
The PINK1-Mediated Crosstalk between Neural Cells and the Underlying Link to Parkinson’s Disease


{kind=link}
Abstract
:1. Introduction
1.1. Parkinson’s Disease
1.2. Mitochondria and Their Role in PD
2. Neurons in PD
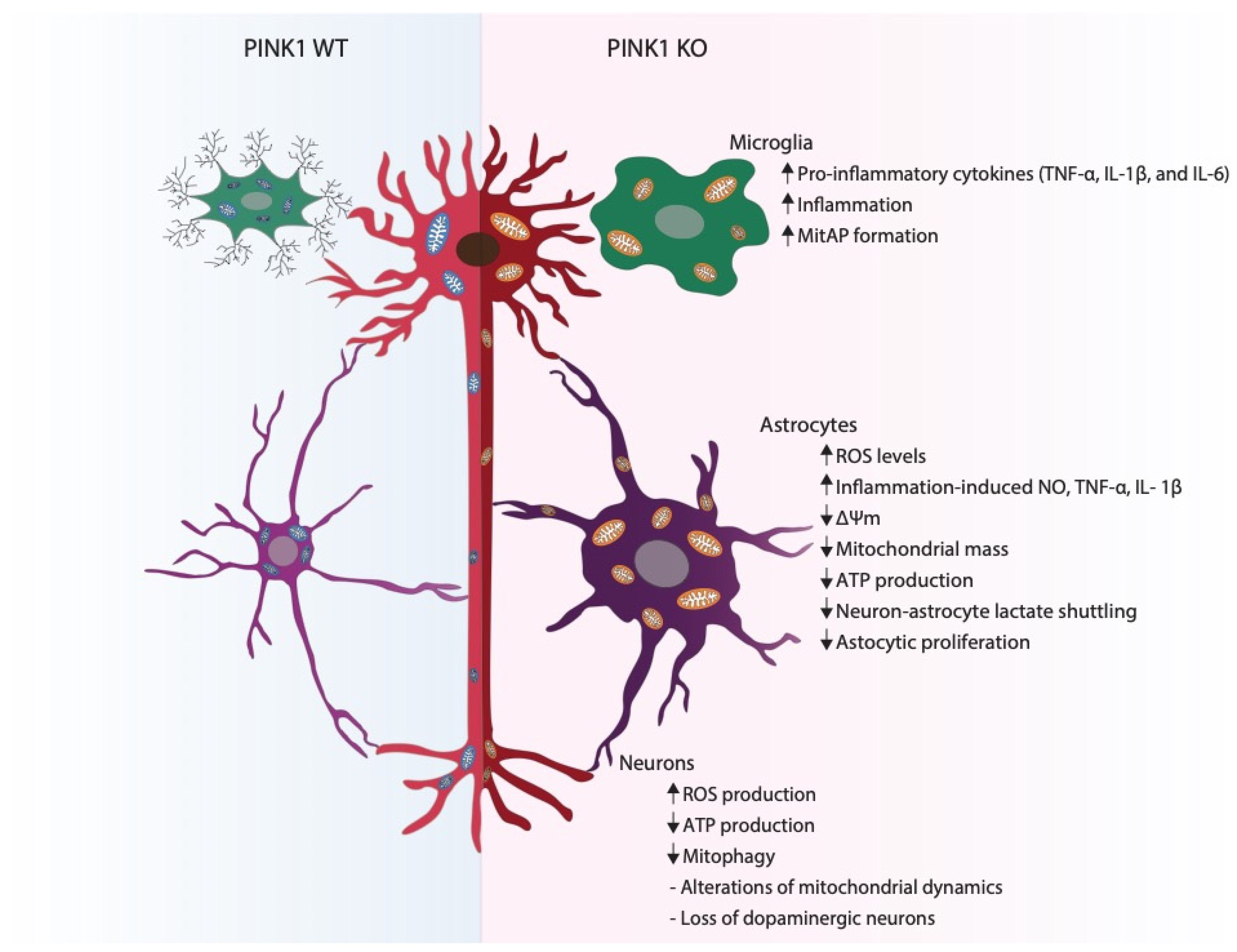
PINK1 in Neurons
3. Astrocytes in PD
PINK1 in Astrocytes
4. Microglia in PD
PINK1 in Microglia
5. PINK1, a Putative Mediator of the Crosstalk between Neural Cells
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kouli, A.; Torsney, K.M.; Kuan, W.-L. Parkinson’s disease: Etiology, neuropathology, and pathogenesis. In Parkinson’s Disease: Pathogenesis and Clinical Aspects; Codon Publications: Brisbane, Australia, 2018; pp. 3–26. ISBN 9780994438164. [Google Scholar]
- De Lau, L.M.; Breteler, M.M. Epidemiology of Parkinson’s disease. Lancet Neurol. 2006, 5, 525–535. [Google Scholar] [CrossRef]
- Mappin-Kasirer, B.; Pan, H.; Lewington, S.; Kizza, J.; Gray, R.; Clarke, R.; Peto, R. Tobacco smoking and the risk of Parkinson disease. Neurology 2020. [Google Scholar] [CrossRef]
- Wertz, M.H.; Heiman, M. Genome-wide genetic screening in the mammalian CNS. In Research and Perspectives in Neurosciences; Springer: Berlin/Heidelberg, Germany, 2017; pp. 31–39. [Google Scholar]
- Morais, V.A.; de Strooper, B. Mitochondria dysfunction and neurodegenerative disorders: Cause or consequence. J. Alzheimers Dis. 2010, 20, S255–S263. [Google Scholar] [CrossRef] [Green Version]
- Leites, E.P.; Morais, V.A. Mitochondrial quality control pathways: PINK1 acts as a gatekeeper. Biochem. Biophys. Res. Commun. 2018, 500, 45–50. [Google Scholar] [CrossRef]
- Rangaraju, V.; Lewis, T.L.; Hirabayashi, Y.; Bergami, M.; Motori, E.; Cartoni, R.; Kwon, S.K.; Courchet, J. Pleiotropic mitochondria: The influence of mitochondria on neuronal development and disease. J. Neurosci. 2019, 39, 8200–8208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McAvoy, K.; Kawamata, H. Glial mitochondrial function and dysfunction in health and neurodegeneration. Mol. Cell. Neurosci. 2019, 101, 103417. [Google Scholar] [CrossRef]
- Barros, L.F.; Brown, A.; Swanson, R.A. Glia in brain energy metabolism: A perspective. Glia 2018, 66, 1134–1137. [Google Scholar] [CrossRef] [PubMed]
- Rose, J.; Brian, C.; Woods, J.; Pappa, A.; Panayiotidis, M.I.; Powers, R.; Franco, R. Mitochondrial dysfunction in glial cells: Implications for neuronal homeostasis and survival. Toxicology 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shank, R.P.; Bennett, G.S.; Freytag, S.O.; Campbell, G.L.M. Pyruvate carboxylase: An astrocyte-specific enzyme implicated in the replenishment of amino acid neurotransmitter pools. Brain Res. 1985. [Google Scholar] [CrossRef]
- Gamberino, W.C.; Berkich, D.A.; Lynch, C.J.; Xu, B.; LaNoue, K.F. Role of pyruvate carboxylase in facilitation of synthesis of glutamate and glutamine in cultured astrocytes. J. Neurochem. 1997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, Q.R. Transport of glutamate and other amino acids at the blood-brain barrier. J. Nutr. 2000, 130, 1016S–1022S. [Google Scholar] [CrossRef] [PubMed]
