
A Novel Model for Nephrotic Syndrome Reveals Associated Dysbiosis of the Gut Microbiome and Extramedullary Hematopoiesis

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Cell Culture
2.3. Antibodies
2.4. SEM and TEM Procedures
2.5. Histology, IF and IHC Staining
2.6. IF Staining of Cultured Cells
2.7. Iterative Indirect Immunofluorescence Imaging
2.8. Microscopy
2.9. Quantification of Glomerular Sclerosis
2.10. Quantification of Glomerular Podocytes
2.11. Histological Analysis
2.12. Measurement of Urinary Albumin and Creatinine
2.13. Gel Analysis
2.14. Analysis of Murine Blood Serum
2.15. Microbiome Analysis
2.16. Quantification and Statistical Analyses
3. Results
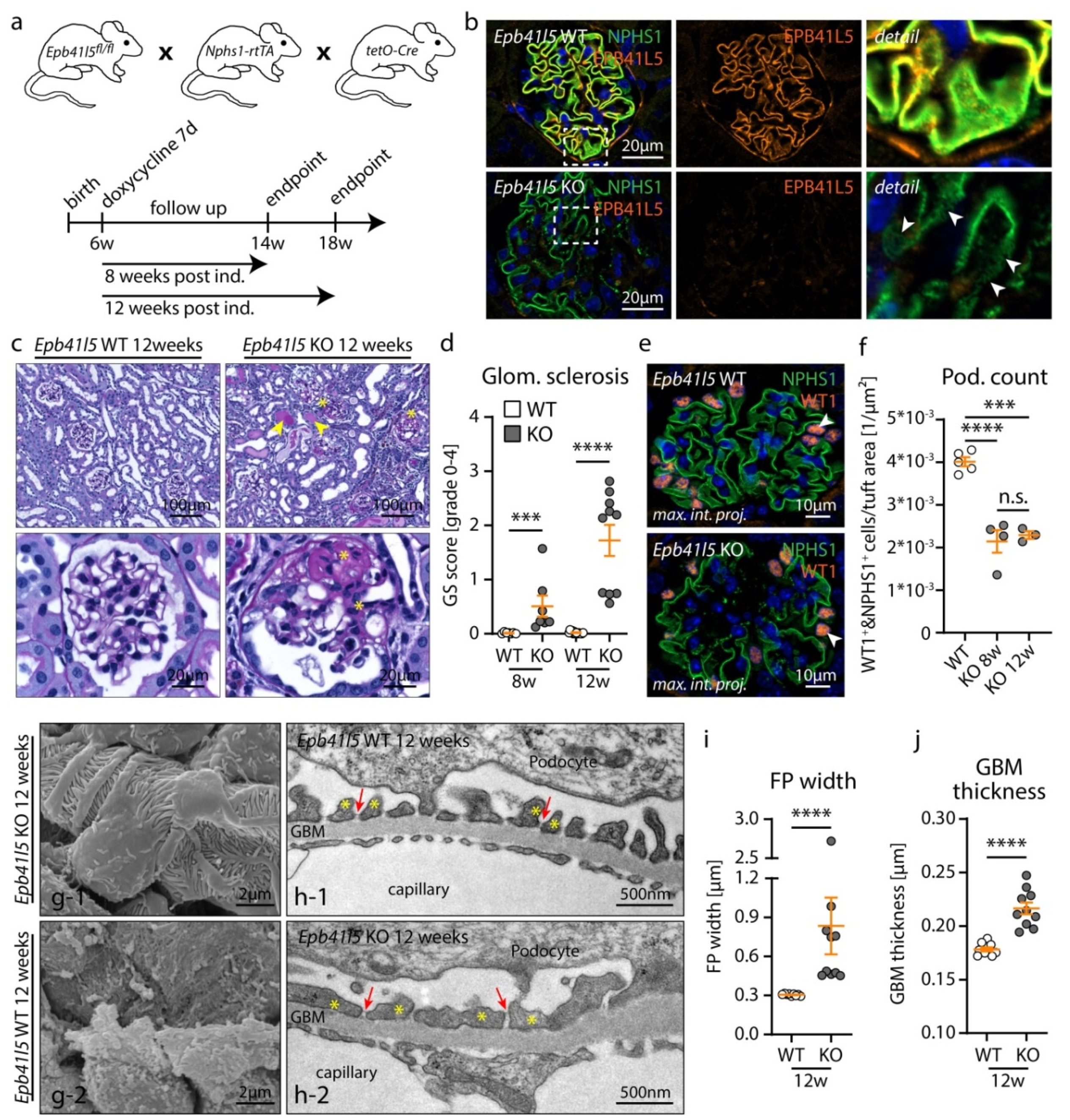
3.1. Inducible, Podocyte-Specific Knockout of Epb41l5 Results in FSGS Manifestation and Nephrotic Syndrome in Adult Mice
