
Multifactorial Pathogenic Processes of Retinal Ganglion Cell Degeneration in Glaucoma towards Multi-Target Strategies for Broader Treatment Effects

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Complex Processes of Glaucomatous Neurodegeneration for Translation into Disease-Modifying Treatments to Preserve Neuron Survival and Visual Function
2.1. Research Approaches to Glaucomatous Neurodegeneration
2.2. Converging Etiological Paths to Evaluate for Unified Treatment Strategies
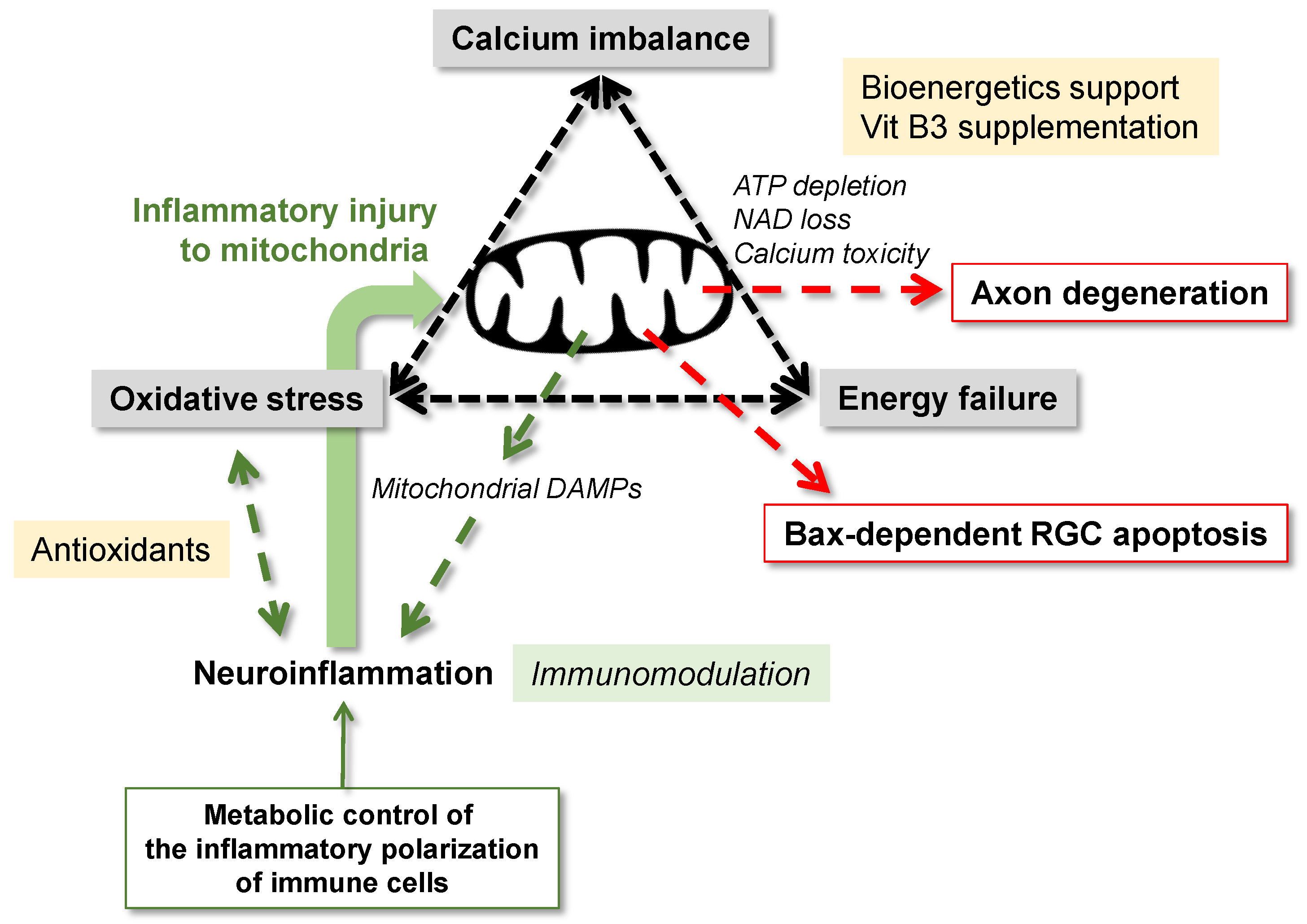
3. Neurodegenerative Consequences of Mitochondrial Dysfunction
3.1. Mitochondrial Dysfunction and Metabolic Failure
3.2. Mitochondrial Dysfunction and Oxidative Stress
3.3. Mitochondrial Dysfunction and Disturbed Calcium Homeostasis
3.4. Mitochondria in RGC Degeneration
3.4.1. Mitochondria in the Regulation of RGC Apoptosis
3.4.2. Mitochondria in the Regulation of Axon Degeneration
3.4.3. MAPKs in Signaling for RGC Degeneration
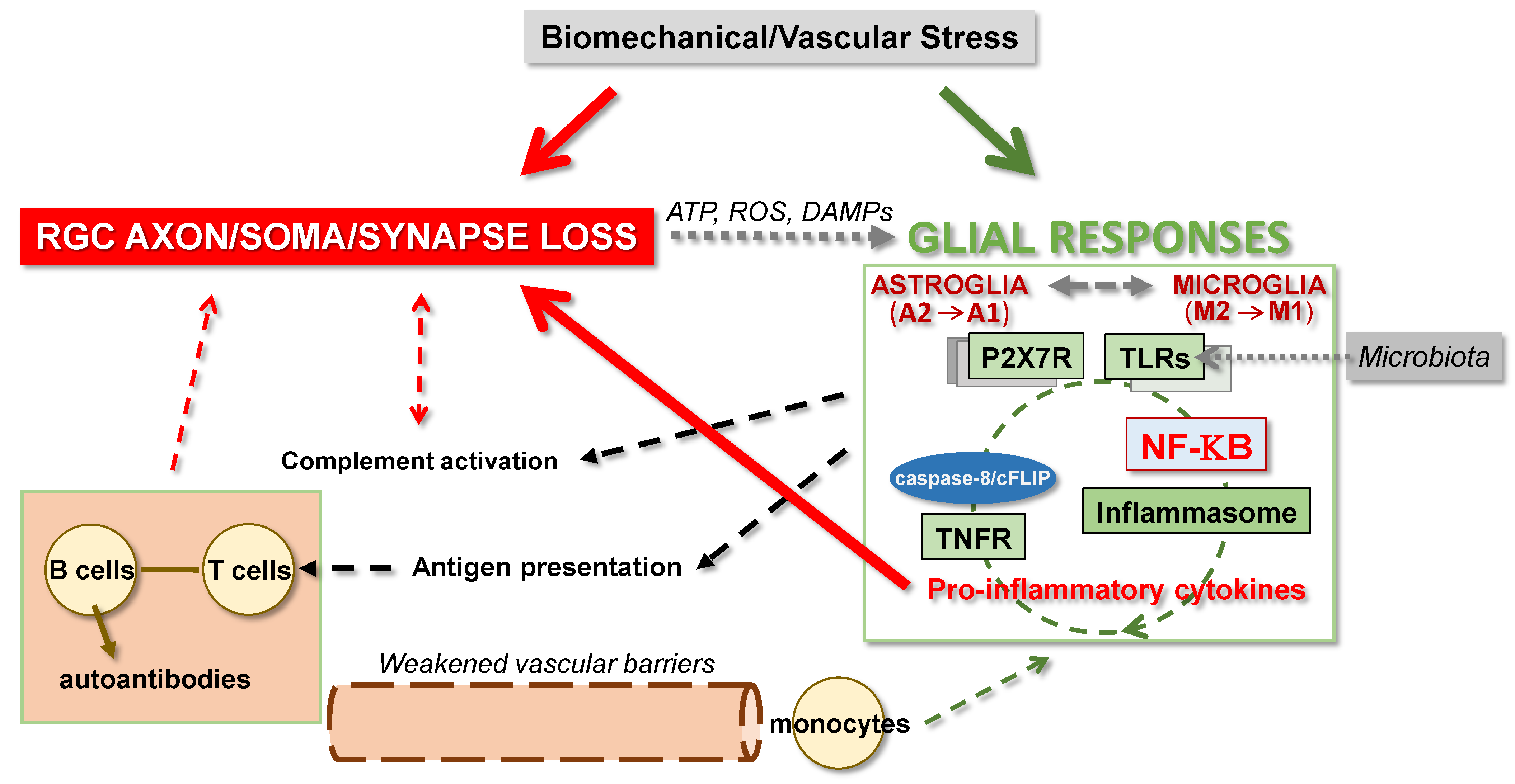
4. Glia-Driven Neuroinflammation in Glaucoma
4.1. Glial Responses to Glaucoma-Related Stress and Injury
4.2. Glial Interactions for Neuroinflammation
4.3. Interplay between Mitochondrial Dysfunction and Neuroinflammation
4.4. Molecular Players of Neuroinflammation in Glaucoma
4.4.1. Transcriptional Regulation of Glia-Driven Neuroinflammation
4.4.2. Other Regulators of Neuroinflammation
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Syc-Mazurek, S.B.; Libby, R.T. Axon injury signaling and compartmentalized injury response in glaucoma. Prog. Retin. Eye Res. 2019, 73, 100769. [Google Scholar] [CrossRef]
- Tezel, G. A broad perspective on the molecular regulation of retinal ganglion cell degeneration in glaucoma. Prog. Brain Res. 2020, 256, 49–77. [Google Scholar]
- Libby, R.T.; Gould, D.B.; Anderson, M.G.; John, S.W. Complex genetics of glaucoma susceptibility. Annu. Rev. Genom. Hum. Genet. 2005, 6, 15–44. [Google Scholar] [CrossRef] [Green Version]
- Wiggs, J.L.; Pasquale, L.R. Genetics of glaucoma. Hum. Mol. Genet. 2017, 26, R21–R27. [Google Scholar] [CrossRef] [PubMed]
- Quigley, H.A.; Broman, A.T. The number of people with glaucoma worldwide in 2010 and 2020. Br. J. Ophthalmol. 2006, 90, 262–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tham, Y.C.; Li, X.; Wong, T.Y.; Quigley, H.A.; Aung, T.; Cheng, C.Y. Global prevalence of glaucoma and projections of glaucoma burden through 2040: A systematic review and meta-analysis. Ophthalmology 2014, 121, 2081–2090. [Google Scholar] [CrossRef]
- Whitmore, A.V.; Libby, R.T.; John, S.W. Glaucoma: Thinking in new ways-a role for autonomous axonal self-destruction and other compartmentalised processes? Prog. Retin. Eye Res. 2005, 24, 639–662. [Google Scholar] [CrossRef] [PubMed]
- Howell, G.R.; Libby, R.T.; Jakobs, T.C.; Smith, R.S.; Phalan, F.C.; Barter, J.W.; Barbay, J.M.; Marchant, J.K.; Manesh, N.; Porciatti, V.; et al. Axons of retinal ganglion cells are insulted in the optic nerve early in DBA/2J glaucoma. J. Cell Biol. 2007, 179, 1523–1537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buckingham, B.P.; Inman, D.M.; Lambert, W.; Oglesby, E.; Calkins, D.J.; Steele, M.R.; Vetter, M.L.; Marsh-Armstrong, N.; Horener, P.J. Progressive ganglion cell degeneration precedes neuronal loss in a mouse model of glaucoma. J. Neurosci. 2008, 28, 2735–2744. [Google Scholar] [CrossRef] [Green Version]
- Libby, R.T.; Li, Y.; Savinova, O.V.; Barter, J.; Smith, R.S.; Nickells, R.W.; John, S.W. Susceptibility to neurodegeneration in a glaucoma is modified by bax gene dosage. PLoS Genet. 2005, 1, e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beirowski, B.; Babetto, E.; Coleman, M.P.; Martin, K.R. The WldS gene delays axonal but not somatic degeneration in a rat glaucoma model. Eur. J. Neurosci. 2008, 28, 1166–1179. [Google Scholar] [CrossRef] [PubMed]
- Ryskamp, D.A.; Witkovsky, P.; Barabas, P.; Huang, W.; Koehler, C.; Akimov, N.P.; Lee, S.H.; Chauhan, S.; Xing, W.; Renteria, R.C.; et al. The polymodal ion channel transient receptor potential vanilloid 4 modulates calcium flux, spiking rate, and apoptosis of mouse retinal ganglion cells. J. Neurosci. 2011, 31, 7089–7101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, J.; Lim, J.C.; Lu, W.; Beckel, J.M.; Macarak, E.J.; Laties, A.M.; Mitchell, C.H. Neurons respond directly to mechanical deformation with pannexin-mediated ATP release and autostimulation of P2X7 receptors. J. Physiol. 2012, 590, 2285–2304. [Google Scholar] [CrossRef] [Green Version]
- Sappington, R.M.; Sidorova, T.; Ward, N.J.; Chakravarthy, R.; Ho, K.W.; Calkins, D.J. Activation of transient receptor potential vanilloid-1 (TRPV1) influences how retinal ganglion cell neurons respond to pressure-related stress. Channels 2015, 9, 102–113. [Google Scholar] [CrossRef]
- Dvoriantchikova, G.; Pronin, A.; Kurtenbach, S.; Toychiev, A.; Chou, T.H.; Yee, C.W.; Prindeville, B.; Tayou, J.; Porciatti, V.; Sagdullaev, B.T.; et al. Pannexin 1 sustains the electrophysiological responsiveness of retinal ganglion cells. Sci. Rep. 2018, 8, 5797. [Google Scholar] [CrossRef] [Green Version]
- Schlamp, C.L.; Johnson, E.C.; Li, Y.; Morrison, J.C.; Nickells, R.W. Changes in Thy1 gene expression associated with damaged retinal ganglion cells. Mol. Vis. 2001, 7, 192–201. [Google Scholar] [PubMed]
- Huang, W.; Fileta, J.; Guo, Y.; Grosskreutz, C.L. Downregulation of Thy1 in retinal ganglion cells in experimental glaucoma. Curr. Eye Res. 2006, 31, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Soto, I.; Oglesby, E.; Buckingham, B.P.; Son, J.L.; Roberson, E.D.; Steele, M.R.; Inman, D.M.; Vetter, M.L.; Horner, P.J.; Marsh-Armstrong, N. Retinal ganglion cells downregulate gene expression and lose their axons within the optic nerve head in a mouse glaucoma model. J. Neurosci. 2008, 28, 548–561. [Google Scholar] [CrossRef] [Green Version]
- Pelzel, H.R.; Schlamp, C.L.; Waclawski, M.; Shaw, M.K.; Nickells, R.W. Silencing of Fem1cR3 gene expression in the DBA/2J mouse precedes retinal ganglion cell death and is associated with histone deacetylase activity. Investig. Ophthalmol. Vis. Sci. 2012, 53, 1428–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, P.A.; Howell, G.R.; Barbay, J.M.; Braine, C.E.; Sousa, G.L.; John, S.W.; Morgan, J.E. Retinal ganglion cell dendritic atrophy in DBA/2J glaucoma. PLoS ONE 2013, 8, e72282. [Google Scholar] [CrossRef] [PubMed]
- Della Santina, L.; Inman, D.M.; Lupien, C.B.; Horner, P.J.; Wong, R.O. Differential progression of structural and functional alterations in distinct retinal ganglion cell types in a mouse model of glaucoma. J. Neurosci. 2013, 33, 17444–17457. [Google Scholar] [CrossRef]
- Pang, J.J.; Frankfort, B.J.; Gross, R.L.; Wu, S.M. Elevated intraocular pressure decreases response sensitivity of inner retinal neurons in experimental glaucoma mice. Proc. Natl. Acad. Sci. USA 2015, 112, 2593–2598. [Google Scholar] [CrossRef] [Green Version]
- Harder, J.M.; Braine, C.E.; Williams, P.A.; Zhu, X.; MacNicoll, K.H.; Sousa, G.L.; Buchanan, R.; Smith, R.S.; Libby, R.T.; Howell, G.R.; et al. Early immune responses are independent of RGC dysfunction in glaucoma with complement component C3 being protective. Proc. Natl. Acad. Sci. USA 2017, 114, E3839–E3848. [Google Scholar] [CrossRef] [Green Version]
- Berry, R.H.; Qu, J.; John, S.W.; Howell, G.R.; Jakobs, T.C. Synapse loss and dendrite remodeling in a mouse model of glaucoma. PLoS ONE 2015, 10, e0144341. [Google Scholar] [CrossRef] [Green Version]
