
Inflammation during Percutaneous Coronary Intervention—Prognostic Value, Mechanisms and Therapeutic Targets


Abstract
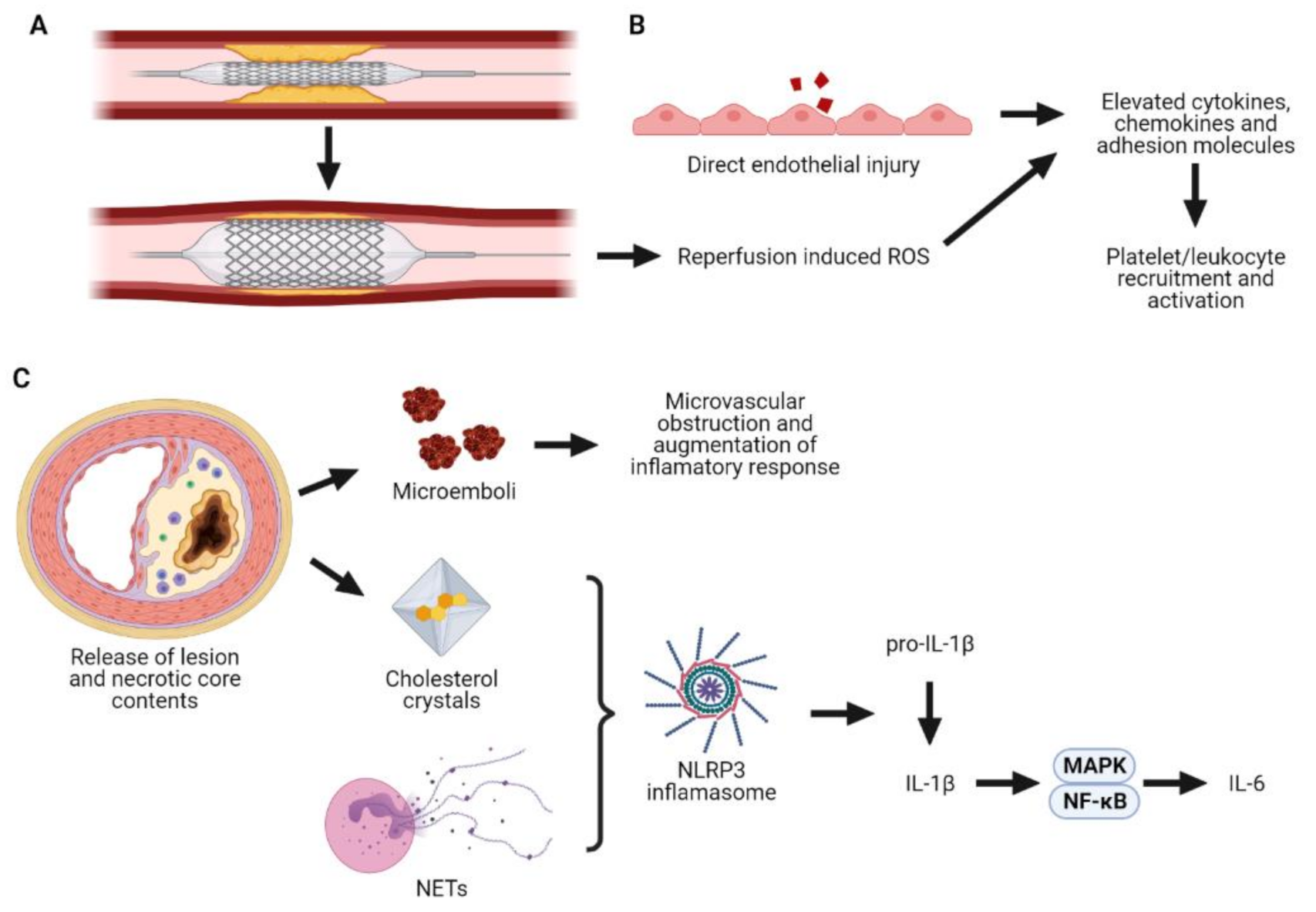
:1. Introduction
2. Periprocedural Myocardial Injury and Infarction
2.1. Definition
2.2. Epidemiology and Prognostic Significance
2.3. Pathophysiology
2.3.1. Proximal Periprocedural MI
2.3.2. Distal Periprocedural MI
3. Preprocedural Inflammation and Prognosis
3.1. C-Reactive Protein
3.2. Haematological Parameters
3.3. Other Inflammatory Markers
4. PCI-Induced Inflammation and Prognosis
5. Postprocedural Inflammation and Prognosis
6. Mechanistic Link between Inflammation and Periprocedural MI
7. Treating Inflammation in the Periprocedural Period
7.1. Colchicine
7.2. Tocilizumab
7.3. Anakinra
7.4. Novel Therapies
8. Challenges to Treating Periprocedural Inflammation
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhu, K.F.; Wang, Y.M.; Zhu, J.Z.; Zhou, Q.Y.; Wang, N.F. National prevalence of coronary heart disease and its relationship with human development index: A systematic review. Eur. J. Prev. Cardiol. 2016, 23, 530–543. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Nidorf, S.M.; Fiolet, A.T.L.; Mosterd, A.; Eikelboom, J.W.; Schut, A.; Opstal, T.S.J.; The, S.H.K.; Xu, X.-F.; Ireland, M.A.; Lenderink, T.; et al. Colchicine in Patients with Chronic Coronary Disease. N. Engl. J. Med. 2020, 383, 1838–1847. [Google Scholar] [CrossRef] [PubMed]
- Tardif, J.C.; Kouz, S.; Waters, D.D.; Bertrand, O.F.; Diaz, R.; Maggioni, A.P.; Pinto, F.J.; Ibrahim, R.; Gamra, H.; Kiwan, G.S.; et al. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction. N. Engl. J. Med. 2019, 381, 2497–2505. [Google Scholar] [CrossRef] [PubMed]
- Lahoud, R.; Dauerman Harold, L. Fall and Rise of Coronary Intervention. J. Am. Heart Assoc. 2020, 9, e016853. [Google Scholar] [CrossRef]
- Gregson, J.; Stone, G.W.; Ben-Yehuda, O.; Redfors, B.; Kandzari, D.E.; Morice, M.C.; Leon, M.B.; Kosmidou, I.; Lembo, N.J.; Brown, W.M., 3rd; et al. Implications of Alternative Definitions of Peri-Procedural Myocardial Infarction After Coronary Revascularization. J. Am. Coll. Cardiol. 2020, 76, 1609–1621. [Google Scholar] [CrossRef]
- Thygesen, K.; Alpert Joseph, S.; Jaffe Allan, S.; Chaitman Bernard, R.; Bax Jeroen, J.; Morrow David, A.; White Harvey, D.; The Executive Group on behalf of the Joint European Society of Cardiology (ESC)/American College of Cardiology (ACC)/American Heart Association (AHA)/World Heart Federation (WHF). Task Force for the Universal Definition of Myocardial Infarction Fourth Universal Definition of Myocardial Infarction. Circulation 2018, 138, e618–e651. [Google Scholar] [CrossRef]
- Moussa, I.D.; Klein, L.W.; Shah, B.; Mehran, R.; Mack, M.J.; Brilakis, E.S.; Reilly, J.P.; Zoghbi, G.; Holper, E.; Stone, G.W. Consideration of a new definition of clinically relevant myocardial infarction after coronary revascularization: An expert consensus document from the Society for Cardiovascular Angiography and Interventions (SCAI). J. Am. Coll. Cardiol. 2013, 62, 1563–1570. [Google Scholar] [CrossRef] [Green Version]
- Maron, D.J.; Hochman, J.S.; Reynolds, H.R.; Bangalore, S.; O’Brien, S.M.; Boden, W.E.; Chaitman, B.R.; Senior, R.; López-Sendón, J.; Alexander, K.P.; et al. Initial Invasive or Conservative Strategy for Stable Coronary Disease. N. Engl. J. Med. 2020, 382, 1395–1407. [Google Scholar] [CrossRef]
- Hara, H.; Serruys, P.W.; Takahashi, K.; Kawashima, H.; Ono, M.; Gao, C.; Wang, R.; Mohr, F.W.; Holmes, D.R.; Davierwala, P.M.; et al. Impact of Peri-Procedural Myocardial Infarction on Outcomes After Revascularization. J. Am. Coll. Cardiol. 2020, 76, 1622–1639. [Google Scholar] [CrossRef]
- Zeitouni, M.; Silvain, J.; Guedeney, P.; Kerneis, M.; Yan, Y.; Overtchouk, P.; Barthelemy, O.; Hauguel-Moreau, M.; Choussat, R.; Helft, G.; et al. Periprocedural myocardial infarction and injury in elective coronary stenting. Eur. Heart J. 2018, 39, 1100–1109. [Google Scholar] [CrossRef]
- Ben-Yehuda, O.; Chen, S.; Redfors, B.; McAndrew, T.; Crowley, A.; Kosmidou, I.; Kandzari, D.E.; Puskas, J.D.; Morice, M.C.; Taggart, D.P.; et al. Impact of large periprocedural myocardial infarction on mortality after percutaneous coronary intervention and coronary artery bypass grafting for left main disease: An analysis from the EXCEL trial. Eur. Heart J. 2019, 40, 1930–1941. [Google Scholar] [CrossRef]
- Herrmann, J. Peri-procedural myocardial injury: 2005 update. Eur. Heart J. 2005, 26, 2493–2519. [Google Scholar] [CrossRef]
- Patel Vishal, G.; Brayton Kimberly, M.; Mintz Gary, S.; Maehara, A.; Banerjee, S.; Brilakis Emmanouil, S. Intracoronary and Noninvasive Imaging for Prediction of Distal Embolization and Periprocedural Myocardial Infarction During Native Coronary Artery Percutaneous Intervention. Circ. Cardiovasc. Imaging 2013, 6, 1102–1114. [Google Scholar] [CrossRef] [Green Version]
- Popma, J.J.; Mauri, L.; O’Shaughnessy, C.; Overlie, P.; McLaurin, B.; Almonacid, A.; Kirtane, A.; Leon, M.B. Frequency and clinical consequences associated with sidebranch occlusion during stent implantation using zotarolimus-eluting and paclitaxel-eluting coronary stents. Circ. Cardiovasc. Interv. 2009, 2, 133–139. [Google Scholar] [CrossRef] [Green Version]
- Jaffe, R.; Dick, A.; Strauss Bradley, H. Prevention and Treatment of Microvascular Obstruction-Related Myocardial Injury and Coronary No-Reflow Following Percutaneous Coronary Intervention. JACC Cardiovasc. Interv. 2010, 3, 695–704. [Google Scholar] [CrossRef] [Green Version]
- Chan, W.; Stub, D.; Clark, D.J.; Ajani, A.E.; Andrianopoulos, N.; Brennan, A.L.; New, G.; Black, A.; Shaw, J.A.; Reid, C.M.; et al. Usefulness of Transient and Persistent No Reflow to Predict Adverse Clinical Outcomes Following Percutaneous Coronary Intervention. Am. J. Cardiol. 2012, 109, 478–485. [Google Scholar] [CrossRef]
- Ndrepepa, G.; Tiroch, K.; Keta, D.; Fusaro, M.; Seyfarth, M.; Pache, J.; Mehilli, J.; Schomig, A.; Kastrati, A. Predictive factors and impact of no reflow after primary percutaneous coronary intervention in patients with acute myocardial infarction. Circ. Cardiovasc. Interv. 2010, 3, 27–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morishima, I.; Sone, T.; Okumura, K.; Tsuboi, H.; Kondo, J.; Mukawa, H.; Matsui, H.; Toki, Y.; Ito, T.; Hayakawa, T. Angiographic no-reflow phenomenon as a predictor of adverse long-term outcome in patients treated with percutaneous transluminal coronary angioplasty for first acute myocardial infarction. J. Am. Coll. Cardiol. 2000, 36, 1202–1209. [Google Scholar] [CrossRef] [Green Version]
- Endo, M.; Hibi, K.; Shimizu, T.; Komura, N.; Kusama, I.; Otsuka, F.; Mitsuhashi, T.; Iwahashi, N.; Okuda, J.; Tsukahara, K.; et al. Impact of Ultrasound Attenuation and Plaque Rupture as Detected by Intravascular Ultrasound on the Incidence of No-Reflow Phenomenon After Percutaneous Coronary Intervention in ST-Segment Elevation Myocardial Infarction. JACC Cardiovasc. Interv. 2010, 3, 540–549. [Google Scholar] [CrossRef] [Green Version]
- Kimura, S.; Kakuta, T.; Yonetsu, T.; Suzuki, A.; Iesaka, Y.; Fujiwara, H.; Isobe, M. Clinical significance of echo signal attenuation on intravascular ultrasound in patients with coronary artery disease. Circ. Cardiovasc. Interv. 2009, 2, 444–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitsuba, N.; Teragawa, H.; Hata, T.; Nishioka, K.; Fujii, Y.; Mikami, S.; Fujimura, N.; Maruhashi, T.; Kurisu, S.; Kihara, Y. Deep echo attenuation without calcification increases the risk of periprocedural myonecrosis after elective percutaneous coronary intervention in patients with coronary artery disease. Intern. Med. 2012, 51, 691–698. [Google Scholar] [CrossRef] [Green Version]
