Abstract
Methodology to access fluorescent 3-amido-1,8-naphthalimides using direct Buchwald–Hartwig amidation is described. The protocol was successfully used to couple a number of substrates (including an alkylamide, an arylamide, a lactam and a carbamate) to 3-bromo-1,8-naphthalimide in good yield. To further exemplify the approach, a set of scriptaid analogues with amide substituents at the 3-position were prepared. The new compounds were more potent than scriptaid at a number of histone deacetylase (HDAC) isoforms including HDAC6. Activity was further confirmed in a whole cell tubulin deacetylation assay where the inhibitors were more active than the established HDAC6 selective inhibitor Tubastatin. The optical properties of these new, highly active, compounds make them amenable to cellular imaging studies and theranostic applications.
1. Introduction
Interest in functionalised 1,8-naphthalimides has primarily focussed on substitution at the 4-position to generate fluorophores suitable for sensing and imaging applications [1,2,3,4]. Examples where sensors have been modified in the 3-position are less common, with notable examples including those reported by Zhang et al., Guo et al. and Elmes et al. for the detection of CO2, ClO− and reductive stress, respectively [5,6,7]. Examples that incorporate a 3-amido substituent are particularly rare [5,8,9,10], likely due to the multistep nature of the synthetic protocols required to access them. We have recently demonstrated that in the synthesis of 4-amido-1,8-naphthalimides, the usual three-step approach can be avoided using a Buchwald–Hartwig cross-coupling protocol in which a range of amides as well as lactams, carbamates and urea can be introduced in a single step [11]. Nicotinamides were also successfully coupled and the resultant probes shown to act as reversible indicators of the cellular redox state [12]. This direct coupling approach has not yet been evaluated for introducing substituents at the 3-position.
The 1,8-naphthalimide core has also been employed in medicinal chemistry [13,14]. Relevant examples (Figure 1) include (i) scriptaid [15] (an inhibitor of histone deacetylases (HDAC)) and amonafide [16,17,18,19] (a DNA intercalator and topoisomerase II inhibitor). Amonafide is converted in vivo into the bioactive N-acetyl-amonafide.
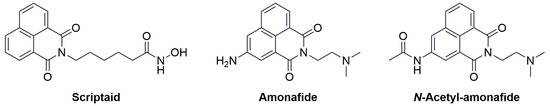
Figure 1.
Examples of 1,8-naphthalimides in medicinal chemistry.
The HDAC family has become well-studied due to their roles in a number of disease states [20,21,22]. Indeed, a number of HDAC inhibitors are FDA-approved for clinical use as treatments for T-cell lymphoma or myeloma [23,24,25,26,27]. In an effort to mitigate side effects, the next generation of HDAC inhibitors have been developed to be selective for specific HDAC classes or isoforms [28,29,30]. In this context, HDAC6 (Class IIb) has emerged as a valuable target as it has a clear role in the progression of a number of cancer types, and, unlike other isoforms, mouse models in which this isoform has been deleted are viable [31,32].
The entrance to the HDAC6 active site is slightly larger [33,34] than that of the other isoforms, and therefore a successful strategy for enhancing selectivity for this isoform is the modification of the pharmacophoric capping group. Amides offer not just a means to introduce additional size; they present both an H-bond donor and acceptor to maximise potential interactions with residues at the active site periphery [35,36,37].
There are currently only three published examples of scriptaid analogues with substitution at the 3-position, and only compound 3 has full HDAC IC50 activity data recorded (Figure 2) [38,39,40]. There are no scriptaid analogues with 3-amido substituents described in the literature. As such, the generation of a small set of 3-amido-substituted scriptaid analogues presents the opportunity to (i) test the Buchwald–Hartwig amidation methodology beyond the 4-position and to (ii) further explore the structure-activity relationship of functionalised scriptaid analogues. Our own recent work (CF010 and CF011, Figure 2) has identified that, for the 4-position, relatively small substituents can dramatically influence isoform selectivity and fluorescence properties [41]. While our efforts have focussed on modifying scriptaid to develop highly fluorescent anticancer agents, related recent examples exist for identifying biofilms and detecting influenza [42,43].
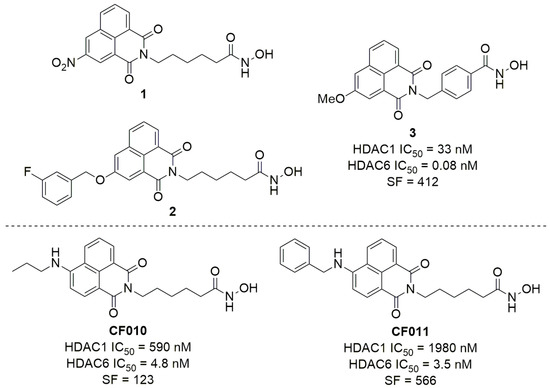
Figure 2.