- El-Danaf, R.N.; Huberman, A.D. Characteristic patterns of dendritic remodeling in early-stage glaucoma: Evidence from genetically identified retinal ganglion cell types. J. Neurosci. 2015, 35, 2329–2343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tribble, J.R.; Vasalauskaite, A.; Redmond, T.; Young, R.D.; Hassan, S.; Fautsch, M.P.; Sengpiel, F.; Williams, P.A.; Morgan, J.E. Midget retinal ganglion cell dendritic and mitochondrial degeneration is an early feature of human glaucoma. Brain Commun. 2019, 1, fcz035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aydin, R.; Baris, M.; Durmaz-Engin, C.; Al-Aswad, L.A.; Blumberg, D.M.; Cioffi, G.A.; Liebmann, J.M.; Tezel, T.H.; Tezel, G. Early localized alterations of the retinal inner plexiform layer in association with visual field worsening in glaucoma patients. PLoS ONE 2021, 16, e0247401. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, M.; Rodriguez, F.D.; Sharma, S.C.; Vecino, E. Immunohistochemical changes in rat retinas at various time periods of elevated intraocular pressure. Mol. Vis. 2009, 15, 2696–2709. [Google Scholar] [PubMed]
- Akopian, A.; Kumar, S.; Ramakrishnan, H.; Viswanathan, S.; Bloomfield, S.A. Amacrine cells coupled to ganglion cells via gap junctions are highly vulnerable in glaucomatous mouse retinas. J. Comp. Neurol. 2019, 527, 159–173. [Google Scholar] [CrossRef]
- Gupta, N.; Ang, L.C.; Noel de Tilly, L.; Bidaisee, L.; Yucel, Y.H. Human glaucoma and neural degeneration in intracranial optic nerve, lateral geniculate nucleus, and visual cortex. Br. J. Ophthalmol. 2006, 90, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Ly, T.; Gupta, N.; Weinreb, R.N.; Kaufman, P.L.; Yucel, Y.H. Dendrite plasticity in the lateral geniculate nucleus in primate glaucoma. Vis. Res. 2011, 51, 243–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.Y.; Jeong, H.J.; Lee, J.H.; Kim, Y.J.; Kim, E.Y.; Kim, Y.Y.; Ryu, T.; Cho, Z.H.; Kim, Y.B. An investigation of lateral geniculate nucleus volume in patients with primary open-angle glaucoma using 7 tesla magnetic resonance imaging. Investig. Ophthalmol. Vis. Sci. 2014, 55, 3468–3476. [Google Scholar] [CrossRef] [Green Version]
- Schlamp, C.L.; Li, Y.; Dietz, J.A.; Janssen, K.T.; Nickells, R.W. Progressive ganglion cell loss and optic nerve degeneration in DBA/2J mice is variable and asymmetric. BMC Neurosci. 2006, 7, 66. [Google Scholar] [CrossRef] [Green Version]
- Crish, S.D.; Sappington, R.M.; Inman, D.M.; Horner, P.J.; Calkins, D.J. Distal axonopathy with structural persistence in glaucomatous neurodegeneration. Proc. Natl. Acad. Sci. USA 2010, 107, 5196–5201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tezel, G. Immune regulation toward immunomodulation for neuroprotection in glaucoma. Curr. Opin. Pharmacol. 2013, 13, 23–31. [Google Scholar] [CrossRef] [Green Version]
- Mac Nair, C.E.; Nickells, R.W. Neuroinflammation in glaucoma and optic nerve damage. Prog. Mol. Biol. Transl. Sci. 2015, 134, 343–363. [Google Scholar]
- Williams, P.A.; Marsh-Armstrong, N.; Howell, G.R. Lasker/IRRF Initiative on Astrocytes and Glaucomatous Neurodegeneration Participants. Neuroinflammation in glaucoma: A new opportunity. Exp. Eye Res. 2017, 157, 20–27. [Google Scholar] [CrossRef] [Green Version]
- Baris, M.; Tezel, G. Immunomodulation as a neuroprotective strategy for glaucoma treatment. Curr. Ophthalmol. Rep. 2019, 7, 160–169. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.H.; Snook, J.D.; Ostrin, E.J.; Kim, S.; Chen, R.; Frankfort, B.J. Transcriptomic profiles of retinal ganglion cells are defined by the magnitude of intraocular pressure elevation in adult mice. Sci. Rep. 2019, 9, 2594. [Google Scholar] [CrossRef] [Green Version]
- Tribble, J.R.; Harder, J.M.; Williams, P.A.; John, S.W.M. Ocular hypertension suppresses homeostatic gene expression in optic nerve head microglia of DBA/2 J mice. Mol. Brain. 2020, 13, 81. [Google Scholar] [CrossRef] [PubMed]
- Tezel, G. A decade of proteomics studies of glaucomatous neurodegeneration. Proteom. Clin. Appl. 2014, 8, 154–167. [Google Scholar] [CrossRef] [Green Version]
- Leruez, S.; Marill, A.; Bresson, T.; de Saint Martin, G.; Buisset, A.; Muller, J.; Tessier, L.; Gadras, C.; Verny, C.; Gohier, P.; et al. A metabolomics profiling of glaucoma points to mitochondrial dysfunction, senescence, and polyamines deficiency. Investig. Ophthalmol. Vis. Sci. 2018, 59, 4355–4361. [Google Scholar] [CrossRef]
- Chauhan, M.Z.; Valencia, A.K.; Piqueras, M.C.; Enriquez-Algeciras, M.; Bhattacharya, S.K. Optic nerve lipidomics reveal impaired glucosylsphingosine lipids pathway in glaucoma. Investig. Ophthalmol. Vis. Sci. 2019, 60, 1789–1798. [Google Scholar] [CrossRef]
- Bader, V.; Winklhofer, K.F. Mitochondria at the interface between neurodegeneration and neuroinflammation. Semin. Cell Dev. Biol. 2020, 99, 163–171. [Google Scholar] [CrossRef] [PubMed]
- van Horssen, J.; van Schaik, P.; Witte, M. Inflammation and mitochondrial dysfunction: A vicious circle in neurodegenerative disorders? Neurosci. Lett. 2019, 710, 132931. [Google Scholar] [CrossRef]
- Baltan, S.; Inman, D.M.; Danilov, C.A.; Morrison, R.S.; Calkins, D.J.; Horner, P.J. Metabolic vulnerability disposes retinal ganglion cell axons to dysfunction in a model of glaucomatous degeneration. J. Neurosci. 2010, 30, 5644–5652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrientos, S.A.; Martinez, N.W.; Yoo, S.; Jara, J.S.; Zamorano, S.; Hetz, C.; Twiss, J.L.; Alvarez, J.; Court, F.A. Axonal degeneration is mediated by the mitochondrial permeability transition pore. J. Neurosci. 2011, 31, 966–978. [Google Scholar] [CrossRef] [PubMed]
- Chidlow, G.; Wood, J.P.M.; Casson, R.J. Investigations into hypoxia and oxidative stress at the optic nerve head in a rat model of glaucoma. Front. Neurosci. 2017, 11, 478. [Google Scholar] [CrossRef] [PubMed]
- Pease, M.E.; McKinnon, S.J.; Quigley, H.A.; Kerrigan-Baumrind, L.A.; Zack, D.J. Obstructed axonal transport of BDNF and its receptor TrkB in experimental glaucoma. Investig. Ophthalmol. Vis. Sci. 2000, 41, 764–774. [Google Scholar]
- Johnson, E.C.; Guo, Y.; Cepurna, W.O.; Morrison, J.C. Neurotrophin roles in retinal ganglion cell survival: Lessons from rat glaucoma models. Exp. Eye Res. 2009, 88, 808–815. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.T.; Medress, Z.A.; Barres, B.A. Axon degeneration: Molecular mechanisms of a self-destruction pathway. J. Cell Biol. 2012, 196, 7–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maes, M.E.; Schlamp, C.L.; Nickells, R.W. BAX to basics: How the BCL2 gene family controls the death of retinal ganglion cells. Prog. Retin. Eye Res. 2017, 57, 1–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pease, M.E.; Zack, D.J.; Berlinicke, C.; Bloom, K.; Cone, F.; Wang, Y.; Klein, R.L.; Hauswirth, W.W.; Quigley, H.A. Effect of CNTF on retinal ganglion cell survival in experimental glaucoma. Investig. Ophthalmol. Vis. Sci. 2009, 50, 2194–2200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambiase, A.; Aloe, L.; Centofanti, M.; Parisi, V.; Bao, S.N.; Mantelli, F.; Colafrancesco, V.; Manni, G.L.; Bucci, M.G.; Bonini, S.; et al. Experimental and clinical evidence of neuroprotection by nerve growth factor eye drops: Implications for glaucoma. Proc. Natl. Acad. Sci. USA 2009, 106, 13469–13474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colafrancesco, V.; Parisi, V.; Sposato, V.; Rossi, S.; Russo, M.A.; Coassin, M.; Lambiase, A.; Aloe, L. Ocular application of nerve growth factor protects degenerating retinal ganglion cells in a rat model of glaucoma. J. Glaucoma 2011, 20, 100–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrari, M.P.; Mantelli, F.; Sacchetti, M.; Antonangeli, M.I.; Cattani, F.; D’Anniballe, G.; Sinigaglia, F.; Ruffini, P.A.; Lambiase, A. Safety and pharmacokinetics of escalating doses of human recombinant nerve growth factor eye drops in a double-masked, randomized clinical trial. BioDrugs 2014, 28, 275–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osborne, N.N.; Nunez-Alvarez, C.; Del Olmo-Aguado, S.; Merrayo-Lloves, J. Visual light effects on mitochondria: The potential implications in relation to glaucoma. Mitochondrion 2017, 36, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Casson, R.J.; Chidlow, G.; Crowston, J.G.; Williams, P.A.; Wood, J.P.M. Retinal energy metabolism in health and glaucoma. Prog. Retin. Eye Res. 2020, 10, 100881. [Google Scholar] [CrossRef] [PubMed]
