
The Heritability of Replication Problems


{kind=link}
Abstract
:1. Introduction
2. Progression of Replication Problems into Mitosis
2.1. Stimulation of Mitotic DNA Synthesis in Early Mitosis
2.2. Generation of UFB in Mitosis
3. Consequences of the Inheritability of Under-Replicated DNA in Daughter Cells
3.1. Transmitted DNA Damage Are Protected within 53BP1 NBs in G1 Phase and Resolved in S Phase of Daughter Cells
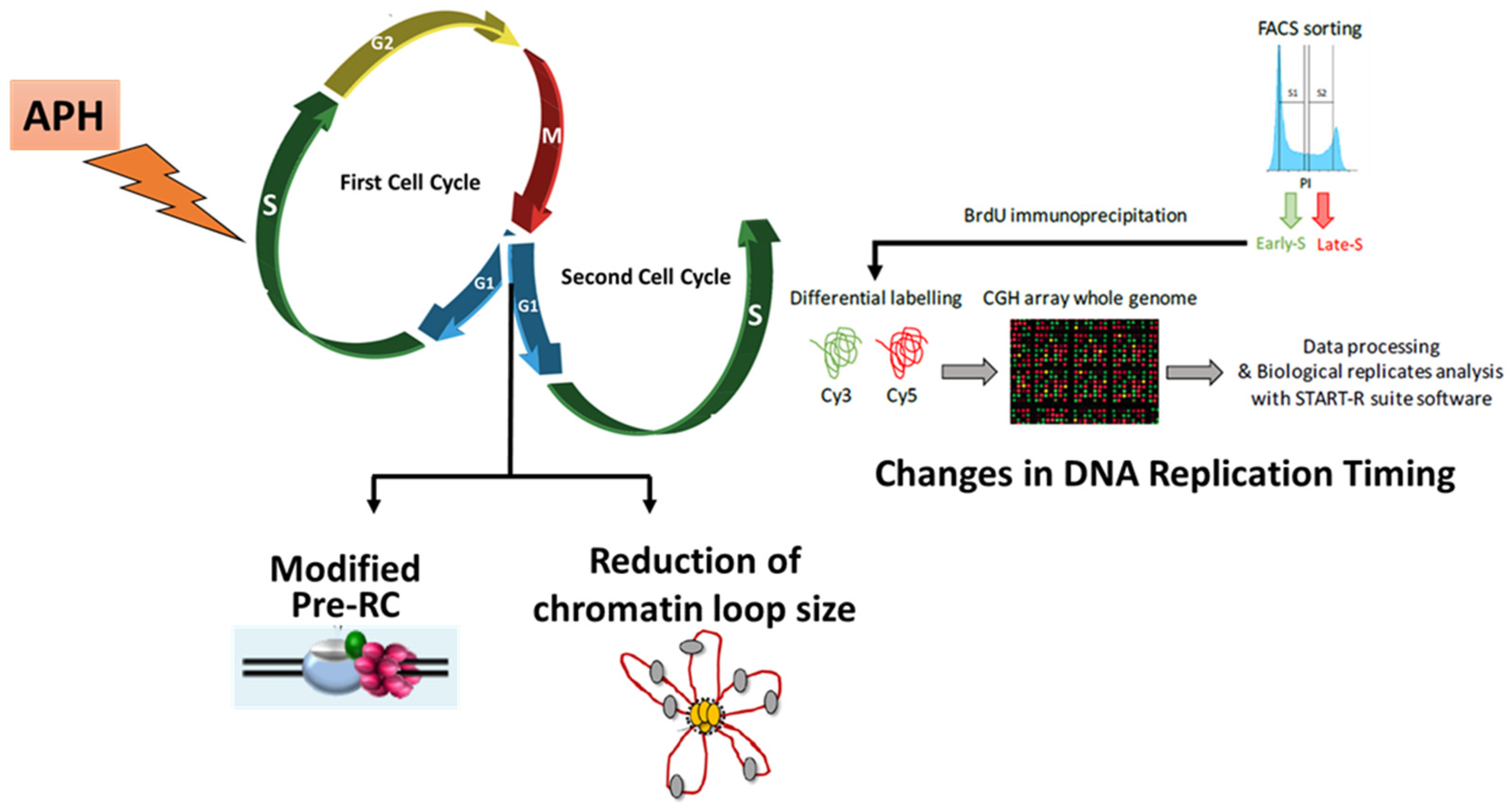
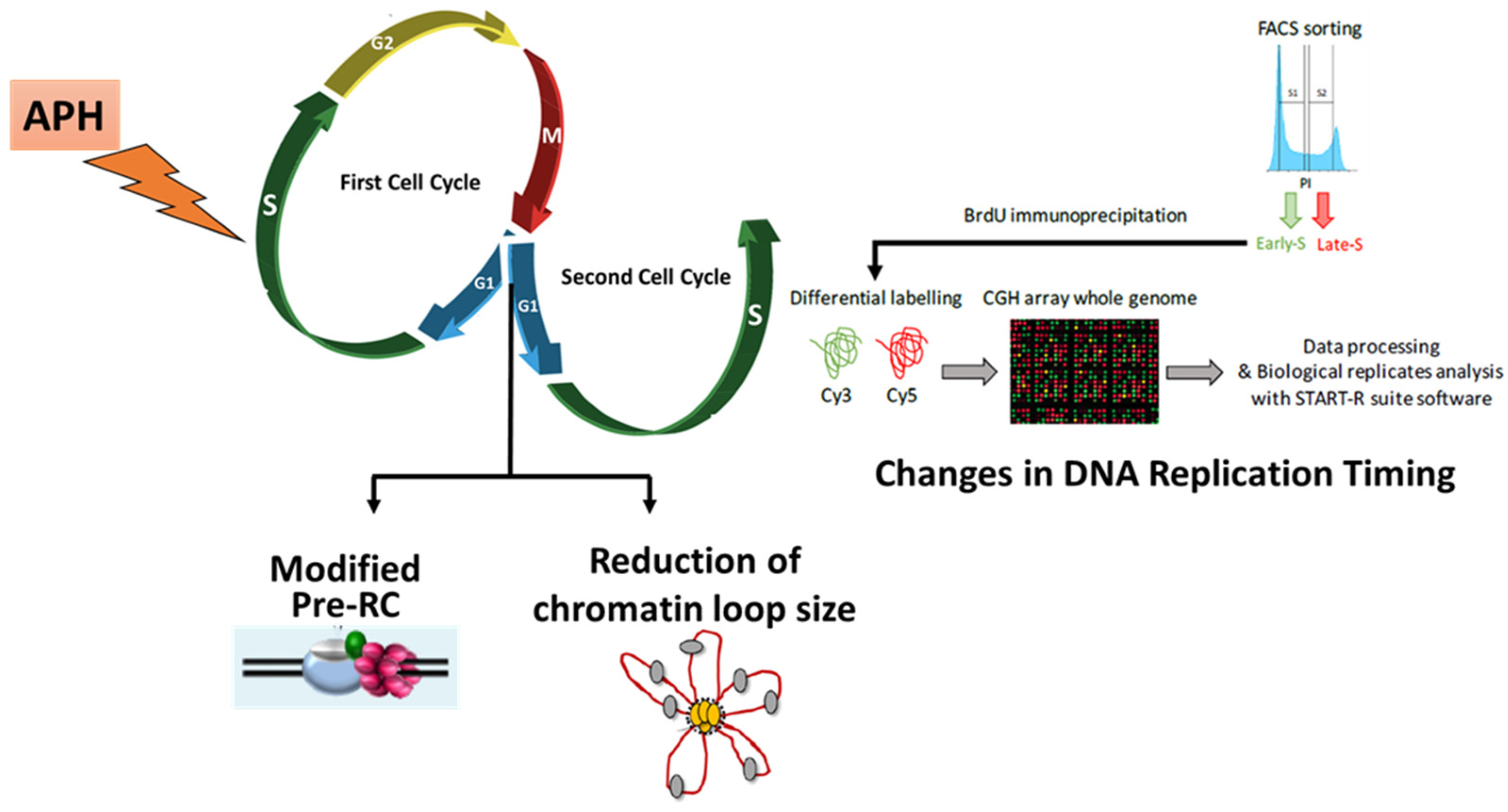
3.2. RS modifies Pre-RC Formation and Spatial Organization of Replication Origins in the Next Generation of Cells
4. RS Modifies the Replication Timing at a Fraction of Chromosomal Domains in Unstressed Daughter Cells
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Yeeles, J.T.P.; Janska, A.; Early, A.; Diffley, J.F.X. How the eukaryotic replisome achieves rapid and efficient DNA replication. Mol. Cell 2017, 65, 105–116. [Google Scholar] [CrossRef] [Green Version]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Courtot, L.; Hoffmann, J.-S.; Bergoglio, V. The protective role of dormant origins in response to replicative stress. Int. J. Mol. Sci. 2018, 19, 3569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertolin, A.P.; Hoffmann, J.-S.; Gottifredi, V. Under-Replicated DNA: The byproduct of large genomes? Cancers 2020, 12, 2764. [Google Scholar] [CrossRef] [PubMed]
- Boyer, A.-S.; Grgurevic, S.; Cazaux, C.; Hoffmann, J.-S. The human specialized DNA polymerases and Non-B DNA: Vital relationships to preserve genome integrity. J. Mol. Biol. 2013, 425, 4767–4781. [Google Scholar] [CrossRef] [PubMed]
- Hecht, F.; Glover, T.W. Cancer chromosome breakpoints and common fragile sites induced by aphidicolin. Cancer Genet. Cytogenet. 1984, 13, 185–188. [Google Scholar] [CrossRef]
- Gaillard, H.; García-Muse, T.; Aguilera, A. Replication stress and cancer. Nat. Rev. Cancer 2015, 15, 276–289. [Google Scholar] [CrossRef]
