
With-No-Lysine Kinase 1 (WNK1) Augments TRPV4 Function in the Aldosterone-Sensitive Distal Nephron

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Animals
2.2. Isolation and Split-Opening of Collecting Ducts
2.3. Cell Culture
2.4. [Ca2+]i Imaging
2.5. Isolation of TRPV4 Membrane Fraction
2.6. Western Blotting
2.7. Over-Expression of TRPV4 in Chinese Hamster Ovary (CHO) Cells
2.8. Data Analysis and Presentation
3. Results
3.1. WNK Blockade Decreases TRPV4 Activity and Expression in the ASDN
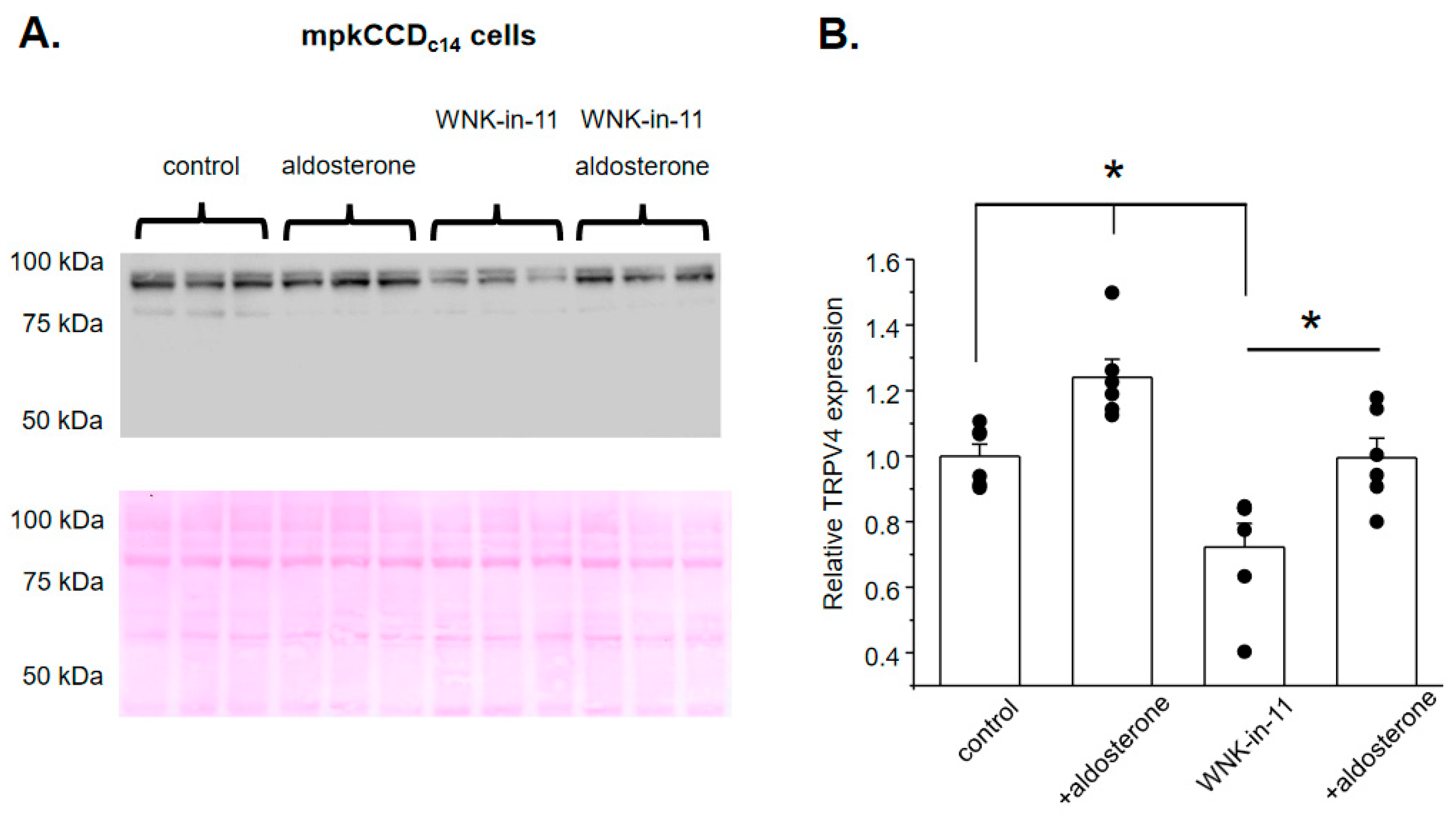
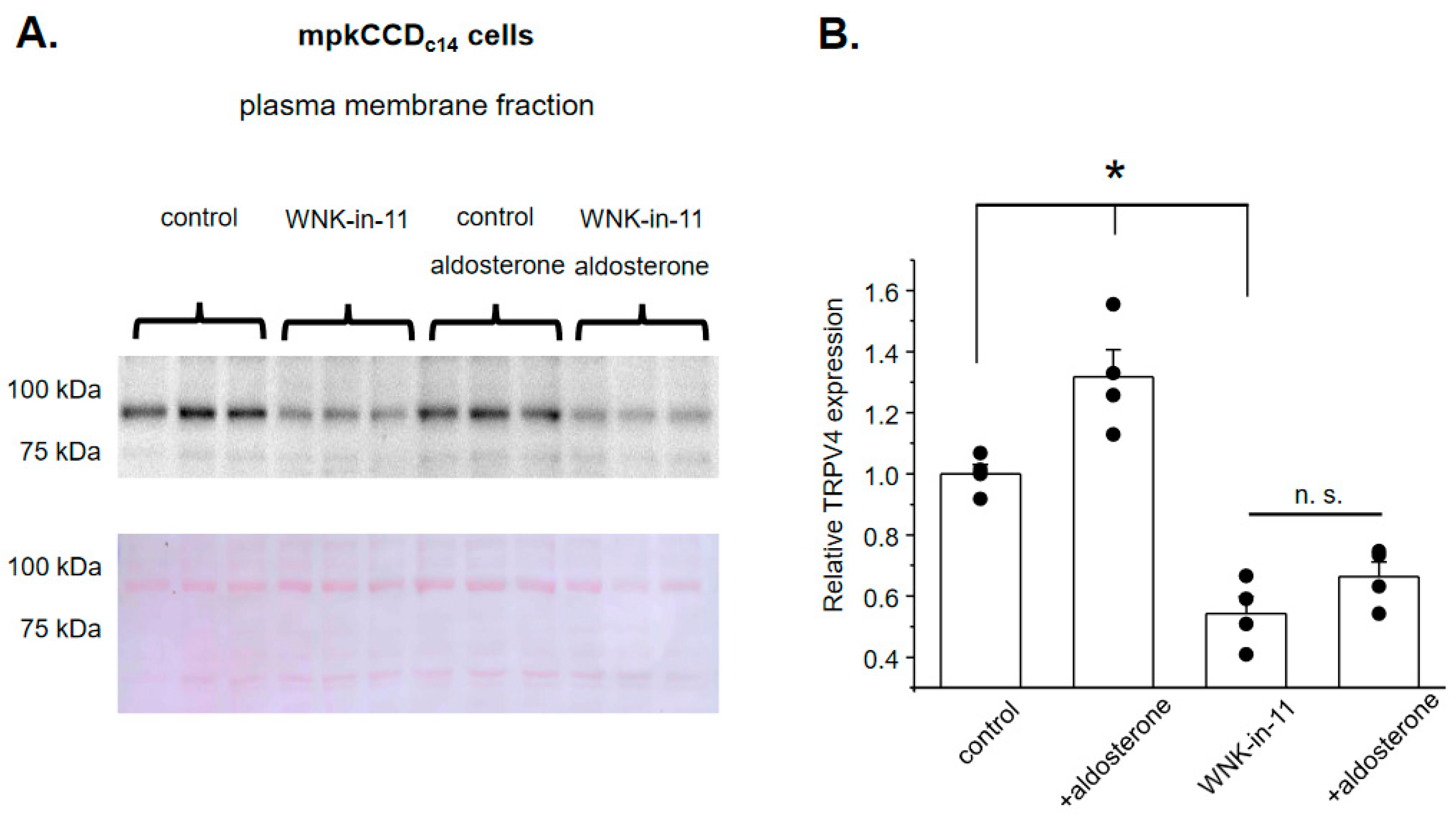
3.2. WNK1 Is Critical for Stimulation of TRPV4 by Aldosterone in Cultured mpkCCDc14 Cells
3.3. WNK1 Increases TRPV4 Activity in a Kinase-Dependent Manner
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Everaerts, W.; Nilius, B.; Owsianik, G. The vanilloid transient receptor potential channel trpv4: From structure to disease. Prog. Biophys. Mol. Biol. 2010, 103, 2–17. [Google Scholar] [CrossRef]
- Guilak, F.; Leddy, H.A.; Liedtke, W. Transient receptor potential vanilloid 4: The sixth sense of the musculoskeletal system? Ann. N. Y. Acad. Sci. 2010, 1192, 404–409. [Google Scholar] [CrossRef] [Green Version]
- O’Neil, R.G.; Heller, S. The mechanosensitive nature of trpv channels. Pflug. Arch. Eur. J. Physiol. 2005, 451, 193–203. [Google Scholar] [CrossRef]
- Liedtke, W.; Choe, Y.; Marti-Renom, M.A.; Bell, A.M.; Denis, C.S.; Sali, A.; Hudspeth, A.J.; Friedman, J.M.; Heller, S. Vanilloid receptor-related osmotically activated channel (vr-oac), a candidate vertebrate osmoreceptor. Cell 2000, 103, 525–535. [Google Scholar] [CrossRef] [Green Version]
- Berrout, J.; Jin, M.; Mamenko, M.; Zaika, O.; Pochynyuk, O.; O’Neil, R.G. Function of trpv4 as a mechanical transducer in flow-sensitive segments of the renal collecting duct system. J. Biol. Chem. 2012, 287, 8782–8791. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, J.; Tsuruoka, S.; Mizuno, A.; Sato, J.; Fujimura, A.; Suzuki, M. Trpv4 as a flow sensor in flow-dependent k+ secretion from the cortical collecting duct. Am. J. Physiol. Renal. Physiol. 2007, 292, F667–F673. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Wong, W.Y.; Sun, L.; Huang, Y.; Yao, X. Protein kinase g inhibits flow-induced Ca2+ entry into collecting duct cells. J. Am. Soc. Nephrol. 2012, 23, 1172–1180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomilin, V.N.; Mamenko, M.; Zaika, O.; Ren, G.; Marrelli, S.P.; Birnbaumer, L.; Pochynyuk, O. Trpc3 determines osmosensitive [Ca2+]i signaling in the collecting duct and contributes to urinary concentration. PLoS ONE 2019, 14, e0226381. [Google Scholar] [CrossRef] [Green Version]