- Takihara, Y.; Inatani, M.; Eto, K.; Inoue, T.; Kreymerman, A.; Miyake, S.; Ueno, S.; Nagaya, M.; Nakanishi, A.; Iwao, K.; et al. In vivo imaging of axonal transport of mitochondria in the diseased and aged mammalian CNS. Proc. Natl. Acad. Sci. USA 2015, 112, 10515–10520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ju, W.K.; Kim, K.Y.; Duong-Polk, K.X.; Lindsey, J.D.; Ellisman, M.H.; Weinreb, R.N. Increased optic atrophy type 1 expression protects retinal ganglion cells in a mouse model of glaucoma. Mol. Vis. 2010, 16, 1331–1342. [Google Scholar]
- Kim, K.Y.; Perkins, G.A.; Shim, M.S.; Bushong, E.; Alcasid, N.; Ju, S.; Ellisman, M.H.; Weinreb, R.N.; Ju, W.K. DRP1 inhibition rescues retinal ganglion cells and their axons by preserving mitochondrial integrity in a mouse model of glaucoma. Cell Death Dis. 2015, 6, e1839. [Google Scholar] [CrossRef] [PubMed]
- Coughlin, L.; Morrison, R.S.; Horner, P.J.; Inman, D.M. Mitochondrial morphology differences and mitophagy deficit in murine glaucomatous optic nerve. Investig. Ophthalmol. Vis. Sci. 2015, 56, 1437–1446. [Google Scholar] [CrossRef] [Green Version]
- Flammer, J.; Mozaffarieh, M. What is the present pathogenetic concept of glaucomatous optic neuropathy? Surv. Ophthalmol. 2007, 52 (Suppl. 2), S162–S173. [Google Scholar] [CrossRef]
- Schmidl, D.; Garhofer, G.; Schmetterer, L. The complex interaction between ocular perfusion pressure and ocular blood flow-relevance for glaucoma. Exp. Eye Res. 2011, 93, 141–155. [Google Scholar] [CrossRef] [PubMed]
- Wareham, L.K.; Calkins, D.J. The neurovascular unit in glaucomatous neurodegeneration. Front. Cell Dev. Biol. 2020, 8, 452. [Google Scholar] [CrossRef] [PubMed]
- Tekkok, S.B.; Brown, A.M.; Westenbroek, R.; Pellerin, L.; Ransom, B.R. Transfer of glycogen-derived lactate from astrocytes to axons via specific monocarboxylate transporters supports mouse optic nerve activity. J. Neurosci. Res. 2005, 81, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Carreras, F.J.; Aranda, C.J.; Porcel, D.; Rodriguez-Hurtado, F.; Martinez-Agustin, O.; Zarzuelo, A. Expression of glucose transporters in the prelaminar region of the optic-nerve head of the pig as determined by immunolabeling and tissue culture. PLoS ONE 2015, 10, e0128516. [Google Scholar] [CrossRef] [PubMed]
- Saab, A.S.; Tzvetavona, I.D.; Trevisiol, A.; Baltan, S.; Dibaj, P.; Kusch, K.; Mobius, W.; Goetze, B.; Jahn, H.M.; Huang, W.; et al. Oligodendroglial NMDA receptors regulate glucose import and axonal energy metabolism. Neuron 2016, 91, 119–132. [Google Scholar] [CrossRef] [Green Version]
- Vohra, R.; Aldana, B.I.; Bulli, G.; Skytt, D.M.; Waagepetersen, H.; Bergersen, L.H.; Kolko, M. Lactate-mediated protection of retinal ganglion cells. J. Mol. Biol. 2019, 431, 1878–1888. [Google Scholar] [CrossRef] [Green Version]
- Chidlow, G.; Wood, J.P.M.; Sia, P.I.; Casson, R.J. Distribution and activity of mitochondrial proteins in vascular and avascular retinas: Implications for retinal metabolism. Investig. Ophthalmol. Vis. Sci. 2019, 60, 331–344. [Google Scholar] [CrossRef] [Green Version]
- Harun-Or-Rashid, M.; Pappenhagen, N.; Palmer, P.G.; Smith, M.A.; Gevorgyan, V.; Wilson, G.N.; Crish, S.D.; Inman, D.M. Structural and functional rescue of chronic metabolically stressed optic nerves through respiration. J. Neurosci. 2018, 38, 5122–5139. [Google Scholar] [CrossRef]
- Harun-Or-Rashid, M.; Pappenhagen, N.; Zubricky, R.; Coughlin, L.; Jassim, A.H.; Inman, D.M. MCT2 overexpression rescues metabolic vulnerability and protects retinal ganglion cells in two models of glaucoma. Neurobiol. Dis. 2020, 141, 104944. [Google Scholar] [CrossRef]
- Ebneter, A.; Chidlow, G.; Wood, J.P.; Casson, R.J. Protection of retinal ganglion cells and the optic nerve during short-term hyperglycemia in experimental glaucoma. Arch. Ophthalmol. 2011, 129, 1337–1344. [Google Scholar] [CrossRef] [Green Version]
- Shibeeb, O.; Chidlow, G.; Han, G.; Wood, J.P.; Casson, R.J. Effect of subconjunctival glucose on retinal ganglion cell survival in experimental retinal ischaemia and contrast sensitivity in human glaucoma. Clin. Exp. Ophthalmol. 2016, 44, 24–32. [Google Scholar] [CrossRef] [Green Version]
- Casson, R.J.; Han, G.; Ebneter, A.; Chidlow, G.; Glihotra, J.; Newland, H.; Wood, J.P. Glucose-induced temporary visual recovery in primary open-angle glaucoma: A double-blind, randomized study. Ophthalmology 2014, 121, 1203–1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeiffer, T.; Schuster, S.; Bonhoeffer, S. Cooperation and competition in the evolution of ATP-producing pathways. Science 2001, 292, 504–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Hondur, G.; Li, M.; Cai, J.; Klein, J.B.; Kuehn, M.H.; Tezel, G. Proteomics analysis of molecular risk factors in the ocular hypertensive human retina. Investig. Ophthalmol. Vis. Sci. 2015, 56, 5816–5830. [Google Scholar] [CrossRef] [PubMed]
- Tezel, G.; Wax, M.B. Hypoxia-inducible factor 1alpha in the glaucomatous retina and optic nerve head. Arch. Ophthalmol. 2004, 122, 1348–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, P.A.; Harder, J.M.; Foxworth, N.E.; Cochran, K.E.; Philip, V.M.; Porciatti, V.; Smithies, O.; John, S.W. Vitamin B3 modulates mitochondrial vulnerability and prevents glaucoma in aged mice. Science 2017, 355, 756–760. [Google Scholar] [CrossRef] [Green Version]
- Jassim, A.H.; Inman, D.M. Evidence of hypoxic glial cells in a model of ocular hypertension. Investig. Ophthalmol. Vis. Sci. 2019, 60, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Jassim, A.H.; Coughlin, L.; Harun-Or-Rashid, M.; Kang, P.T.; Chen, Y.R.; Inman, D.M. Higher reliance on glycolysis limits glycolytic responsiveness in degenerating glaucomatous optic nerve. Mol. Neurobiol. 2019, 56, 7097–7112. [Google Scholar] [CrossRef] [Green Version]
- Tezel, G. Oxidative stress in glaucomatous neurodegeneration: Mechanisms and consequences. Prog. Retin. Eye Res. 2006, 25, 490–513. [Google Scholar] [CrossRef] [Green Version]
- Harder, J.M.; Guymer, C.; Wood, J.P.M.; Daskalaki, E.; Chidlow, G.; Zhang, C.; Balasubramanian, R.; Cardozo, B.H.; Foxworth, N.E.; Deering, K.E.; et al. Disturbed glucose and pyruvate metabolism in glaucoma with neuroprotection by pyruvate or rapamycin. Proc. Natl. Acad. Sci. USA 2020, 117, 33619–33627. [Google Scholar] [CrossRef]
- Chrysostomou, V.; Rezania, F.; Trounce, I.A.; Crowston, J.G. Oxidative stress and mitochondrial dysfunction in glaucoma. Curr. Opin. Pharmacol. 2013, 13, 12–15. [Google Scholar] [CrossRef]
- Kanamori, A.; Catrinescu, M.M.; Kanamori, N.; Mears, K.A.; Beaubien, R.; Levin, L.A. Superoxide is an associated signal for apoptosis in axonal injury. Brain 2010, 133, 2612–2625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tezel, G.; Yang, X. Caspase-independent component of retinal ganglion cell death, in vitro. Investig. Ophthalmol. Vis. Sci. 2004, 45, 4049–4059. [Google Scholar] [CrossRef] [PubMed]
- Tezel, G.; Yang, X.; Cai, J. Proteomic identification of oxidatively modified retinal proteins in a chronic pressure-induced rat model of glaucoma. Investig. Ophthalmol. Vis. Sci. 2005, 46, 3177–3187. [Google Scholar] [CrossRef] [PubMed]
- Tezel, G.; Yang, X.; Luo, C.; Peng, Y.; Sun, S.L.; Sun, D. Mechanisms of immune system activation in glaucoma: Oxidative stress-stimulated antigen presentation by the retina and optic nerve head glia. Investig. Ophthalmol. Vis. Sci. 2007, 48, 705–714. [Google Scholar] [CrossRef]
- Tezel, G. The immune response in glaucoma: A perspective on the roles of oxidative stress. Exp. Eye Res. 2011, 93, 178–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inman, D.M.; Lambert, W.S.; Calkins, D.J.; Horner, P.J. alpha-Lipoic acid antioxidant treatment limits glaucoma-related retinal ganglion cell death and dysfunction. PLoS ONE 2013, 8, e65389. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Hondur, G.; Tezel, G. Antioxidant treatment limits neuroinflammation in experimental glaucoma. Investig. Ophthalmol. Vis. Sci. 2016, 57, 2344–2354. [Google Scholar] [CrossRef] [Green Version]
