Dying Cell Clearance and Immune Tolerance. Immunity 2016, 44, 287–302. [Google Scholar] [CrossRef] [Green Version]
- Elliott, M.R.; Chekeni, F.B.; Trampont, P.C.; Lazarowski, E.R.; Kadl, A.; Walk, S.F.; Park, D.; Woodson, R.I.; Sharma, P.; Lysiak, J.J.; et al. Nucleotides Released by Apoptotic Cells Act as a Find-Me Signal for Phagocytic Clearance. Nature 2009, 461, 282–286. [Google Scholar] [CrossRef] [Green Version]
- Chiu, Y.; Jin, X.; Medina, C.B.; Leonhardt, S.A.; Kiessling, V.; Bennett, B.C.; Shu, S.; Tamm, L.K.; Yeager, M.; Ravichandran, K.S.; et al. A Quantized Mechanism for Activation of Pannexin Channels. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Chekeni, F.B.; Elliott, M.R.; Sandilos, J.K.; Walk, S.F.; Kinchen, M.; Lazarowski, E.R.; Armstrong, A.J.; Penuela, S.; Laird, W.; Salvesen, G.S.; et al. Pannexin 1 Channels Mediate “find-Me” Signal Release and Membrane Permeability during Apoptosis. Nature 2011, 467, 863–867. [Google Scholar] [CrossRef] [Green Version]
- Kopp, R.; Krautloher, A.; Ramírez-fernández, A.; Nicke, A. P2X7 Interactions and Signaling – Making Head or Tail of It. Front. Mol. Neurosci. 2019, 12, 1–25. [Google Scholar] [CrossRef]
- Wang, Q.; Ju, X.; Zhou, Y.; Chen, K. Necroptotic Cells Release Find-Me Signal and Are Engulfed without Proinflammatory Cytokine Production. In Vitro Cell. Dev. Biol. Anim. 2015, 51, 1033–1039. [Google Scholar] [CrossRef]
- Peng, K.; Liu, L.; Wei, D.; Lv, Y.; Wang, G.; Xiong, W.; Wang, X.; Altaf, A.; Wang, L.; He, D.A.N.; et al. P2X7R Is Involved in the Progression of Atherosclerosis by Promoting NLRP3 Inflammasome Activation. Int. J. Mol. Med. 2015, 35, 1179–1188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segawa, K.; Kurata, S.; Yanagihashi, Y.; Brummelkamp, T.R.; Matsuda, F.; Nagata, S. Caspase-Mediated Cleavage of Phospholipid Flippase for Apoptotic Phosphatidylserine Exposure. Science 2014, 344, 1164–1169. [Google Scholar] [CrossRef] [Green Version]
- Sakuragi, T.; Kosako, H.; Nagata, S. Phosphorylation-Mediated Activation of Mouse Xkr8 Scramblase for Phosphatidylserine Exposure. Proc. Natl. Acad. Sci. USA 2019, 116, 2907–2912. [Google Scholar] [CrossRef] [Green Version]
- Daleke, D.L. Regulation of Transbilayer Plasma Membrane Phospholipid Asymmetry. J. Lipid Res. 2003, 44, 233–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldridge, R.D.; Graham, T.R. Identification of Residues Defining Phospholipid Flippase Substrate Specificity of Type IV P-Type ATPases. Proc. Natl. Acad. Sci. USA 2012, 109, 290–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henson, P.M. Cell Removal: Efferocytosis. Annu. Rev. Cell. Dev. Biol. 2017, 33, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, J.; Umeda, M.; Sims, P.J.; Nagata, S. Calcium-Dependent Phospholipid Scrambling by TMEM16F. Nature 2010, 468, 834–840. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, J.; Denning, D.P.; Imanishi, E.; Horvitz, H.R.; Nagata, S. Xk-Related Protein 8 and CED-8 Promote Phosphatidylserine Exposure in Apoptotic Cells. Science 2013, 341, 403–407. [Google Scholar] [CrossRef] [Green Version]
- Kawano, M.; Nagata, S. Lupus-like Autoimmune Disease Caused by a Lack of Xkr8, a Caspase-Dependent Phospholipid Scramblase. Proc. Natl. Acad. Sci. USA 2018, 115, 2132–2137. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, J.; Fujii, T.; Imao, T.; Ishihara, K.; Kuba, H.; Nagata, S. Calcium-Dependent Phospholipid Scramblase Activity of TMEM16 Protein Family Members. J. Biol. Chem. 2013, 288, 13305–13316. [Google Scholar] [CrossRef] [Green Version]
- Fujii, T.; Sakata, A.; Nishimura, S.; Eto, K.; Nagata, S. TMEM16F Is Required for Phosphatidylserine Exposure and Microparticle Release in Activated Mouse Platelets. Proc. Natl. Acad. Sci. USA 2015, 112, 12800–12805. [Google Scholar] [CrossRef] [Green Version]
- Garg, A.D.; Krysko, D.V.; Verfaillie, T.; Kaczmarek, A.; Ferreira, G.B.; Marysael, T.; Rubio, N.; Firczuk, M.; Mathieu, C.; Roebroek, A.J.M.; et al. A Novel Pathway Combining Calreticulin Exposure and ATP Secretion in Immunogenic Cancer Cell Death. EMBO J. 2012, 31, 1062–1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, M.T.; do Vale, A.; dos Santos, N.M. Secondary Necrosis in Multicellular Animals: An Outcome of Apoptosis with Pathogenic Implications. Apoptosis 2008, 13, 463–482. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.L.; Caricchio, R.; Abraham, V.; Camenisch, T.D.; Jennette, J.C.; Roubey, R.A.S.; Earp, H.S.; Matsushima, G.; Reap, E.A. Delayed Apoptotic Cell Clearance and Lupus-like Autoimmunity in Mice Lacking the c-Mer Membrane Tyrosine Kinase. J. Exp. Med. 2002, 196, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Hanayama, R.; Tanaka, M.; Miyasaka, K.; Aozasa, K.; Koike, M.; Uchiyama, Y.; Nagata, S. Autoimmune Disease and Impaired Uptake of Apoptotic Cells in MFG-E8 – Deficient Mice. Science 2004, 304, 1147–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kojima, Y.; Weissman, I.L.; Leeper, N.J. The Role of Efferocytosis in Atherosclerosis. Circulation 2017, 135, 476–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinger, J.G.; Omari, K.M.; Marsden, K.; Raine, C.S.; Shafit-zagardo, B. Up-Regulation of Soluble Axl and Mer Receptor Tyrosine Kinases Negatively Correlates with Gas6 in Established Multiple Sclerosis Lesions. Am. J. Pathol. 2009, 175, 283–293. [Google Scholar] [CrossRef] [Green Version]
- Zaza, R.; Ibayyan, L.; El-khateeb, M.; Bahou, Y.G.; Khreisat, E.; Al-Khateeb, W.; Ahram, M. Association of Genetic Polymorphisms of MERTK with Multiple Sclerosis among Jordanians. Biomed. Res. 2017, 28, 399–404. [Google Scholar]
- International Multiple Sclerosis Genetics Consortium; Wellcome Trust Case Control Consortium 2. Genetic Risk and a Primary Role for Cell-Mediated Immune Mechanisms in Multiple Sclerosis. Nature 2011, 476, 214–219. [Google Scholar] [CrossRef]
- Barth, N.D.; Marwick, J.A.; Vendrell, M.; Rossi, A.G.; Dransfield, I. The “Phagocytic Synapse” and Clearance of Apoptotic Cells. Front. Immunol. 2017, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Lemke, G. Biology of the TAM Receptors. Cold Spring Harb. Perspect. Biol. 2013, 5, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Kourtzelis, I.; Hajishengallis, G.; Chavakis, T. Phagocytosis of Apoptotic Cells in Resolution of Inflammation. Front. Immunol. 2020, 11, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Yurdagul, A., Jr.; Doran, A.C.; Cai, B.; Fredman, G.; Tabas, I.A. Mechanisms and Consequences of Defective Efferocytosis in Atherosclerosis. Front. Cardiovas. Med. 2018, 4, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Boada-Romero, E.; Martinez, J.; Heckmann, B.L.; Green, D.R. The Clearance of Dead Cells by Efferocytosis. Nat. Rev. Mol. Cell Biol. 2020, 21, 398–414. [Google Scholar] [CrossRef] [PubMed]