- Böse, D.; von Birgelen, C.; Zhou, X.Y.; Schmermund, A.; Philipp, S.; Sack, S.; Konorza, T.; Möhlenkamp, S.; Leineweber, K.; Kleinbongard, P.; et al. Impact of atherosclerotic plaque composition on coronary microembolization during percutaneous coronary interventions. Basic Res. Cardiol. 2008, 103, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, R.; Oshima, S.; Jingu, M.; Tsurugaya, H.; Toyama, T.; Hoshizaki, H.; Taniguchi, K. Usefulness of virtual histology intravascular ultrasound to predict distal embolization for ST-segment elevation myocardial infarction. J. Am. Coll. Cardiol. 2007, 50, 1641–1646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakakura, K.; Nakano, M.; Otsuka, F.; Ladich, E.; Kolodgie, F.D.; Virmani, R. Pathophysiology of Atherosclerosis Plaque Progression. HeartLung Circ. 2013, 22, 399–411. [Google Scholar] [CrossRef] [Green Version]
- Lee Sung, Y.; Mintz Gary, S.; Kim, S.-Y.; Hong Young, J.; Kim Sang, W.; Okabe, T.; Pichard Augusto, D.; Satler Lowell, F.; Kent Kenneth, M.; Suddath William, O.; et al. Attenuated Plaque Detected by Intravascular Ultrasound. JACC: Cardiovasc. Interv. 2009, 2, 65–72. [Google Scholar] [CrossRef] [Green Version]
- Bibek, S.-b.; Xie, Y.; Gao, J.-j.; Wang, Z.; Wang, J.-f.; Geng, D.-f. Role of Pre-procedural C-reactive Protein Level in the Prediction of Major Adverse Cardiac Events in Patients Undergoing Percutaneous Coronary Intervention: A Meta-analysisof Longitudinal Studies. Inflammation 2015, 38, 159–169. [Google Scholar] [CrossRef]
- Patti, G.; Mangiacapra, F.; Ricottini, E.; Cannatà, A.; Cavallari, I.; Vizzi, V.; D’Ambrosio, A.; Dicuonzo, G.; Di Sciascio, G. Correlation of Platelet Reactivity and C-Reactive Protein Levels to Occurrence of Peri-Procedural Myocardial Infarction in Patients Undergoing Percutaneous Coronary Intervention (from the ARMYDA-CRP Study). Am. J. Cardiol. 2013, 111, 1739–1744. [Google Scholar] [CrossRef]
- Ellis Stephen, G.; Chew, D.; Chan, A.; Whitlow Patrick, L.; Schneider Jakob, P.; Topol Eric, J. Death Following Creatine Kinase-MB Elevation After Coronary Intervention. Circulation 2002, 106, 1205–1210. [Google Scholar] [CrossRef] [Green Version]
- Niccoli, G.; Sgueglia, G.A.; Latib, A.; Crea, F.; Colombo, A.; on behalf of the CACTUS Study Group. Association of baseline C-reactive protein levels with periprocedural myocardial injury in patients undergoing percutaneous bifurcation intervention: A CACTUS study subanalysis. Catheter. Cardiovasc. Interv. 2014, 83, E37–E44. [Google Scholar] [CrossRef]
- Saadeddin, S.M.; Habbab, M.d.A.; Sobki, S.H.; Ferns, G.A. Association of systemic inflammatory state with troponin I elevation after elective uncomplicated percutaneous coronary intervention. Am. J. Cardiol. 2002, 89, 981–983. [Google Scholar] [CrossRef]
- Yao, M.; Zhao, L.; Wu, L.; Zhang, W.; Luan, Y.; Song, J.; Fu, G.; Zhu, J. Predictive value of baseline C-reactive protein for periprocedural myocardial infraction of higher risk stratifications: A retrospective cohort clinical study. Anatol. J. Cardiol. 2018, 20, 310–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zairis, M.N.; Ambrose, J.A.; Ampartzidou, O.; Lyras, A.G.; Manousakis, S.J.; Makrygiannis, S.S.; Beldekos, D.J.; Devoe, M.C.; Fakiolas, C.N.; Prekates, A.A.; et al. Preprocedural plasma C-reactive protein levels, postprocedural creatine kinase-MB release, and long-term prognosis after successful coronary stenting (four-year results from the GENERATION study). Am. J. Cardiol. 2005, 95, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Li, Y.; Xu, T.; Luan, Y.; Lv, Q.; Wang, Y.; Lv, X.; Fu, G.; Zhang, W. Impact of increased inflammation biomarkers on periprocedural myocardial infarction in patients undergoing elective percutaneous coronary intervention: A cohort study. J. Thorac. Dis. 2020, 12, 5398–5410. [Google Scholar] [CrossRef]
- Karabağ, Y.; Çağdaş, M.; Rencuzogullari, I.; Karakoyun, S.; Artaç, İ.; İliş, D.; Yesin, M.; Çağdaş, Ö.S.; Altıntaş, B.; Burak, C.; et al. Usefulness of The C-Reactive Protein/Albumin Ratio for Predicting No-Reflow in ST-elevation myocardial infarction treated with primary percutaneous coronary intervention. Eur. J. Clin. Investig. 2018, 48, e12928. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, H.-W.; Wang, C.-F.; Zhang, X.-J.; Tao, J.; Cui, C.-S.; Meng, Q.-K.; Zhu, Y.; Luo, D.-F.; Hou, A.-J.; et al. Incidence, Predictors, and Prognosis of Coronary Slow-Flow and No-Reflow Phenomenon in Patients with Chronic Total Occlusion Who Underwent Percutaneous Coronary Intervention. Clin. Risk Manag. 2020, 16, 95–101. [Google Scholar] [CrossRef] [Green Version]
- Ishii, H.; Toriyama, T.; Aoyama, T.; Takahashi, H.; Amano, T.; Hayashi, M.; Tanaka, M.; Kawamura, Y.; Yasuda, Y.; Yuzawa, Y.; et al. Prognostic values of C-reactive protein levels on clinical outcome after implantation of sirolimus-eluting stents in patients on hemodialysis. Circ. Cardiovasc. Interv. 2009, 2, 513–518. [Google Scholar] [CrossRef] [Green Version]
- Delhaye, C.; Maluenda, G.; Wakabayashi, K.; Ben-Dor, I.; Lemesle, G.; Collins, S.D.; Syed, A.I.; Torguson, R.; Kaneshige, K.; Xue, Z.; et al. Long-term prognostic value of preprocedural C-reactive protein after drug-eluting stent implantation. Am. J. Cardiol. 2010, 105, 826–832. [Google Scholar] [CrossRef]
- Park, D.-W.; Lee, C.W.; Yun, S.-C.; Kim, Y.-H.; Hong, M.-K.; Kim, J.-J.; Park, S.-W.; Park, S.-J. Prognostic impact of preprocedural C reactive protein levels on 6-month angiographic and 1-year clinical outcomes after drug-eluting stent implantation. Heart 2007, 93, 1087–1092. [Google Scholar] [CrossRef] [Green Version]
- Aronow, H.D.; Quinn, M.J.; Gurm, H.S.; Lauer, M.S.; Brennan, D.M.; Topol, E.J.; Lincoff, A.M. Preprocedure inflammatory state predicts periprocedural myocardial infarction after elective percutaneous coronary intervention: An EPIC substudy. J. Am. Coll. Cardiol. 2003, 41, 17. [Google Scholar] [CrossRef] [Green Version]
- Aronow Herbert, D.; Shishehbor, M.; Davis DonaLee, A.; Katzan Irene, L.; Bhatt Deepak, L.; Bajzer Christopher, T.; Abou-Chebl, A.; Derk Krieger, W.; Whitlow Patrick, L.; Yadav Jay, S. Leukocyte Count Predicts Microembolic Doppler Signals During Carotid Stenting. Stroke 2005, 36, 1910–1914. [Google Scholar] [CrossRef] [Green Version]
- Abdi, S.; Rafizadeh, O.; Peighambari, M.; Basiri, H.; Bakhshandeh, H. Evaluation of the Clinical and Procedural Predictive Factors of no-Reflow Phenomenon Following Primary Percutaneous Coronary Intervention. Res. Cardiovasc. Med. 2015, 4, e25414. [Google Scholar] [CrossRef]
- Gurm, H.S.; Bhatt, D.L.; Gupta, R.; Ellis, S.G.; Topol, E.J.; Lauer, M.S. Preprocedural white blood cell count and death after percutaneous coronary intervention. Am. Heart J. 2003, 146, 692–698. [Google Scholar] [CrossRef]
- Palmerini, T.; Marzocchi, A.; Marrozzini, C.; Ortolani, P.; Saia, F.; Bacchi-Reggiani, L.; Virzì, S.; Gianstefani, S.; Branzi, A. Preprocedural Levels of C-Reactive Protein and Leukocyte Counts Predict 9-Month Mortality After Coronary Angioplasty for the Treatment of Unprotected Left Main Coronary Artery Stenosis. Circulation 2005, 112, 2332–2338. [Google Scholar] [CrossRef] [Green Version]
- Toor, I.S.; Jaumdally, R.; Lip, G.Y.; Millane, T.; Varma, C. Eosinophil count predicts mortality following percutaneous coronary intervention. Thromb. Res. 2012, 130, 607–611. [Google Scholar] [CrossRef]
- Verdoia, M.; Schaffer, A.; Barbieri, L.; Sinigaglia, F.; Marino, P.; Suryapranata, H.; De Luca, G. Eosinophils count and periprocedural myocardial infarction in patients undergoing percutaneous coronary interventions. Atherosclerosis 2014, 236, 169–174. [Google Scholar] [CrossRef]
- Akpek, M.; Kaya, M.G.; Lam, Y.Y.; Sahin, O.; Elcik, D.; Celik, T.; Ergin, A.; Gibson, C.M. Relation of neutrophil/lymphocyte ratio to coronary flow to in-hospital major adverse cardiac events in patients with ST-elevated myocardial infarction undergoing primary coronary intervention. Am. J. Cardiol. 2012, 110, 621–627. [Google Scholar] [CrossRef]
- Tian, J.; Liu, Y.; Liu, Y.; Song, X.; Zhang, M.; Xu, F.; Yuan, F.; Lyu, S. Prognostic Association of Circulating Neutrophil Count with No-Reflow in Patients with ST-Segment Elevation Myocardial Infarction following Successful Primary Percutaneous Intervention. Dis. Markers 2017, 2017, 8458492. [Google Scholar] [CrossRef] [Green Version]