- Luo, C.; Yang, X.; Kain, A.D.; Powell, D.W.; Kuehn, M.H.; Tezel, G. Glaucomatous tissue stress and the regulation of immune response through glial toll-like receptor signaling. Investig. Ophthalmol. Vis. Sci. 2010, 51, 5697–5707. [Google Scholar] [CrossRef]
- Lee, D.; Shim, M.S.; Kim, K.Y.; Noh, Y.H.; Kim, H.; Kim, S.Y.; Weinreb, R.N.; Ju, W.K. Coenzyme Q10 inhibits glutamate excitotoxicity and oxidative stress-mediated mitochondrial alteration in a mouse model of glaucoma. Investig. Ophthalmol. Vis. Sci. 2014, 55, 993–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, B.M.; Tian, K.; Pahlitzsch, M.; Brenton, J.; Ravindran, N.; Butt, G.; Malaguarnera, G.; Normando, E.M.; Guo, L.; Cordeiro, M.F. Topical Coenzyme Q10 demonstrates mitochondrial-mediated neuroprotection in a rodent model of ocular hypertension. Mitochondrion 2017, 36, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Medina, J.J.; Rubio-Velazquez, E.; Lopez-Bernal, M.D.; Cobo-Martinez, A.; Zanon-Moreno, V.; Pinazo-Duran, M.D.; Del-Rio-Vellosillo, M. Glaucoma and antioxidants: Review and update. Antioxidants 2020, 9, 1031. [Google Scholar] [CrossRef] [PubMed]
- Tribble, J.R.; Hui, F.; Joe, M.; Bell, K.; Chrysostomou, V.; Crowston, J.G.; Williams, P.A. Targeting diet and exercise for neuroprotection and neurorecovery in glaucoma. Cells 2021, 10, 295. [Google Scholar] [CrossRef]
- Verdin, E. NAD(+) in aging, metabolism, and neurodegeneration. Science 2015, 350, 1208–1213. [Google Scholar] [CrossRef]
- Lautrup, S.; Sinclair, D.A.; Mattson, M.P.; Fang, E.F. NAD(+) in brain aging and neurodegenerative disorders. Cell Metab. 2019, 30, 630–655. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhang, L.; Sasaki, Y.; Milbrandt, J.; Gidday, J.M. Protection of mouse retinal ganglion cell axons and soma from glaucomatous and ischemic injury by cytoplasmic overexpression of Nmnat1. Investig. Ophthalmol. Vis. Sci. 2013, 54, 25–36. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, Y.; Vohra, B.P.; Baloh, R.H.; Milbrandt, J. Transgenic mice expressing the Nmnat1 protein manifest robust delay in axonal degeneration in vivo. J. Neurosci. 2009, 29, 6526–6534. [Google Scholar] [CrossRef] [Green Version]
- Williams, P.A.; Harder, J.M.; Foxworth, N.E.; Cardozo, B.H.; Cochran, K.E.; John, S.W.M. Nicotinamide and WLD(S) act together to prevent neurodegeneration in glaucoma. Front. Neurosci. 2017, 11, 232. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Hyrc, K.L.; Goldberg, M.P. Maintaining energy homeostasis is an essential component of Wld(S)-mediated axon protection. Neurobiol. Dis. 2013, 59, 69–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, K.W.; Milbrandt, J.; DiAntonio, A. SARM1 acts downstream of neuroinflammatory and necroptotic signaling to induce axon degeneration. J. Cell Biol. 2020, 219, e201912047. [Google Scholar] [CrossRef]
- Hui, F.; Tang, J.; Williams, P.A.; McGuinness, M.B.; Hadoux, X.; Casson, R.J.; Coote, M.; Trounce, I.A.; Martin, K.R.; van Wijngaarden, P.; et al. Improvement in inner retinal function in glaucoma with nicotinamide (vitamin B3) supplementation: A crossover randomized clinical trial. Clin. Exp. Ophthalmol. 2020, 48, 903–914. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, B.N.; Thackray, J.K.; Serrano, L. Sirtuins and DNA damage repair: SIRT7 comes to play. Nucleus 2017, 8, 107–115. [Google Scholar] [CrossRef] [Green Version]
- Cao, K.; Ishida, T.; Fang, Y.; Shinohara, K.; Li, X.; Nagaoka, N.; Ohno-Matsui, K.; Yoshida, T. Protection of the retinal ganglion cells: Intravitreal injection of resveratrol in mouse model of ocular hypertension. Investig. Ophthalmol. Vis. Sci. 2020, 61, 13. [Google Scholar] [CrossRef] [Green Version]
- Niittykoski, M.; Kalesnykas, G.; Larsson, K.P.; Kaarniranta, K.; Akerman, K.E.; Uusitalo, H. Altered calcium signaling in an experimental model of glaucoma. Investig. Ophthalmol. Vis. Sci. 2010, 51, 6387–6393. [Google Scholar] [CrossRef] [Green Version]
- Ma, M.; Ferguson, T.A.; Schoch, K.M.; Li, J.; Qian, Y.; Shofer, F.S.; Saatman, K.E.; Neumar, R.W. Calpains mediate axonal cytoskeleton disintegration during Wallerian degeneration. Neurobiol. Dis. 2013, 56, 34–46. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Luo, C.; Cai, J.; Powell, D.W.; Yu, D.; Kuehn, M.H.; Tezel, G. Neurodegenerative and inflammatory pathway components linked to TNF-alpha/TNFR1 signaling in the glaucomatous human retina. Investig. Ophthalmol. Vis. Sci. 2011, 52, 8442–8454. [Google Scholar] [CrossRef]
- Huang, W.; Fileta, J.; Rawe, I.; Qu, J.; Grosskreutz, C.L. Calpain activation in experimental glaucoma. Investig. Ophthalmol. Vis. Sci. 2010, 51, 3049–3054. [Google Scholar] [CrossRef]
- Deng, S.; Wang, M.; Yan, Z.; Tian, Z.; Chen, H.; Yang, X.; Zhuo, Y. Autophagy in retinal ganglion cells in a rhesus monkey chronic hypertensive glaucoma model. PLoS ONE 2013, 8, e77100. [Google Scholar]
- Wakatsuki, S.; Tokunaga, S.; Shibata, M.; Araki, T. GSK3B-mediated phosphorylation of MCL1 regulates axonal autophagy to promote Wallerian degeneration. J. Cell Biol. 2017, 216, 477–493. [Google Scholar] [CrossRef] [Green Version]
- Park, H.L.; Kim, J.H.; Park, C.K. Different contributions of autophagy to retinal ganglion cell death in the diabetic and glaucomatous retinas. Sci. Rep. 2018, 8, 13321. [Google Scholar] [CrossRef] [Green Version]
- Hirt, J.; Porter, K.; Dixon, A.; McKinnon, S.; Liton, P.B. Contribution of autophagy to ocular hypertension and neurodegeneration in the DBA/2J spontaneous glaucoma mouse model. Cell Death Discov. 2018, 4, 14. [Google Scholar] [CrossRef]
- Kleesattel, D.; Crish, S.D.; Inman, D.M. Decreased energy capacity and increased autophagic activity in optic nerve axons with defective anterograde transport. Investig. Ophthalmol. Vis. Sci. 2015, 56, 8215–8227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, Y.; Hu, X.; Sun, X. Overexpression of parkin protects retinal ganglion cells in experimental glaucoma. Cell Death Dis. 2018, 9, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hass, D.T.; Barnstable, C.J. Mitochondrial uncoupling protein 2 knock-out promotes mitophagy to decrease retinal ganglion cell death in a mouse model of glaucoma. J. Neurosci. 2019, 39, 3582–3596. [Google Scholar]
- Shpilka, T.; Haynes, C.M. The mitochondrial UPR: Mechanisms, physiological functions and implications in ageing. Nat. Rev. Mol. Cell Biol. 2018, 19, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Doh, S.H.; Kim, J.H.; Lee, K.M.; Park, H.Y.; Park, C.K. Retinal ganglion cell death induced by endoplasmic reticulum stress in a chronic glaucoma model. Brain Res. 2010, 1308, 158–166. [Google Scholar] [CrossRef]
- Zode, G.S.; Bugge, K.E.; Mohan, K.; Grozdanic, S.D.; Peters, J.C.; Koehn, D.R.; Anderson, M.G.; Kardon, R.H.; Stone, E.M.; Sheffield, V.C. Topical ocular sodium 4-phenylbutyrate rescues glaucoma in a myocilin mouse model of primary open-angle glaucoma. Investig. Ophthalmol. Vis. Sci. 2012, 53, 1557–1565. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Li, S.; Miao, L.; Huang, H.; Liang, F.; Teng, X.; Xu, L.; Wang, Q.; Xiao, W.; Ridder, W.H.; et al. Rescue of glaucomatous neurodegeneration by differentially modulating neuronal endoplasmic reticulum stress molecules. J. Neurosci. 2016, 36, 5891–5903. [Google Scholar] [CrossRef] [Green Version]
- Marola, O.J.; Syc-Mazurek, S.B.; Libby, R.T. DDIT3 (CHOP) contributes to retinal ganglion cell somal loss but not axonal degeneration in DBA/2J mice. Cell Death Discov. 2019, 5, 140. [Google Scholar] [CrossRef] [Green Version]
- Donahue, R.J.; Maes, M.E.; Grosser, J.A.; Nickells, R.W. BAX-depleted retinal ganglion cells survive and become quiescent following optic nerve damage. Mol. Neurobiol. 2019, 57, 1070–1084. [Google Scholar] [CrossRef]
- Tezel, G.; Wax, M.B. Increased production of tumor necrosis factor-alpha by glial cells exposed to simulated ischemia or elevated hydrostatic pressure induces apoptosis in cocultured retinal ganglion cells. J. Neurosci. 2000, 20, 8693–8700. [Google Scholar] [CrossRef]