- Ley, K.; Pramod, A.B.; Croft, M.; Ravichandran, K.S.; Ting, J.P. How Mouse Macrophages Sense What Is Going On. Front. Immunol. 2016, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, K.; Valdez, P.A.; Tan, C.; Yeh, S.; Hongo, J.-A.; Ouyang, W. Phosphatidylserine Receptor Tim-4 Is Essential for the Maintenance of the Homeostatic State of Resident Peritoneal Macrophages. Proc. Natl. Acad. Sci. USA 2010, 107, 8712–8717. [Google Scholar] [CrossRef] [Green Version]
- Yanagihashi, Y.; Segawa, K.; Maeda, R.; Nabeshima, Y.; Nagata, S. Mouse Macrophages Show Different Requirements for Phosphatidylserine Receptor Tim4 in Efferocytosis. Proc. Natl. Acad. Sci. USA 2017, 114, 8800–8805. [Google Scholar] [CrossRef] [Green Version]
- Rantakari, P.; Patten, D.A.; Valtonen, J.; Karikoski, M.; Gerke, H.; Dawes, H.; Laurila, J.; Ohlmeier, S.; Elima, K.; Hübscher, S.G.; et al. Stabilin-1 Expression Defines a Subset of Macrophages That Mediate Tissue Homeostasis and Prevent Fibrosis in Chronic Liver Injury. Proc. Natl. Acad. Sci. USA 2016, 113, 9298–9303. [Google Scholar] [CrossRef] [Green Version]
- Patten, D.A.; Shetty, S. The Role of Stabilin-1 in Lymphocyte Trafficking and Macrophage Scavenging in the Liver Microenvironment. Biomolecules 2019, 9, 283. [Google Scholar] [CrossRef] [Green Version]
- Kumawat, A.K.; Yu, C.; Mann, E.A.; Schridde, A.; Finnemann, S.C.; Mowat, A.M. Expression and Characterization of Avβ5 Integrin on Intestinal Macrophages. Eur. J. Immunol. 2018, 48, 1181–1187. [Google Scholar] [CrossRef]
- Summers, K.M.; Bush, S.J.; Hume, D.A. Network Analysis of Transcriptomic Diversity amongst Resident Tissue Macrophages and Dendritic Cells in the Mouse Mononuclear Phagocyte System. PLoS Biol. 2020, 18, e3000859. [Google Scholar] [CrossRef] [PubMed]
- Patten, D.A.; Kamarajah, S.K.; Rose, J.M.; Tickle, J.; Shepherd, E.L.; Adams, D.H.; Weston, C.J.; Shetty, S. SCARF-1 Promotes Adhesion of CD4+ T Cells to Human Hepatic Sinusoidal Endothelium under Conditions of Shear Stress. Sci. Rep. 2017, 7, 17600. [Google Scholar] [CrossRef] [Green Version]
- Greenberg, M.E.; Sun, M.; Zhang, R.; Febbraio, M.; Silverstein, R.; Hazen, S.L. Oxidized Phosphatidylserine-CD36 Interactions Play an Essential Role in Macrophage-Dependent Phagocytosis of Apoptotic Cells. J. Exp. Med. 2006, 203, 2613–2625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yancey, P.G.; Blakemore, J.; Ding, L.; Fan, D.; Overton, C.D.; Zhang, Y.; Linton, M.F.; Fazio, S. Macrophage LRP-1 Controls Plaque Cellularity by Regulating Efferocytosis and Akt Activation. Arter. Thromb. Vasc. Biol. 2010, 30, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Yin, C.; Vrieze, A.M.; Rosoga, M.; Akingbasote, J.; Pawlak, E.N.; Jacob, R.A.; Hu, J.; Sharma, N.; Dikeakos, J.D.; Barra, L.; et al. Efferocytic Defects in Early Atherosclerosis Are Driven by GATA2 Overexpression in Macrophages. Front. Immunol. 2020, 11, 594136. [Google Scholar] [CrossRef]
- Zagórska, A.; Través, P.G.; Lew, E.D.; Dransfield, I.; Lemke, G. Diversification of TAM Receptor Function. Nat. Immunol. 2014, 15, 920–928. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Jung, M.; Kim, H.; Lee, S.; Kim, S.; Lee, B.; Kwon, T.; Park, R.; Kim, I. Rapid Cell Corpse Clearance by Stabilin-2, a Membrane Phosphatidylserine Receptor. Cell Death Differ. 2008, 15, 192–201. [Google Scholar] [CrossRef] [Green Version]
- David, C.; Nance, J.P.; Hubbard, J.; Hsu, M.; Binder, D.; Wilson, E.H. Stabilin-1 Expression in Tumor Associated Macrophages. Brain Res. 2012, 1481, 71–78. [Google Scholar] [CrossRef] [Green Version]
- Ramirez-Ortiz, Z.G.; Iii, W.F.P.; Prasad, A.; Byrne, M.H.; Iram, T.; Blanchette, C.J.; Luster, A.D.; Hacohen, N.; El Khoury, J.; Means, T.K. The Scavenger Receptor SCARF1 Mediates the Clearance of Apoptotic Cells and Prevents Autoimmunity. Nat. Immunol. 2013, 14, 917–928. [Google Scholar] [CrossRef] [Green Version]
- Heit, B.; Kim, H.; Cosío, G.; Castaño, D.; Collins, R.; Lowell, C.A.; Kain, K.C.; Trimble, W.S.; Grinstein, S. Multimolecular Signaling Complexes Enable Syk-Mediated Signaling of CD36 Internalization. Dev. Cell 2013, 24, 372–383. [Google Scholar] [CrossRef] [Green Version]
- Blackburn, J.W.D.; Lau, D.H.C.; Liu, E.Y.; Ellins, J.; Vrieze, A.M.; Pawlak, E.N.; Dikeakos, J.D.; Heit, B. Soluble CD93 Is an Apoptotic Cell Opsonin Recognized by Axβ2. Eur. J. Immunol. 2019, 49, 600–610. [Google Scholar] [CrossRef]
- Gardai, S.J.; McPhillips, K.A.; Frasch, S.C.; Janssen, W.J.; Starefeldt, A.; Murphy-Ullrich, J.E.; Bratton, D.L.; Oldenborg, P.-A.; Michalak, M.; Henson, P.M. Cell-Surface Calreticulin Initiates Clearance of Viable or Apoptotic Cells through Trans-Activation of LRP on the Phagocyte. Cell 2005, 123, 321–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seitz, H.M.; Camenisch, T.D.; Lemke, G.; Earp, S.; Matsushima, G.K.; Seitz, H.M.; Camenisch, T.D.; Lemke, G.; Earp, H.S.; Matsushima, G.K. Macrophages and Dendritic Cells Use Different Axl/Mertk/Tyro3 Receptors in Clearance of Apoptotic Cells. J. Immunol. 2007, 178, 5635–5642. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, Q.; Wang, J.; Li, G.; Weiland, M.; Yu, F.-S.; Mi, Q.-S.; Gu, J.; Zhou, L. TIM-4 Is Differentially Expressed in the Distinct Subsets of Dendritic Cells in Skin and Skin-Draining Lymph Nodes and Controls Skin Langerhans Cell Homeostasis. Oncotarget 2016, 7, 37498–37512. [Google Scholar] [CrossRef]
- Zimmer, A.; Bouley, J.; Le Mignon, M.; Pliquet, E.; Horiot, S.; Turfkruyer, M.; Baron-Bodo, V.; Horak, F.; Nony, E.; Louise, A.; et al. A Regulatory Dendritic Cell Signature Correlates with the Clinical Efficacy of Allergen-Specific Sublingual Immunotherapy. J. Allergy Clin. Immunol. 2012, 129, 1020–1030. [Google Scholar] [CrossRef]
- Matthew, L.A.; Kim, J.-I.; Birge, R.B. Avβ5 Integrin Recruits the CrkII–Dock180–Rac1 Complex for Phagocytosis of Apoptotic Cells. Nat. Cell Biol. 2000, 2, 899–905. [Google Scholar]
- Kim, D.; Lee, S.-A.; Moon, H.; Kim, K.; Park, D. The Tim Gene Family in Efferocytosis. Genes Genomics 2020, 42, 979–986. [Google Scholar] [CrossRef]
- Fourgeaud, L.; Través, P.G.; Tufail, Y.; Leal-Bailey, H.; Lew, E.D.; Burrola, P.G.; Callaway, P.; Zagórska, A.; Rothlin, C.V.; Nimmerjahn, A.; et al. TAM Receptors Regulate Multiple Features of Microglial Physiology. Nature 2016, 532, 240–244. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, K.; El Khoury, J. Microglial Scavenger Receptors and Their Roles in the Pathogenesis of Alzheimer’s Disease. Int. J. Alzheimers Dis. 2012, 2012, 489456. [Google Scholar] [CrossRef] [Green Version]