Top: All published examples of 3-substituted scriptaid analogues [38,39,40], and Bottom: selected examples of 4-substituted scriptaid analogues from our recent work [41]. No activity data for 1 and 2 has been published. Selectivity factor (SF) = HDAC1 IC50/HDAC6 IC50.
Herein we outline the methodology for the direct synthesis of 3-amido-1,8-naphthalimides and the use of this approach to construct a set of scriptaid analogues. The results of the photophysical and biological assessment (IC50 and in cell tubulin acetylation) are also presented.
2. Results and Discussion
2.1. Methodology
To trial the Buchwald–Hartwig amidation methodology, 3-bromo-1,8-naphthalimide 4 was first required. The treatment of 1,8-naphthalic anhydride with 1.1 equiv. N-bromosuccinimide in H2SO4 provided the desired brominated compound 4 in >90% purity after trituration with EtOH [44]. The bromoanhydride was then converted to the corresponding methoxyethyl imide 5 using microwave irradiation (Scheme 1, see supplementary material for full details).
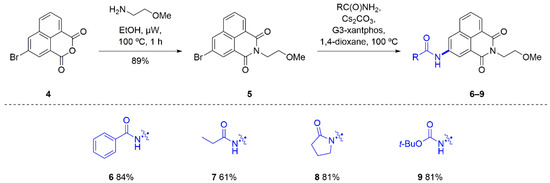
Scheme 1.
Direct synthesis of 3-amido and 3-carbamato-1,8-naphthalimides.
Using the reported conditions to introduce amides to the 4-position as a guide [11], a test reaction was performed in which 3-bromo-1,8-naphthalimide 5 was reacted with benzamide, Cs2CO3 and G3-xantphos at 100 °C (Scheme 1). Monitoring the reaction progress using thin layer chromatography (TLC) indicated the consumption of the starting material and the appearance of a new blue spot (using UV visualisation) within 2.5 h, whereupon the reaction mixture was poured over H2O to provide a yellow solid. The analysis of the solid using 1H NMR spectroscopy revealed new aryl resonances centred at 8.08 and 7.60 ppm and a new broad singlet at 10.81 ppm that was assigned to the amide N-H proton. Evaluation using HRMS further confirmed that the desired 3-benzamido-1,8-naphthalimide 6 had been synthesised. The product was obtained in good yield (84%) and as such several other substrates—an aliphatic amide (propionamide), a lactam (pyrrolidinone) and a carbamate (tert-butyl carbamate)—were also trialled as reaction partners. These additional reactions also provided the desired 3-substituted 1,8-naphthalimide in under 3 h and in good yields (61–81%, Scheme 1). The substrates were well tolerated with yields and reaction times comparable to those observed for the coupling reactions that used 4-bromo-1,8-naphthalimide [11]. In terms of reactivity, 3-bromo-1,8-naphthalimides are an excellent, readily accessible substrate for palladium-mediated amidation reactions.
The new compounds showed an absorption maximum and a secondary maximum (~340 and ~380 nm, respectively, Table 1 and Figure 3) and a single emission (~440 nm, Table 1 and Figure 3). Consistent with the decreased extent of intramolecular charge transfer (as compared to the amino substituted examples), the quantum yields for the new compounds ranged from 0.02 to 0.06 (Table 1). For comparison, the 4-benzamido isomer of 6 has Φf = 0.33 and for the 4-fluorobenzamido Φf = 0.05 [11]. It is our experience that even with low values the compounds are amenable to imaging applications [45].

Table 1.
Photophysical properties of 3-amido-1,8-naphthalimides in DMSO.
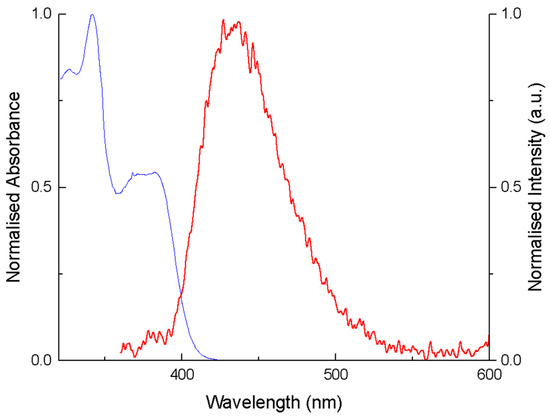
Figure 3.