- Hertz, L.; Rothman, D.L. Glutamine-glutamate cycle flux is similar in cultured astrocytes and brain and both glutamate production and oxidation are mainly catalyzed by aspartate aminotransferase. Biology 2017, 6, 17. [Google Scholar] [CrossRef] [Green Version]
- Yu, A.C.H.; Drejer, J.; Hertz, L.; Schousboe, A. Pyruvate carboxylase activity in primary cultures of astrocytes and neurons. J. Neurochem. 1983. [Google Scholar] [CrossRef] [PubMed]
- Bélanger, M.; Allaman, I.; Magistretti, P.J. Brain energy metabolism: Focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011, 14, 724–738. [Google Scholar] [CrossRef] [Green Version]
- Fecher, C.; Trovò, L.; Müller, S.A.; Snaidero, N.; Wettmarshausen, J.; Heink, S.; Ortiz, O.; Wagner, I.; Kühn, R.; Hartmann, J.; et al. Cell-type-specific profiling of brain mitochondria reveals functional and molecular diversity. Nat. Neurosci. 2019. [Google Scholar] [CrossRef]
- Orihuela, R.; McPherson, C.A.; Harry, G.J. Microglial M1/M2 polarization and metabolic states. Br. J. Pharmacol. 2016, 173, 649–665. [Google Scholar] [CrossRef]
- Gao, F.; Chen, D.; Hu, Q.; Wang, G. Rotenone directly induces BV2 cell activation via the p38 MAPK pathway. PLoS ONE 2013, 8, e72046. [Google Scholar] [CrossRef]
- Park, J.; Choi, H.; Min, J.S.; Park, S.J.; Kim, J.H.; Park, H.J.; Kim, B.; Chae, J., II; Yim, M.; Lee, D.S. Mitochondrial dynamics modulate the expression of pro-inflammatory mediators in microglial cells. J. Neurochem. 2013. [Google Scholar] [CrossRef] [PubMed]
- Tanner, C.M.; Kame, F.; Ross, G.W.; Hoppin, J.A.; Goldman, S.M.; Korell, M.; Marras, C.; Bhudhikanok, G.S.; Kasten, M.; Chade, A.R.; et al. Rotenone, paraquat, and Parkinson’s disease. Environ. Health Perspect. 2011, 119, 866–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuter, K.; Nowak, P.; Gołembiowska, K.; Ossowska, K. Increased reactive oxygen species production in the brain after repeated low-dose pesticide paraquat exposure in rats. A comparison with peripheral tissues. Neurochem. Res. 2010. [Google Scholar] [CrossRef] [PubMed]
- Krueger, M.J.; Singer, T.P.; Casida, J.E.; Ramsay, R.R. Evidence that the blockade of mitochondrial respiration by the neurotoxin 1-methyl-4-phenylpyridinium (MPP+) involves binding at the same site as the respiratory inhibitor, rotenone. Biochem. Biophys. Res. Commun. 1990, 169, 123–128. [Google Scholar] [CrossRef]
- Lin, M.T.; Cantuti-Castelvetri, I.; Zheng, K.; Jackson, K.E.; Tan, Y.B.; Arzberger, T.; Lees, A.J.; Betensky, R.A.; Beal, M.F.; Simon, D.K. Somatic mitochondrial DNA mutations in early Parkinson and incidental lewy body disease. Ann. Neurol. 2012. [Google Scholar] [CrossRef] [Green Version]