- Neniskyte, U.; Neher, J.J.; Brown, G.C. Neuronal Death Induced by Nanomolar Amyloid β Is Mediated by Primary Phagocytosis of Neurons by Microglia. J. Biol. Chem. 2011, 286, 39904–39913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazaheri, F.; Breus, O.; Durdu, S.; Haas, P.; Wittbrodt, J.; Gilmour, D.; Peri, F. Distinct Roles for BAI1 and TIM-4 in the Engulfment of Dying Neurons by Microglia. Nat. Commun. 2014, 5, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Ichimura, T.; Asseldonk, E.J.P.; Humphreys, B.D.; Gunaratnam, L.; Duffield, J.S.; Bonventre, J.V. Kidney Injury Molecule – 1 Is a Phosphatidylserine Receptor That Confers a Phagocytic Phenotype on Epithelial Cells. J. Clin. Investig. 2008, 118, 1657–1668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finnemann, S.C.; Nandrot, E.F. MerTK Activation during RPE Phagocytosis In Vivo Requires AlphaVbeta5 Integrin. Adv. Exp. Med. Biol. 2006, 572, 499–503. [Google Scholar] [CrossRef] [Green Version]
- Shelby, S.J.; Colwill, K.; Dhe-Paganon, S.; Pawson, T.; Thompson, D. a MERTK Interactions with SH2-Domain Proteins in the Retinal Pigment Epithelium. PLoS ONE 2013, 8, e53964. [Google Scholar] [CrossRef] [Green Version]
- Hochreiter-Hufford, A.E.; Lee, C.S.; Kinchen, J.M.; Sokolowski, J.D.; Arandjelovic, S.; Call, J.A.; Klibanov, A.L.; Yan, Z.; Mandell, J.W.; Ravichandran, K.S. Phosphatidylserine Receptor BAI1 and Apoptotic Cells as New Promoters of Myoblast Fusion. Nature 2013, 497, 263–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, J.-H.; Ko, H.-M.; Han, G.-D.; Lee, S.-Y.; Moon, J.-S.; Kim, M.-S.; Koh, J.-T.; Kim, S.-H. Dual Role of Phosphatidylserine and Its Receptors in Osteoclastogenesis. Cell Death Dis. 2020, 11, 497. [Google Scholar] [CrossRef] [PubMed]
- Lv, Z.; Bian, Z.; Shi, L.; Niu, S.; Ha, B.; Tremblay, A.; Li, L.; Zhang, X. Loss of Cell Surface CD47 Clustering Formation and Binding Avidity to SIRP α Facilitate Apoptotic Cell Clearance by Macrophages. J. Immunol. 2015, 195, 661–671. [Google Scholar] [CrossRef] [Green Version]
- Lopes, F.B.; Bálint, Š.; Valvo, S.; Felce, J.H.; Hessel, E.M.; Dustin, M.L.; Davis, D.M. Membrane Nanoclusters of Fc γ RI Segregate from Inhibitory SIRP α upon Activation of Human Macrophages. J. Cell Biol. 2017, 216, 1123–1141. [Google Scholar] [CrossRef] [PubMed]
- Mcdonald, J.F.; Zheleznyak, A.; Frazier, W.A. Cholesterol-Independent Interactions with CD47 Enhance v 3 Avidity*. J. Biol. Chem. 2004, 279, 17301–17311. [Google Scholar] [CrossRef] [Green Version]
- Green, J.M.; Zhele, A.; Chung, J.; Lindberg, F.P.; Sarfati, M.; Frazier, W.A.; Brown, E.J. Role of Cholesterol in Formation and Function of a Signaling Complex Involving Avß3, Integrin-Associted Protein (CD47), and Heterotrimeric G Proteins. J. Cell Biol. 1999, 146, 673–682. [Google Scholar] [CrossRef] [PubMed]
- Borisenko, G.G.; Matsura, T.; Liu, S.; Tyurin, V.A.; Jianfei, J.; Serinkan, F.B.; Kagan, V.E. Macrophage Recognition of Externalized Phosphatidylserine and Phagocytosis of Apoptotic Jurkat Cells—Existence of a Threshold. Arch. Biochem. Biophys. 2003, 413, 41–52. [Google Scholar] [CrossRef]
- Albacker, L.A.; Karisola, P.; Chang, Y.-J.; Umetsu, S.E.; Zhou, M.; Akbari, O.; Kobayashi, N.; Baumgarth, N.; Freeman, G.J.; Umetsu, D.T.; et al. TIM-4, a Receptor for Phosphatidylserine, Controls Adaptive Immunity by Regulating the Removal of Antigen-Specific T Cells. J. Immunol. 2010, 185, 6839–6849. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, N.; Karisola, P.; Peña-cruz, V.; Dorfman, D.M.; Umetsu, S.E.; Butte, M.J.; Nagumo, H.; Chernova, I.; Sharpe, A.H.; Ito, S.; et al. T Cell Immunoglobulin Mucin Protein (TIM)-4 Binds Phosphatidylserien and Mediates Uptake of Apoptotic Cells. Immunity 2009, 27, 927–940. [Google Scholar] [CrossRef] [Green Version]
- Park, D.; Hochreiter-hufford, A. The Phosphatidylserine Receptor TIM-4 Does Not Mediate Direct Signaling. Curr. Biol. 2009, 19, 346–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santiago, C.; Ballesteros, A.; Martinez-muñoz, L.; Mellado, M.; Gerardo, G.; Freeman, G.J.; Casasnovas, J.M. TIM-4 Structures Identify a Metal Ion-Dependent Ligand Binding Site Where Phosphatidylserine Binds. Immunity 2008, 27, 941–951. [Google Scholar] [CrossRef] [Green Version]
- Gielen, A.W.; Lobell, A.; Lidman, O.; Khademi, M.; Olsson, T.; Piehl, F. Expression of T Cell Immunoglobulin- and Mucin-Domain-Containing Molecules-1 and -3 (TIM-1 and -3) in the Rat Nervous and Immune Systems. J. Neuroimmunol. 2005, 164, 93–104. [Google Scholar] [CrossRef]
- Nakayama, M.; Akiba, H.; Takeda, K.; Kojima, Y.; Hashiguchi, M.; Azuma, M.; Yagita, H.; Okumura, K. Tim-3 Mediates Phagocytosis of Apoptotic Cells and Cross-Presentation. Blood 2009, 113, 3821–3830. [Google Scholar] [CrossRef]
- Nishi, C.; Toda, S.; Segawa, K.; Nagata, S. Tim4- and MerTK-Mediated Engulfment of Apoptotic Cells by Mouse Resident Peritoneal Macrophages. Mol. Cell. Biol. 2014, 34, 1512–1520. [Google Scholar] [CrossRef] [Green Version]
- Flannagan, R.S.; Canton, J.; Furuya, W.; Glogauer, M. The Phosphatidylserine Receptor TIM4 Utilizes Integrins as Coreceptors to Effect Phagocytosis. Mol. Biol. Cell 2014, 25, 1511–1522. [Google Scholar] [CrossRef]
- Park, D.; Elliott, M.R.; Lu, M.; Haney, L.B.; Ma, Z.; Klibanov, A.L.; Mandell, J.W.; Ravichandran, K.S. BAI1 Is an Engulfment Receptor for Apoptotic Cells Upstream of the ELMO / Dock180 / Rac Module. Nature 2007, 450, 430–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penberthy, K.K.; Ravichandran, K.S. Apoptotic Cell Recognition Receptors and Scavenger Receptors. Immunol. Rev. 2016, 269, 44–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, S.; Owen, K.A.; Ly, K.T.; Park, D.; Black, S.G.; Wilson, J.M.; Sifri, C.D.; Ravichandran, K.S.; Ernst, P.B.; Casanova, J.E. Brain Angiogenesis Inhibitor 1 (BAI1) Is a Pattern Recognition Receptor That Mediates Macrophage Binding and Engulfment of Gram-Negative Bacteria. Proc. Natl. Acad. Sci. USA 2011, 108, 2136–2141. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Kim, S.; Jung, M.; Bae, D.; Kim, I. Epidermal Growth Factor-Like Domain Repeat of Stabilin-2 Recognizes Phosphatidylserine during Cell Corpse Clearance. Mol. Cell. Biol. 2008, 28, 5288–5298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.; Kang, K.; Thapa, N.; Kim, S.; Lee, S.; Kim, I. Requirement of Adaptor Protein GULP during Stabilin-2-Mediated Cell Corpse Engulfment. J. Biol. Chem. 2008, 283, 10593–10600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, Y.; Adachi, H.; Osuga, J.; Ohashi, K.; Yahagi, N.; Sekiya, M.; Okazaki, H.; Tomita, S.; Iizuka, Y.; Shimano, H.; et al. FEEL-1 and FEEL-2 Are Endocytic Receptors for Advanced Glycation End Products. J. Biol. Chem. 2003, 278, 12613–12617. [Google Scholar] [CrossRef] [Green Version]