- Macheret, M.; Halazonetis, T.D. DNA replication stress as a hallmark of cancer. Annu. Rev. Pathol. Mech. Dis. 2015, 10, 425–448. [Google Scholar] [CrossRef] [Green Version]
- Saldivar, J.C.; Cortez, D.; Cimprich, K.A. The essential kinase ATR: Ensuring faithful duplication of a challenging genome. Nat. Rev. Mol. Cell Biol. 2017, 18, 622–636. [Google Scholar] [CrossRef] [Green Version]
- Franchet, C.; Hoffmann, J.-S. When RAD52 allows mitosis to accept unscheduled DNA synthesis. Cancers 2020, 12, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergoglio, V.; Boyer, A.S.; Walsh, E.; Naim, V.; Legube, G.; Lee, M.Y.; Rey, L.; Rosselli, F.; Cazaux, C.; Eckert, K.A.; et al. DNA synthesis by Pol eta promotes fragile site stability by preventing under-replicated DNA in mitosis. J. Cell Biol. 2013, 201, 395–408. [Google Scholar] [CrossRef] [Green Version]
- Minocherhomji, S.; Ying, S.; Bjerregaard, V.A.; Bursomanno, S.; Aleliunaite, A.; Wu, W.; Mankouri, H.W.; Shen, H.; Liu, Y.; Hickson, I.D. Replication stress activates DNA repair synthesis in mitosis. Nature 2015, 528, 286–290. [Google Scholar] [CrossRef]
- Lai, X.; Broderick, R.; Bergoglio, V.; Zimmer, J.; Badie, S.; Niedzwiedz, W.; Hoffmann, J.S.; Tarsounas, M. MUS81 nuclease activity is essential for replication stress tolerance and chromosome segregation in BRCA2-deficient cells. Nat. Commun. 2017, 8, 15983. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Exposito, L.; Bournique, E.; Bergoglio, V.; Bose, A.; Barroso-Gonzalez, J.; Zhang, S.; Roncaioli, J.L.; Lee, M.; Wallace, C.T.; Watkins, S.C.; et al. Proteomic profiling reveals a specific role for translesion DNA polymerase eta in the alternative lengthening of telomeres. Cell Rep. 2016, 17, 1858–1871. [Google Scholar] [CrossRef] [Green Version]
- Ozer, O.; Bhowmick, R.; Liu, Y.; Hickson, I.D. Human cancer cells utilize mitotic DNA synthesis to resist replication stress at telomeres regardless of their telomere maintenance mechanism. Oncotarget 2018, 9, 15836–15846. [Google Scholar] [CrossRef] [PubMed]