Normalised absorption and emission spectra of 6 in DMSO.
2.2. Scriptaid Analogues
The most potent of the previously synthesised 4-amino series were CF010 and CF011 (Figure 2) possessing propyl and benzyl substituents, respectively [41]. As such, two aromatic and one aliphatic amide were chosen as suitable substituents for the new 3-amido analogues (KNH019, 020 and 021, respectively, Figure 4).
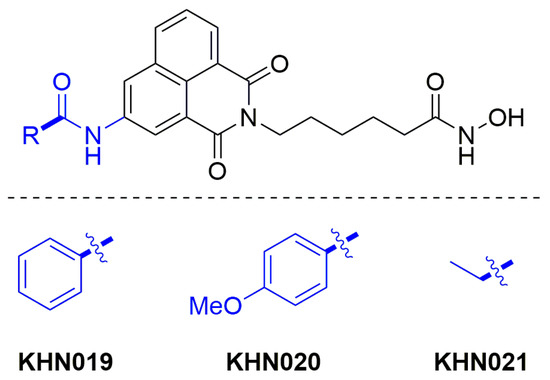
Figure 4.
Target 3-amido scriptaid analogues.
The strategy reported by Fleming et al. for the construction of 4-substituted scriptaid analogues was followed here for the synthesis of the 3-substituted relatives [41]. First, 3-bromo-1,8-naphthalic anhydride 4 was transformed into imide 11 in an excellent yield (96% over two steps, see supplementary material for details). Next, the key Buchwald–Hartwig reaction was used to introduce the amido-substituent (Scheme 2). For example, in the synthesis of KNH019 a solution of 3-bromo-1,8-naphthalimide 11, benzamide, Cs2CO3 and G3-xantphos in 1,4-dioxane were heated at 100 °C with the reaction progress being monitored using TLC. Following reaction completion (3.5 h), the desired product 12 was isolated as a light brown solid (63% yield). The methyl ester was carefully removed (1.7 equiv. of LiOH in THF/H2O), and the resultant carboxylic acid 13 was converted to the desired hydroxamic acid via the mixed anhydride (formed using ethyl chloroformate and an in-situ treatment with a solution of freshly prepared NH2OH in MeOH). After 24 h, the solvent was removed and the residue was triturated with H2O to provide the desired hydroxamic acid KNH019 in a good yield (76% over two steps). In the 1H NMR spectrum, two broad singlets at 10.34 and 8.68 ppm (each integrating for one proton) were characteristic of the newly installed hydroxamic acid N-H and O-H protons, respectively. The singlet corresponding to the benzamide N-H proton at 10.85 ppm was also present, indicating the successful formation of the 3-amido scriptaid analogue.
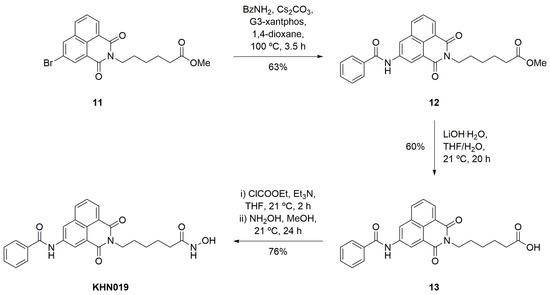
Scheme 2.
Synthesis of 3-benzamido scriptaid analogue KNH019.
The remaining scriptaid analogues KNH020 and KNH021 were accessed in a good yield using the same reaction sequence (see supplementary material for full details). As with the 3-amido-1,8-naphthalimides synthesised at the outset (6–9), the quantum yield of the new inhibitors ranged from 0.03 to 0.05 (Table 2) with emission at λem ~437 nm.
Table 2.
Photophysical properties of 3-amido scriptaid analogues in DMSO.
2.3. HDAC Inhibition
The new scriptaid analogues were initially assessed using a single point assay where enzyme activity is reported as a percentage of full enzyme activity (Table 3). For HDAC isoforms 1, 3, 8 and 11, a concentration of 10 µM of the inhibitor was used. Against HDAC6, inhibition was determined using 0.01 µM of the inhibitor. All 3-amido-1,8-naphthalimide analogues were significantly more effective inhibitors of HDAC6 than the control compound scriptaid. However, no substantial difference in HDAC6 enzyme activity was observed between the benzamide, p-methoxybenzamide and propionamide analogues. Furthermore, when the 3-amido scriptaid analogues were evaluated at the “off-target” HDAC isoforms (Table 3), considerable activity was noted; again, they were more effective than scriptaid.