- Adachi, H.; Tsujimoto, M. FEEL-1, a Novel Scavenger Receptor with in Vitro Bacteria-Binding and Angiogenesis-Modulating Activities. J. Biol. Chem. 2002, 277, 34264–34270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Païdassi, H.; Tacnet-delorme, P.; Garlatti, V.; Darnault, C.; Ghebrehiwet, B.; Gaboriaud, C.; Arlaud, G.J.; Frachet, P.; Tacnet-delorme, P.; Garlatti, V.; et al. C1q Binds Phosphatidylserine and Likely Acts as a Multiligand-Bridging Molecule in Apoptotic Cell Recognition. J. Immunol. 2008, 180, 2329–2338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wicker-planquart, C.; Dufour, S.; Tacnet-delorme, P.; Bally, I.; Delneste, Y.; Frachet, P.; Housset, D.; Thielens, N.M. Molecular and Cellular Interactions of Scavenger Receptor SR-F1 With Complement C1q Provide Insights Into Its Role in the Clearance of Apoptotic Cells. Front. Immunol. 2020, 11, 1–15. [Google Scholar] [CrossRef]
- Akakura, S.; Singh, S.; Spataro, M.; Akakura, R.; Kim, J.; Albert, M.L.; Birge, R.B. The Opsonin MFG-E8 Is a Ligand for the Avß5 Integrin and Triggers DOCK180-Dependent Rac1 Activation for the Phagocytosis of Apoptotic Cells. Exp. Cell Res. 2004, 292, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Singh, S.; Georgescu, M.; Birge, R.B. A Role for Mer Tyrosine Kinase in α v β 5 Integrin- Mediated Phagocytosis of Apoptotic Cells. J. Cell Sci. 2005, 118, 539–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanayama, R.; Tanaka, M.; Miwa, K.; Shinohara, A.; Iwamatsu, A.; Nagata, S. Identification of a Factor That Links Apoptotic Cells to Phagocytes. Nature 2002, 417, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zhang, Z.; Zhuang, Z.; Lu, Y.; Chen, C.; Li, W.; Hang, C. Recombinant Milk Fat Globule-EGF Factor-8 Reduces Apoptosis via Integrin Β3/FAK/PI3K/AKT Signaling Pathway in Rats after Traumatic Brain Injury. Cell Death Dis. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Arienti, S.; Barth, N.D.; Dorward, D.A.; Rossi, A.G.; Dransfield, I. Regulation of Apoptotic Cell Clearance During Resolution of Inflammation. Front. Pharmacol. 2019, 10, 891. [Google Scholar] [CrossRef] [Green Version]
- Dransfield, I.; Zagórska, A.; Lew, E.D.; Michail, K.; Lemke, G. Mer Receptor Tyrosine Kinase Mediates Both Tethering and Phagocytosis of Apoptotic Cells. Cell Death Dis. 2015, 6, e1646. [Google Scholar] [CrossRef] [PubMed]
- Lemke, G.; Rothlin, C.V. Immunobiology of the TAM Receptors. Nat. Rev. Immunol. 2008, 8, 327–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz, P.M.D.; Yasumura, D.; Weir, J.; Matthes, M.T.; Abderrahim, H.; Lavail, M.M.; Vollrath, D. Mutation of the Receptor Tyrosine Kinase Gene Mertk in the Retinal Dystrophic RCS Rat. Hum. Mol. Genet. 2000, 9, 645–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gal, A.; Li, Y.; Debra, A.; Weir, J.; Orth, U.; Vollrath, D. Mutations in MERTK, the Human Orthologue of the RCS Rat Retinal Dystrophy Gene, Cause Retinitis Pigmentosa. Nat. Genet. 2000, 26, 270–271. [Google Scholar] [CrossRef] [PubMed]
- Duncan, J.L.; Lavail, M.M.; Yasumura, D.; Matthes, M.T.; Yang, H.; Trautmann, N.; Chappelow, A.V.; Feng, W.; Earp, H.S.; Matsushima, G.K.; et al. An RCS-Like Retinal Dystrophy Phenotype in Mer Knockout Mice. IOVS 2003, 44, 826–838. [Google Scholar] [CrossRef] [Green Version]
- Burstyn-Cohen, T.; Lew, E.D.; Través, P.G.; Burrola, P.G.; Hash, J.C.; Lemke, G. Genetic Dissection of TAM Receptor-Ligand Interaction in Retinal Pigment Epithelial Cell Phagocytosis. Neuron 2013, 76, 1123–1132. [Google Scholar] [CrossRef] [Green Version]
- Lu, Q.; Gore, M.; Zhang, Q.; Camenisch, T.; Boast, S.; Casagranda, F.; Klein, È.; Lai, C.; Klein, R.; Matsushima, G.K.; et al. Tyro-3 Family Receptors Are Essential Regulators of Mammalian Spermatogenesis. Nature 1999, 398, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Angelillo-Scherrer, A.; de Frutos, P.G.; Aparicio, C.; Melis, E.; Savi, P.; Lupu, F.; Arnout, J.; Dewerchin, M.; Hoylaerts, M.F.; Herbert, J.-M.; et al. Deficiency or Inhibition of Gas6 Causes Platelet Dysfunction and Protects Mice against Thrombosis. Nat. Med. 2001, 7, 215–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Meer, J.H.M.; Van Der Poll, T.; Van Veer, C. TAM Receptors, Gas6, and Protein S: Roles in Inflammation and Hemostasis. Blood 2014, 123, 2460–2469. [Google Scholar] [CrossRef] [PubMed]
- Caberoy, N.B.; Alvarado, G.; Bigcas, J.-L.; Li, W. Galectin-3 Is a New MerTK-Specific Eat-Me Signal. J. Cell Physiol. 2013, 227, 401–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caberoy, N.B.; Zhou, Y. Tubby and Tubby-like Protein 1 Are New MerTK Ligands for Phagocytosis. EMBO J. 2010, 29, 3898–3910. [Google Scholar] [CrossRef] [Green Version]
- Uehara, H.; Shacter, E. Auto-Oxidation and Oligomerization of Protein S on the Apoptotic Cell Surface Is Required for Mer Tyrosine Kinase-Mediated Phagocytosis of Apoptotic Cells. J. Immunol. 2008, 180, 2522–2530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rezende, S.M.; Lane, D.A.; Mille-baker, B.; Samama, M.M.; Conard, J.; Simmonds, R.E. Protein S Gla-Domain Mutations Causing Impaired Ca2+-Induced Phospholipid Binding and Severe Functional Protein S Deficiency. Blood 2002, 100, 2812–2819. [Google Scholar] [CrossRef]
- Lemke, G.; Jolla, L. Phosphatidylserine Is the Signal for TAM Receptors and Their Ligands. Trends Biochem. Sci. 2017, 42, 738–748. [Google Scholar] [CrossRef]
- Tsou, W.; Nguyen, K.N.; Calarese, D.A.; Garforth, S.J.; Antes, A.L.; Smirnov, S.V.; Almo, S.C.; Birge, R.B.; Kotenko, S.V. Receptor Tyrosine Kinases, TYRO3, AXL, and MER, Demonstrate Distinct Patterns and Complex Regulation of Ligand-Induced Activation. J. Biol. Chem. 2014, 289, 25750–25763. [Google Scholar] [CrossRef] [Green Version]
- Manfioletti, G.; Bancolini, C.; Avanzi, G.; Schneider, C. The Protein Encoded by a Growth Arrest-Specific Gene (Gas6) Is a New Member of the Vitamin K-Dependent Proteins Related to Protein S, a Negative Coregulator in the Blood Coagulation Cascade. Mol. Cell Biol. 1993, 13, 4976–4985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lew, E.D.; Oh, J.; Burrola, P.G.; Lax, I.; Zagórska, A.; Través, P.G.; Schlessinger, J.; Lemke, G. Differential TAM Receptor – Ligand – Phospholipid Interactions Delimit Differential TAM Bioactivities. eLife 2014, 3, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Fujimori, T.; Grabiec, A.M.; Kaur, M.; Bell, T.J.; Fujino, N.; Cook, P.C.; Svedberg, F.R.; Macdonald, A.S.; Maciewicz, R.A.; Singh, D.; et al. The Axl Receptor Tyrosine Kinase Is a Discriminator of Macrophage Function in the Inflamed Lung. Mucosal Immunol. 2015, 8, 1021–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stitt, T.N.; Conn, G.; Gore, M.; Lai, C.; Bruno, J.; Radziejewski, C.; Mattsson, K.; Fisher, J.; Gies, D.R.; Jones, P.F.; et al. The Anticoagulation Factor Protein S and Its Relative, Gas6, Are Ligands for the Tyro 3/Axl Family of Receptor Tyrosine Kinases. Cell 1995, 80, 661–670. [Google Scholar] [CrossRef] [Green Version]
- Al Kafri, N.; Hafizi, S. Galectin-3 Stimulates Tyro3 Receptor Tyrosine Kinase and Erk Signalling, Cell Survival and Migration in Human Cancer Cells. Biomolecules 2020, 10, 1035. [Google Scholar] [CrossRef] [PubMed]