- Vakili, H.; Khaheshi, I.; Sharifi, A.; Nickdoost, N.; Namazi, M.H.; Safi, M.; Saadat, H.; Parsa, S.A.; Akbarzadeh, M.A.; Naderian, M.; et al. Assessment of Admission Time Cell Blood Count (CBC) Parameters in Predicting Post-primary Percutaneous Coronary Intervention TIMI Frame Count in Patients with ST-segment Elevation Myocardial Infarction. Cardiovasc. Hematol. Disord. Drug Targets 2020, 20, 191–197. [Google Scholar] [CrossRef]
- Sen, N.; Afsar, B.; Ozcan, F.; Buyukkaya, E.; Isleyen, A.; Akcay, A.B.; Yuzgecer, H.; Kurt, M.; Karakas, M.F.; Basar, N.; et al. The neutrophil to lymphocyte ratio was associated with impaired myocardial perfusion and long term adverse outcome in patients with ST-elevated myocardial infarction undergoing primary coronary intervention. Atherosclerosis 2013, 228, 203–210. [Google Scholar] [CrossRef]
- Kalyoncuoglu, M.; Biter, H.I.; Ozturk, S.; Belen, E.; Can, M.M. Predictive accuracy of lymphocyte-to-monocyte ratio and monocyte-to-high-density-lipoprotein-cholesterol ratio in determining the slow flow/no-reflow phenomenon in patients with non–ST-elevated myocardial infarction. Coron. Artery Dis. 2020, 31. [Google Scholar] [CrossRef] [PubMed]
- Kurtul, A.; Yarlioglues, M.; Celik, I.E.; Duran, M.; Elcik, D.; Kilic, A.; Oksuz, F.; Murat, S.N. Association of lymphocyte-to-monocyte ratio with the no-reflow phenomenon in patients who underwent a primary percutaneous coronary intervention for ST-elevation myocardial infarction. Coron. Artery Dis. 2015, 26, 706–712. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Ma, J.; Jiang, Z.; Wu, F.; Ping, J.; Ming, L. Association of lymphocyte-to-monocyte ratio with in-hospital and long-term major adverse cardiac and cerebrovascular events in patients with ST-elevated myocardial infarction. Medicine 2017, 96, e7897. [Google Scholar] [CrossRef] [PubMed]
- Ren, F.; Mu, N.; Zhang, X.; Tan, J.; Li, L.; Zhang, C.; Dong, M. Increased Platelet-leukocyte Aggregates Are Associated With Myocardial No-reflow in Patients With ST Elevation Myocardial Infarction. Am. J. Med Sci. 2016, 352, 261–266. [Google Scholar] [CrossRef]
- Funayama, H.; Ishikawa, S.E.; Sugawara, Y.; Kubo, N.; Momomura, S.; Kawakami, M. Myeloperoxidase may contribute to the no-reflow phenomenon in patients with acute myocardial infarction. Int. J. Cardiol. 2010, 139, 187–192. [Google Scholar] [CrossRef]
- Stamboul, K.; Zeller, M.; Rochette, L.; Cottin, Y.; Cochet, A.; Leclercq, T.; Porot, G.; Guenancia, C.; Fichot, M.; Maillot, N.; et al. Relation between high levels of myeloperoxidase in the culprit artery and microvascular obstruction, infarct size and reverse remodeling in ST-elevation myocardial infarction. PLoS ONE 2017, 12, e0179929. [Google Scholar] [CrossRef] [Green Version]
- Buyukkaya, E.; Poyraz, F.; Karakas, M.F.; Kurt, M.; Akcay, A.B.; Akpinar, I.; Motor, S.; Turak, O.; Ozturk, O.H.; Sen, N.; et al. Usefulness of Monocyte Chemoattractant Protein-1 to Predict No-Reflow and Three-Year Mortality in Patients With ST-Segment Elevation Myocardial Infarction Undergoing Primary Percutaneous Coronary Intervention. Am. J. Cardiol. 2013, 112, 187–193. [Google Scholar] [CrossRef]
- Yin, Y.-J.; Chen, Y.-C.; Xu, L.; Zhao, X.-H.; Song, Y. Relationship of lipoprotein-associated phospholipase A2(Lp-PLA2) and periprocedural myocardial injury in patients undergoing elective percutaneous coronary intervention. Int. J. Cardiol. Heart Vasc. 2020, 28, 100541. [Google Scholar] [CrossRef]
- Lagrand Wim, K.; Visser Cees, A.; Hermens Willem, T.; Niessen Hans, W.M.; Verheugt Freek, W.A.; Wolbink, G.-J.; Hack, C.E. C-Reactive Protein as a Cardiovascular Risk Factor. Circulation 1999, 100, 96–102. [Google Scholar] [CrossRef] [Green Version]
- Briguori, C.; Visconti, G.; Focaccio, A.; Golia, B.; Chieffo, A.; Castelli, A.; Mussardo, M.; Montorfano, M.; Ricciardelli, B.; Colombo, A. Novel Approaches for Preventing or Limiting Events (Naples) II Trial. J. Am. Coll. Cardiol. 2009, 54, 2157–2163. [Google Scholar] [CrossRef] [Green Version]
- Zhang, E.; Gao, M.; Gao, J.; Xiao, J.; Li, X.; Zhao, H.; Wang, J.; Zhang, N.; Wang, S.; Liu, Y. Inflammatory and Hematological Indices as Simple, Practical Severity Predictors of Microdysfunction Following Coronary Intervention: A Systematic Review and Meta-Analysis. Angiology 2020, 71, 349–359. [Google Scholar] [CrossRef]
- Ray Kausik, K.; Cannon Christopher, P.; Cairns, R.; Morrow David, A.; Ridker Paul, M.; Braunwald, E. Prognostic Utility of ApoB/AI, Total Cholesterol/HDL, Non-HDL Cholesterol, or hs-CRP as Predictors of Clinical Risk in Patients Receiving Statin Therapy After Acute Coronary Syndromes. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 424–430. [Google Scholar] [CrossRef] [Green Version]
- Swiatkiewicz, I.; Kozinski, M.; Magielski, P.; Fabiszak, T.; Sukiennik, A.; Navarese, E.P.; Odrowaz-Sypniewska, G.; Kubica, J. Value of C-reactive protein in predicting left ventricular remodelling in patients with a first ST-segment elevation myocardial infarction. Mediat. Inflamm. 2012, 2012, 250867. [Google Scholar] [CrossRef]
- Świątkiewicz, I.; Magielski, P.; Kubica, J. C-Reactive Protein as a Risk Marker for Post-Infarct Heart Failure over a Multi-Year Period. Int. J. Mol. Sci. 2021, 22, 3169. [Google Scholar] [CrossRef]
- Świątkiewicz, I.; Magielski, P.; Kubica, J.; Zadourian, A.; DeMaria, A.N.; Taub, P.R. Enhanced Inflammation is a Marker for Risk of Post-Infarct Ventricular Dysfunction and Heart Failure. Int. J. Mol. Sci. 2020, 21, 807. [Google Scholar] [CrossRef] [Green Version]
- Welsh, C.; Welsh, P.; Mark Patrick, B.; Celis-Morales Carlos, A.; Lewsey, J.; Gray Stuart, R.; Lyall Donald, M.; Iliodromiti, S.; Gill Jason, M.R.; Pell, J.; et al. Association of Total and Differential Leukocyte Counts With Cardiovascular Disease and Mortality in the UK Biobank. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1415–1423. [Google Scholar] [CrossRef] [Green Version]
- Madjid, M.; Awan, I.; Willerson, J.T.; Casscells, S.W. Leukocyte count and coronary heart disease: Implications for risk assessment. J. Am. Coll. Cardiol. 2004, 44, 1945–1956. [Google Scholar] [CrossRef] [Green Version]
- Bhat, T.; Teli, S.; Rijal, J.; Bhat, H.; Raza, M.; Khoueiry, G.; Meghani, M.; Akhtar, M.; Costantino, T. Neutrophil to lymphocyte ratio and cardiovascular diseases: A review. Expert Rev. Cardiovasc. Ther. 2013, 11, 55–59. [Google Scholar] [CrossRef]
- Botto, N.; Sbrana, S.; Trianni, G.; Andreassi, M.G.; Ravani, M.; Rizza, A.; Al-Jabri, A.; Palmieri, C.; Berti, S. An increased platelet–leukocytes interaction at the culprit site of coronary artery occlusion in acute myocardial infarction: A pathogenic role for “no-reflow” phenomenon? Int. J. Cardiol. 2007, 117, 123–130. [Google Scholar] [CrossRef]
- Nicholls Stephen, J.; Hazen Stanley, L. Myeloperoxidase and Cardiovascular Disease. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1102–1111. [Google Scholar] [CrossRef] [Green Version]
- Ali, M.; Pulli, B.; Courties, G.; Tricot, B.; Sebas, M.; Iwamoto, Y.; Hilgendorf, I.; Schob, S.; Dong, A.; Zheng, W.; et al. Myeloperoxidase Inhibition Improves Ventricular Function and Remodeling After Experimental Myocardial Infarction. JACC Basic Transl. Sci. 2016, 1, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, J.; Lekstrom-Himes, J.; Donaldson, D.; Lee, Y.; Hu, M.; Xu, J.; Wyant, T.; Davidson, M. Effect of CC Chemokine Receptor 2 CCR2 Blockade on Serum C-Reactive Protein in Individuals at Atherosclerotic Risk and With a Single Nucleotide Polymorphism of the Monocyte Chemoattractant Protein-1 Promoter Region. Am. J. Cardiol. 2011, 107, 906–911. [Google Scholar] [CrossRef] [PubMed]
- Tucker, B.; Kurup, R.; Barraclough, J.; Henriquez, R.; Cartland, S.; Arnott, C.; Misra, A.; Martínez, G.; Kavurma, M.; Patel, S. Colchicine as a Novel Therapy for Suppressing Chemokine Production in Patients With an Acute Coronary Syndrome: A Pilot Study. Clin. Ther. 2019, 41, 2172–2181. [Google Scholar] [CrossRef]