- Gu, G.; Reyes, P.F.; Golden, G.T.; Woltjer, R.L.; Hulette, C.; Montine, T.J.; Zhang, J. Mitochondrial DNA deletions/rearrangements in Parkinson disease and related neurodegenerative disorders. J. Neuropathol. Exp. Neurol. 2002. [Google Scholar] [CrossRef] [Green Version]
- Paillusson, S.; Gomez-Suaga, P.; Stoica, R.; Little, D.; Gissen, P.; Devine, M.J.; Noble, W.; Hanger, D.P.; Miller, C.C.J. α-Synuclein binds to the ER–mitochondria tethering protein VAPB to disrupt Ca2+ homeostasis and mitochondrial ATP production. Acta Neuropathol. 2017, 134, 129–149. [Google Scholar] [CrossRef] [Green Version]
- Bonello, F.; Hassoun, S.M.; Mouton-Liger, F.; Shin, Y.S.; Muscat, A.; Tesson, C.; Lesage, S.; Beart, P.M.; Brice, A.; Krupp, J.; et al. LRRK2 impairs PINK1/Parkin-dependent mitophagy via its kinase activity: Pathologic insights into Parkinson’s disease. Hum. Mol. Genet. 2019, 28, 1645–1660. [Google Scholar] [CrossRef] [PubMed]
- Shimura, H.; Hattori, N.; Kubo, S.I.; Mizuno, Y.; Asakawa, S.; Minoshima, S.; Shimizu, N.; Iwai, K.; Chiba, T.; Tanaka, K.; et al. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat. Genet. 2000, 25, 302–305. [Google Scholar] [CrossRef]
- Dolgacheva, L.P.; Berezhnov, A.V.; Fedotova, E.I.; Zinchenko, V.P.; Abramov, A.Y. Role of DJ-1 in the mechanism of pathogenesis of Parkinson’s disease. J. Bioenerg. Biomembr. 2019, 51, 175–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aerts, L.; de Strooper, B.; Morais, V.A. PINK1 activation—Turning on a promiscuous kinase. Biochem. Soc. Trans. 2015. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.M.; Lazarou, M.; Wang, C.; Kane, L.A.; Narendra, D.P.; Youle, R.J. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 2010, 191, 933–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greene, A.W.; Grenier, K.; Aguileta, M.A.; Muise, S.; Farazifard, R.; Haque, M.E.; McBride, H.M.; Park, D.S.; Fon, E.A. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep. 2012. [Google Scholar] [CrossRef] [PubMed]
- Yamano, K.; Youle, R.J. PINK1 is degraded through the N-end rule pathway. Autophagy 2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morais, V.A.; Verstreken, P.; Roethig, A.; Smet, J.; Snellinx, A.; Vanbrabant, M.; Haddad, D.; Frezza, C.; Mandemakers, W.; Vogt-Weisenhorn, D.; et al. Parkinson’s disease mutations in PINK1 result in decreased complex I activity and deficient synaptic function. EMBO Mol. Med. 2009. [Google Scholar] [CrossRef] [PubMed]
- Pridgeon, J.W.; Olzmann, J.A.; Chin, L.S.; Li, L. PINK1 protects against oxidative stress by phosphorylating mitochondrial chaperone TRAP1. PLoS Biol. 2007, 5, 1494–1503. [Google Scholar] [CrossRef]
- Plun-Favreau, H.; Klupsch, K.; Moisoi, N.; Gandhi, S.; Kjaer, S.; Frith, D.; Harvey, K.; Deas, E.; Harvey, R.J.; McDonald, N.; et al. The mitochondrial protease HtrA2 is regulated by Parkinson’s disease-associated kinase PINK1. Nat. Cell Biol. 2007, 9, 1243–1252. [Google Scholar] [CrossRef]