- Sonneville, R.; Bhowmick, R.; Hoffmann, S.; Mailand, N.; Hickson, I.D.; Labib, K. TRAIP drives replisome disassembly and mitotic DNA repair synthesis at sites of incomplete DNA replication. eLife 2019, 8, e48686. [Google Scholar] [CrossRef]
- Chan, K.L.; Hickson, I.D. New insights into the formation and resolution of ultra-fne anaphase bridges. Sem. Cell Dev. Biol. 2011, 22, 906–912. [Google Scholar] [CrossRef]
- Liu, Y.; Nielsen, C.F.; Yao, Q.; Hickson, I.D. The origins and processing of ultra fne anaphase DNA bridges. Curr. Opin. Genet. Dev. 2014, 26, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Mankouri, H.W.; Huttner, D.; Hickson, I.D. How unfinished business from S-phase affects mitosis and beyond. EMBO J. 2013, 32, 2661–2671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukas, C.; Savic, V.; Bekker-Jensen, S.; Doil, C.; Neumann, B.; Pedersen, R.S.; Grøfte, M.; Chan, K.L.; Hickson, I.D.; Bartek, J.; et al. 53BP1 nuclear bodies form around DNA lesions generated by mitotic. Nat. Cell Biol. 2011, 13, 243–253. [Google Scholar] [CrossRef]
- Harrigan, J.A.; Belotserkovskaya, R.; Coates, J.; Dimitrova, D.S.; Polo, S.E.; Bradshaw, C.R.; Fraser, P.; Jackson, S.P. Replication stress induces 53BP1-containing OPT domains in G1 cells. J. Cell Biol. 2011, 193, 97–108. [Google Scholar] [CrossRef] [Green Version]
- Bothmer, A.; Robbiani, D.F.; Feldhahn, N.; Gazumyan, A.; Nussenzweig, A.; Nussenzweig, M.C. 53BP1 regulates DNA resection and the choice between classical and alternative end joining during class switch recombination. J. Exp. Med. 2010, 207, 855–865. [Google Scholar] [CrossRef] [Green Version]
- Mansilla, S.F.; Bertolin, A.P.; Bergoglio, V.; Pillaire, M.J.; Besteiro, M.A.G.; Luzzani, C.; Miriuka, S.G.; Cazaux, C.; Hoffmann, J.S.; Gottifredi, V. Cyclin Kinase-independent role of p21CDKN1A in the promotion of nascent DNA elongation in unstressed cells. eLife 2016, 5, e18020. [Google Scholar] [CrossRef]