- Khuseyinova, N.; Imhof, A.; Rothenbacher, D.; Trischler, G.; Kuelb, S.; Scharnagl, H.; Maerz, W.; Brenner, H.; Koenig, W. Association between Lp-PLA2 and coronary artery disease: Focus on its relationship with lipoproteins and markers of inflammation and hemostasis. Atherosclerosis 2005, 182, 181–188. [Google Scholar] [CrossRef] [PubMed]
- De Stefano, A.; Mannucci, L.; Tamburi, F.; Cardillo, C.; Schinzari, F.; Rovella, V.; Nisticò, S.; Bennardo, L.; Di Daniele, N.; Tesauro, M. Lp-PLA(2), a new biomarker of vascular disorders in metabolic diseases. Int. J. Immunopathol. Pharm. 2019, 33, 2058738419827154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gach, O.; Legrand, V.; Biessaux, Y.; Chapelle, J.P.; Vanbelle, S.; Pierard, L.A. Long-Term Prognostic Significance of High-Sensitivity C-Reactive Protein Before and After Coronary Angioplasty in Patients With Stable Angina Pectoris. Am. J. Cardiol. 2007, 99, 31–35. [Google Scholar] [CrossRef]
- Saleh, N.; Svane, B.; Hansson, L.-O.; Jensen, J.; Nilsson, T.; Danielsson, O.; Tornvall, P. Response of Serum C-Reactive Protein to Percutaneous Coronary Intervention Has Prognostic Value. Clin. Chem. 2005, 51, 2124–2130. [Google Scholar] [CrossRef] [Green Version]
- Gaspardone, A.; Versaci, F.; Tomai, F.; Citone, C.; Proietti, I.; Gioffrè, G.; Skossyreva, O. C-Reactive Protein, Clinical Outcome, and Restenosis Rates After Implantation of Different Drug-Eluting Stents. Am. J. Cardiol. 2006, 97, 1311–1316. [Google Scholar] [CrossRef]
- Gottsauner-Wolf, M.; Zasmeta, G.; Hornykewycz, S.; Nikfardjam, M.; Stepan, E.; Wexberg, P.; Zorn, G.; Glogar, D.; Probst, P.; Maurer, G.; et al. Plasma levels of C-reactive protein after coronary stent implantation. Eur. Heart J. 2000, 21, 1152–1158. [Google Scholar] [CrossRef]
- Kang, W.C.; Il Moon, C.; Lee, K.; Han, S.H.; Suh, S.Y.; Moon, J.; Shin, M.S.; Ahn, T.; Shin, E.K. Comparison of inflammatory markers for the prediction of neointimal hyperplasia after drug-eluting stent implantation. Coron. Artery Dis. 2011, 22, 526–532. [Google Scholar] [CrossRef]
- Li, C.; Zhang, F.; Shen, Y.; Xu, R.; Chen, Z.; Dai, Y.; Lu, H.; Chang, S.; Qian, J.; Wang, X.; et al. Impact of Neutrophil to Lymphocyte Ratio (NLR) Index and Its Periprocedural Change (NLRΔ) for Percutaneous Coronary Intervention in Patients With Chronic Total Occlusion. Angiology 2016, 68, 640–646. [Google Scholar] [CrossRef]
- Cole, J.; Htun, N.; Lew, R.; Freilich, M.; Quinn, S.; Layland, J. Colchicine to Prevent Periprocedural Myocardial Injury in Percutaneous Coronary Intervention: The COPE-PCI Pilot Trial. Circ. Cardiovasc. Interv. 2021, 14, e009992. [Google Scholar] [CrossRef]
- Nakachi, T.; Kosuge, M.; Hibi, K.; Ebina, T.; Hashiba, K.; Mitsuhashi, T.; Endo, M.; Umemura, S.; Kimura, K. C-reactive protein elevation and rapid angiographic progression of nonculprit lesion in patients with non-ST-segment elevation acute coronary syndrome. Circ. J. 2008, 72, 1953–1959. [Google Scholar] [CrossRef] [Green Version]
- Inoue, K.; Kodama, T.; Daida, H. Pentraxin 3: A Novel Biomarker for Inflammatory Cardiovascular Disease. Int. J. Vasc. Med. 2012, 2012, 657025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, S.; Sugiyama, T.; Hishikari, K.; Nakagama, S.; Nakamura, S.; Misawa, T.; Mizusawa, M.; Hayasaka, K.; Yamakami, Y.; Sagawa, Y.; et al. Relationship of systemic pentraxin-3 values with coronary plaque components on optical coherence tomography and post-percutaneous coronary intervention outcomes in patients with stable angina pectoris. Atherosclerosis 2020, 292, 127–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Husebye, T.; Eritsland, J.; Arnesen, H.; Bjørnerheim, R.; Mangschau, A.; Seljeflot, I.; Andersen, G.Ø. Association of Interleukin 8 and Myocardial Recovery in Patients with ST-Elevation Myocardial Infarction Complicated by Acute Heart Failure. PLoS ONE 2014, 9, e112359. [Google Scholar] [CrossRef] [PubMed]
- Stumpf, C.; Sheriff, A.; Zimmermann, S.; Schaefauer, L.; Schlundt, C.; Raaz, D.; Garlichs, C.D.; Achenbach, S. C-reactive protein levels predict systolic heart failure and outcome in patients with first ST-elevation myocardial infarction treated with coronary angioplasty. Arch. Med Sci. 2017, 13, 1086–1093. [Google Scholar] [CrossRef] [Green Version]
- Everett, B.M.; Cornel, J.H.; Lainscak, M.; Anker, S.D.; Abbate, A.; Thuren, T.; Libby, P.; Glynn, R.J.; Ridker, P.M. Anti-Inflammatory Therapy With Canakinumab for the Prevention of Hospitalization for Heart Failure. Circulation 2019, 139, 1289–1299. [Google Scholar] [CrossRef]
- Fanola, C.L.; Morrow, D.A.; Cannon, C.P.; Jarolim, P.; Lukas, M.A.; Bode, C.; Hochman, J.S.; Goodrich, E.L.; Braunwald, E.; O’Donoghue, M.L. Interleukin-6 and the Risk of Adverse Outcomes in Patients After an Acute Coronary Syndrome: Observations From the SOLID-TIMI 52 (Stabilization of Plaque Using Darapladib-Thrombolysis in Myocardial Infarction 52) Trial. J. Am. Heart Assoc. 2017, 6. [Google Scholar] [CrossRef] [Green Version]
- Ridker, P.M.; MacFadyen, J.G.; Everett, B.M.; Libby, P.; Thuren, T.; Glynn, R.J. Relationship of C-reactive protein reduction to cardiovascular event reduction following treatment with canakinumab: A secondary analysis from the CANTOS randomised controlled trial. Lancet 2018, 391, 319–328. [Google Scholar] [CrossRef]
- Guedeney, P.; Claessen, B.E.; Kalkman, D.N.; Aquino, M.; Sorrentino, S.; Giustino, G.; Farhan, S.; Vogel, B.; Sartori, S.; Montalescot, G.; et al. Residual Inflammatory Risk in Patients With Low LDL Cholesterol Levels Undergoing Percutaneous Coronary Intervention. J. Am. Coll. Cardiol. 2019, 73, 2401–2409. [Google Scholar] [CrossRef]
- Kalkman, D.N.; Aquino, M.; Claessen, B.E.; Baber, U.; Guedeney, P.; Sorrentino, S.; Vogel, B.; de Winter, R.J.; Sweeny, J.; Kovacic, J.C.; et al. Residual inflammatory risk and the impact on clinical outcomes in patients after percutaneous coronary interventions. Eur. Heart J. 2018, 39, 4101–4108. [Google Scholar] [CrossRef] [PubMed]
- Martínez Gonzalo, J.; Robertson, S.; Barraclough, J.; Xia, Q.; Mallat, Z.; Bursill, C.; Celermajer David, S.; Patel, S. Colchicine Acutely Suppresses Local Cardiac Production of Inflammatory Cytokines in Patients With an Acute Coronary Syndrome. J. Am. Heart Assoc. 2015, 4, e002128. [Google Scholar] [CrossRef] [Green Version]
- Bahrmann, P.; Werner Gerald, S.; Heusch, G.; Ferrari, M.; Poerner Tudor, C.; Voss, A.; Figulla Hans, R. Detection of Coronary Microembolization by Doppler Ultrasound in Patients With Stable Angina Pectoris Undergoing Elective Percutaneous Coronary Interventions. Circulation 2007, 115, 600–608. [Google Scholar] [CrossRef] [Green Version]
- Charron, T.; Jaffe, R.; Segev, A.; Bang, K.W.A.; Qiang, B.; Sparkes, J.D.; Butany, J.; Dick, A.J.; Freedman, J.; Strauss, B.H. Effects of distal embolization on the timing of platelet and inflammatory cell activation in interventional coronary no-reflow. Thromb. Res. 2010, 126, 50–55. [Google Scholar] [CrossRef]
- Boos, C.J.; Balakrishnan, B.; Jessani, S.; Blann, A.D.; Lip, G.Y. Effects of percutaneous coronary intervention on peripheral venous blood circulating endothelial cells and plasma indices of endothelial damage/dysfunction. Chest 2007, 132, 1920–1926. [Google Scholar] [CrossRef]
- Cornelissen, A.; Vogt, F.J. The effects of stenting on coronary endothelium from a molecular biological view: Time for improvement? J. Cell. Mol. Med. 2019, 23, 39–46. [Google Scholar] [CrossRef]
- Munk, P.S.; Breland, U.M.; Aukrust, P.; Skadberg, O.; Ueland, T.; Larsen, A.I. Inflammatory response to percutaneous coronary intervention in stable coronary artery disease. J. Thromb. Thrombolysis 2011, 31, 92–98. [Google Scholar] [CrossRef]
- Tousoulis, D.; Charakida, M.; Stefanadis, C. Endothelial function and inflammation in coronary artery disease. Heart 2006, 92, 441–444. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Yellon, D.M. Myocardial ischemia-reperfusion injury: A neglected therapeutic target. J. Clin. Investig. 2013, 123, 92–100. [Google Scholar] [CrossRef]