- Arena, G.; Gelmetti, V.; Torosantucci, L.; Vignone, D.; Lamorte, G.; de Rosa, P.; Cilia, E.; Jonas, E.A.; Valente, E.M. PINK1 protects against cell death induced by mitochondrial depolarization, by phosphorylating Bcl-xL and impairing its pro-apoptotic cleavage. Cell Death Differ. 2013, 20, 920–930. [Google Scholar] [CrossRef] [Green Version]
- Morais, V.A.; Haddad, D.; Craessaerts, K.; de Bock, P.J.; Swerts, J.; Vilain, S.; Aerts, L.; Overbergh, L.; Grünewald, A.; Seibler, P.; et al. PINK1 loss-of-function mutations affect mitochondrial complex I activity via NdufA10 ubiquinone uncoupling. Science 2014, 344, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Okatsu, K.; Oka, T.; Iguchi, M.; Imamura, K.; Kosako, H.; Tani, N.; Kimura, M.; Go, E.; Koyano, F.; Funayama, M.; et al. PINK1 autophosphorylation upon membrane potential dissipation is essential for Parkin recruitment to damaged mitochondria. Nat. Commun. 2012, 3. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y.; Nagano, Y.; Taylor, J.P.; Lim, K.L.; Yao, T.P. Disease-causing mutations in Parkin impair mitochondrial ubiquitination, aggregation, and HDAC6-dependent mitophagy. J. Cell Biol. 2010, 189, 671–679. [Google Scholar] [CrossRef] [Green Version]
- Aerts, L.; Craessaerts, K.; de Strooper, B.; Morais, V.A. PINK1 kinase catalytic activity is regulated by phosphorylation on serines 228 and 402. J. Biol. Chem. 2015, 290, 2798–2811. [Google Scholar] [CrossRef] [Green Version]
- Kondapalli, C.; Kazlauskaite, A.; Zhang, N.; Woodroof, H.I.; Campbell, D.G.; Gourlay, R.; Burchell, L.; Walden, H.; MacArtney, T.J.; Deak, M.; et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2012. [Google Scholar] [CrossRef] [Green Version]
- McLelland, G.L.; Soubannier, V.; Chen, C.X.; McBride, H.M.; Fon, E.A. Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J. 2014, 33, 282–295. [Google Scholar] [CrossRef]
- Soubannier, V.; Rippstein, P.; Kaufman, B.A.; Shoubridge, E.A.; McBride, H.M. Reconstitution of mitochondria derived vesicle formation demonstrates selective enrichment of oxidized cargo. PLoS ONE 2012, 7, e52830. [Google Scholar] [CrossRef] [Green Version]
- Braak, H.; Rüb, U.; Gai, W.P.; del Tredici, K. Idiopathic Parkinson’s disease: Possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J. Neural. Transm. 2003, 110, 517–536. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E. Pathoanatomy of Parkinson’s disease. J. Neurol. 2000. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Qian, L.; Xiong, H.; Liu, J.; Neckameyer, W.S.; Oldham, S.; Xia, K.; Wang, J.; Bodmer, R.; Zhang, Z. Antioxidants protect PINK1-dependent dopaminergic neurons in drosophila. Proc. Natl. Acad. Sci. USA 2006, 103, 13520–13525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.K.; Harvey, K.; Gispert, S.; Ali, Z.; del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [Green Version]