- Mamenko, M.V.; Boukelmoune, N.; Tomilin, V.N.; Zaika, O.L.; Jensen, V.B.; O’Neil, R.G.; Pochynyuk, O.M. The renal trpv4 channel is essential for adaptation to increased dietary potassium. Kidney Int. 2017, 91, 1398–1409. [Google Scholar] [CrossRef]
- Zaika, O.; Mamenko, M.; Berrout, J.; Boukelmoune, N.; O’Neil, R.G.; Pochynyuk, O. Trpv4 dysfunction promotes renal cystogenesis in autosomal recessive polycystic kidney disease. J. Am. Soc. Nephrol. JASN 2013, 24, 604–616. [Google Scholar] [CrossRef] [Green Version]
- Tomilin, V.; Reif, G.A.; Zaika, O.; Wallace, D.P.; Pochynyuk, O. Deficient transient receptor potential vanilloid type 4 function contributes to compromised [Ca2+]i homeostasis in human autosomal-dominant polycystic kidney disease cells. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2018, 32, 4612–4623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, L.G.; Schnermann, J. Integrated control of na transport along the nephron. Clin. J. Am. Soc. Nephrol. CJASN 2015, 10, 676–687. [Google Scholar] [CrossRef] [Green Version]
- Palmer, B.F. Regulation of potassium homeostasis. Clin. J. Am. Soc. Nephrol. CJASN 2015, 10, 1050–1060. [Google Scholar] [CrossRef] [Green Version]
- Pearce, D.; Soundararajan, R.; Trimpert, C.; Kashlan, O.B.; Deen, P.M.; Kohan, D.E. Collecting duct principal cell transport processes and their regulation. Clin. J. Am. Soc. Nephrol. CJASN 2014, 10, 135–146. [Google Scholar] [CrossRef] [Green Version]
- Arroyo, J.P.; Ronzaud, C.; Lagnaz, D.; Staub, O.; Gamba, G. Aldosterone paradox: Differential regulation of ion transport in distal nephron. Physiol. Bethesda 2011, 26, 115–123. [Google Scholar] [CrossRef] [Green Version]
- Shekarabi, M.; Zhang, J.; Khanna, A.R.; Ellison, D.H.; Delpire, E.; Kahle, K.T. Wnk kinase signaling in ion homeostasis and human disease. Cell Metab. 2017, 25, 285–299. [Google Scholar] [CrossRef] [Green Version]
- Hadchouel, J.; Ellison, D.H.; Gamba, G. Regulation of renal electrolyte transport by wnk and spak-osr1 kinases. Annu. Rev. Physiol. 2016, 78, 367–389. [Google Scholar] [CrossRef]