Table 3.
HDAC activity (percentage of full enzyme activity) for 3-amido scriptaid analogues.
The IC50 for the three compounds against the same isoform panel was then measured (Table 4). In agreement with the results of the single point assay, all compounds were potent inhibitors of HDAC6, with similar IC50 values (ranging from 0.58 nM to 1.0 nM). Compared to our previously reported 4-amino analogues CF010 and CF011 [41], the new compounds were more effective inhibitors of HDAC6 (by an order of magnitude) and compared favourably with the 3-methoxy analogue 3 reported by Ho [39]. Based on the inhibition of other isoforms, the selectivity of the new compounds was considered fair to good (ranging from 38 to 150 for HDAC6 over HDAC1); however, when the 3-substituted series were compared to the previous 4-amino analogues CF010 and CF011 (up to 566-fold selectivity for HDAC6, Table 4), they were universally less selective. While the set of compounds produced for this study was not extensive, it would appear that amido substitution at the 3-position of the 1,8-naphthalimide core leads to a slightly enhanced activity at all HDAC isoforms to ultimately provide highly potent but only moderately selective HDAC6 inhibitors.
Table 4.
HDAC activity (IC50) and isoform selectivity factor for 3-amido scriptaid analogues.
To confirm the observed activities of the selected test compounds against HDAC6, the acetylation status of tubulin in the human lung cancer cell line A549 was investigated by immunostaining combined with automated high content image analysis (see supplementary material for full details). In addition to the previously published 4-benzylamino scriptaid analogue CF011 (HDAC6 IC50 = 3.5 nM, SF = 566, Table 4), the structurally related 3-benzamido scriptaid KNH019 (HDAC 6 IC50 = 1.0 nM, SF = 91) was tested with regards to dose response and time dependency, where both acetylated tubulin and acetylated histone were detected (Figure 5A–E). Compared to untreated cells, both CF011 and KNH019 rapidly increased tubulin acetylation in a dose-dependent manner by up to ∼1.5-fold from 10−9 mM and 10−6 mM, respectively. In contrast, the reference compound tubastatin only demonstrated a significantly increased tubulin acetylation from 10−3 mM in the same test system (Figure 5A). When acetylated histone was detected for both test compounds, CF011 showed a significant increase from 10−6 mM, while KNH019 only demonstrated a significant activity at 10−2 mM. Surprisingly, the reference compound tubastatin increased histone acetylation from 10−3 mM, similar to its activity against tubulin acetylation (Figure 5A,B). The apparent drop in histone acetylation by both compounds above 10 μM is indicative of a potential cytotoxicity at higher concentrations (data not shown). Why this effect did not also manifest as reduced tubulin acetylation is unclear at this point.
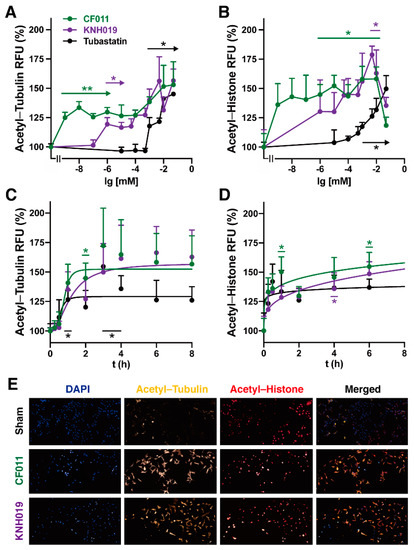
Figure 5.
Cell-based assessment of HDAC activity and selectivity. Dose and time-dependency of tubulin and histone acetylation by test compounds. A549 cells were treated with (A,B) different concentrations (0–50 µM) of CF011, KNH019 and tubastatin for 24 h or with (C,D) 1 µM over different time intervals before (A,C) tubulin acetylation or (B,D) histone acetylation were automatically quantified using high content imaging. Data represent the average of four independent experiments with four replicate wells/experiments. The statistical significances of effects compared to the untreated control were analysed using Brown–Forsythe and Welch one-way ANOVA analyses using Graph Pad Prism. The significance was set as * 0.05 > p ≥ 0.01, ** 0.01 > p ≥ 0.002. Error bars represent the Standard Error of Mean (SEM). (E) Representative fluorescence images show tubulin acetylation (yellow), histone acetylation (red) and nuclear counterstain (DAPI, blue) after 24 h of exposure to test compounds. Secondary antibodies were detected using excitation/emission filters for CY3 (excitation: 542/27 nm, emission: 597/45 nm), CY5 (excitation: 632/22 nm, emission: 684/25 nm) and DAPI (excitation: 390/18 nm, emission: 435/48 nm), respectively.