- Valente, E.M.; Salvi, S.; Ialongo, T.; Marongiu, R.; Elia, A.E.; Caputo, V.; Romito, L.; Albanese, A.; Dallapiccola, B.; Bentivoglio, A.R. PINK1 mutations are associated with sporadic early-onset Parkinsonism. Ann. Neurol. 2004, 56, 336–341. [Google Scholar] [CrossRef]
- Liu, X.L.; di Wang, Y.; Yu, X.M.; Li, D.W.; Li, G.R. Mitochondria-mediated damage to dopaminergic neurons in Parkinson’s disease (review). Int. J. Mol. Med. 2018, 41, 615–623. [Google Scholar] [CrossRef] [Green Version]
- Birsa, N.; Norkett, R.; Higgs, N.; Lopez-Domenech, G.; Kittler, J.T. Mitochondrial trafficking in neurons and the role of the Miro family of GTPase proteins. Biochem. Soc. Trans. 2013, 41, 1525–1531. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Winter, D.; Ashrafi, G.; Schlehe, J.; Wong, Y.L.; Selkoe, D.; Rice, S.; Steen, J.; Lavoie, M.J.; Schwarz, T.L. PINK1 and Parkin target miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 2011. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Dorn, G.W. PINK1-phosphorylated mitofusin 2 is a parkin receptor for culling damaged mitochondria. Science 2013, 340, 471–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pryde, K.R.; Smith, H.L.; Chau, K.Y.; Schapira, A.H.V. PINK1 disables the anti-fission machinery to segregate damaged mitochondria for mitophagy. J. Cell Biol. 2016, 213, 163–171. [Google Scholar] [CrossRef]
- Otera, H.; Mihara, K. Molecular mechanisms and physiologic functions of mitochondrial dynamics. J. Biochem. 2011, 149, 241–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, H.; Tan, J.; Wang, R.; Wan, H.; He, Y.; Yan, X.; Guo, J.; Gao, Q.; Li, J.; Shang, S.; et al. PINK 1 phosphorylates Drp1 S616 to regulate mitophagy-independent mitochondrial dynamics. EMBO Rep. 2020, 21. [Google Scholar] [CrossRef] [PubMed]
- Wood-Kaczmar, A.; Gandhi, S.; Yao, Z.; Abramov, A.S.Y.; Miljan, E.A.; Keen, G.; Stanyer, L.; Hargreaves, I.; Klupsch, K.; Deas, E.; et al. PINK1 is necessary for long term survival and mitochondrial function in human dopaminergic neurons. PLoS ONE 2008, 3, e2455. [Google Scholar] [CrossRef]
- Pirooznia, S.K.; Yuan, C.; Khan, M.R.; Karuppagounder, S.S.; Wang, L.; Xiong, Y.; Kang, S.U.; Lee, Y.; Dawson, V.L.; Dawson, T.M. PARIS induced defects in mitochondrial biogenesis drive dopamine neuron loss under conditions of parkin or PINK1 deficiency. Mol. Neurodegener. 2020, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandhi, S.; Wood-Kaczmar, A.; Yao, Z.; Plun-Favreau, H.; Deas, E.; Klupsch, K.; Downward, J.; Latchman, D.S.; Tabrizi, S.J.; Wood, N.W.; et al. PINK1-associated Parkinson’s disease is caused by neuronal vulnerability to calcium-induced cell death. Mol. Cell 2009. [Google Scholar] [CrossRef] [Green Version]
- Gandhi, S.; Muqit, M.M.K.; Stanyer, L.; Healy, D.G.; Abou-Sleiman, P.M.; Hargreaves, I.; Heales, S.; Ganguly, M.; Parsons, L.; Lees, A.J.; et al. PINK1 protein in normal human brain and Parkinson’s disease. Brain 2006, 129, 1720–1731. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Li, S.; Jiang, N.; Shao, X.; Zhang, M.; Jin, H.; Zhang, Z.; Shen, J.; Zhou, Y.; Zhou, W.; et al. PINK1-parkin pathway of mitophagy protects against contrast-induced acute kidney injury via decreasing mitochondrial ROS and NLRP3 inflammasome activation. Redox Biol. 2019. [Google Scholar] [CrossRef]