- Terker, A.S.; Zhang, C.; McCormick, J.A.; Lazelle, R.A.; Zhang, C.; Meermeier, N.P.; Siler, D.A.; Park, H.J.; Fu, Y.; Cohen, D.M.; et al. Potassium modulates electrolyte balance and blood pressure through effects on distal cell voltage and chloride. Cell Metab. 2015, 21, 39–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webb, T.N.; Carrisoza-Gaytan, R.; Montalbetti, N.; Rued, A.; Roy, A.; Socovich, A.M.; Subramanya, A.R.; Satlin, L.M.; Kleyman, T.R.; Carattino, M.D. Cell-specific regulation of l-wnk1 by dietary k. Am. J. physiology. Ren. Physiol. 2016, 310, F15–F26. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Song, X.; Shi, Y.; Shi, Z.; Niu, W.; Feng, X.; Gu, D.; Bao, H.F.; Ma, H.P.; Eaton, D.C.; et al. Wnk1 activates large-conductance Ca2+-activated k+ channels through modulation of erk1/2 signaling. J. Am. Soc. Nephrol. JASN 2015, 26, 844–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rieg, T.; Vallon, V.; Sausbier, M.; Sausbier, U.; Kaissling, B.; Ruth, P.; Osswald, H. The role of the bk channel in potassium homeostasis and flow-induced renal potassium excretion. Kidney Int. 2007, 72, 566–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woda, C.B.; Bragin, A.; Kleyman, T.R.; Satlin, L.M. Flow-dependent k+ secretion in the cortical collecting duct is mediated by a maxi-k channel. Am. J. Physiol. Renal. Physiol. 2001, 280, F786–F793. [Google Scholar] [CrossRef]
- Shibata, S.; Zhang, J.; Puthumana, J.; Stone, K.L.; Lifton, R.P. Kelch-like 3 and cullin 3 regulate electrolyte homeostasis via ubiquitination and degradation of wnk4. Proc. Natl. Acad. Sci. USA 2013, 110, 7838–7843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyden, L.M.; Choi, M.; Choate, K.A.; Nelson-Williams, C.J.; Farhi, A.; Toka, H.R.; Tikhonova, I.R.; Bjornson, R.; Mane, S.M.; Colussi, G.; et al. Mutations in kelch-like 3 and cullin 3 cause hypertension and electrolyte abnormalities. Nature 2012, 482, 98–102. [Google Scholar] [CrossRef]
- Wilson, F.H.; Disse-Nicodeme, S.; Choate, K.A.; Ishikawa, K.; Nelson-Williams, C.; Desitter, I.; Gunel, M.; Milford, D.V.; Lipkin, G.W.; Achard, J.M.; et al. Human hypertension caused by mutations in wnk kinases. Science 2001, 293, 1107–1112. [Google Scholar] [CrossRef] [Green Version]
- Yamada, K.; Park, H.M.; Rigel, D.F.; DiPetrillo, K.; Whalen, E.J.; Anisowicz, A.; Beil, M.; Berstler, J.; Brocklehurst, C.E.; Burdick, D.A.; et al. Small-molecule wnk inhibition regulates cardiovascular and renal function. Nat. Chem. Biol. 2016, 12, 896–898. [Google Scholar] [CrossRef] [PubMed]