- Tezel, G.; Li, L.Y.; Patil, R.V.; Wax, M.B. Tumor necrosis factor-alpha and its receptor-1 in the retina of normal and glaucomatous eyes. Investig. Ophthalmol. Vis. Sci. 2001, 42, 1787–1794. [Google Scholar]
- Krishnan, A.; Kocab, A.J.; Zacks, D.N.; Marshak-Rothstein, A.; Gregory-Ksander, M. A small peptide antagonist of the Fas receptor inhibits neuroinflammation and prevents axon degeneration and retinal ganglion cell death in an inducible mouse model of glaucoma. J. Neuroinflamm. 2019, 16, 184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.; Dobberfuhl, A.; Filippopoulos, T.; Ingelsson, M.; Fileta, J.B.; Poulin, N.R.; Grosskreutz, C.L. Transcriptional up-regulation and activation of initiating caspases in experimental glaucoma. Am. J. Pathol. 2005, 167, 673–681. [Google Scholar] [CrossRef] [Green Version]
- McKinnon, S.J.; Lehman, D.M.; Kerrigan-Baumrind, L.A.; Merges, C.A.; Pease, M.E.; Kerrigan, D.F.; Ransom, N.L.; Tahzib, N.G.; Reitsamer, H.A.; Levkovitch-Verbin, H.; et al. Caspase activation and amyloid precursor protein cleavage in rat ocular hypertension. Investig. Ophthalmol. Vis. Sci. 2002, 43, 1077–1087. [Google Scholar]
- Tezel, G.; Yang, X.; Luo, C.; Cai, J.; Powell, D.W. An astrocyte-specific proteomic approach to inflammatory responses in experimental rat glaucoma. Investig. Ophthalmol. Vis. Sci. 2012, 53, 4220–4233. [Google Scholar] [CrossRef] [PubMed]
- Tezel, G.; Wax, M.B. Inhibition of caspase activity in retinal cell apoptosis induced by various stimuli in vitro. Investig. Ophthalmol. Vis. Sci. 1999, 40, 2660–2667. [Google Scholar]
- McKinnon, S.J.; Lehman, D.M.; Tahzib, N.G.; Ransom, N.L.; Reitsamer, H.A.; Liston, P.; LaCasse, E.; Li, Q.; Korneluk, R.G.; Hauswirth, W.W. Baculoviral IAP repeat-containing-4 protects optic nerve axons in a rat glaucoma model. Mol. Ther. 2002, 5, 780–787. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Luo, C.; Cai, J.; Pierce, W.M.; Tezel, G. Phosphorylation-dependent interaction with 14-3-3 in the regulation of bad trafficking in retinal ganglion cells. Investig. Ophthalmol. Vis. Sci. 2008, 49, 2483–2494. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Wu, Z.; Renier, N.; Simon, D.J.; Uryu, K.; Park, D.S.; Greer, P.A.; Tournier, C.; Davis, R.J.; Tessier-Lavigne, M. Pathological axonal death through a MAPK cascade that triggers a local energy deficit. Cell 2015, 160, 161–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandes, K.A.; Mitchell, K.L.; Patel, A.; Marola, O.J.; Shrager, P.; Zack, D.J.; Libby, R.T.; Welsbie, D.S. Role of SARM1 and DR6 in retinal ganglion cell axonal and somal degeneration following axonal injury. Exp. Eye Res. 2018, 171, 54–61. [Google Scholar] [CrossRef]
- You, Y.; Joseph, C.; Wang, C.; Gupta, V.; Liu, S.; Yiannikas, C.; Chua, B.E.; Chitranshi, N.; Shen, T.; Dheer, Y.; et al. Demyelination precedes axonal loss in the transneuronal spread of human neurodegenerative disease. Brain 2019, 142, 426–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikolaev, A.; McLaughlin, T.; O’Leary, D.D.; Tessier-Lavigne, M. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature 2009, 457, 981–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandes, K.A.; Harder, J.M.; Fornarola, L.B.; Freeman, R.S.; Clark, A.F.; Pang, I.H.; John, S.W.; Libby, R.T. JNK2 and JNK3 are major regulators of axonal injury-induced retinal ganglion cell death. Neurobiol. Dis. 2012, 46, 393–401. [Google Scholar] [CrossRef] [Green Version]
- Harder, J.M.; Williams, P.A.; Soto, I.; Foxworth, N.E.; Fernandes, K.A.; Freeburg, N.F.; Libby, R.T.; John, S.W.M. Jnk2 deficiency increases the rate of glaucomatous neurodegeneration in ocular hypertensive DBA/2J mice. Cell Death Dis. 2018, 9, 705. [Google Scholar] [CrossRef]
- Fernandes, K.A.; Harder, J.M.; Kim, J.; Libby, R.T. JUN regulates early transcriptional responses to axonal injury in retinal ganglion cells. Exp. Eye Res. 2013, 112, 106–117. [Google Scholar] [CrossRef] [Green Version]
- Syc-Mazurek, S.B.; Fernandes, K.A.; Libby, R.T. JUN is important for ocular hypertension-induced retinal ganglion cell degeneration. Cell Death Dis. 2017, 8, e2945. [Google Scholar] [CrossRef] [Green Version]
- Syc-Mazurek, S.B.; Fernandes, K.A.; Wilson, M.P.; Shrager, P.; Libby, R.T. Together JUN and DDIT3 (CHOP) control retinal ganglion cell death after axonal injury. Mol. Neurodegener. 2017, 12, 71. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, K.A.; Harder, J.M.; John, S.W.; Shrager, P.; Libby, R.T. DLK-dependent signaling is important for somal but not axonal degeneration of retinal ganglion cells following axonal injury. Neurobiol. Dis. 2014, 69, 108–116. [Google Scholar] [CrossRef] [Green Version]
- Welsbie, D.S.; Mitchell, K.L.; Jaskula-Ranga, V.; Sluch, V.M.; Yang, Z.; Kim, J.; Buehler, E.; Patel, A.; Martin, S.E.; Zhang, P.W.; et al. Enhanced functional genomic screening identifies novel mediators of dual leucine zipper kinase-dependent injury signaling in neurons. Neuron 2017, 94, 1142–1154.e1146. [Google Scholar] [CrossRef] [Green Version]
- Syc-Mazurek, S.B.; Rausch, R.L.; Fernandes, K.A.; Wilson, M.P.; Libby, R.T. Mkk4 and Mkk7 are important for retinal development and axonal injury-induced retinal ganglion cell death. Cell Death Dis. 2018, 9, 1095. [Google Scholar] [CrossRef] [Green Version]
- Watkins, T.A.; Wang, B.; Huntwork-Rodriguez, S.; Yang, J.; Jiang, Z.; Eastham-Anderson, J.; Modrusan, Z.; Kaminker, J.S.; Tessier-Lavigne, M.; Lewcock, J.W. DLK initiates a transcriptional program that couples apoptotic and regenerative responses to axonal injury. Proc. Natl. Acad. Sci. USA 2013, 110, 4039–4044. [Google Scholar] [CrossRef] [Green Version]
- Tezel, G.; Chauhan, B.C.; LeBlanc, R.P.; Wax, M.B. Immunohistochemical assessment of the glial mitogen-activated protein kinase activation in glaucoma. Investig. Ophthalmol. Vis. Sci. 2003, 44, 3025–3033. [Google Scholar] [CrossRef] [Green Version]
- Tezel, G.; Yang, X.; Yang, J.; Wax, M.B. Role of tumor necrosis factor receptor-1 in the death of retinal ganglion cells following optic nerve crush injury in mice. Brain Res. 2004, 996, 202–212. [Google Scholar] [CrossRef]
- Kwong, J.M.; Caprioli, J. Expression of phosphorylated c-Jun N-terminal protein kinase (JNK) in experimental glaucoma in rats. Exp. Eye Res. 2006, 82, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Dvoriantchikova, G.; Ivanov, D. Tumor necrosis factor-alpha mediates activation of NF-kappaB and JNK signaling cascades in retinal ganglion cells and astrocytes in opposite ways. Eur. J. Neurosci. 2014, 40, 3171–3178. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zeng, Q.; Tezel, G. Regulation of distinct caspase-8 functions in retinal ganglion cells and astroglia in experimental glaucoma. Neurobiol. Dis. 2021, 150, 105258. [Google Scholar] [CrossRef] [PubMed]
- Marola, O.J.; Syc-Mazurek, S.B.; Howell, G.R.; Libby, R.T. Endothelin 1-induced retinal ganglion cell death is largely mediated by JUN activation. Cell Death Dis. 2020, 11, 811. [Google Scholar] [CrossRef] [PubMed]
- Song, N.; Liu, Z.S.; Xue, W.; Bai, Z.F.; Wang, Q.Y.; Dai, J.; Liu, X.; Huang, Y.J.; Cai, H.; Zhan, X.Y.; et al. NLRP3 phosphorylation is an essential priming event for inflammasome activation. Mol. Cell. 2017, 68, 185–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez, M.R.; Miao, H.; Lukas, T. Astrocytes in glaucomatous optic neuropathy. Prog. Brain Res. 2008, 173, 353–373. [Google Scholar] [PubMed]
- Qu, J.; Jakobs, T.C. The time course of gene expression during reactive gliosis in the optic nerve. PLoS ONE 2013, 8, e67094. [Google Scholar] [CrossRef]
- Lye-Barthel, M.; Sun, D.; Jakobs, T.C. Morphology of astrocytes in a glaucomatous optic nerve. Investig. Ophthalmol. Vis. Sci. 2013, 54, 909–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tehrani, S.; Davis, L.; Cepurna, W.O.; Choe, T.E.; Lozano, D.C.; Monfared, A.; Cooper, L.; Cheng, J.; Johnson, E.C.; Morrison, J.C. Astrocyte structural and molecular response to elevated intraocular pressure occurs rapidly and precedes axonal tubulin rearrangement within the optic nerve head in a rat model. PLoS ONE 2016, 11, e0167364. [Google Scholar] [CrossRef] [Green Version]