In A549 cells, all test and reference compounds rapidly increased tubulin and histone acetylation within only a few hours (Figure 5C,D). After 4 h, the increases in tubulin acetylation reached a plateau that was sustained for up to 24 h (data not shown) (Figure 5C,D). Representative immunostaining images also demonstrate that both CF011 and KNH019 at 10 µM strongly induce tubulin and histone acetylation. This illustrates that the antibodies used can differentiate between nuclear and cytoplasmic targets and that at this concentration both compounds therefore do not seem to be HDAC6 selective, which predominantly resides in the cytoplasm (Figure 5E). Similar results have been generated in the hepatocarcinoma cell line HepG2 (See supplementary material Figure S24 for details), which highlights that the observed effects are not cell line-specific but are likely relevant for other cells and tissues.
Overall, the data clearly indicate that the new compounds retain HDAC6 activity in cells with a significantly higher potency compared to the reference compound tubastatin. A broader HDAC activity was confirmed for the test and reference compounds when measuring both tubulin and histone acetylation, which reflected the residual activity of isoforms other than HDAC6. The discrepancy between the cell-free and cellular activity observed in this study was reported for tubastatin before and should be considered when using tubastatin as a reference compound [46].
3. Conclusions
In summary, the direct amidation of 3-bromo-1,8-naphthalimides was readily achieved using the Buchwald–Hartwig methodology to give amido, lactam and carbamato products. The method was used to produce a set of novel fluorescent scriptaid analogues, and, as identified using direct IC50 measurements and whole cell tubulin deacetylation assays, the analogues were potent (but less selective) inhibitors of histone deacetylase enzymes. The fluorescent nature of these compounds makes them well-suited as tools for additional cell-based studies.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/cells10061505/s1, Pages S2–S11: Experimental procedures, Figures S1–S16: 1H and 13C NMR of synthesised compounds, Table S1: Photophysical properties, Figures S17–S24: UV/Vis and fluorescence data, Figure S24: Cell-based assessment of HDAC activity and selectivity in HepG2 cells.
Author Contributions
Conceptualisation, T.D.A. and F.M.P.; methodology, K.N.H., T.D.A., N.G. and F.M.P.; investigation, K.N.H., R.A., Z.F. and N.G.; writing—original draft preparation, K.N.H., N.G. and F.M.P.; writing—review and editing, K.N.H., T.D.A., N.G. and F.M.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data has been presented in this article or associated supplementary material.
Acknowledgments
The authors would like to acknowledge the assistance of Shane Hickey (University of South Australia) in the characterisation of new compounds using high-resolution mass spectrometry.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Duke, R.M.; Veale, E.B.; Pfeffer, F.M.; Kruger, P.E.; Gunnlaugsson, T. Colorimetric and Fluorescent Anion Sensors: An Overview of Recent Developments in the Use of 1,8-Naphthalimide-Based Chemosensors. Chem. Soc. Rev. 2010, 39, 3936–3953. [Google Scholar] [CrossRef]
- Ashton, T.D.; Jolliffe, K.A.; Pfeffer, F.M. Luminescent Probes for the Bioimaging of Small Anionic Species in Vitro and in Vivo. Chem. Soc. Rev. 2015, 44, 4547–4595. [Google Scholar] [CrossRef] [PubMed]