- Liedtke, W.; Friedman, J.M. Abnormal osmotic regulation in trpv4-/- mice. Proc. Natl. Acad. Sci. USA 2003, 100, 13698–13703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mironova, E.; Bugay, V.; Pochynyuk, O.; Staruschenko, A.; Stockand, J.D. Recording ion channels in isolated, split-opened tubules. Methods Mol. Biol. 2013, 998, 341–353. [Google Scholar]
- Mamenko, M.; Zaika, O.; O’Neil, R.G.; Pochynyuk, O. Ca2+ imaging as a tool to assess trp channel function in murine distal nephrons. Methods Mol. Biol. 2013, 998, 371–384. [Google Scholar] [PubMed]
- Bens, M.; Vallet, V.; Cluzeaud, F.; Pascual-Letallec, L.; Kahn, A.; Rafestin-Oblin, M.E.; Rossier, B.C.; Vandewalle, A. Corticosteroid-dependent sodium transport in a novel immortalized mouse collecting duct principal cell line. J. Am. Soc. Nephrol. JASN 1999, 10, 923–934. [Google Scholar] [CrossRef]
- Mamenko, M.; Zaika, O.L.; Boukelmoune, N.; Berrout, J.; O’Neil, R.G.; Pochynyuk, O. Discrete control of trpv4 channel function in the distal nephron by protein kinases a and c. J. Biol. Chem. 2013, 288, 20306–20314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grynkiewicz, G.; Poenie, M.; Tsien, R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985, 260, 3440–3450. [Google Scholar] [CrossRef]
- Levental, K.R.; Levental, I. Isolation of giant plasma membrane vesicles for evaluation of plasma membrane structure and protein partitioning. Methods Mol. Biol. 2015, 1232, 65–77. [Google Scholar]
- Tomilin, V.N.; Zaika, O.; Subramanya, A.R.; Pochynyuk, O. Dietary k(+) and cl(-) independently regulate basolateral conductance in principal and intercalated cells of the collecting duct. Pflugers. Archiv. Eur. J. Physiol. 2018, 470, 339–353. [Google Scholar] [CrossRef]
- Roy, A.; Al-Qusairi, L.; Donnelly, B.F.; Ronzaud, C.; Marciszyn, A.L.; Gong, F.; Chang, Y.P.; Butterworth, M.B.; Pastor-Soler, N.M.; Hallows, K.R.; et al. Alternatively spliced proline-rich cassettes link wnk1 to aldosterone action. J. Clin. Investig. 2015, 125, 3433–3448. [Google Scholar] [CrossRef] [Green Version]
- Yamada, K.; Levell, J.; Yoon, T.; Kohls, D.; Yowe, D.; Rigel, D.F.; Imase, H.; Yuan, J.; Yasoshima, K.; DiPetrillo, K.; et al. Optimization of allosteric with-no-lysine (wnk) kinase inhibitors and efficacy in rodent hypertension models. J. Med. Chem. 2017, 60, 7099–7107. [Google Scholar] [CrossRef] [PubMed]