- Quillen, S.; Schaub, J.; Quigley, H.; Pease, M.; Korneva, A.; Kimball, E. Astrocyte responses to experimental glaucoma in mouse optic nerve head. PLoS ONE 2020, 15, e0238104. [Google Scholar] [CrossRef]
- Neufeld, A.H. Microglia in the optic nerve head and the region of parapapillary chorioretinal atrophy in glaucoma. Arch. Ophthalmol. 1999, 117, 1050–1056. [Google Scholar] [CrossRef] [Green Version]
- Ebneter, A.; Casson, R.J.; Wood, J.P.; Chidlow, G. Microglial activation in the visual pathway in experimental glaucoma: Spatiotemporal characterization and correlation with axonal injury. Investig. Ophthalmol. Vis. Sci. 2010, 51, 6448–6460. [Google Scholar] [CrossRef] [Green Version]
- Bosco, A.; Steele, M.R.; Vetter, M.L. Early microglia activation in a mouse model of chronic glaucoma. J. Comp. Neurol. 2011, 519, 599–620. [Google Scholar] [CrossRef] [Green Version]
- Taylor, S.; Calder, C.J.; Albon, J.; Erichsen, J.T.; Boulton, M.E.; Morgan, J.E. Involvement of the CD200 receptor complex in microglia activation in experimental glaucoma. Exp. Eye Res. 2011, 92, 338–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howell, G.R.; Macalinao, D.G.; Sousa, G.L.; Walden, M.; Soto, I.; Kneeland, S.C.; Barbay, J.M.; King, B.L.; Marchant, J.K.; Hibbs, M.; et al. Molecular clustering identifies complement and endothelin induction as early events in a mouse model of glaucoma. J. Clin. Investig. 2011, 121, 1429–1444. [Google Scholar] [CrossRef] [PubMed]
- Seitz, R.; Ohlmann, A.; Tamm, E.R. The role of Muller glia and microglia in glaucoma. Cell Tissue Res. 2013, 353, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Irnaten, M.; Zhdanov, A.; Brennan, D.; Crotty, T.; Clark, A.; Papkovsky, D.; O’Brien, C. Activation of the NFAT-calcium signaling pathway in human lamina cribrosa cells in glaucoma. Investig. Ophthalmol. Vis. Sci. 2018, 59, 831–842. [Google Scholar] [CrossRef] [Green Version]
- Son, J.L.; Soto, I.; Oglesby, E.; Lopez-Roca, T.; Pease, M.E.; Quigley, H.A.; Marsh-Armstrong, N. Glaucomatous optic nerve injury involves early astrocyte reactivity and late oligodendrocyte loss. Glia 2010, 58, 780–789. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, K.; Ver Hoeve, J.N.; Teixeira, L.B.C.; Snyder, K.C.; Kiland, J.A.; Ellinwood, N.M.; McLennan, G.J. Sub-region-specific optic nerve head glial activation in glaucoma. Mol. Neurobiol. 2020, 57, 2620–2638. [Google Scholar] [CrossRef] [PubMed]
- Howell, G.R.; MacNicoll, K.H.; Braine, C.E.; Soto, I.; Macalinao, D.G.; Sousa, G.L.; John, S.W. Combinatorial targeting of early pathways profoundly inhibits neurodegeneration in a mouse model of glaucoma. Neurobiol. Dis. 2014, 71, 44–52. [Google Scholar] [CrossRef] [Green Version]
- Williams, P.A.; Tribble, J.R.; Pepper, K.W.; Cross, S.D.; Morgan, B.P.; Morgan, J.E.; John, S.W.; Howell, G.R. Inhibition of the classical pathway of the complement cascade prevents early dendritic and synaptic degeneration in glaucoma. Mol. Neurodegener. 2016, 11, 26–39. [Google Scholar] [CrossRef] [Green Version]
- Bosco, A.; Anderson, S.R.; Breen, K.T.; Romero, C.O.; Steele, M.R.; Chiodo, V.A.; Boye, S.L.; Hauswirth, W.W.; Tomlinson, S.; Vetter, M.L. Complement C3-targeted gene therapy restricts onset and progression of neurodegeneration in chronic mouse glaucoma. Mol. Ther. 2018, 26, 2379–2396. [Google Scholar] [CrossRef] [Green Version]
- Harder, J.M.; Williams, P.A.; Braine, C.E.; Yang, H.S.; Thomas, J.M.; Foxworth, N.E.; John, S.W.M.; Howell, G.R. Complement peptide C3a receptor 1 promotes optic nerve degeneration in DBA/2J mice. J. Neuroinflamm. 2020, 17, 336. [Google Scholar] [CrossRef] [PubMed]
- Howell, G.R.; Soto, I.; Zhu, X.; Ryan, M.; Macalinao, D.G.; Sousa, G.L.; Caddle, L.B.; MacNicoll, K.H.; Barbay, J.M.; Porciatti, V.; et al. Radiation treatment inhibits monocyte entry into the optic nerve head and prevents neuronal damage in a mouse model of glaucoma. J. Clin. Investig. 2012, 122, 1246–1261. [Google Scholar] [CrossRef] [Green Version]
- Williams, P.A.; Braine, C.E.; Kizhatil, K.; Foxworth, N.E.; Tolman, N.G.; Harder, J.M.; Scott, R.A.; Sousa, G.L.; Panitch, A.; Howell, G.R.; et al. Inhibition of monocyte-like cell extravasation protects from neurodegeneration in DBA/2J glaucoma. Mol. Neurodegener. 2019, 14, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beckel, J.M.; Argall, A.J.; Lim, J.C.; Xia, J.; Lu, W.; Coffey, E.E.; Macarak, E.J.; Shahidulah, M.; Delamere, N.A.; Zode, G.S.; et al. Mechanosensitive release of adenosine 5′-triphosphate through pannexin channels and mechanosensitive upregulation of pannexin channels in optic nerve head astrocytes: A mechanism for purinergic involvement in chronic strain. Glia 2014, 62, 1486–1501. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Sun, D.; Jakobs, T.C. Astrocytes in the optic nerve head express putative mechanosensitive channels. Mol. Vis. 2015, 21, 749–766. [Google Scholar] [PubMed]
- Krizaj, D.; Ryskamp, D.A.; Tian, N.; Tezel, G.; Mitchell, C.H.; Slepak, V.Z.; Shestopalov, V.I. From mechanosensitivity to inflammatory responses: New players in the pathology of glaucoma. Curr. Eye Res. 2014, 39, 105–119. [Google Scholar] [CrossRef]
- Lu, W.; Hu, H.; Sevigny, J.; Gabelt, B.T.; Kaufman, P.L.; Johnson, E.C.; Morrison, J.C.; Zode, G.S.; Sheffield, V.C.; Zhang, X.; et al. Rat, mouse, and primate models of chronic glaucoma show sustained elevation of extracellular ATP and altered purinergic signaling in the posterior eye. Investig. Ophthalmol. Vis. Sci. 2015, 56, 3075–3083. [Google Scholar] [CrossRef] [Green Version]
- Albalawi, F.; Lu, W.; Beckel, J.M.; Lim, J.C.; McCaughey, S.A.; Mitchell, C.H. The P2X7 receptor primes IL-1beta and the NLRP3 inflammasome in astrocytes exposed to mechanical strain. Front. Cell Neurosci. 2017, 11, 227. [Google Scholar] [CrossRef] [PubMed]
- Astafurov, K.; Elhawy, E.; Ren, L.; Dong, C.Q.; Igboin, C.; Hyman, L.; Griffen, A.; Mittag, T.; Danias, J. Oral microbiome link to neurodegeneration in glaucoma. PLoS ONE 2014, 9, e104416. [Google Scholar] [CrossRef]
- Chen, H.; Cho, K.S.; Vu, T.H.K.; Shen, C.H.; Kaur, M.; Chen, G.; Mathew, R.; McHam, M.L.; Fazelat, A.; Lashkari, K.; et al. Commensal microflora-induced T cell responses mediate progressive neurodegeneration in glaucoma. Nat. Commun. 2018, 9, 3209. [Google Scholar] [CrossRef]
- Mac Nair, C.E.; Schlamp, C.L.; Montgomery, A.D.; Shestopalov, V.I.; Nickells, R.W. Retinal glial responses to optic nerve crush are attenuated in Bax-deficient mice and modulated by purinergic signaling pathways. J. Neuroinflamm. 2016, 13, 93. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, A.I.; Salazar, J.J.; de Hoz, R.; Rojas, B.; Gallego, B.I.; Salinas-Navarro, M.; Alarcon-Martinez, L.; Ortin-Martinez, A.; Aviles-Trigueros, M.; Vidal-Sanz, M.; et al. Quantification of the effect of different levels of IOP in the astroglia of the rat retina ipsilateral and contralateral to experimental glaucoma. Investig. Ophthalmol. Vis. Sci. 2010, 51, 5690–5696. [Google Scholar] [CrossRef] [Green Version]
- Gallego, B.I.; Salazar, J.J.; de Hoz, R.; Rojas, B.; Ramirez, A.I.; Salinas-Navarro, M.; Ortin-Martinez, A.; Valiente-Soriano, F.J.; Aviles-Trigueros, M.; Villegas-Perez, M.P.; et al. IOP induces upregulation of GFAP and MHC-II and microglia reactivity in mice retina contralateral to experimental glaucoma. J. Neuroinflamm. 2012, 9, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramirez, A.I.; de Hoz, R.; Fernandez-Albarral, J.A.; Salobrar-Garcia, E.; Rojas, B.; Valiente-Soriano, F.J.; Aviles-Trigueros, M.; Villegas-Perez, P.; Vidal-Sanz, M.; Trivino, A.; et al. Time course of bilateral microglial activation in a mouse model of laser-induced glaucoma. Sci. Rep. 2020, 10, 4890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sapienza, A.; Raveu, A.L.; Reboussin, E.; Roubeix, C.; Boucher, C.; Degardin, J.; Godefroy, D.; Rostene, W.; Reaux-Le Goazigo, A.; Baudouin, C.; et al. Bilateral neuroinflammatory processes in visual pathways induced by unilateral ocular hypertension in the rat. J. Neuroinflamm. 2016, 13, 44. [Google Scholar] [CrossRef] [PubMed]