- Fleming, C.L.; Nalder, T.D.; Doeven, E.H.; Barrow, C.J.; Pfeffer, F.M.; Ashton, T.D. Synthesis of N-Substituted 4-Hydroxynaphthalimides Using Palladium-Catalysed Hydroxylation. Dye Pigment. 2016, 126, 118–120. [Google Scholar] [CrossRef]
- Nalder, T.D.; Ashton, T.D.; Pfeffer, F.M.; Marshall, S.N.; Barrow, C.J. 4-Hydroxy-N-Propyl-1,8-Naphthalimide Esters: New Fluorescence-Based Assay for Analysing Lipase and Esterase Activity. Biochimie 2016, 128–129, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Song, Y.; Liu, M.; Li, H.; Sun, H.; Sun, M.; Yu, H. Visual Sensing of CO2 in Air with a 3-Position Modified Naphthalimide-Derived Organogelator Based on a Fluoride Ion-Induced Strategy. Dye Pigment. 2019, 160, 799–805. [Google Scholar] [CrossRef]
- Guo, T.; Cui, L.; Shen, J.; Wang, R.; Zhu, W.; Xu, Y.; Qian, X. A Dual-Emission and Large Stokes Shift Fluorescence Probe for Real-Time Discrimination of ROS/RNS and Imaging in Live Cells. Chem. Commun. 2013, 49, 1862–1864. [Google Scholar] [CrossRef] [PubMed]
- Ao, X.; Bright, S.A.; Taylor, N.C.; Elmes, R.B.P. 2-Nitroimidazole Based Fluorescent Probes for Nitroreductase; Monitoring Reductive Stress: In Cellulo. Org. Biomol. Chem. 2017, 15, 6104–6108. [Google Scholar] [CrossRef]
- Wang, K.R.; Qian, F.; Wang, X.M.; Tan, G.H.; Rong, R.X.; Cao, Z.R.; Chen, H.; Zhang, P.Z.; Li, X.L. Cytotoxic Activity and DNA Binding of Naphthalimide Derivatives with Amino Acid and Dichloroacetamide Functionalizations. Chin. Chem. Lett. 2014, 25, 1087–1093. [Google Scholar] [CrossRef]
- Xie, L.; Xu, Y.; Wang, F.; Liu, J.; Qian, X.; Cui, J. Synthesis of New Amonafide Analogues via Coupling Reaction and Their Cytotoxic Evaluation and DNA-Binding Studies. Bioorg. Med. Chem. 2009, 17, 804–810. [Google Scholar] [CrossRef]
- Van Quaquebeke, E.; Mahieu, T.; Dumont, P.; Dewelle, J.; Ribaucour, F.; Simon, G.; Sauvage, S.; Gaussin, J.F.; Tuti, J.; El Yazidi, M.; et al. 2,2,2-Trichloro-N-({2-[2-(Dimethylamino)Ethyl]-1,3-Dioxo-2, 3-Dihydro-1H-Benzo[de]Isoquinolin-5-Yl}carbamoyl)Acetamide (UNBS3157), a Novel Nonhematotoxic Naphthalimide Derivative with Potent Antitumor Activity. J. Med. Chem. 2007, 50, 4122–4134. [Google Scholar] [CrossRef]
- Hearn, K.N.; Nalder, T.D.; Cox, R.P.; Maynard, H.D.; Bell, T.D.M.; Pfeffer, F.M.; Ashton, T.D. Modular Synthesis of 4-Aminocarbonyl Substituted 1,8-Naphthalimides and Application in Single Molecule Fluorescence Detection. Chem. Commun. 2017, 53, 12298–12301. [Google Scholar] [CrossRef] [PubMed]
- Sharma, H.; Tan, N.K.; Trinh, N.; Yeo, J.H.; New, E.J.; Pfeffer, F.M. A Fluorescent Naphthalimide NADH Mimic for Continuous and Reversible Sensing of Cellular Redox State. Chem. Commun. 2020, 56, 2240–2243. [Google Scholar] [CrossRef] [PubMed]
- Kamal, A.; Bolla, N.R.; Srikanth, P.S.; Srivastava, A.K. Naphthalimide Derivatives with Therapeutic Characteristics: A Patent Review. Expert Opin. Ther. Pat. 2013, 23, 299–317. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Veale, E.B.; Phelan, C.M.; Murphy, S.A.; Tocci, G.M.; Gillespie, L.J.; Frimannsson, D.O.; Kelly, J.M.; Gunnlaugsson, T. Recent Advances in the Development of 1,8-Naphthalimide Based DNA Targeting Binders, Anticancer and Fluorescent Cellular Imaging Agents. Chem. Soc. Rev. 2013, 42, 1601–1618. [Google Scholar] [CrossRef]
- Kern, S.E.; Su, G.H.; Sohn, T.A.; Ryu, B. A Novel Histone Deacetylase Inhibitor Identified by High-Throughput Transcriptional Screening of a Compound Library. Cancer Res. 2000, 60, 3137–3142. [Google Scholar]