- Wu, M.Y.; Yiang, G.T.; Liao, W.T.; Tsai, A.P.Y.; Cheng, Y.L.; Cheng, P.W.; Li, C.Y.; Li, C.J. Current Mechanistic Concepts in Ischemia and Reperfusion Injury. Cell. Physiol. Biochem. 2018, 46, 1650–1667. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Chemokines in ischemia and reperfusion. Thromb Haemost 2007, 97, 738–747. [Google Scholar] [CrossRef] [PubMed]
- Gordon, J.W.; Shaw, J.A.; Kirshenbaum, L.A. Multiple Facets of NF-κB in the Heart. Circ. Res. 2011, 108, 1122–1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinten-Johansen, J. Involvement of neutrophils in the pathogenesis of lethal myocardial reperfusion injury. Cardiovasc. Res. 2004, 61, 481–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arslan, F.; Smeets, M.B.; O’Neill, L.A.; Keogh, B.; McGuirk, P.; Timmers, L.; Tersteeg, C.; Hoefer, I.E.; Doevendans, P.A.; Pasterkamp, G.; et al. Myocardial ischemia/reperfusion injury is mediated by leukocytic toll-like receptor-2 and reduced by systemic administration of a novel anti-toll-like receptor-2 antibody. Circulation 2010, 121, 80–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liehn, E.A.; Piccinini, A.-M.; Koenen, R.R.; Soehnlein, O.; Adage, T.; Fatu, R.; Curaj, A.; Popescu, A.; Zernecke, A.; Kungl, A.J.; et al. A New Monocyte Chemotactic Protein-1/Chemokine CC Motif Ligand-2 Competitor Limiting Neointima Formation and Myocardial Ischemia/Reperfusion Injury in Mice. J. Am. Coll. Cardiol. 2010, 56, 1847–1857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayasaki, T.; Kaikita, K.; Okuma, T.; Yamamoto, E.; Kuziel, W.A.; Ogawa, H.; Takeya, M. CC chemokine receptor-2 deficiency attenuates oxidative stress and infarct size caused by myocardial ischemia-reperfusion in mice. Circ. J. 2006, 70, 342–351. [Google Scholar] [CrossRef] [Green Version]
- Maekawa, N.; Wada, H.; Kanda, T.; Niwa, T.; Yamada, Y.; Saito, K.; Fujiwara, H.; Sekikawa, K.; Seishima, M. Improved myocardial ischemia/reperfusion injury in mice lacking tumor necrosis factor-α. J. Am. Coll. Cardiol. 2002, 39, 1229–1235. [Google Scholar] [CrossRef] [Green Version]
- Toldo, S.; Marchetti, C.; Mauro, A.G.; Chojnacki, J.; Mezzaroma, E.; Carbone, S.; Zhang, S.; Van Tassell, B.; Salloum, F.N.; Abbate, A. Inhibition of the NLRP3 inflammasome limits the inflammatory injury following myocardial ischemia-reperfusion in the mouse. Int. J. Cardiol. 2016, 209, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Döring, Y.; Drechsler, M.; Soehnlein, O.; Weber, C. Neutrophils in Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 288–295. [Google Scholar] [CrossRef] [Green Version]
- Martínez, G.J.; Barraclough, J.Y.; Nakhla, S.; Kienzle, V.; Robertson, S.; Mallat, Z.; Celermajer, D.S.; Patel, S. Neutrophil-derived microparticles are released into the coronary circulation following percutaneous coronary intervention in acute coronary syndrome patients. Biosci. Rep. 2017, 37, BSR20160430. [Google Scholar] [CrossRef] [Green Version]
- Mangold, A.; Alias, S.; Scherz, T.; Hofbauer Thomas, M.; Jakowitsch, J.; Panzenböck, A.; Simon, D.; Laimer, D.; Bangert, C.; Kammerlander, A.; et al. Coronary Neutrophil Extracellular Trap Burden and Deoxyribonuclease Activity in ST-Elevation Acute Coronary Syndrome Are Predictors of ST-Segment Resolution and Infarct Size. Circ. Res. 2015, 116, 1182–1192. [Google Scholar] [CrossRef] [Green Version]
- Vaidya, K.; Tucker, B.; Kurup, R.; Khandkar, C.; Pandzic, E.; Barraclough, J.; Machet, J.; Misra, A.; Kavurma, M.; Martinez, G.; et al. Colchicine Inhibits Neutrophil Extracellular Trap Formation in Patients With Acute Coronary Syndrome After Percutaneous Coronary Intervention. J. Am. Heart Assoc. 2021, 10, e018993. [Google Scholar] [CrossRef]
- García-Méndez, R.C.; Almeida-Gutierrez, E.; Serrano-Cuevas, L.; Sánchez-Díaz, J.S.; Rosas-Peralta, M.; Ortega-Ramirez, J.A.; Palomo-Villada, J.A.; Isordia-Salas, I.; Alonso-Bravo, R.M.; Borrayo-Sanchez, G. Reduction of No Reflow with a Loading Dose of Atorvastatin before Primary Angioplasty in Patients with Acute ST Myocardial Infarction. Arch. Med. Res. 2018, 49, 620–629. [Google Scholar] [CrossRef]
- Thålin, C.; Hisada, Y.; Lundström, S.; Mackman, N.; Wallén, H. Neutrophil Extracellular Traps. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1724–1738. [Google Scholar] [CrossRef]
- Ge, L.; Zhou, X.; Ji, W.-J.; Lu, R.-Y.; Zhang, Y.; Zhang, Y.-D.; Ma, Y.-Q.; Zhao, J.-H.; Li, Y.-M. Neutrophil extracellular traps in ischemia-reperfusion injury-induced myocardial no-reflow: Therapeutic potential of DNase-based reperfusion strategy. Am. J. Physiol.-Heart Circ. Physiol. 2014, 308, H500–H509. [Google Scholar] [CrossRef] [Green Version]
- Parodi, G.; Marcucci, R.; Valenti, R.; Gori, A.M.; Migliorini, A.; Giusti, B.; Buonamici, P.; Gensini, G.F.; Abbate, R.; Antoniucci, D. High Residual Platelet Reactivity After Clopidogrel Loading and Long-term Cardiovascular Events Among Patients With Acute Coronary Syndromes Undergoing PCI. JAMA 2011, 306, 1215–1223. [Google Scholar] [CrossRef] [Green Version]
- Price, M.J.; Angiolillo, D.J.; Teirstein, P.S.; Lillie, E.; Manoukian, S.V.; Berger, P.B.; Tanguay, J.-F.; Cannon, C.P.; Topol, E.J. Platelet Reactivity and Cardiovascular Outcomes After Percutaneous Coronary Intervention. Circulation 2011, 124, 1132–1137. [Google Scholar] [CrossRef] [Green Version]
- Alexopoulos, D.; Xenogiannis, I.; Vlachakis, P.; Tantry, U.; Gurbel, P.A. Peri-Procedural Platelet Reactivity in Percutaneous Coronary Intervention. Thromb. Haemost. 2018, 118, 1131–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, M.R.; Storey, R.F. The role of platelets in inflammation. Thromb. Haemost. 2015, 114, 449–458. [Google Scholar] [CrossRef]
- Gleissner, C.A.; von Hundelshausen, P.; Ley, K. Platelet chemokines in vascular disease. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1920–1927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasper, B.; Petersen, F. Molecular pathways of platelet factor 4/CXCL4 signaling. Eur. J. Cell Biol. 2011, 90, 521–526. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Yasuoka, H.; Yoshimoto, K.; Suzuki, K.; Takeuchi, T. Platelet CXCL4 mediates neutrophil extracellular traps formation in ANCA-associated vasculitis. Sci. Rep. 2021, 11, 222. [Google Scholar] [CrossRef]
- Michelson, A.D.; Barnard, M.R.; Krueger, L.A.; Valeri, C.R.; Furman, M.I. Circulating Monocyte-Platelet Aggregates Are a More Sensitive Marker of In Vivo Platelet Activation Than Platelet Surface P-Selectin. Circulation 2001, 104, 1533–1537. [Google Scholar] [CrossRef] [Green Version]
- Lisman, T. Platelet-neutrophil interactions as drivers of inflammatory and thrombotic disease. Cell Tissue Res. 2018, 371, 567–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badrnya, S.; Schrottmaier, W.C.; Kral, J.B.; Yaiw, K.C.; Volf, I.; Schabbauer, G.; Söderberg-Nauclér, C.; Assinger, A. Platelets mediate oxidized low-density lipoprotein-induced monocyte extravasation and foam cell formation. Arter. Thromb. Vasc. Biol. 2014, 34, 571–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, M.; von Ungern-Sternberg, S.N.; Seizer, P.; Schlegel, F.; Büttcher, M.; Sindhu, N.A.; Müller, S.; Mack, A.; Gawaz, M. Platelet-derived CXCL12 regulates monocyte function, survival, differentiation into macrophages and foam cells through differential involvement of CXCR4-CXCR7. Cell Death Dis. 2015, 6, e1989. [Google Scholar] [CrossRef] [Green Version]
- Passacquale, G.; Vamadevan, P.; Pereira, L.; Hamid, C.; Corrigall, V.; Ferro, A. Monocyte-Platelet Interaction Induces a Pro-Inflammatory Phenotype in Circulating Monocytes. PLoS ONE 2011, 6, e25595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grebe, A.; Hoss, F.; Latz, E. NLRP3 Inflammasome and the IL-1 Pathway in Atherosclerosis. Circ. Res. 2018, 122, 1722–1740. [Google Scholar] [CrossRef]
- Tsimikas, S.; Lau, H.K.; Han, K.R.; Shortal, B.; Miller, E.R.; Segev, A.; Curtiss, L.K.; Witztum, J.L.; Strauss, B.H. Percutaneous coronary intervention results in acute increases in oxidized phospholipids and lipoprotein(a): Short-term and long-term immunologic responses to oxidized low-density lipoprotein. Circulation 2004, 109, 3164–3170. [Google Scholar] [CrossRef] [Green Version]