- Barth, N.D.; Marwick, J.A.; Heeb, M.J.; Gale, A.J.; Rossi, A.G. Augmentation of Human Monocyte Responses to LPS by the Protein S and Mer / Tyro3 Receptor Tyrosine Kinase Axis. J. Immunol. 2018, 201, 2602–2611. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, M.; Hayes, C.D.; Thome, J.J.; Thorp, E.; Matsushima, G.K.; Herz, J.; Farber, D.L.; Liu, K.; Lakshmana, M.; Tabas, I. An AXL/LRP-1/RANBP9 Complex Mediates DC Efferocytosis and Antigen Cross-Presentation in Vivo. J. Clin. Investig. 2014, 124, 1296–1308. [Google Scholar] [CrossRef] [Green Version]
- Rothlin, C.V.; Ghosh, S.; Zuniga, E.I.; Oldstone, M.B.A.; Lemke, G. TAM Receptors Are Pleiotropic Inhibitors of the Innate Immune Response. Cell 2007, 131, 1124–1136. [Google Scholar] [CrossRef] [Green Version]
- Mahajan, N.P.; Earp, H.S. An SH2 Domain-Dependent, Phosphotyrosine-Independent Interaction between Vav1 and the Mer Receptor Tyrosine Kinase. J. Biol. Chem. 2003, 278, 42596–42603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemke, G. How Macrophages Deal with Death. Nat. Rev. Immunol. 2020, 19, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Dustin, M.L. The Immunological Synapse. Cancer Immunol. Res. 2014, 2, 1023–1033. [Google Scholar] [CrossRef] [Green Version]
- Ostrowski, P.P.; Grinstein, S.; Freeman, S.A. Review Diffusion Barriers, Mechanical Forces, and the Biophysics of Phagocytosis. Dev. Cell 2016, 38, 135–146. [Google Scholar] [CrossRef] [Green Version]
- Freeman, S.A.; Goyette, J.; Furuya, W.; Woods, E.C.; Bertozzi, C.R.; Bergmeier, W.; Hinz, B.; van der Merwe, P.A.; Das, R.; Grinstein, S. Integrins Form an Expanding Diffusional Barrier That Coordinates Phagocytosis. Cell 2016, 164, 128–140. [Google Scholar] [CrossRef] [Green Version]
- Freeman, S.A.; Grinstein, S. Phagocytosis: Receptors, Signal Integration, and the Cytoskeleton. Immunol. Rev. 2014, 262, 193–215. [Google Scholar] [CrossRef]
- Flannagan, R.S.; Harrison, R.E.; Yip, C.M.; Jaqaman, K.; Grinstein, S. Dynamic Macrophage “Probing” Is Required for the Efficient Capture of Phagocytic Targets. J. Cell Biol. 2010, 191, 1205–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupuy, A.G.; Caron, E. Integrin-Dependent Phagocytosis – Spreading from Microadhesion to New Concepts. J. Cell Sci. 2008, 121, 1773–1783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castellano, F.; Montcourrier, P.; Chavrier, P. Membrane Recruitment of Rac1 Triggers Phagocytosis. J. Cell Sci. 2000, 113, 2955–2961. [Google Scholar] [CrossRef] [PubMed]
- Nakaya, M.; Kitano, M.; Matsuda, M.; Nagata, S. Spatiotemporal Activation of Rac1 for Engulfment of Apoptotic Cells. Proc. Natl. Acad. Sci. USA 2008, 105, 9198–9203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, Y.; Finnemann, S.C. Essential Diurnal Rac1 Activation during Retinal Phagocytosis Requires α v β 5 Integrin but Not Tyrosine Kinases Focal Adhesion Kinase or Mer Tyrosine Kinase. Mol. Biol. Cell 2012, 23, 1104–1114. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kim, S.; Bae, D.; Park, S.; Lee, Y.; Park, G.; Kim, I. Coordinated Balance of Rac1 and RhoA Plays Key Roles in Determining Phagocytic Appetite. PLoS ONE 2017, 12, e0174603. [Google Scholar] [CrossRef] [PubMed]
- Nakaya, M.; Tanaka, M.; Okabe, Y.; Hanayama, R.; Nagata, S. Opposite Effects of Rho Family GTPases on Engulfment of Apoptotic Cells by Macrophages. J. Biol. Chem. 2006, 281, 8836–8842. [Google Scholar] [CrossRef] [Green Version]
- Cook, D.R.; Rossman, K.L.; Der, C.J. Rho Guanine Nucleotide Exhange Factors: Regulators of Rho GTPase Activity in Development and Disease. Oncogene 2014, 33, 4021–4035. [Google Scholar] [CrossRef] [Green Version]
- Kinchen, J.M.; Cabello, J.; Klingele, D.; Wong, K.; Feichtinger, R.; Schnabel, H.; Schnabel, R.; Hengartner, M.O. Two Pathways Converge at CED-10 to Mediate Actin Rearrangement and Corpse Removal in C. Elegans. Nature 2005, 434, 93–99. [Google Scholar] [CrossRef]
- Reddien, P.W.; Horvitz, H.R. CED-2/CrkII and CED-10/Rac Control Phagocytosis and Cell Migration in Caenorhabditis Elegans. Nat. Cell Biol. 2000, 2, 131–136. [Google Scholar] [CrossRef]
- Osada, Y.; Sunatani, T.; Kim, I.; Nakanishi, Y. Signalling Pathway Involving GULP, MAPK and Rac1 for SR-BI-Induced Phagocytosis of Apoptotic Cells. J. Biochem. 2009, 145, 387–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tosello-Trampont, A.-C.; Kinchen, J.M.; Brugnera, E.; Haney, L.B.; Hengartner, M.O.; Ravichandran, K.S. Identification of Two Signaling Submodules within the CrkII / ELMO / Dock180 Pathway Regulating Engulfment of Apoptotic Cells. Cell Death Diff. 2007, 14, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Kiss, R.S.; Elliott, M.R.; Ma, Z.; Marcel, Y.L.; Ravichandran, K.S. Apoptotic Cells Induce a Homeostatic Response from Phagocytes. Curr. Biol. 2006, 16, 2252–2258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, M.; Ravichandran, K.S. Dock180–ELMO Cooperation in Rac Activation. Methods Enzymol. 2004, 406, 388–402. [Google Scholar] [CrossRef]
- Kim, S.; Park, S.; Kim, S.; Bae, D.; Pyo, J.; Hong, M.; Kim, I. Cross Talk between Engulfment Receptors Stabilin-2 and Integrin Avß5 Orchestrates Engulfment of Phosphatidylserine-Exposed. Mol. Cell. Biol. 2012, 32, 2698–2708. [Google Scholar] [CrossRef] [Green Version]
- Morimoto, K.; Janssen, W.J.; Fessler, M.B.; McPhillips, K.A.; Borges, V.M.; Bowler, R.P.; Xiao, Y.-Q.; Kench, J.A.; Henson, P.M.; Vandivier, R.W. Lovastatin Enhances Clearance of Apoptotic Cells (Efferocytosis) with Implications for Chronic Obstructive Pulmonary Disease. J. Immunol. 2006, 176, 7657–7665. [Google Scholar] [CrossRef] [Green Version]
- Martinez, J.; Almendinger, J.; Oberst, A.; Ness, R.; Dillon, C.P.; Fitzgerald, P.; Hengartner, M.O.; Green, D.R. Microtubule-Associated Protein 1 Light Chain 3 Alpha (LC3)-Associated Phagocytosis Is Required for the Ef Fi Cient Clearance of Dead Cells. Proc. Natl. Acad. Sci. USA 2011, 108, 17396–17401. [Google Scholar] [CrossRef] [Green Version]
- Wan, J.; Weiss, E.; Ben Mkaddem, S.; Mabire, M.; Choinier, P.; Picq, O.; Thibault-sogorb, T.; Hegde, P.; Pishvaie, D.; Bens, M.; et al. LC3-Associated Phagocytosis Protects against Inflammation and Liver Fibrosis via Immunoreceptor Inhibitory Signaling. Sci. Transl. Med. 2020, 12, eaaw8523. [Google Scholar] [CrossRef] [PubMed]
- Flannagan, R.S.; Jaumouillé, V.; Grinstein, S. The Cell Biology of Phagocytosis. Ann. Rev. Pathol. 2011, 7, 61–98. [Google Scholar] [CrossRef]