- Li, X.; Shi, Z.; Zhu, Y.; Shen, T.; Wang, H.; Shui, G.; Loor, J.J.; Fang, Z.; Chen, M.; Wang, X.; et al. Cyanidin-3-O-glucoside improves non-alcoholic fatty liver disease by promoting PINK1-mediated mitophagy in mice. Br. J. Pharmacol. 2020. [Google Scholar] [CrossRef]
- Wang, B.; Nie, J.; Wu, L.; Hu, Y.; Wen, Z.; Dong, L.; Zou, M.H.; Chen, C.; Wang, D.W. AMPKa2 protects against the development of heart failure by enhancing mitophagy via PINK1 phosphorylation. Circ. Res. 2018. [Google Scholar] [CrossRef]
- Matheoud, D.; Cannon, T.; Voisin, A.; Penttinen, A.M.; Ramet, L.; Fahmy, A.M.; Ducrot, C.; Laplante, A.; Bourque, M.J.; Zhu, L.; et al. Intestinal infection triggers Parkinson’s disease-like symptoms in Pink1−/− mice. Nature 2019, 571, 565–569. [Google Scholar] [CrossRef] [PubMed]
- Benarroch, E.E. Astrocyte signaling and synaptic homeostasis II: Astrocyte-neuron interactions and clinical correlations. Neurology 2016, 87, 726–735. [Google Scholar] [CrossRef] [PubMed]
- Herculano-Houzel, S. The human brain in numbers: A linearly scaled-up primate brain. Front. Hum. Neurosci. 2009, 3, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, L.F.H.; Doherty, D.H.; Lile, J.D.; Bektesh, S.; Collins, F. GDNF: A glial cell line—Derived neurotrophic factor for midbrain dopaminergic neurons. Science 1993, 260, 1130–1132. [Google Scholar] [CrossRef]
- Kwon, H.S.; Koh, S.H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Miklossy, J.; Doudet, D.D.; Schwab, C.; Yu, S.; McGeer, E.G.; McGeer, P.L. Role of ICAM-1 in persisting inflammation in Parkinson disease and MPTP monkeys. Exp. Neurol. 2006, 197, 275–283. [Google Scholar] [CrossRef]
- Koprich, J.B.; Reske-Nielsen, C.; Mithal, P.; Isacson, O. Neuroinflammation mediated by IL-1β increases susceptibility of dopamine neurons to degeneration in an animal model of Parkinson’s disease. J. Neuroinflamm. 2008, 5. [Google Scholar] [CrossRef] [Green Version]
- Di Domenico, A.; Carola, G.; Calatayud, C.; Pons-Espinal, M.; Muñoz, J.P.; Richaud-Patin, Y.; Fernandez-Carasa, I.; Gut, M.; Faella, A.; Parameswaran, J.; et al. Patient-specific iPSC-derived astrocytes contribute to non-cell-autonomous neurodegeneration in Parkinson’s disease. Stem Cell Rep. 2019, 12, 213–229. [Google Scholar] [CrossRef] [Green Version]
- Lindström, V.; Gustafsson, G.; Sanders, L.H.; Howlett, E.H.; Sigvardson, J.; Kasrayan, A.; Ingelsson, M.; Bergström, J.; Erlandsson, A. Extensive uptake of α-synuclein oligomers in astrocytes results in sustained intracellular deposits and mitochondrial damage. Mol. Cell. Neurosci. 2017, 82, 143–156. [Google Scholar] [CrossRef]