- Spies, J.; Lukas, C.; Somyajit, K.; Rask, M.B.; Lukas, J.; Neelsen, K.J. 53BP1 nuclear bodies enforce replication timing at under-replicated DNA to limit heritable DNA damage. Nat. Cell Biol. 2019, 21, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Mechali, M. Eukaryotic DNA replication origins: Many choices for appropriate answers. Nat. Rev. Mol. Cell Biol. 2010, 11, 728–738. [Google Scholar] [CrossRef] [PubMed]
- Courtot, L.; Bournique, E.; Maric, C.; Guitton-Sert, L.; Madrid-Mencía, M.; Pancaldi, V.; Cadoret, J.-C.; Hoffmann, J.-S.; Bergoglio, V. Low replicative stress triggers cell-type specific inheritable advanced replication timing. Int. J. Mol. Sci. 2021, 22, 4959. [Google Scholar] [CrossRef] [PubMed]
- Guillou, E.; Ibarra, A.; Coulon, V.; Casado-Vela, J.; Rico, D.; Casal, I.; Schwob, E.; Losada, A.; Méndez, J. Cohesin organizes chromatin loops at DNA replication factories. Genes Dev. 2010, 24, 2812–2822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Vidal, A.; Guitton-Sert, L.; Cadoret, J.C.; Drac, M.; Schwob, E.; Baldacci, J.; Cazaux, C.; Hoffmann, J.S. A role for DNA polymerase theta in the timing of DNA replication. Nat. Commun. 2014, 5, 4285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kermi, C.; Aze, A.; Maiorano, D. Preserving genome integrity during the early embryonic DNA replication cycles. Genes 2019, 10, 398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoffmann, J.-S. The Heritability of Replication Problems. Cells 2021, 10, 1464. https://doi.org/10.3390/cells10061464
Hoffmann J-S. The Heritability of Replication Problems. Cells. 2021; 10(6):1464. https://doi.org/10.3390/cells10061464
Chicago/Turabian StyleHoffmann, Jean-Sébastien. 2021. "The Heritability of Replication Problems" Cells 10, no. 6: 1464. https://doi.org/10.3390/cells10061464
APA StyleHoffmann, J.-S. (2021). The Heritability of Replication Problems. Cells, 10(6), 1464. https://doi.org/10.3390/cells10061464