- Kinchen, J.M.; Ravichandran, K.S. Identification of Two Evolutionarily Conserved Genes Regulating Processing of Engulfed Apoptotic Cells. Nature 2010, 464, 778–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, Y.; Zhao, Q.; Gao, C.; Ding, Y.; Zeng, Y.; Ueda, T.; Nakano, A.; Jiang, L. Activation of the Rab7 GTPase by the MON1-CCZ1 Complex Is Essential for PVC-to-Vacuole Trafficking and Plant Growth in Arabidopsis. Plant. Cell 2014, 26, 2080–2097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohdanowicz, M.; Balkin, D.M.; De Camilli, P.; Grinstein, S. Recruitment of OCRL and Inpp5B to Phagosomes by Rab5 and APPL1 Depletes Phosphoinositides and Attenuates Akt Signaling. Mol. Biol. Cell 2012, 23, 176–187. [Google Scholar] [CrossRef]
- Rupper, A.; Grove, B.; Cardelli, J. Rab7 Regulates Phagosome Maturation in Dictyostelium. J. Cell Sci. 2001, 114, 2449–2460. [Google Scholar] [CrossRef]
- Rocha, N.; Neefjes, J. MHC Class II Molecules on the Move for Successful Antigen Presentation. EMBO J. 2008, 27, 1–5. [Google Scholar] [CrossRef]
- Mintern, J.D.; Macri, C.; Villadangos, J.A. Modulation of Antigen Presentation by Intracellular Trafficking. Curr. Opin. Immunol. 2015, 34, 16–21. [Google Scholar] [CrossRef]
- Yin, C.; Kim, Y.; Argintaru, D.; Heit, B. Rab17 Mediates Differential Antigen Sorting Following Efferocytosis and Phagocytosis. Cell Death Dis. 2016, 7, e2529-12. [Google Scholar] [CrossRef]
- Yin, C.; Argintaru, D.; Heit, B. Rab17 Mediates Intermixing of Phagocytosed Apoptotic Cells with Recycling Endosomes. Small GTPases 2017, 10, 1–9. [Google Scholar] [CrossRef]
- Hamon, Y.; Broccardo, C.; Chambenoit, O.; Luciani, M.; Toti, F.; Chaslin, S.; Freyssinet, J.; Devaux, P.F.; Mcneish, J.; Marguet, D.; et al. ABC1 Promotes Engulfment of Apoptotic Cells and Transbilayer Redistribution of Phosphatidylserine. Nat. Cell Biol. 2000, 2, 399–406. [Google Scholar] [CrossRef]
- Luciani, M.; Chiminil, G. The ATP Binding Cassette Transporter ABC1, Is Required for the Engulfment of Corpses Generated by Apoptotic Cell Death. EMBO J. 1996, 15, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Fond, A.M.; Kiss, R.S.; Ravichandran, K.S.; Fond, A.M.; Lee, C.S.; Schulman, I.G.; Kiss, R.S.; Ravichandran, K.S. Apoptotic Cells Trigger a Membrane-Initiated Pathway to Increase ABCA1. J. Clin. Investig. 2015, 125, 2748–2758. [Google Scholar] [CrossRef]
- Luo, J.; Jiang, L.; Yang, H.; Song, B.-L. Routes and Mechanisms of Post-Endosomal Cholesterol Trafficking: A Story That Never Ends. Traffic 2017, 18, 209–217. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, K.; Balch, W.E. NPC1/NPC2 Function as a Tag Team Duo to Mobilize Cholesterol. Proc. Natl. Acad. Sci. USA 2008, 105, 15223–15224. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.-H.; Elvington, A.; Randolph, G.J. The Role of the Lymphatic System in Cholesterol Transport. Front. Pharmacol. 2015, 6, 182. [Google Scholar] [CrossRef] [Green Version]
- Pagler, T.A.; Wang, M.; Mondal, M.; Murphy, A.J.; Westerterp, M.; Moore, K.J.; Maxfield, F.R.; Tall, A.R. Deletion of ABCA1 and ABCG1 Impairs Macrophage Migration Because of Increased Rac1 Signaling. Circ. Res. 2011, 194–200. [Google Scholar] [CrossRef] [Green Version]
- Kidani, Y.; Bensinger, S.J. Liver X Receptor and Peroxisome Proliferator-Activated Receptor as Integrators of Lipid Homeostasis and Immunity. Immunol. Rev. 2012, 249, 72–83. [Google Scholar] [CrossRef]
- Kim, S.; Lim, E.; Yoon, Y.; Ahn, Y.; Park, E. Liver X Receptor and STAT1 Cooperate Downstream of Gas6 / Mer to Induce Anti-Inflammatory Arginase 2 Expression in Macrophages. Sci. Rep. 2016, 6, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korns, D.; Frasch, S.C.; Fernandez-boyanapalli, R.; Henson, P.M.; Bratton, D.L. Modulation of Macrophage Efferocytosis in Inflammation. Front. Immunol. 2011, 2, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.; Park, H.; Lee, Y.; Byun, J.; Youn, Y.; Choi, J.H.; Woo, S.; Kang, J.L. Upregulation of Mer Receptor Tyrosine Kinase Signaling Attenuated Lipopolysaccharide-Induced Lung Inflammation. J. Pharmacol. Exp. Ther. 2013, 344, 447–458. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Han, J.; Byun, J.; Park, H.; Park, E.; Chong, Y.H.; Cho, M.-S.; Kang, J.L. Inhibiting Mer Receptor Tyrosine Kinase Activation and Enhances Inflammatory Responses in Lipopolysaccharide-Induced Acute Lung Injury. J. Leukoc. Biol. 2012, 91, 921–932. [Google Scholar] [CrossRef]
- Liau, N.P.D.; Laktyushin, A.; Lucet, I.S.; Murphy, J.M.; Yao, S.; Whitlock, E.; Callaghan, K.; Nicola, N.A.; Kershaw, N.J.; Babon, J.J. The Molecular Basis of JAK/STAT Inhibition by SOCS1. Nat. Commun. 2018, 9, 1558. [Google Scholar] [CrossRef] [PubMed]
- Rui, L.; Yuan, M.; Frantz, D.; Shoelson, S.; White, M.F. SOCS-1 and SOCS-3 Block Insulin Signaling by Ubiquitin-Mediated Degradation of IRS1 and IRS2. J. Biol. Chem. 2002, 277, 42394–42398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, B.; Thorp, E.B.; Doran, A.C.; Subramanian, M.; Sansbury, B.E. MerTK Cleavage Limits Proresolving Mediator Biosynthesis and Exacerbates Tissue Inflammation. Proc. Natl. Acad. Sci. USA 2016, 113, 6526–6531. [Google Scholar] [CrossRef] [Green Version]
- Kedage, V.; Ellerman, D.; Chen, Y.; Liang, W.; Borneo, J.; Wu, Y.; Yan, M. Harnessing MerTK Agonism for Targeted Therapeutics. mAbs 2020, 12, e1685832. [Google Scholar] [CrossRef] [Green Version]
- Doran, A.C.; Yurdagul, A.; Tabas, I. Efferocytosis in Health and Disease. Nat. Rev. Immunol. 2020, 20, 254–267. [Google Scholar] [CrossRef]
- Mocarski, E.S.; Upton, J.W.; Kaiser, W.J. Viral Infection and the Evolution of Caspase 8-Regulated Apoptotic and Necrotic Death Pathways. Nat. Rev. Immunol. 2011, 12, 79–88. [Google Scholar] [CrossRef] [Green Version]
- Kaiser, W.J.; Sridharan, H.; Huang, C.; Mandal, P.; Upton, J.W.; Gough, P.J.; Sehon, C.A.; Marquis, R.W.; Bertin, J.; Mocarski, E.S. Toll-like Receptor 3-Mediated Necrosis via TRIF, RIP3, and MLKL. J. Biol. Chem. 2013, 288, 31268–31279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank, D.; Vince, J.E. Pyroptosis versus Necroptosis: Similarities, Differences, and Crosstalk. Cell Death Differ. 2019, 26, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Viganò, E.; Diamond, C.E.; Spreafico, R.; Balachander, A.; Sobota, R.M.; Mortellaro, A. Human Caspase-4 and Caspase-5 Regulate the One-Step Non-Canonical Inflammasome Activation in Monocytes. Nat. Commun. 2015, 6, 8761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baroja-Mazo, A.; Martín-Sánchez, F.; Gomez, A.I.; Martínez, C.M.; Amores-Iniesta, J.; Compan, V.; Barberà-Cremades, M.; Yagüe, J.; Ruiz-Ortiz, E.; Antón, J.; et al. The NLRP3 Inflammasome Is Released as a Particulate Danger Signal That Amplifies the Inflammatory Response. Nat. Immunol. 2014, 15, 738–748. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Wang, K.; Liu, W.; She, Y.; Sun, Q.; Shi, J.; Sun, H.; Wang, D.-C.; Shao, F. Pore-Forming Activity and Structural Autoinhibition of the Gasdermin Family. Nature 2016, 535, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, P.; Bai, T.; Sun, Y. Mechanisms of Ferroptosis and Relations with Regulated Cell Death: A Review. Front. Physiol. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Imamura, R.; Motani, K.; Kushiyama, H.; Nagata, S.; Suda, T. Pyroptotic Cells Externalize Eat-Me and Release Find-Me Signals and Are Efficiently Engulfed by Macrophages. Int. Immunol. 2013, 25, 363–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klöditz, K.; Fadeel, B. Three Cell Deaths and a Funeral: Macrophage Clearance of Cells Undergoing Distinct Modes of Cell Death. Cell Death Discov. 2019, 5, 1–9. [Google Scholar] [CrossRef]