- Robertson, S.; Martínez, G.J.; Payet, C.A.; Barraclough, J.Y.; Celermajer, D.S.; Bursill, C.; Patel, S. Colchicine therapy in acute coronary syndrome patients acts on caspase-1 to suppress NLRP3 inflammasome monocyte activation. Clin. Sci. 2016, 130, 1237–1246. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Wu, S.; Hu, S.; Li, H.; Li, M.; Geng, X.; Wang, H. NLRP3 inflammasome expression in peripheral blood monocytes of coronary heart disease patients and its modulation by rosuvastatin. Mol. Med. Rep. 2019, 20, 1826–1836. [Google Scholar] [CrossRef] [Green Version]
- Martínez, G.J.; Celermajer, D.S.; Patel, S. The NLRP3 inflammasome and the emerging role of colchicine to inhibit atherosclerosis-associated inflammation. Atherosclerosis 2018, 269, 262–271. [Google Scholar] [CrossRef]
- Kahlenberg, J.M.; Carmona-Rivera, C.; Smith, C.K.; Kaplan, M.J. Neutrophil extracellular trap-associated protein activation of the NLRP3 inflammasome is enhanced in lupus macrophages. J Immunol 2013, 190, 1217–1226. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [Green Version]
- Chamberlain, J.; Gunn, J.; Francis, S.; Holt, C.; Crossman, D. Temporal and spatial distribution of interleukin-1β in balloon injured porcine coronary arteries. Cardiovasc. Res. 1999, 44, 156–165. [Google Scholar] [CrossRef] [Green Version]
- Zeng, M.; Yan, H.; Chen, Y.; Zhao, H.J.; Lv, Y.; Liu, C.; Zhou, P.; Zhao, B. Suppression of NF-κB reduces myocardial no-reflow. PLoS ONE 2012, 7, e47306. [Google Scholar] [CrossRef]
- Libby, P.; Rocha, V.Z. All roads lead to IL-6: A central hub of cardiometabolic signaling. Int. J. Cardiol. 2018, 259, 213–215. [Google Scholar] [CrossRef]
- Collaboration, I.R.G.C.E.R.F.; Sarwar, N.; Butterworth, A.S.; Freitag, D.F.; Gregson, J.; Willeit, P.; Gorman, D.N.; Gao, P.; Saleheen, D.; Rendon, A.; et al. Interleukin-6 receptor pathways in coronary heart disease: A collaborative meta-analysis of 82 studies. Lancet 2012, 379, 1205–1213. [Google Scholar] [CrossRef] [Green Version]
- Ridker, P.M. From C-Reactive Protein to Interleukin-6 to Interleukin-1: Moving Upstream To Identify Novel Targets for Atheroprotection. Circ Res 2016, 118, 145–156. [Google Scholar] [CrossRef] [Green Version]
- Hojo, Y.; Ikeda, U.; Katsuki, T.; Mizuno, O.; Fukazawa, H.; Kurosaki, K.; Fujikawa, H.; Shimada, K. Interleukin 6 expression in coronary circulation after coronary angioplasty as a risk factor for restenosis. Heart 2000, 84, 83–87. [Google Scholar] [CrossRef] [Green Version]
- Ramadan, M.M.; Kodama, M.; Mitsuma, W.; Ito, M.; Kashimura, T.; Ikrar, T.; Hirono, S.; Okura, Y.; Aizawa, Y. Impact of percutaneous coronary intervention on the levels of interleukin-6 and C-reactive protein in the coronary circulation of subjects with coronary artery disease. Am. J. Cardiol. 2006, 98, 915–917. [Google Scholar] [CrossRef]
- Aggarwal, A.; Schneider, D.J.; Terrien, E.F.; Gilbert, K.E.; Dauerman, H.L. Increase in Interleukin-6 in the First Hour after Coronary Stenting: An Early Marker of the Inflammatory Response. J. Thromb. Thrombolysis 2003, 15, 25–31. [Google Scholar] [CrossRef]
- Saleh, N.; Svane, B.; Jensen, J.; Hansson, L.O.; Nordin, M.; Tornvall, P. Stent implantation, but not pathogen burden, is associated with plasma C-reactive protein and interleukin-6 levels after percutaneous coronary intervention in patients with stable angina pectoris. Am. Heart J. 2005, 149, 876–882. [Google Scholar] [CrossRef]
- Ritschel, V.N.; Seljeflot, I.; Arnesen, H.; Halvorsen, S.; Weiss, T.; Eritsland, J.; Andersen, G.Ø. IL-6 signalling in patients with acute ST-elevation myocardial infarction. Results Immunol 2013, 4, 8–13. [Google Scholar] [CrossRef] [Green Version]
- Schuett, H.; Oestreich, R.; Waetzig, G.H.; Annema, W.; Luchtefeld, M.; Hillmer, A.; Bavendiek, U.; von Felden, J.; Divchev, D.; Kempf, T.; et al. Transsignaling of interleukin-6 crucially contributes to atherosclerosis in mice. Arter. Thromb. Vasc. Biol. 2012, 32, 281–290. [Google Scholar] [CrossRef] [Green Version]
- Kaminski, K.A.; Kozuch, M.; Bonda, T.; Wojtkowska, I.; Kozieradzka, A.; Dobrzycki, S.; Kralisz, P.; Nowak, K.; Prokopczuk, P.; Winnicka, M.M.; et al. Coronary sinus concentrations of interleukin 6 and its soluble receptors are affected by reperfusion and may portend complications in patients with myocardial infarction. Atherosclerosis 2009, 206, 581–587. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.-B.; Hernández-Reséndiz, S.; Crespo-Avilan, G.E.; Mukhametshina, R.T.; Kwek, X.-Y.; Cabrera-Fuentes, H.A.; Hausenloy, D.J. Inflammation following acute myocardial infarction: Multiple players, dynamic roles, and novel therapeutic opportunities. Pharmacol. Ther. 2018, 186, 73–87. [Google Scholar] [CrossRef] [PubMed]
- Kothari, P.; Pestana, R.; Mesraoua, R.; Elchaki, R.; Khan, K.M.; Dannenberg, A.J.; Falcone, D.J. IL-6-mediated induction of matrix metalloproteinase-9 is modulated by JAK-dependent IL-10 expression in macrophages. J. Immunol. 2014, 192, 349–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polyakova, V.; Loeffler, I.; Hein, S.; Miyagawa, S.; Piotrowska, I.; Dammer, S.; Risteli, J.; Schaper, J.; Kostin, S. Fibrosis in endstage human heart failure: Severe changes in collagen metabolism and MMP/TIMP profiles. Int. J. Cardiol. 2011, 151, 18–33. [Google Scholar] [CrossRef] [PubMed]
- Wagner, D.R.; Delagardelle, C.; Ernens, I.; Rouy, D.; Vaillant, M.; Beissel, J. Matrix Metalloproteinase-9 Is a Marker of Heart Failure After Acute Myocardial Infarction. J. Card. Fail. 2006, 12, 66–72. [Google Scholar] [CrossRef]
- Van Tassell, B.W.; Raleigh, J.M.V.; Abbate, A. Targeting Interleukin-1 in Heart Failure and Inflammatory Heart Disease. Curr. Heart Fail. Rep. 2015, 12, 33–41. [Google Scholar] [CrossRef]
- Harouki, N.; Nicol, L.; Remy-Jouet, I.; Henry, J.-P.; Dumesnil, A.; Lejeune, A.; Renet, S.; Golding, F.; Djerada, Z.; Wecker, D.; et al. The IL-1β Antibody Gevokizumab Limits Cardiac Remodeling and Coronary Dysfunction in Rats With Heart Failure. JACC: Basic Transl. Sci. 2017, 2, 418–430. [Google Scholar] [CrossRef]
- Dibra, A.; Ndrepepa, G.; Mehilli, J.; Dirschinger, J.; Pache, J.; Schühlen, H.; Schömig, A.; Kastrati, A. Comparison of C-Reactive Protein Levels Before and After Coronary Stenting and Restenosis Among Patients Treated With Sirolimus-Eluting Versus Bare Metal Stents. Am. J. Cardiol. 2005, 95, 1238–1240. [Google Scholar] [CrossRef]
- Sardella, G.; Mariani, P.; D’Alessandro, M.; De Luca, L.; Pierro, M.; Mancone, M.; Porretta, A.; Accapezzato, D.; Fedele, F.; Paroli, M. Early elevation of interleukin-1β and interleukin-6 levels after bare or drug-eluting stent implantation in patients with stable angina. Thromb. Res. 2006, 117, 659–664. [Google Scholar] [CrossRef]
- Sakr, S.A.; Ramadan, M.M.; El-Gamal, A. The inflammatory response to percutaneous coronary intervention is related to the technique of stenting and not the type of stent. Egypt. Heart J. 2016, 68, 37–43. [Google Scholar] [CrossRef] [Green Version]
- Niccoli, G.; Montone, R.A.; Ferrante, G.; Crea, F. The Evolving Role of Inflammatory Biomarkers in Risk Assessment After Stent Implantation. J. Am. Coll. Cardiol. 2010, 56, 1783–1793. [Google Scholar] [CrossRef]
- Tesfamariam, B. Bioresorbable vascular scaffolds: Biodegradation, drug delivery and vascular remodeling. Pharmacol. Res. 2016, 107, 163–171. [Google Scholar] [CrossRef]
- Wiviott, S.D.; Braunwald, E.; McCabe, C.H.; Montalescot, G.; Ruzyllo, W.; Gottlieb, S.; Neumann, F.-J.; Ardissino, D.; De Servi, S.; Murphy, S.A.; et al. Prasugrel versus Clopidogrel in Patients with Acute Coronary Syndromes. N. Engl. J. Med. 2007, 357, 2001–2015. [Google Scholar] [CrossRef] [Green Version]
- Shah, B.; Pillinger, M.; Zhong, H.; Cronstein, B.; Xia, Y.; Lorin, J.D.; Smilowitz, N.R.; Feit, F.; Ratnapala, N.; Keller, N.M.; et al. Effects of Acute Colchicine Administration Prior to Percutaneous Coronary Intervention: COLCHICINE-PCI Randomized Trial. Circ. Cardiovasc. Interv. 2020, 13, e008717. [Google Scholar] [CrossRef]