- Wakida, N.M.; Cruz, G.M.S.; Ro, C.C.; Moncada, E.G.; Khatibzadeh, N.; Flanagan, L.A.; Berns, M.W. Phagocytic response of astrocytes to damaged neighboring cells. PLoS ONE 2018, 13, e0196153. [Google Scholar] [CrossRef]
- Morales, I.; Sanchez, A.; Puertas-Avendaño, R.; Rodriguez-Sabate, C.; Perez-Barreto, A.; Rodriguez, M. Neuroglial transmitophagy and Parkinson’s disease. Glia 2020, 68, 2277–2299. [Google Scholar] [CrossRef]
- English, K.; Shepherd, A.; Uzor, N.E.; Trinh, R.; Kavelaars, A.; Heijnen, C.J. Astrocytes rescue neuronal health after cisplatin treatment through mitochondrial transfer. Acta Neuropathol. Commun. 2020, 8. [Google Scholar] [CrossRef] [Green Version]
- Choi, I.; Choi, D.J.; Yang, H.; Woo, J.H.; Chang, M.Y.; Kim, J.Y.; Sun, W.; Park, S.M.; Jou, I.; Lee, S.H.; et al. PINK1 expression increases during brain development and stem cell differentiation, and affects the development of GFAP-positive astrocytes. Mol. Brain 2016, 9. [Google Scholar] [CrossRef] [Green Version]
- Barodia, S.K.; McMeekin, L.J.; Creed, R.B.; Quinones, E.K.; Cowell, R.M.; Goldberg, M.S. PINK1 phosphorylates ubiquitin predominantly in astrocytes. NPJ Park. Dis. 2019, 5. [Google Scholar] [CrossRef] [Green Version]
- Choi, I.; Kim, J.; Jeong, H.K.; Kim, B.; Jou, I.; Park, M.; Chen, L.; Kang, U.J.; Zhuang, X.; Joe, E. Hye PINK1 deficiency attenuates astrocyte proliferation through mitochondrial dysfunction, reduced AKT and increased p38 MAPK activation, and downregulation of EGFR. Glia 2013, 61, 800–812. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Shen, R.; Agnihotri, S.K.; Chen, Y.; Huang, Z.; Büeler, H. Lack of PINK1 alters glia innate immune responses and enhances inflammation-induced, nitric oxide-mediated neuron death. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [Green Version]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009, 32, 638–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, M.S. Microglia in Parkinson’s disease. In Advances in Experimental Medicine and Biology; Springer: Berlin/Heidelberg, Germany, 2019; Volume 1175, pp. 335–353. [Google Scholar]
- Du, L.; Zhang, Y.; Chen, Y.; Zhu, J.; Yang, Y.; Zhang, H.L. Role of microglia in neurological disorders and their potentials as a therapeutic target. Mol. Neurobiol. 2016, 54, 7567–7584. [Google Scholar] [CrossRef] [PubMed]
- Napoli, I.; Neumann, H. Protective effects of microglia in multiple sclerosis. Exp. Neurol. 2010, 225, 24–28. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, T.; Pei, Z.; Miller, D.S.; Wu, X.; Block, M.L.; Wilson, B.; Zhang, W.; Zhou, Y.; Hong, J.-S.; et al. Aggregated α-synuclein activates microglia: A process leading to disease progression in Parkinson’s disease. FASEB J. 2005, 19, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Pósfai, B.; Cserép, C.; Orsolits, B.; Dénes, Á. New insights into microglia-neuron interactions: A neuron’s perspective. Neuroscience 2019, 405, 103–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tremblay, M.E.; Cookson, M.R.; Civiero, L. Glial phagocytic clearance in Parkinson’s disease. Mol. Neurodegener. 2019, 14, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Gerhard, A.; Pavese, N.; Hotton, G.; Turkheimer, F.; Es, M.; Hammers, A.; Eggert, K.; Oertel, W.; Banati, R.B.; Brooks, D.J. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol. Dis. 2006, 21, 404–412. [Google Scholar] [CrossRef]