- Sezgin, E.; Kaiser, H.J.; Baumgart, T.; Schwille, P.; Simons, K.; Levental, I. Elucidating membrane structure and protein behavior using giant plasma membrane vesicles. Nat. Protoc. 2012, 7, 1042–1051. [Google Scholar] [CrossRef] [PubMed]
- Staruschenko, A.; Pochynyuk, O.; Stockand, J.D. Regulation of epithelial na+ channel activity by conserved serine/threonine switches within sorting signals. J. Biol. Chem. 2005, 280, 39161–39167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilius, B.; Vriens, J.; Prenen, J.; Droogmans, G.; Voets, T. Trpv4 calcium entry channel: A paradigm for gating diversity. Am. J. physiology. Cell Physiol. 2004, 286, C195–C205. [Google Scholar] [CrossRef]
- Brooks, C.A.; Barton, L.S.; Behm, D.J.; Eidam, H.S.; Fox, R.M.; Hammond, M.; Hoang, T.H.; Holt, D.A.; Hilfiker, M.A.; Lawhorn, B.G.; et al. Discovery of gsk2798745: A clinical candidate for inhibition of transient receptor potential vanilloid 4 (trpv4). ACS Med. Chem. Lett. 2019, 10, 1228–1233. [Google Scholar] [CrossRef]
- Xu, B.E.; Lee, B.H.; Min, X.; Lenertz, L.; Heise, C.J.; Stippec, S.; Goldsmith, E.J.; Cobb, M.H. Wnk1: Analysis of protein kinase structure, downstream targets, and potential roles in hypertension. Cell Res. 2005, 15, 6–10. [Google Scholar] [CrossRef]
- Min, X.; Lee, B.H.; Cobb, M.H.; Goldsmith, E.J. Crystal structure of the kinase domain of wnk1, a kinase that causes a hereditary form of hypertension. Structure 2004, 12, 1303–1311. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.W.; Ku, S.K.; Han, M.H.; Kim, K.Y.; Kim, S.G.; Kim, G.Y.; Hwang, H.J.; Kim, B.W.; Kim, C.M.; Choi, Y.H. The administration of fructus schisandrae attenuates dexamethasone-induced muscle atrophy in mice. Int. J. Mol. Med. 2015, 36, 29–42. [Google Scholar] [CrossRef] [Green Version]
- Boychuk, C.R.; Zsombok, A.; Tasker, J.G.; Smith, B.N. Rapid glucocorticoid-induced activation of trp and cb1 receptors causes biphasic modulation of glutamate release in gastric-related hypothalamic preautonomic neurons. Front. Neurosci. 2013, 7, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazrak, A.; Liu, Z.; Huang, C.L. Antagonistic regulation of romk by long and kidney-specific wnk1 isoforms. Proc. Natl. Acad. Sci. USA 2006, 103, 1615–1620. [Google Scholar] [CrossRef] [Green Version]