- Lu, J.; Shi, W.; Liang, B.; Chen, C.; Wu, R.; Lin, H.; Zhang, Y.; Han, J. Efficient Engulfment of Necroptotic and Pyroptotic Cells by Nonprofessional and Professional Phagocytes. Cell Discov. 2019, 5, 39. [Google Scholar] [CrossRef]
- Rymut, N.; Heinz, J.; Sadhu, S.; Hosseini, Z.; Riley, C.O.; Marinello, M.; Maloney, J.; MacNamara, K.C.; Spite, M.; Fredman, G. Resolvin D1 Promotes Efferocytosis in Aging by Limiting Senescent Cell-Induced MerTK Cleavage. FASEB J. 2020, 34, 597–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.-Y.; Zhen, Z.-D.; Fan, D.-Y.; Qin, C.-F.; Han, D.-S.; Zhou, H.-N.; Wang, P.-G.; An, J. Axl Deficiency Promotes the Neuroinvasion of Japanese Encephalitis Virus by Enhancing IL-1α Production from Pyroptotic Macrophages. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [PubMed]
- Kaczmarek, A.; Vandenabeele, P.; Krysko, D.V. Necroptosis: The Release of Damage-Associated Molecular Patterns and Its Physiological Relevance. Immunity 2013, 38, 209–223. [Google Scholar] [CrossRef] [Green Version]
- Fink, S.L.; Cookson, B.T. Caspase-1-Dependent Pore Formation during Pyroptosis Leads to Osmotic Lysis of Infected Host Macrophages. Cell Microbiol. 2006, 8, 1812–1825. [Google Scholar] [CrossRef] [PubMed]
- Man, S.M.; Karki, R.; Kanneganti, T.-D. Molecular Mechanisms and Functions of Pyroptosis, Inflammatory Caspases and Inflammasomes in Infectious Diseases. Immunol. Rev. 2017, 277, 61–75. [Google Scholar] [CrossRef] [Green Version]
- Codo, A.C.; Saraiva, A.C.; Dos Santos, L.L.; Visconde, M.F.; Gales, A.C.; Zamboni, D.S.; Medeiros, A.I. Inhibition of Inflammasome Activation by a Clinical Strain of Klebsiella Pneumoniae Impairs Efferocytosis and Leads to Bacterial Dissemination. Cell Death Dis. 2018, 9, 1182. [Google Scholar] [CrossRef]
- Jondle, C.N.; Gupta, K.; Mishra, B.B.; Sharma, J. Klebsiella Pneumoniae Infection of Murine Neutrophils Impairs Their Efferocytic Clearance by Modulating Cell Death Machinery. PLoS Pathog. 2018, 14, e1007338. [Google Scholar] [CrossRef] [Green Version]
- Zheng, D.J.; Abou Taka, M.; Heit, B. Role of Apoptotic Cell Clearance in Pneumonia and Inflammatory Lung Disease. Pathogens 2021, 10, 134. [Google Scholar] [CrossRef]
- Cai, B.; Fredman, G.; Tabas, I.; Cai, B.; Thorp, E.B.; Doran, A.C.; Sansbury, B.E.; Daemen, M.J.A.P.; Dorweiler, B.; Spite, M.; et al. MerTK Receptor Cleavage Promotes Plaque Necrosis and Defective Resolution in Atherosclerosis MerTK Receptor Cleavage Promotes Plaque Necrosis and Defective Resolution in Atherosclerosis. J. Clin. Investig. 2017, 127, 564–568. [Google Scholar] [CrossRef]
- Wootla, B.; Eriguchi, M.; Rodriguez, M. Is Multiple Sclerosis an Autoimmune Disease? Autoimmune Dis. 2012, 2012. [Google Scholar] [CrossRef]
- Hurtado, B.; Abasolo, N.; Muñoz, X.; García, N.; Benavente, Y.; Rubio, F.; De Frutos, P.G.; Krupinski, J.; Sala, N. Association Study between Polymorphims in GAS6-TAM Genes and Carotid Atherosclerosis. Thromb. Haemost. 2010, 104, 592–598. [Google Scholar] [CrossRef]
- Lampron, A.; Larochelle, A.; Laflamme, N.; Préfontaine, P.; Plante, M.; Sánchez, M.G.; Yong, V.W.; Stys, P.K.; Tremblay, M.-è.; Rivest, S. Inefficient Clearance of Myelin Debris by Microglia Impairs Remyelinating Processes. J. Exp. Med. 2015, 212, 481–495. [Google Scholar] [CrossRef] [Green Version]
- Binder, M.D.; Fox, A.D.; Merlo, D.; Johnson, L.J.; Perera, A.A.; Gresle, M.M.; Laverick, L.; Foo, G.; Fabis-Pedrini, M.J.; Butzkueven, H.; et al. Common and Low Frequency Variants in MERTK Are Independently Associated with Multiple Sclerosis Susceptibility with Discordant Association Dependent Upon. PLoS Genet. 2016, 12, 1–25. [Google Scholar] [CrossRef] [Green Version]
- Ma, G.Z.M.; Stankovich, J.; ANZgene; Kilpatrick, T.J.; Binder, M.D.; Field, J. Polymorphisms in the Receptor Tyrosine Kinase MERTK Gene Are Associated with Multiple Sclerosis Susceptibility. PLoS ONE 2011, 6, e16964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Healy, L.M.; Yan, J.H.; Won, S.; Lin, Y.H.; Touil, H.; Aljarallah, S.; Bar-Or, A.; Antel, J.P. MerTK-Mediated Regulation of Myelin Phagocytosis by Macrophages Generated from Patients with MS. Neurol. Neuroimmunol. Neuroinflamm. 2017, 4, e402. [Google Scholar] [CrossRef] [Green Version]
- Healy, L.M.; Perron, G.; Won, S.; Rezk, A.; Ludwin, S.K.; Moore, C.S.; Hall, J.A.; Bar-or, A.; Jack, P.A. MerTK Is a Functional Regulator of Myelin Phagocytosis by Human Myeloid Cells. J. Immunol. 2016, 196, 3375–3384. [Google Scholar] [CrossRef] [Green Version]
- Williamson, J.M.; Lyons, D.A. Myelin Dynamics Throughout Life: An Ever-Changing Landscape? Front. Cell. Neurosci. 2018, 12, 424. [Google Scholar] [CrossRef]
- Shen, K.; Reichelt, M.; Kyauk, R.V.; Ngu, H.; Shen, Y.-A.A.; Foreman, O.; Modrusan, Z.; Friedman, B.A.; Sheng, M.; Yuen, T.J. Article Multiple Sclerosis Risk Gene Mertk Is Required for Microglial Activation and Subsequent Remyelination Ll Ll Multiple Sclerosis Risk Gene Mertk Is Required for Microglial Activation and Subsequent Remyelination. Cell Rep. 2021, 34, 108835. [Google Scholar] [CrossRef]
- Hamaguchi, M.; Muramatsu, R.; Fujimura, H. Circulating Transforming Growth Factor-SS1 Facilitates Remyelination in the Adult Central Nervous System. eLife 2019, 8, e41869. [Google Scholar] [CrossRef]
- Mirshafiey, A.; Mohsenzadegan, M. TGF-ß as a Promising Option in the Treatment of Multiple Sclerosis. Neuropharmacology 2009, 56, 929–936. [Google Scholar] [CrossRef]
- Natrajan, M.S.; Komori, M.; Kosa, P.; Johnson, K.R.; Wu, T.; Robin, J.M.; Bielekova, B. Pioglitazone Regulates Myelin Phagocytosis and Multiple Sclerosis Monocytes. Ann. Clin. Transl. Neurol. 2015, 2, 1071–1084. [Google Scholar] [CrossRef]
- Linton, M.F.; Babaev, V.R.; Huang, J.; Linton, E.F.; Tao, H.; Yancey, P.G. Macrophage Apoptosis and Efferocytosis in the Pathogenesis of Atherosclerosis. Circ. J. 2017, 80, 2259–2268. [Google Scholar] [CrossRef] [Green Version]
- Pulanco, M.C.; Cosman, J.; Ho, M.; Huynh, J.; Fing, K.; Fraser, D.A. Complement Protein C1q Enhances Macrophage Foam Cell Survival and Efferocytosis. J. Immunol. 2017, 198, 472–480. [Google Scholar] [CrossRef] [Green Version]
- Yurdagul Jr, A.; Finney, A.C.; Woolard, M.D.; Orr, A.W. The Arterial Microenvironment: The Where and Why of Atherosclerosis. Biochem. J. 2016, 473, 1281–1295. [Google Scholar] [CrossRef] [Green Version]