- Vivekananthan, D.P.; Bhatt, D.L.; Chew, D.P.; Zidar, F.J.; Chan, A.W.; Moliterno, D.J.; Ellis, S.G.; Topol, E.J. Effect of clopidogrel pretreatment on periprocedural rise in C-reactive protein after percutaneous coronary intervention. Am. J. Cardiol. 2004, 94, 358–360. [Google Scholar] [CrossRef]
- Schnorbus, B.; Daiber, A.; Jurk, K.; Warnke, S.; Koenig, J.; Lackner, K.J.; Münzel, T.; Gori, T. Effects of clopidogrel vs. prasugrel vs. ticagrelor on endothelial function, inflammatory parameters, and platelet function in patients with acute coronary syndrome undergoing coronary artery stenting: A randomized, blinded, parallel study. Eur. Heart J. 2020, 41, 3144–3152. [Google Scholar] [CrossRef] [PubMed]
- Deftereos, S.; Giannopoulos, G.; Angelidis, C.; Alexopoulos, N.; Filippatos, G.; Papoutsidakis, N.; Sianos, G.; Goudevenos, J.; Alexopoulos, D.; Pyrgakis, V.; et al. Anti-Inflammatory Treatment With Colchicine in Acute Myocardial Infarction. Circulation 2015, 132, 1395–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleveland, O.; Kunszt, G.; Bratlie, M.; Ueland, T.; Broch, K.; Holte, E.; Michelsen, A.E.; Bendz, B.; Amundsen, B.H.; Espevik, T.; et al. Effect of a single dose of the interleukin-6 receptor antagonist tocilizumab on inflammation and troponin T release in patients with non-ST-elevation myocardial infarction: A double-blind, randomized, placebo-controlled phase 2 trial. Eur. Heart J. 2016, 37, 2406–2413. [Google Scholar] [CrossRef]
- Broch, K.; Anstensrud Anne, K.; Woxholt, S.; Sharma, K.; Tøllefsen Ingvild, M.; Bendz, B.; Aakhus, S.; Ueland, T.; Amundsen Brage, H.; Damås Jan, K.; et al. Randomized Trial of Interleukin-6 Receptor Inhibition in Patients With Acute ST-Segment Elevation Myocardial Infarction. J. Am. Coll. Cardiol. 2021, 77, 1845–1855. [Google Scholar] [CrossRef] [PubMed]
- Holte, E.; Kleveland, O.; Ueland, T.; Kunszt, G.; Bratlie, M.; Broch, K.; Michelsen, A.E.; Bendz, B.; Amundsen, B.H.; Aakhus, S.; et al. Effect of interleukin-6 inhibition on coronary microvascular and endothelial function in myocardial infarction. Heart 2017, 103, 1521. [Google Scholar] [CrossRef] [PubMed]
- George, M.J.; Kleveland, O.; Garcia-Hernandez, J.; Palmen, J.; Lovering, R.; Wiseth, R.; Aukrust, P.; Engmann, J.; Damas, J.K.; Hingorani, A.D.; et al. Novel Insights Into the Effects of Interleukin 6 Antagonism in Non-ST-Segment-Elevation Myocardial Infarction Employing the SOMAscan Proteomics Platform. J. Am. Heart Assoc. 2020, 9, e015628. [Google Scholar] [CrossRef] [PubMed]
- Vaidya, K.; Martínez, G.; Patel, S. The Role of Colchicine in Acute Coronary Syndromes. Clin. Ther. 2019, 41, 11–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalbeth, N.; Lauterio, T.J.; Wolfe, H.R. Mechanism of Action of Colchicine in the Treatment of Gout. Clin. Ther. 2014, 36, 1465–1479. [Google Scholar] [CrossRef] [Green Version]
- Martínez, G.J.; Bailey, B.P.; Celermajer, D.S.; Patel, S. A safe and easy technique to sample the coronary sinus--facilitating a closer look at cardiac disease. Int. J. Cardiol. 2014, 176, 1321–1322. [Google Scholar] [CrossRef]
- Hennessy, T.; Soh, L.; Bowman, M.; Kurup, R.; Schultz, C.; Patel, S.; Hillis, G.S. The Low Dose Colchicine after Myocardial Infarction (LoDoCo-MI) study: A pilot randomized placebo controlled trial of colchicine following acute myocardial infarction. Am. Heart J. 2019, 215, 62–69. [Google Scholar] [CrossRef]
- Akodad, M.; Lattuca, B.; Nagot, N.; Georgescu, V.; Buisson, M.; Cristol, J.P.; Leclercq, F.; Macia, J.C.; Gervasoni, R.; Cung, T.T.; et al. COLIN trial: Value of colchicine in the treatment of patients with acute myocardial infarction and inflammatory response. Arch. Cardiovasc. Dis. 2017, 110, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Bresson, D.; Roubille, F.; Prieur, C.; Biere, L.; Ivanes, F.; Bouleti, C.; Dubreuil, O.; Rioufol, G.; Boutitie, F.; Sideris, G.; et al. Colchicine for Left Ventricular Infarct Size Reduction in Acute Myocardial Infarction: A Phase II, Multicenter, Randomized, Double-Blinded, Placebo-Controlled Study Protocol—The COVERT-MI Study. Cardiology 2021, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Bouabdallaoui, N.; Tardif, J.-C.; Waters, D.D.; Pinto, F.J.; Maggioni, A.P.; Diaz, R.; Berry, C.; Koenig, W.; Lopez-Sendon, J.; Gamra, H.; et al. Time-to-treatment initiation of colchicine and cardiovascular outcomes after myocardial infarction in the Colchicine Cardiovascular Outcomes Trial (COLCOT). Eur. Heart J. 2020, 41, 4092–4099. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Tanaka, T.; Narazaki, M.; Kishimoto, T. Targeting Interleukin-6 Signaling in Clinic. Immunity 2019, 50, 1007–1023. [Google Scholar] [CrossRef] [PubMed]
- Anstensrud, A.K.; Woxholt, S.; Sharma, K.; Broch, K.; Bendz, B.; Aakhus, S.; Ueland, T.; Amundsen, B.H.; Damås, J.K.; Hopp, E.; et al. Rationale for the ASSAIL-MI-trial: A randomised controlled trial designed to assess the effect of tocilizumab on myocardial salvage in patients with acute ST-elevation myocardial infarction (STEMI). Open Heart 2019, 6, e001108. [Google Scholar] [CrossRef] [PubMed]
- Abbate, A.; Kontos, M.C.; Grizzard, J.D.; Biondi-Zoccai, G.G.L.; Van Tassell, B.W.; Robati, R.; Roach, L.M.; Arena, R.A.; Roberts, C.S.; Varma, A.; et al. Interleukin-1 Blockade With Anakinra to Prevent Adverse Cardiac Remodeling After Acute Myocardial Infarction (Virginia Commonwealth University Anakinra Remodeling Trial [VCU-ART] Pilot Study). Am. J. Cardiol. 2010, 105, 1371–1377.e1. [Google Scholar] [CrossRef] [Green Version]
- Abbate, A.; Van Tassell, B.W.; Biondi-Zoccai, G.; Kontos, M.C.; Grizzard, J.D.; Spillman, D.W.; Oddi, C.; Roberts, C.S.; Melchior, R.D.; Mueller, G.H.; et al. Effects of Interleukin-1 Blockade With Anakinra on Adverse Cardiac Remodeling and Heart Failure After Acute Myocardial Infarction [from the Virginia Commonwealth University-Anakinra Remodeling Trial (2) (VCU-ART2) Pilot Study]. Am. J. Cardiol. 2013, 111, 1394–1400. [Google Scholar] [CrossRef] [Green Version]
- Abbate, A.; Kontos, M.C.; Abouzaki, N.A.; Melchior, R.D.; Thomas, C.; Van Tassell, B.W.; Oddi, C.; Carbone, S.; Trankle, C.R.; Roberts, C.S.; et al. Comparative Safety of Interleukin-1 Blockade With Anakinra in Patients With ST-Segment Elevation Acute Myocardial Infarction (from the VCU-ART and VCU-ART2 Pilot Studies). Am. J. Cardiol. 2015, 115, 288–292. [Google Scholar] [CrossRef]
- Abbate, A.; Trankle Cory, R.; Buckley Leo, F.; Lipinski Michael, J.; Appleton, D.; Kadariya, D.; Canada Justin, M.; Carbone, S.; Roberts Charlotte, S.; Abouzaki, N.; et al. Interleukin-1 Blockade Inhibits the Acute Inflammatory Response in Patients With ST-Segment–Elevation Myocardial Infarction. J. Am. Heart Assoc. 2020, 9, e014941. [Google Scholar] [CrossRef]
- Morton, A.C.; Rothman, A.M.K.; Greenwood, J.P.; Gunn, J.; Chase, A.; Clarke, B.; Hall, A.S.; Fox, K.; Foley, C.; Banya, W.; et al. The effect of interleukin-1 receptor antagonist therapy on markers of inflammation in non-ST elevation acute coronary syndromes: The MRC-ILA Heart Study. Eur. Heart J. 2015, 36, 377–384. [Google Scholar] [CrossRef] [Green Version]
- Sheriff, A.; Schindler, R.; Vogt, B.; Abdel-Aty, H.; Unger, J.K.; Bock, C.; Gebauer, F.; Slagman, A.; Jerichow, T.; Mans, D.; et al. Selective apheresis of C-reactive protein: A new therapeutic option in myocardial infarction? J. Clin. Apher. 2015, 30, 15–21. [Google Scholar] [CrossRef]
- Ries, W.; Torzewski, J.; Heigl, F.; Pfluecke, C.; Kelle, S.; Darius, H.; Ince, H.; Mitzner, S.; Nordbeck, P.; Butter, C.; et al. C-Reactive Protein Apheresis as Anti-inflammatory Therapy in Acute Myocardial Infarction: Results of the CAMI-1 Study. Front Cardiovasc Med 2021, 8, 591714. [Google Scholar] [CrossRef]
- Hara, T.; Fukuda, D.; Tanaka, K.; Higashikuni, Y.; Hirata, Y.; Nishimoto, S.; Yagi, S.; Yamada, H.; Soeki, T.; Wakatsuki, T.; et al. Rivaroxaban, a novel oral anticoagulant, attenuates atherosclerotic plaque progression and destabilization in ApoE-deficient mice. Atherosclerosis 2015, 242, 639–646. [Google Scholar] [CrossRef]