- Tribble, J.R.; Kokkali, E.; Otmani, A.; Plastino, F.; Lardner, E.; Vohra, R.; Kolko, M.; Andre, H.; Morgan, J.E.; Williams, P.A. When is a control not a control? Reactive microglia occur throughout the control contralateral pathway of retinal ganglion cell projections in experimental glaucoma. Transl. Vis. Sci. Technol. 2021, 10, 22. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.L.; Pasini, S.; Lambert, W.S.; D’Alessandro, K.B.; Yao, V.; Risner, M.L.; Calkins, D.J. Redistribution of metabolic resources through astrocyte networks mitigates neurodegenerative stress. Proc. Natl. Acad. Sci. USA 2020, 117, 18810–18821. [Google Scholar] [CrossRef]
- Burgoyne, C.F. A biomechanical paradigm for axonal insult within the optic nerve head in aging and glaucoma. Exp. Eye Res. 2011, 93, 120–132. [Google Scholar] [CrossRef] [Green Version]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Clarke, L.E.; Liddelow, S.A.; Chakraborty, C.; Munch, A.E.; Heiman, M.; Barres, B.A. Normal aging induces A1-like astrocyte reactivity. Proc. Natl. Acad. Sci. USA 2018, 115, E1896–E1905. [Google Scholar] [CrossRef] [Green Version]
- Guttenplan, K.A.; Stafford, B.K.; El-Danaf, R.N.; Adler, D.I.; Munch, A.E.; Weigel, M.K.; Huberman, A.D.; Liddelow, S.A. Neurotoxic reactive astrocytes drive neuronal death after retinal injury. Cell Rep. 2020, 31, 107776. [Google Scholar] [CrossRef] [PubMed]
- Sterling, J.K.; Adetunji, M.O.; Guttha, S.; Bargoud, A.R.; Uyhazi, K.E.; Ross, A.G.; Dunaief, J.L.; Cui, Q.N. GLP-1 receptor agonist NLY01 reduces retinal inflammation and neuron death secondary to ocular hypertension. Cell Rep. 2020, 33, 108271. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zeng, Q.; Baris, M.; Tezel, G. Transgenic inhibition of astroglial NF-kappaB restrains the neuroinflammatory and neurodegenerative outcomes of experimental mouse glaucoma. J. Neuroinflamm. 2020, 17, 252. [Google Scholar] [CrossRef] [PubMed]
- Wax, M.B. Serum autoantibodies to heat shock proteins in glaucoma patients from Japan and the United States. Investig. Ophthalmol. Vis. Sci. 2001, 108, 296–302. [Google Scholar] [CrossRef]
- Wax, M.B.; Tezel, G.; Yang, J.; Peng, G.; Patil, R.V.; Agarwal, N.; Sappington, R.M.; Calkins, D.J. Induced autoimmunity to heat shock proteins elicits glaucomatous loss of retinal ganglion cell neurons via activated T-cell-derived fas-ligand. J. Neurosci. 2008, 28, 12085–12096. [Google Scholar] [CrossRef] [Green Version]
- Tezel, G.; Thornton, I.L.; Tong, M.G.; Luo, C.; Yang, X.; Cai, J.; Powell, D.W.; Soltau, J.B.; Liebmann, J.M.; Ritch, R. Immunoproteomic analysis of potential serum biomarker candidates in human glaucoma. Investig. Ophthalmol. Vis. Sci. 2012, 53, 8222–8231. [Google Scholar] [CrossRef] [Green Version]
- Tezel, G.; Luo, C.; Yang, X. Accelerated aging in glaucoma: Immunohistochemical assessment of advanced glycation end products in the human retina and optic nerve head. Investig. Ophthalmol. Vis. Sci. 2007, 48, 1201–1211. [Google Scholar] [CrossRef]
- Tezel, G.; Yang, X.; Luo, C.; Kain, A.D.; Powell, D.W.; Kuehn, M.H.; Kaplan, H.J. Oxidative stress and the regulation of complement activation in human glaucoma. Investig. Ophthalmol. Vis. Sci. 2010, 51, 5071–5082. [Google Scholar] [CrossRef]
- Harun-Or-Rashid, M.; Inman, D.M. Reduced AMPK activation and increased HCAR activation drive anti-inflammatory response and neuroprotection in glaucoma. J. Neuroinflamm. 2018, 15, 313. [Google Scholar] [CrossRef]
- Jha, A.K.; Huang, S.C.; Sergushichev, A.; Lampropoulou, V.; Ivanova, Y.; Loginicheva, E.; Chmielewski, K.; Stewart, K.M.; Ashall, J.; Everts, B.; et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 2015, 42, 419–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baik, S.H.; Kang, S.; Lee, W.; Choi, H.; Chung, S.; Kim, J.I.; Mook-Jung, I. A breakdown in metabolic reprogramming causes microglia dysfunction in Alzheimer’s Disease. Cell Metab. 2019, 30, 493–507. [Google Scholar] [CrossRef]
- Yan, X.; Tezel, G.; Wax, M.B.; Edward, D.P. Matrix metalloproteinases and tumor necrosis factor alpha in glaucomatous optic nerve head. Arch. Ophthalmol. 2000, 118, 666–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tezel, G. TNF-alpha signaling in glaucomatous neurodegeneration. Prog. Brain Res. 2008, 173, 409–421. [Google Scholar] [PubMed] [Green Version]
- Nakazawa, T.; Nakazawa, C.; Matsubara, A.; Noda, K.; Hisatomi, T.; She, H.; Michaud, N.; Hafezi-Moghadam, A.; Miller, J.W.; Benowitz, L.I. Tumor necrosis factor-alpha mediates oligodendrocyte death and delayed retinal ganglion cell loss in a mouse model of glaucoma. J. Neurosci. 2006, 26, 12633–12641. [Google Scholar] [CrossRef] [Green Version]
- Roh, M.; Zhang, Y.; Murakami, Y.; Thanos, A.; Lee, S.C.; Vavvas, D.G.; Benowitz, L.I.; Miller, J.W. Etanercept, a widely used inhibitor of tumor necrosis factor-alpha (TNF-alpha), prevents retinal ganglion cell loss in a rat model of glaucoma. PLoS ONE 2012, 7, e40065. [Google Scholar] [CrossRef] [PubMed]
- Levkovitch-Verbin, H.; Kalev-Landoy, M.; Habot-Wilner, Z.; Melamed, S. Minocycline delays death of retinal ganglion cells in experimental glaucoma and after optic nerve transection. Arch. Ophthalmol. 2006, 124, 520–526. [Google Scholar] [CrossRef] [Green Version]
- Bosco, A.; Inman, D.M.; Steele, M.R.; Wu, G.; Soto, I.; Marsh-Armstrong, N.; Hubbard, W.C.; Calkins, D.J.; Horner, P.J.; Vetter, M.L. Reduced retina microglial activation and improved optic nerve integrity with minocycline treatment in the DBA/2J mouse model of glaucoma. Investig. Ophthalmol. Vis. Sci. 2008, 49, 1437–1446. [Google Scholar] [CrossRef]
- Bordone, M.P.; Gonzalez Fleitas, M.F.; Pasquini, L.A.; Bosco, A.; Sande, P.H.; Rosenstein, R.E.; Dorfman, D. Involvement of microglia in early axoglial alterations of the optic nerve induced by experimental glaucoma. J. Neurochem. 2017, 142, 323–337. [Google Scholar] [CrossRef] [PubMed]
- Pronin, A.; Pham, D.; An, W.; Dvoriantchikova, G.; Reshetnikova, G.; Qiao, J.; Kozhekbaeva, Z.; Reiser, A.E.; Slepak, V.Z.; Shestopalov, V.I. Inflammasome activation induces pyroptosis in the retina exposed to ocular hypertension injury. Front. Mol. Neurosci. 2019, 12, 36. [Google Scholar] [CrossRef] [Green Version]
- Steele, M.R.; Inman, D.M.; Calkins, D.J.; Horner, P.J.; Vetter, M.L. Microarray analysis of retinal gene expression in the DBA/2J model of glaucoma. Investig. Ophthalmol. Vis. Sci. 2006, 47, 977–985. [Google Scholar] [CrossRef]
- Nikolskaya, T.; Nikolsky, Y.; Serebryiskaya, T.; Zvereva, S.; Sviridov, E.; Dezso, Z.; Rahkmatulin, E.; Brennan, R.J.; Yankovsky, N.; Bhattacharya, S.K.; et al. Network analysis of human glaucomatous optic nerve head astrocytes. BMC Med. Genom. 2009, 2, 24. [Google Scholar] [CrossRef] [Green Version]
- Neufeld, A.H.; Sawada, A.; Becker, B. Inhibition of nitric-oxide synthase 2 by aminoguanidine provides neuroprotection of retinal ganglion cells in a rat model of chronic glaucoma. Proc. Natl. Acad. Sci. USA 1999, 96, 9944–9948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Yang, P.; Tezel, G.; Patil, R.V.; Hernandez, M.R.; Wax, M.B. Induction of HLA-DR expression in human lamina cribrosa astrocytes by cytokines and simulated ischemia. Investig. Ophthalmol. Vis. Sci. 2001, 42, 365–371. [Google Scholar]
- Pena, J.D.; Varela, H.J.; Ricard, C.S.; Hernandez, M.R. Enhanced tenascin expression associated with reactive astrocytes in human optic nerve heads with primary open angle glaucoma. Exp. Eye Res. 1999, 68, 29–40. [Google Scholar] [CrossRef]
- Zuliani-Alvarez, L.; Marzeda, A.M.; Deligne, C.; Schwenzer, A.; McCann, F.E.; Marsden, B.D.; Piccinini, A.M.; Midwood, K.S. Mapping tenascin-C interaction with toll-like receptor 4 reveals a new subset of endogenous inflammatory triggers. Nat. Commun. 2017, 8, 1595. [Google Scholar] [CrossRef] [PubMed]