- Zee-Cheng, R.K.Y.; Cheng, C.C. N-(Aminoalkyl)Imide Antineoplastic Agents. Synthesis and Biological Activity. J. Med. Chem. 1985, 28, 1216–1222. [Google Scholar] [CrossRef]
- Tomczyk, M.D.; Walczak, K.Z. 1,8-Naphthalimide Based DNA Intercalators and Anticancer Agents. A Systematic Review from 2007 to 2017. Eur. J. Med. Chem. 2018, 159, 393–422. [Google Scholar] [CrossRef]
- Brider, T.; Redko, B.; Grynszpan, F.; Gellerman, G. Three Overlooked Chemical Approaches toward 3-Naphthalimide Amonafide N-Derivatives. Tetrahedron Lett. 2014, 55, 6675–6679. [Google Scholar] [CrossRef]
- Gellerman, G. Recent Developments in the Synthesis and Applications of Anticancer Amonafide Derivatives. A Mini Review. Lett. Drug Des. Discov. 2016, 13, 47–63. [Google Scholar] [CrossRef]
- Bassett, S.A.; Barnett, M.P.G. The Role of Dietary Histone Deacetylases (HDACs) Inhibitors in Health and Disease. Nutrients 2014, 6, 4273–4301. [Google Scholar] [CrossRef]
- Kim, H.; Bae, S. Histone Deacetylase Inhibitors: Molecular Mechanisms of Action and Clinical Trials as Anti-Cancer Drugs. Am. J. Transl. Res. 2011, 3, 166–179. [Google Scholar]
- Dokmanovic, M.; Clarke, C.; Marks, P.A. Histone Deacetylase Inhibitors: Overview and Perspectives. Mol. Cancer Res. 2007, 5, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Marks, P.A.; Breslow, R. Dimethyl Sulfoxide to Vorinostat: Development of This Histone Deacetylase Inhibitor as an Anticancer Drug. Nat. Biotechnol. 2007, 25, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, S.S.; Belani, C.P.; Ruel, C.; Frankel, P.; Gitlitz, B.; Koczywas, M.; Espinoza-Delgado, I.; Gandara, D. Phase II Study of Belinostat (PXD101), a Histone Deacetylase Inhibitor, for Second Line Therapy of Advanced Malignant Pleural Mesothelioma. J. Thorac. Oncol. 2009, 4, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Ellis, L.; Pan, Y.; Smyth, G.K.; George, D.J.; McCormack, C.; Williams-Truax, R.; Mita, M.; Beck, J.; Burris, H.; Ryan, G.; et al. Histone Deacetylase Inhibitor Panobinostat Induces Clinical Responses with Associated Alterations in Gene Expression Profiles in Cutaneous T-Cell Lymphoma. Clin. Cancer Res. 2008, 14, 4500–4510. [Google Scholar] [CrossRef] [PubMed]
- Prince, H.M.; Dickinson, M.; Khot, A. Romidepsin for Cutaneous T-Cell Lymphoma. Future Oncol. 2013, 9, 1819–1827. [Google Scholar] [CrossRef]
- Manal, M.; Chandrasekar, M.J.N.; Gomathi Priya, J.; Nanjan, M.J. Inhibitors of Histone Deacetylase as Antitumor Agents: A Critical Review. Bioorg. Chem. 2016, 67, 18–42. [Google Scholar] [CrossRef]
- Bertrand, P.; Roche, J. Inside HDACs with More Selective HDAC Inhibitors. Eur. J. Med. Chem. 2016, 121, 451–483. [Google Scholar] [CrossRef]
- Thaler, F.; Mercurio, C. Towards Selective Inhibition of Histone Deacetylase Isoforms: What Has Been Achieved, Where We Are and What Will Be Next. ChemMedChem 2014, 9, 523–536. [Google Scholar] [CrossRef]
- Yang, F.; Zhao, N.; Ge, D.; Chen, Y. Next-Generation of Selective Histone Deacetylase Inhibitors. RSC Adv. 2019, 9, 19571–19583. [Google Scholar] [CrossRef]
- Wang, X.X.; Wan, R.Z.; Liu, Z.P. Recent Advances in the Discovery of Potent and Selective HDAC6 Inhibitors. Eur. J. Med. Chem. 2018, 143, 1406–1418. [Google Scholar] [CrossRef]
- Witt, O.; Deubzer, H.E.; Milde, T.; Oehme, I. HDAC Family: What Are the Cancer Relevant Targets? Cancer Lett. 2009, 277, 8–21. [Google Scholar] [CrossRef]