- Zhou, Q.X.; Bea, F.; Preusch, M.; Wang, H.J.; Isermann, B.; Shahzad, K.; Katus, H.A.; Blessing, E. Evaluation of Plaque Stability of Advanced Atherosclerotic Lesions in Apo E-Deficient Mice after Treatment with the Oral Factor Xa Inhibitor Rivaroxaban. Mediat. Inflamm. 2011, 2011, 9. [Google Scholar] [CrossRef] [Green Version]
- Ellinghaus, P.; Perzborn, E.; Hauenschild, P.; Gerdes, C.; Heitmeier, S.; Visser, M.; Summer, H.; Laux, V. Expression of pro-inflammatory genes in human endothelial cells: Comparison of rivaroxaban and dabigatran. Thromb. Res. 2016, 142, 44–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
| Population | Findings | Endpoint | Reference |
|---|---|---|---|
| CRP | |||
| n = 500 SA, UA and NSTEMI | Preprocedural CRP > 3 mg/L associated with 2.4-fold higher incidence of PMI | PMI: CK-MB or troponin I > 3× URL | [28] |
| n = 1337 UA and NSTEMI | ↑ Preprocedural hsCRP associated with ↑ risk of PMI and death | PMI: CK-MB > 5× URL | [29] |
| n = 96 SA and UA undergoing percutaneous bifurcation intervention | Positive linear relationship of preprocedural CRP with post procedural CK-MB | PMI: CK-MB > 3× URL | [30] |
| n = 85 SA | Preprocedural CRP > 6 mg/L associated with 2.5-fold higher incidence of PMI | PMI: Troponin I > 2 ng/mL | [31] |
| n = 4426 SA and UA | Preprocedural CRP > 3 mg/L associated with increased risk of PMI regardless of definition used | PMI: 2007 and 2012 universal definitions and SCAI definition | [32] |
| n = 463 SA, NSTEMI and STEMI | Positive linear relationship of preprocedural CRP with CK-MB ↑ CRP associated with 3-year MACE | PMI: CK-MB ≥ 3× URL | [33] |
| Retrospective: n = 7413 Prospective: n = 1189 SA and UA | Preprocedural CRP ≥ 3 mg/L, leukocyte count ≥ 7.3 × 109/L and NLR ≥ 2.2 associated with increased risk of PMI | PMI: Troponin I > 5× URL | [34] |
| n = 1140 STEMI | ↑ Preprocedural CRP independently associated with ↑ risk of no-reflow | Coronary no-reflow | [18] |
| n = 1217 STEMI | Preprocedural CRP, CRP to albumin ratio, leukocyte count and NLR were all independent predictors of no-reflow | Coronary no-reflow | [35] |
| n = 552 All with CTO | ↑ Preprocedural hsCRP independently associated with ↑ risk of slow- and no-reflow | Coronary no-reflow | [36] |
| n = 167 SA on haemodialysis | ↑ Preprocedural CRP associated with higher rates of MACE at 4 years and restenosis at 8 months | MACE and restenosis | [37] |
| n = 936 SA and UA | ↑ Preprocedural CRP associated with higher incidence of primary endpoint at 2 years | Composite of death and Q-wave MI | [38] |
| n = 1650 SA and UA | ↑ Preprocedural CRP associated with higher incidence of the primary outcome at 1 year | Composite of cardiac death and Q-wave MI | [39] |
| Haematological parameters | |||
| n = 880 SA | ↑ Preprocedural leukocyte count associated with ↑ incidence of PMI | PMI: CK-MB > 3× URL | [40] |
| n = 43 Patients undergoing carotid stenting | Preprocedural leukocyte count positively correlated with degree of intra-procedural microembolisation | Extent of distal embolisation | [41] |
| n = 99 STEMI | ↑ Preprocedural leukocyte count associated with greater risk of no-reflow | Coronary no-reflow | [42] |
| n = 4450 SA and UA | ↑ Preprocedural leukocyte count associated with greater 4-year mortality | All-cause mortality | [43] |
| n = 83 SA, UA and NSTEMI | Preprocedural leukocyte count and CRP were independent predictors of death and MI at 9 months | Composite of death and non-fatal MI | [44] |
| n = 909 SA, UA and NSTEMI | ↑ Preprocedural eosinophil count favourable in short-term, yet detrimental in long-term | All-cause mortality | [45] |
| n = 1543 SA, UA and NSTEMI | No relationship of eosinophil count with occurrence of PMI | PMI: CK-MB ≥ 3× URL or an increase ≥ 50% if already elevated | [46] |
| n = 418 STEMI | Inverse relationship of TIMI flow grade with N/L ratio. ↑ Preprocedural N/L ratio associated with ↑ in-hospital MACE. | Coronary no-reflow and in-hospital MACE | [47] |
| n = 361 STEMI | ↑ Preprocedural neutrophil count associated with higher incidence of no-reflow | Coronary no-reflow | [48] |
| n = 208 STEMI | TIMI frame count positively correlated with preprocedural neutrophil and platelet count, yet negatively correlated with lymphocyte count | Coronary no-reflow | [49] |
| n = 204 STEMI | ↑ Preprocedural N/L ratio associated with no ST-resolution and greater 3-year mortality | Coronary no-reflow and 3-year all-cause mortality | [50] |
| n = 426 NSTEMI | ↑ Preprocedural M/HDL ratio and ↓ L/M ratio associated with higher incidence of slow flow/no-reflow | Coronary no-reflow | [51] |
| n = 857 STEMI | ↓ Preprocedural L/M ratio associated with higher incidence of no-reflow | Coronary no-reflow | [52] |
| n = 306 STEMI | ↓ Preprocedural L/M ratio associated with greater short- and long-term MACCE | In-hospital and long-term MACCE | [53] |
| n = 83 STEMI | ↑ Preprocedural platelet–neutrophil and platelet–monocyte aggregates associated with higher incidence of no-reflow | Coronary no-reflow | [54] |
| Others | |||
| n = 50 STEMI | Preprocedural MPO concentration at culprit lesion directly correlated with TIMI frame count | Coronary no-reflow | [55] |
| n = 40 STEMI | ↑ Preprocedural coronary MPO associated with greater post procedure microvascular obstruction | Microvascular obstruction | [56] |
| n = 192 STEMI | ↑ Preprocedural MCP-1 associated with greater risk of no-reflow and 3-year mortality | Coronary no-reflow and all-cause mortality | [57] |
| n = 265 SA | Positive linear relationship between preprocedural Lp-PLA2 and postprocedural troponin T | PMI: troponin T > 20% baseline value and within 5× baseline value | [58] |
| Population | Dose | Effect of anti-inflammatory | Reference |
|---|---|---|---|
| Colchicine | |||
| n = 73 SA and ACS | 1.5 mg pre-PCI | ↓ Transcoronary concentration of IL-1β, IL-18 and IL-6 in ACS but not SA patients | [93] |
| n = 38 SA and ACS | 1.5 mg pre-PCI | ↓ Transcoronary concentration of MCP-1, CCL5 and fractalkine in ACS but not SA patients | [73] |
| n = 60 SA and ACS | 1.5 mg pre-PCI | ↓ Intra-procedural NET release within the coronary circulation | [113] |
| n = 400 SA and ACS | 1.8 mg pre-PCI | No effect on incidence of PMI No effect on incidence of composite endpoint of death, non-fatal MI and target vessel revascularisation Suppressed post-procedural CRP and IL-6 elevation | [160] |
| n = 75 SA and NSTEMI | 1.5 mg pre-PCI | ↓ Periprocedural myocardial injury in NSTEMI patients | [82] |
| n = 151 STEMI | 2 mg pre-PCI plus 0.5 mg twice daily for 5 days | ↓ Area under the CK-MB curve during admission ↓ Infarct size | [163] |
| Tocilizumab | |||
| n = 117 NSTEMI | 280 mg prior to coronary angiography | ↓ Area under the hsCRP curve during admission ↓ Area under the hsTnT curve during admission | [164] |
| n = 199 STEMI | 280 mg prior to coronary angiography | ↑ Myocardial salvage index at 3–7 post-PCI ↓ Microvascular obstruction | [165] |
| n = 42 NSTEMI | 280 mg prior to coronary angiography | No effect on coronary flow reserve | [166] |
| n = 48 NSTEMI | 280 mg prior to coronary angiography | ↓ Lipopolysaccharide-binding protein, hepcidin, IGF-binding protein 4 and CCL23 ↑ Proteinase 3 | [167] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tucker, B.; Vaidya, K.; Cochran, B.J.; Patel, S. Inflammation during Percutaneous Coronary Intervention—Prognostic Value, Mechanisms and Therapeutic Targets. Cells 2021, 10, 1391. https://doi.org/10.3390/cells10061391
Tucker B, Vaidya K, Cochran BJ, Patel S. Inflammation during Percutaneous Coronary Intervention—Prognostic Value, Mechanisms and Therapeutic Targets. Cells. 2021; 10(6):1391. https://doi.org/10.3390/cells10061391
Chicago/Turabian StyleTucker, Bradley, Kaivan Vaidya, Blake J. Cochran, and Sanjay Patel. 2021. "Inflammation during Percutaneous Coronary Intervention—Prognostic Value, Mechanisms and Therapeutic Targets" Cells 10, no. 6: 1391. https://doi.org/10.3390/cells10061391