- Yun, S.P.; Kam, T.I.; Panicker, N.; Kim, S.; Oh, Y.; Park, J.S.; Kwon, S.H.; Park, Y.J.; Karuppagounder, S.S.; Park, H.; et al. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat. Med. 2018, 24, 931–938. [Google Scholar] [CrossRef] [PubMed]
- Rojanathammanee, L.; Murphy, E.J.; Combs, C.K. Expression of mutant alpha-synuclein modulates microglial phenotype in vitro. J. Neuroinflamm. 2011, 8. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Byun, J.-W.; Choi, I.; Kim, B.; Jeong, H.-K.; Jou, I.; Joe, E. PINK1 deficiency enhances inflammatory cytokine release from acutely prepared brain slices. Exp. Neurobiol. 2013, 22, 38–44. [Google Scholar] [CrossRef] [Green Version]
- Matheoud, D.; Sugiura, A.; Bellemare-Pelletier, A.; Laplante, A.; Rondeau, C.; Chemali, M.; Fazel, A.; Bergeron, J.J.; Trudeau, L.E.; Burelle, Y.; et al. Parkinson’s disease-related proteins PINK1 and parkin repress mitochondrial antigen presentation. Cell 2016, 166, 314–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deeb, W.; Nozile-Firth, K.; Okun, M.S. Parkinson’s disease: Diagnosis and appreciation of comorbidities. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2019; Volume 167, pp. 257–277. [Google Scholar]
- Ntetsika, T.; Papathoma, P.E.; Markaki, I. Novel targeted therapies for Parkinson’s disease. Mol. Med. 2021, 27, 1–20. [Google Scholar] [CrossRef]
- George, S.; Rey, N.L.; Tyson, T.; Esquibel, C.; Meyerdirk, L.; Schulz, E.; Pierce, S.; Burmeister, A.R.; Madaj, Z.; Steiner, J.A.; et al. Microglia affect α-synuclein cell-to-cell transfer in a mouse model of Parkinson’s disease. Mol. Neurodegener. 2019, 14, 1–22. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leites, E.P.; Morais, V.A. The PINK1-Mediated Crosstalk between Neural Cells and the Underlying Link to Parkinson’s Disease. Cells 2021, 10, 1395. https://doi.org/10.3390/cells10061395
Leites EP, Morais VA. The PINK1-Mediated Crosstalk between Neural Cells and the Underlying Link to Parkinson’s Disease. Cells. 2021; 10(6):1395. https://doi.org/10.3390/cells10061395
Chicago/Turabian StyleLeites, Elvira Pequeno, and Vanessa Alexandra Morais. 2021. "The PINK1-Mediated Crosstalk between Neural Cells and the Underlying Link to Parkinson’s Disease" Cells 10, no. 6: 1395. https://doi.org/10.3390/cells10061395
APA StyleLeites, E. P., & Morais, V. A. (2021). The PINK1-Mediated Crosstalk between Neural Cells and the Underlying Link to Parkinson’s Disease. Cells, 10(6), 1395. https://doi.org/10.3390/cells10061395