- Thorp, E.; Cui, D.; Schrijvers, D.M.; Kuriakose, G.; Tabas, I. Mertk Receptor Mutation Reduces Efferocytosis Efficiency and Promotes Apoptotic Cell Accumulation and Plaque Necrosis in Atherosclerotic Lesions of Apoe−/− Mice. Arterioscler. Thromb. Vasc. Biol. 2009, 28, 1421–1428. [Google Scholar] [CrossRef] [Green Version]
- Ait-Oufella, H.; Pouresmail, V.; Simon, T.; Blanc-Brude, O.; Kinugawa, K.; Merval, R.; Offenstadt, G.; Lese, G.; Cohen, P.L.; Tedgui, A.; et al. Defective Mer Receptor Tyrosine Kinase Signaling in Bone Marrow Cells Promotes Apoptotic Cell Accumulation and Accelerates Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1429–1431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kojima, Y.; Volkmer, J.; Mckenna, K.; Civelek, M.; Lusis, A.J.; Miller, C.L.; Direnzo, D.; Nanda, V.; Ye, J.; Connolly, A.J.; et al. CD47-Blocking Antibodies Restore Phagocytosis and Prevent Atherosclerosis. Nature 2016, 536, 86–90. [Google Scholar] [CrossRef] [Green Version]
- Alhakamy, N.A.; Ahmed, O.A.A.; Aldawsari, H.M.; Alfaifi, M.Y.; Eid, B.G.; Abdel-Naim, A.B.; Fahmy, U.A. Encapsulation of Lovastatin in Zein Nanoparticles Exhibits Enhanced Apoptotic Activity in HepG2 Cells. Int. J. Mol. Sci. 2019, 20, 5788. [Google Scholar] [CrossRef] [Green Version]
- Burstyn-cohen, T.; Maimon, A. TAM Receptors, Phosphatidylserine, Inflammation, and Cancer. Cell Commun. Signal. 2019, 17, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sather, S.; Kenyon, K.D.; Lefkowitz, J.B.; Liang, X.; Varnum, B.C.; Henson, P.M.; Graham, D.K. A Soluble Form of the Mer Receptor Tyrosine Kinase Inhibits Macrophage Clearance of Apoptotic Cells and Platelet Aggregation. Blood 2007, 109, 1026–1033. [Google Scholar] [CrossRef]
- Myers, K.V.; Amend, S.R.; Pienta, K.J. Targeting Tyro3, Axl and MerTK (TAM Receptors): Implications for Macrophages in the Tumor Microenvironment. Mol. Cancer 2019, 18, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Linger, R.M.; Keating, A.K.; Earp, H.S.; Graham, D.K. TAM Receptor Tyrosine Kinases: Biologic Functions, Signaling, and Potential Therapeutic Targeting in Human Cancer. Adv. Cancer Res. 2008, 100, 35–83. [Google Scholar] [CrossRef] [PubMed]
- Hucthagowder, V.; Meyer, R.; Mullins, C.; Nagarajan, R.; DiPersio, J.F.; Vij, R.; Tomasson, M.H.; Kulkarni, C. Resequencing Analysis of the Candidate Tyrosine Kinase and RAS Pathway Gene Families in Multiple Myeloma. Cancer Genet. 2013, 205, 474–478. [Google Scholar] [CrossRef] [Green Version]
- Greenman, C.; Stephens, P.; Smith, R.; Dalgliesh, G.L.; Hunter, C.; Bignell, G.; Davies, H.; Teague, J.; Butler, A.; Edkins, S.; et al. Patterns of Somatic Mutation in Human Cancer Genomes. Nature 2007, 446, 153–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, R.S.; Jacobsen, K.M.; Wofford, A.M.; Deryckere, D.; Stanford, J.; Prieto, A.L.; Redente, E.; Sandahl, M.; Hunter, D.M.; Strunk, K.E.; et al. MerTK Inhibition in Tumor Leukocytes Decreases Tumor Growth and Metastasis. J. Clin. Investig. 2013, 123, 3231–3242. [Google Scholar] [CrossRef] [Green Version]
- Sinik, L.; Minson, K.A.; Tentler, J.J.; Carrico, J.; Bagby, S.M.; Robinson, W.A.; Kami, R.; Burstyn-cohen, T.; Eckhardt, S.G.; Wang, X.; et al. Inhibition of MERTK Promotes Suppression of Tumor Growth in BRAF Mutant and BRAF Wild-Type Melanoma. Mol. Cancer Therapy 2019, 18, 278–289. [Google Scholar] [CrossRef] [Green Version]
- Lotsberg, M.L.; Wnuk-lipinska, K.; Terry, S.; Tan, T.Z.; Lu, N.; Trachsel-moncho, L.; Røsland, G.V.; Siraji, M.I.; Hellesøy, M.; Rayford, A.; et al. AXL Targeting Abrogates Autophagic Flux and Induces Immunogenic Cell Death in Drug-Resistant Cancer Cells. J. Thorac. Oncol. 2020, 15, 973–999. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Fei, M.; Zhang, G.; Liang, W.; Lin, W.; Wu, Y.; Piskol, R.; Ridgway, J. Blockade of the Phagocytic Receptor MerTK on Tumor- Associated Macrophages Enhances P2X7R-Dependent STING Activation by Tumor-Derived CGAMP Article Blockade of the Phagocytic Receptor MerTK on Tumor-Associated Macrophages Enhances P2X7R- Dependent STING A. Immunity 2020, 52, 1–17. [Google Scholar] [CrossRef]
- Sancho, D.; Mourão-Sá, D.; Joffre, O.P.; Schulz, O.; Rogers, N.C.; Pennington, D.J.; Carlyle, J.R.; Reis e Sousa, C. Tumor Therapy in Mice via Antigen Targeting to a Novel, DC-Restricted C-Type Lectin. J. Clin. Investig. 2008, 118, 2098–2110. [Google Scholar] [CrossRef] [PubMed]
- Canton, J.; Blees, H.; Henry, C.M.; Buck, M.D.; Schulz, O.; Rogers, N.C.; Childs, E.; Zelenay, S.; Rhys, H.; Domart, M.-C.; et al. The Receptor DNGR-1 Signals for Phagosomal Rupture to Promote Cross-Presentation of Dead-Cell-Associated Antigens. Nat. Immunol. 2021, 22, 140–153. [Google Scholar] [CrossRef]
- Sancho, D.; Joffre, O.P.; Keller, A.M.; Rogers, N.C.; Martínez, D.; Hernanz-Falcón, P.; Rosewell, I.; Reis e Sousa, C. Identification of a Dendritic Cell Receptor That Couples Sensing of Necrosis to Immunity. Nature 2009, 458, 899–903. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, D.; Stashko, M.A.; DeRyckere, D.; Hunter, D.; Kireev, D.; Miley, M.J.; Cummings, C.; Lee, M.; Norris-Drouin, J.; et al. Pseudo-Cyclization through Intramolecular Hydrogen Bond Enables Discovery of Pyridine Substituted Pyrimidines as New Mer Kinase Inhibitors. J. Med. Chem. 2013, 56, 9683–9692. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Type | Receptors | Citations |
|---|---|---|
| Tissue-Resident Macrophages 1 | MERTK 2, Axl 2, TIM-4, Stabilin 1, Stabilin 2 2, αvβ5, αxβ2, CD36, SCARF-1, LRP-1 | [66,67,68,69,70,71,72,73,74,75,76] |
| Bone Marrow Derived Macrophages | MERTK 2, Axl 2, TIM-4, Stabilin 1, Stabilin 2, αvβ5, αvβ3, αxβ2, CD36, SCARF-1, LRP-1 | [66,67,77,78,79,80,81,82,83] |
| Dendritic cells | Tyro3, Axl, MERTK 2, TIM-4 2, Stabilin 1, αvβ5, SCARF-1 | [80,84,85,86,87] |
| Microglia | Axl, MERTK, TIM-4, Stabilin 1, αvβ5, αvβ3, BAI-1 | [66,79,88,89,90,91,92] |
| Kidney Tubule Epithelial Cells | TIM-1 | [93] |
| Retinal Pigment Epithelium Cells | MERTK, αvβ5 | [94,95] |
| Myoblasts 3 | BAI-1 | [96] |
| Osteoclasts | BAI-1, TIM-4, Stabilin 1 | [97] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lam, A.L.; Heit, B. Having an Old Friend for Dinner: The Interplay between Apoptotic Cells and Efferocytes. Cells 2021, 10, 1265. https://doi.org/10.3390/cells10051265
Lam AL, Heit B. Having an Old Friend for Dinner: The Interplay between Apoptotic Cells and Efferocytes. Cells. 2021; 10(5):1265. https://doi.org/10.3390/cells10051265
Chicago/Turabian StyleLam, Austin Le, and Bryan Heit. 2021. "Having an Old Friend for Dinner: The Interplay between Apoptotic Cells and Efferocytes" Cells 10, no. 5: 1265. https://doi.org/10.3390/cells10051265
APA StyleLam, A. L., & Heit, B. (2021). Having an Old Friend for Dinner: The Interplay between Apoptotic Cells and Efferocytes. Cells, 10(5), 1265. https://doi.org/10.3390/cells10051265