- Hai, Y.; Christianson, D.W. Histone Deacetylase 6 Structure and Molecular Basis of Catalysis and Inhibition. Nat. Chem. Biol. 2016, 12, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, L.; Min, J. Structural Biology: HDAC6 Finally Crystal Clear. Nat. Chem. Biol. 2016, 12, 660–661. [Google Scholar] [CrossRef] [PubMed]
- Haggarty, S.J.; Koeller, K.M.; Wong, J.C.; Grozinger, C.M.; Schreiber, S.L. Domain-Selective Small-Molecule Inhibitor of Histone Deacetylase 6 (HDAC6)-Mediated Tubulin Deacetylation. Proc. Natl. Acad. Sci. USA 2003, 100, 4389–4394. [Google Scholar] [CrossRef]
- Santo, L.; Hideshima, T.; Kung, A.L.; Tseng, J.C.; Tamang, D.; Yang, M.; Jarpe, M.; Van Duzer, J.H.; Mazitschek, R.; Ogier, W.C.; et al. Preclinical Activity, Pharmacodynamic, and Pharmacokinetic Properties of a Selective HDAC6 Inhibitor, ACY-1215, in Combination with Bortezomib in Multiple Myeloma. Blood 2012, 119, 2579–2589. [Google Scholar] [CrossRef]
- Kalin, J.H.; Butler, K.V.; Kozikowski, A.P. Creating Zinc Monkey Wrenches in the Treatment of Epigenetic Disorders. Curr. Opin. Chem. Biol. 2009, 13, 263–271. [Google Scholar] [CrossRef]
- Gillet, N.; Vandermeers, F.; de Brogniez, A.; Florins, A.; Nigro, A.; François, C.; Bouzar, A.-B.; Verlaeten, O.; Stern, E.; Lambert, D.M.; et al. Chemoresistance to Valproate Treatment of Bovine Leukemia Virus-Infected Sheep; Identification of Improved HDAC Inhibitors. Pathogens 2012, 1, 65–82. [Google Scholar] [CrossRef]
- Ho, Y.H.; Wang, K.J.; Hung, P.Y.; Cheng, Y.S.; Liu, J.R.; Fung, S.T.; Liang, P.H.; Chern, J.W.; Yu, C.W. A Highly HDAC6-Selective Inhibitor Acts as a Fluorescent Probe. Org. Biomol. Chem. 2018, 16, 7820–7832. [Google Scholar] [CrossRef]
- Rudebeck, E.E.; Cox, R.P.; Bell, T.D.M.; Acharya, R.; Feng, Z.; Gueven, N.; Ashton, T.D.; Pfeffer, F.M. Mixed Alkoxy/Hydroxy 1,8-Naphthalimides: Expanded Fluorescence Colour Palette and in Vitro Bioactivity. Chem. Commun. 2020, 56, 6866–6869. [Google Scholar] [CrossRef] [PubMed]
- Fleming, C.L.; Natoli, A.; Schreuders, J.; Devlin, M.; Yoganantharajah, P.; Gibert, Y.; Leslie, K.G.; New, E.J.; Ashton, T.D.; Pfeffer, F.M. Highly Fluorescent and HDAC6 Selective Scriptaid Analogues. Eur. J. Med. Chem. 2019, 162, 321–333. [Google Scholar] [CrossRef]
- Dou, W.-T.; Qin, Z.-Y.; Li, J.; Zhou, D.-M.; He, X.-P. Self-Assembled Sialyllactosyl Probes with Aggregation-Enhanced Properties for Ratiometric Detection and Blocking of Influenza Viruses. Sci. Bull. 2019, 64, 1902–1909. [Google Scholar] [CrossRef]
- Sedgwick, A.C.; Yan, K.-C.; Mangel, D.N.; Shang, Y.; Steinbrueck, A.; Han, H.-H.; Brewster, J.T.; Hu, X.-L.; Snelson, D.W.; Lynch, V.M.; et al. Deferasirox (ExJade): An FDA-Approved AIEgen Platform with Unique Photophysical Properties. J. Am. Chem. Soc. 2021, 143, 1278–1283. [Google Scholar] [CrossRef] [PubMed]
- Moseley, J.D.; Moss, W.O.; Welham, M.J.; Ancell, C.L.; Banister, J.; Bowden, S.A.; Norton, G.; Young, M.J. A New Approach to Rapid Parallel Development of Four Neurokinin Antagonists. Part 2. Synthesis of ZD6021 Cyano Acid. Org. Process Res. Dev. 2003, 7, 58–66. [Google Scholar] [CrossRef]
- Fleming, C.L.; Ashton, T.D.; Nowell, C.; Devlin, M.; Natoli, A.; Schreuders, J.; Pfeffer, F.M. A Fluorescent Histone Deacetylase (HDAC) Inhibitor for Cellular Imaging. Chem. Commun. 2015, 51, 7827–7830. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Kang, M.H.; Hong, K.; Kim, J.H. Tubastatin A Inhibits HDAC and Sirtuin Activity Rather than Being a HDAC6-Specific Inhibitor in Mouse Oocytes. Aging 2019, 11, 1759–1777. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).