- Wade, J.B.; Fang, L.; Liu, J.; Li, D.; Yang, C.L.; Subramanya, A.R.; Maouyo, D.; Mason, A.; Ellison, D.H.; Welling, P.A. Wnk1 kinase isoform switch regulates renal potassium excretion. Proc. Natl. Acad. Sci. USA 2006, 103, 8558–8563. [Google Scholar] [CrossRef] [Green Version]
- Grimm, P.R.; Irsik, D.L.; Settles, D.C.; Holtzclaw, J.D.; Sansom, S.C. Hypertension of kcnmb1-/- is linked to deficient k secretion and aldosteronism. Proc. Natl. Acad. Sci. USA 2009, 106, 11800–11805. [Google Scholar] [CrossRef] [Green Version]
- Mayan, H.; Vered, I.; Mouallem, M.; Tzadok-Witkon, M.; Pauzner, R.; Farfel, Z. Pseudohypoaldosteronism type ii: Marked sensitivity to thiazides, hypercalciuria, normomagnesemia, and low bone mineral density. J. Clin. Endocrinol. Metab. 2002, 87, 3248–3254. [Google Scholar] [CrossRef]
- Fu, Y.; Subramanya, A.; Rozansky, D.; Cohen, D.M. Wnk kinases influence trpv4 channel function and localization. Am. J. physiology. Ren. Physiol. 2006, 290, F1305–F1314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, H.C.; Zhang, X.; McNaughton, P.A. Activation of the trpv4 ion channel is enhanced by phosphorylation. J. Biol. Chem. 2009, 284, 27884–27891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, S.H.; Lee, E.J.; Hyun, S.; Chun, J.; Kim, Y.; Kang, S.S. Phosphorylation on the ser 824 residue of trpv4 prefers to bind with f-actin than with microtubules to expand the cell surface area. Cell. Signal. 2012, 24, 641–651. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tomilin, V.N.; Pyrshev, K.; Khayyat, N.H.; Zaika, O.; Pochynyuk, O. With-No-Lysine Kinase 1 (WNK1) Augments TRPV4 Function in the Aldosterone-Sensitive Distal Nephron. Cells 2021, 10, 1482. https://doi.org/10.3390/cells10061482
Tomilin VN, Pyrshev K, Khayyat NH, Zaika O, Pochynyuk O. With-No-Lysine Kinase 1 (WNK1) Augments TRPV4 Function in the Aldosterone-Sensitive Distal Nephron. Cells. 2021; 10(6):1482. https://doi.org/10.3390/cells10061482
Chicago/Turabian StyleTomilin, Viktor N., Kyrylo Pyrshev, Naghmeh Hassanzadeh Khayyat, Oleg Zaika, and Oleh Pochynyuk. 2021. "With-No-Lysine Kinase 1 (WNK1) Augments TRPV4 Function in the Aldosterone-Sensitive Distal Nephron" Cells 10, no. 6: 1482. https://doi.org/10.3390/cells10061482
APA StyleTomilin, V. N., Pyrshev, K., Khayyat, N. H., Zaika, O., & Pochynyuk, O. (2021). With-No-Lysine Kinase 1 (WNK1) Augments TRPV4 Function in the Aldosterone-Sensitive Distal Nephron. Cells, 10(6), 1482. https://doi.org/10.3390/cells10061482