- Brambilla, R.; Dvoriantchikova, G.; Barakat, D.; Ivanov, D.; Bethea, J.R.; Shestopalov, V.I. Transgenic inhibition of astroglial NF-kappaB protects from optic nerve damage and retinal ganglion cell loss in experimental optic neuritis. J. Neuroinflamm. 2012, 9, 213. [Google Scholar] [CrossRef] [Green Version]
- Haenold, R.; Weih, F.; Herrmann, K.H.; Schmidt, K.F.; Krempler, K.; Engelmann, C.; Nave, K.A.; Reichenbach, J.R.; Lowel, S.; Witte, O.W.; et al. NF-kappaB controls axonal regeneration and degeneration through cell-specific balance of RelA and p50 in the adult CNS. J. Cell Sci. 2014, 127, 3052–3065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lian, H.; Yang, L.; Cole, A.; Sun, L.; Chiang, A.C.; Fowler, S.W.; Shim, D.J.; Rodriguez-Rivera, J.; Taglialatela, G.; Jankowsky, J.L.; et al. NFkappaB-activated astroglial release of complement C3 compromises neuronal morphology and function associated with Alzheimer’s disease. Neuron 2015, 85, 101–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Antwerp, D.J.; Martin, S.J.; Kafri, T.; Green, D.R.; Verma, I.M. Suppression of TNF-alpha-induced apoptosis by NF-kappaB. Science 1996, 274, 787–789. [Google Scholar] [CrossRef]
- Karin, M.; Lin, A. NF-kappaB at the crossroads of life and death. Nat. Immunol. 2002, 3, 221–227. [Google Scholar] [CrossRef]
- Meffert, M.K.; Chang, J.M.; Wiltgen, B.J.; Fanselow, M.S.; Baltimore, D. NF-kappa B functions in synaptic signaling and behavior. Nat. Neurosci. 2003, 6, 1072–1078. [Google Scholar] [CrossRef]
- Boersma, M.C.; Dresselhaus, E.C.; De Biase, L.M.; Mihalas, A.B.; Bergles, D.E.; Meffert, M.K. A requirement for nuclear factor-kappaB in developmental and plasticity-associated synaptogenesis. J. Neurosci. 2011, 31, 5414–5425. [Google Scholar] [CrossRef]
- Koerber, J.T.; Klimczak, R.; Jang, J.H.; Dalkara, D.; Flannery, J.G.; Schaffer, D.V. Molecular evolution of adeno-associated virus for enhanced glial gene delivery. Mol. Ther. 2009, 17, 2088–2095. [Google Scholar] [CrossRef] [PubMed]
- Dalkara, D.; Kolstad, K.D.; Guerin, K.I.; Hoffmann, N.V.; Visel, M.; Klimczak, R.R.; Schaffer, D.V.; Flannery, J.G. AAV mediated GDNF secretion from retinal glia slows down retinal degeneration in a rat model of retinitis pigmentosa. Mol. Ther. 2011, 19, 1602–1608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prentice, H.M.; Biswal, M.R.; Dorey, C.K.; Blanks, J.C. Hypoxia-regulated retinal glial cell-specific promoter for potential gene therapy in disease. Investig. Ophthalmol. Vis. Sci. 2011, 52, 8562–8570. [Google Scholar] [CrossRef] [PubMed]
- Biswal, M.R.; Prentice, H.M.; Dorey, C.K.; Blanks, J.C. A hypoxia-responsive glial cell-specific gene therapy vector for targeting retinal neovascularization. Investig. Ophthalmol. Vis. Sci. 2014, 55, 8044–8053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, X.; Yang, F.; Ouyang, T.; Liu, B.; Wu, C.; Jiang, W. Specific gene expression in mouse cortical astrocytes is mediated by a 1740bp-GFAP promoter-driven combined adeno-associated virus 2/5/7/8/9. Neurosci. Lett. 2015, 593, 45–50. [Google Scholar] [CrossRef]
- Liu, Y.; Miao, Q.; Yuan, J.; Han, S.; Zhang, P.; Li, S.; Rao, Z.; Zhao, W.; Ye, Q.; Geng, J.; et al. Ascl1 converts dorsal midbrain astrocytes into functional neurons in vivo. J. Neurosci. 2015, 35, 9336–9355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.; Parry, M.; Hou, X.Y.; Liu, M.H.; Wang, H.; Cain, R.; Pei, Z.F.; Chen, Y.C.; Guo, Z.Y.; Abhijeet, S.; et al. Gene therapy conversion of striatal astrocytes into GABAergic neurons in mouse models of Huntington’s disease. Nat. Commun. 2020, 11, 1105. [Google Scholar] [CrossRef]
- Tummers, B.; Green, D.R. Caspase-8: Regulating life and death. Immunol. Rev. 2017, 277, 76–89. [Google Scholar] [CrossRef] [Green Version]
- Oberst, A.; Dillon, C.P.; Weinlich, R.; McCormick, L.L.; Fitzgerald, P.; Pop, C.; Hakem, R.; Salvesen, G.S.; Green, D.R. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 2011, 471, 363–367. [Google Scholar] [CrossRef] [Green Version]
- Burguillos, M.A.; Deierborg, T.; Kavanagh, E.; Persson, A.; Hajji, N.; Garcia-Quintanilla, A.; Cano, J.; Brundin, P.; Englund, E.; Venero, J.L.; et al. Caspase signalling controls microglia activation and neurotoxicity. Nature 2011, 472, 319–324. [Google Scholar] [CrossRef]
- Lemmers, B.; Salmena, L.; Bidere, N.; Su, H.; Matysiak-Zablocki, E.; Murakami, K.; Ohashi, P.S.; Jurisicova, A.; Lenardo, M.; Hakem, R.; et al. Essential role for caspase-8 in Toll-like receptors and NFkappaB signaling. J. Biol. Chem. 2007, 282, 7416–7423. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.J.; Li, J. Caspase blockade induces RIP3-mediated programmed necrosis in Toll-like receptor-activated microglia. Cell Death Dis. 2013, 4, e716. [Google Scholar] [CrossRef] [Green Version]
- Shenderov, K.; Riteau, N.; Yip, R.; Mayer-Barber, K.D.; Oland, S.; Hieny, S.; Fitzgerald, P.; Oberst, A.; Dillon, C.P.; Green, D.R.; et al. Cutting edge: Endoplasmic reticulum stress licenses macrophages to produce mature IL-1beta in response to TLR4 stimulation through a caspase-8- and TRIF-dependent pathway. J. Immunol. 2014, 192, 2029–2033. [Google Scholar] [CrossRef] [PubMed]
- Philip, N.H.; DeLaney, A.; Peterson, L.W.; Santos-Marrero, M.; Grier, J.T.; Sun, Y.; Wynosky-Dolfi, M.A.; Zwack, E.E.; Hu, B.; Olsen, T.M.; et al. Activity of uncleaved caspase-8 controls anti-bacterial immune defense and TLR-induced cytokine production independent of cell death. PLoS Pathog. 2016, 12, e1005910. [Google Scholar] [CrossRef] [PubMed]
- Monie, T.P.; Bryant, C.E. Caspase-8 functions as a key mediator of inflammation and pro-IL-1beta processing via both canonical and non-canonical pathways. Immunol. Rev. 2015, 265, 181–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chi, W.; Li, F.; Chen, H.; Wang, Y.; Zhu, Y.; Yang, X.; Zhu, J.; Wu, F.; Ouyang, H.; Ge, J.; et al. Caspase-8 promotes NLRP1/NLRP3 inflammasome activation and IL-1beta production in acute glaucoma. Proc. Natl. Acad. Sci. USA 2014, 111, 11181–11186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurung, P.; Kanneganti, T.D. Novel roles for caspase-8 in IL-1beta and inflammasome regulation. Am. J. Pathol. 2015, 185, 17–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feltham, R.; Vince, J.E.; Lawlor, K.E. Caspase-8: Not so silently deadly. Clin. Transl. Immunol. 2017, 6, e124. [Google Scholar] [CrossRef]
- Irmler, M.; Thome, M.; Hahne, M.; Schneider, P.; Hofmann, K.; Steiner, V.; Bodmer, J.L.; Schroter, M.; Burns, K.; Mattmann, C.; et al. Inhibition of death receptor signals by cellular FLIP. Nature 1997, 388, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.W.; Xing, Z.; Pan, Y.; Algeciras-Schimnich, A.; Barnhart, B.C.; Yaish-Ohad, S.; Peter, M.E.; Yang, X. c-FLIP(L) is a dual function regulator for caspase-8 activation and CD95-mediated apoptosis. EMBO J. 2002, 21, 3704–3714. [Google Scholar] [CrossRef] [PubMed]
- Vandenabeele, P.; Melino, G. The flick of a switch: Which death program to choose? Cell Death Differ. 2012, 19, 1093–1095. [Google Scholar] [CrossRef]
- Silke, J.; Strasser, A. The FLIP Side of Life. Sci. Signal. 2013, 258, pe2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golks, A.; Brenner, D.; Krammer, P.H.; Lavrik, I.N. The c-FLIP-NH2 terminus (p22-FLIP) induces NF-kappaB activation. J. Exp. Med. 2006, 203, 1295–1305. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tezel, G. Multifactorial Pathogenic Processes of Retinal Ganglion Cell Degeneration in Glaucoma towards Multi-Target Strategies for Broader Treatment Effects. Cells 2021, 10, 1372. https://doi.org/10.3390/cells10061372
Tezel G. Multifactorial Pathogenic Processes of Retinal Ganglion Cell Degeneration in Glaucoma towards Multi-Target Strategies for Broader Treatment Effects. Cells. 2021; 10(6):1372. https://doi.org/10.3390/cells10061372
Chicago/Turabian StyleTezel, Gülgün. 2021. "Multifactorial Pathogenic Processes of Retinal Ganglion Cell Degeneration in Glaucoma towards Multi-Target Strategies for Broader Treatment Effects" Cells 10, no. 6: 1372. https://doi.org/10.3390/cells10061372