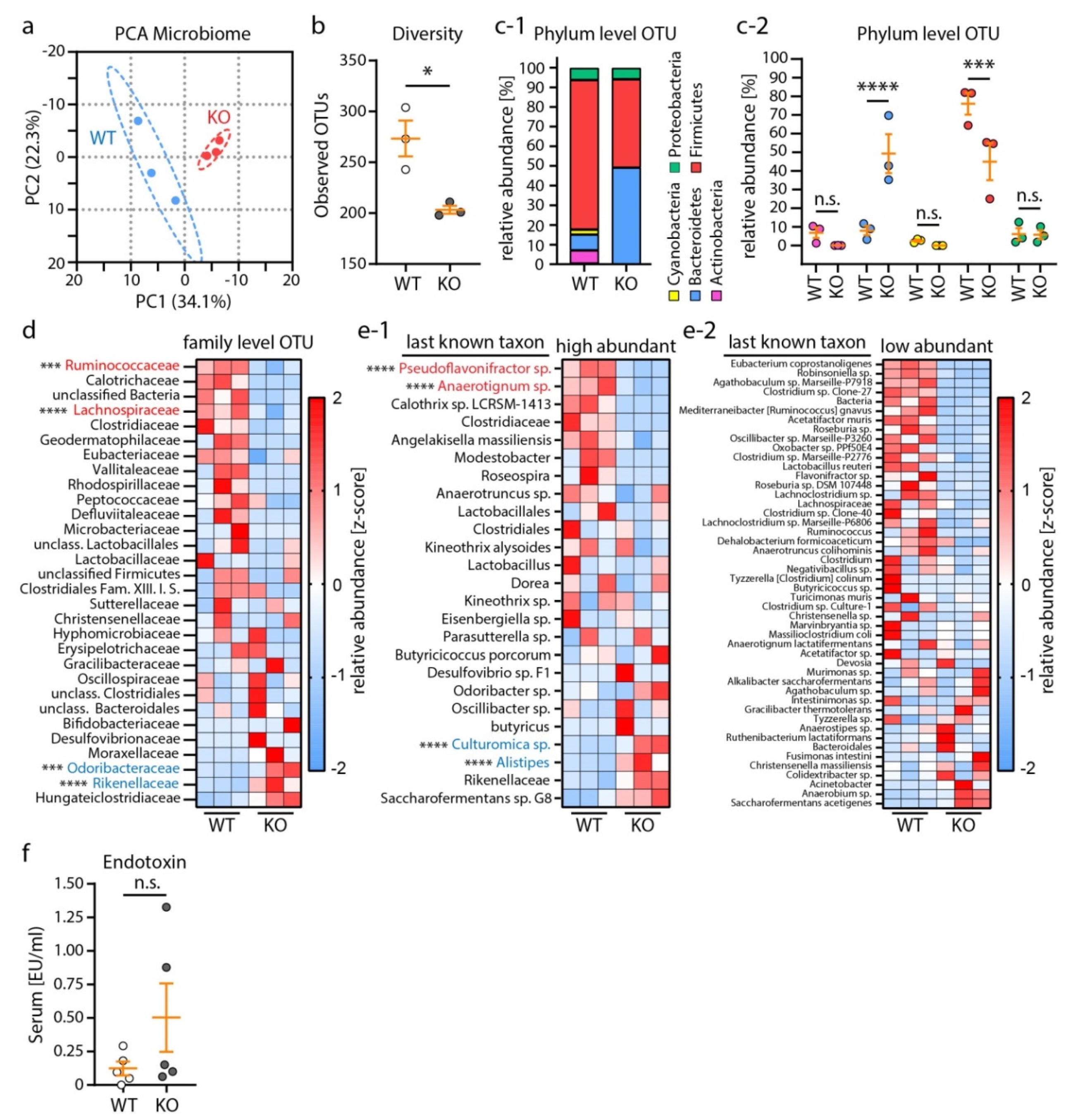
3.2. Nephrotic Syndrome Was Accompanied by Alterations of the Microbiome Composition
3.3. Nephrotic Syndrome Leads to Multiple Organ Pathologies in Mice, Including Reactive Hepatomegaly
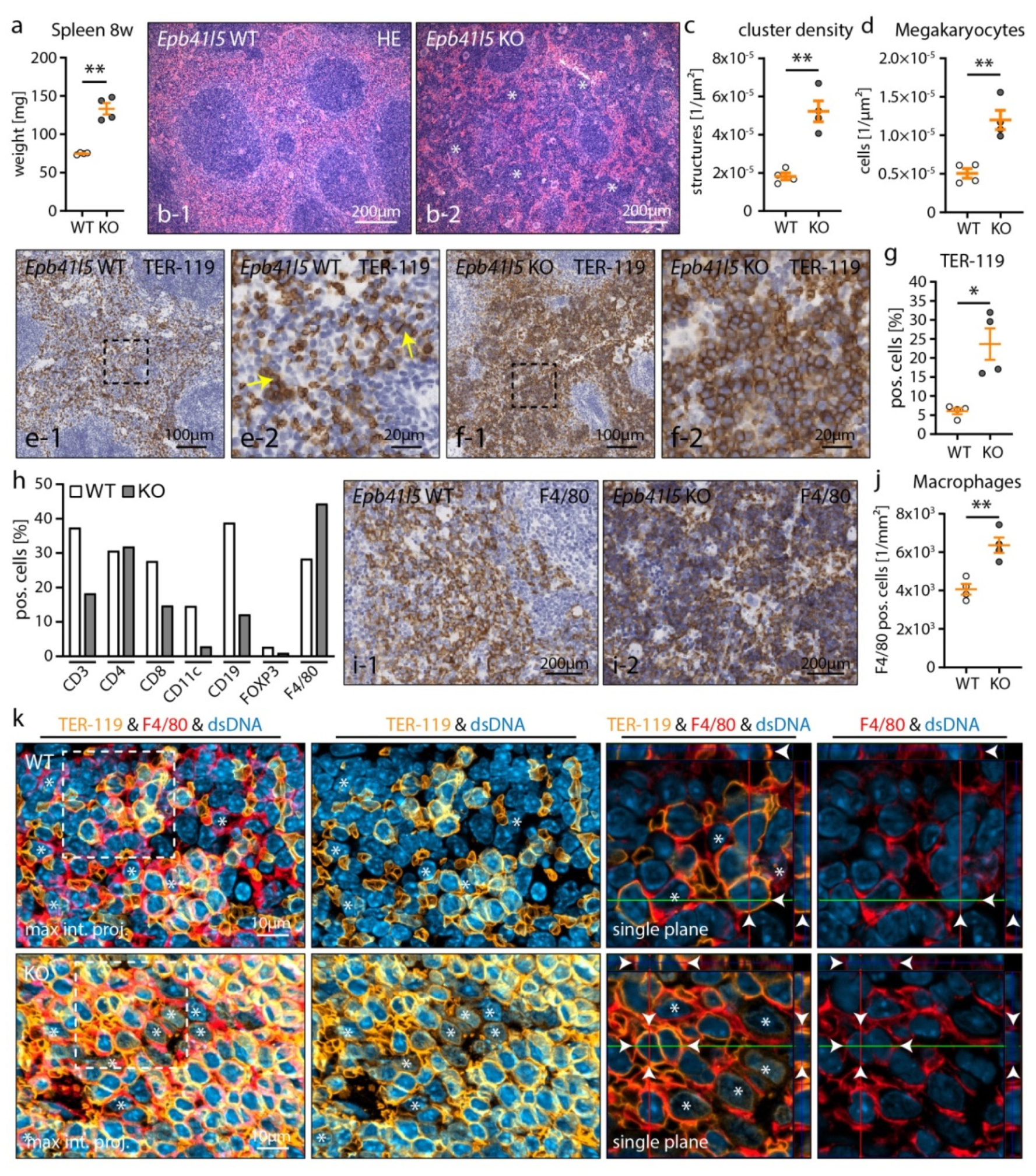
3.4. Nephrotic Syndrome Causes Extramedullary Hematopoiesis and Red Pulp Macrophage (RpMΦ) Expansion
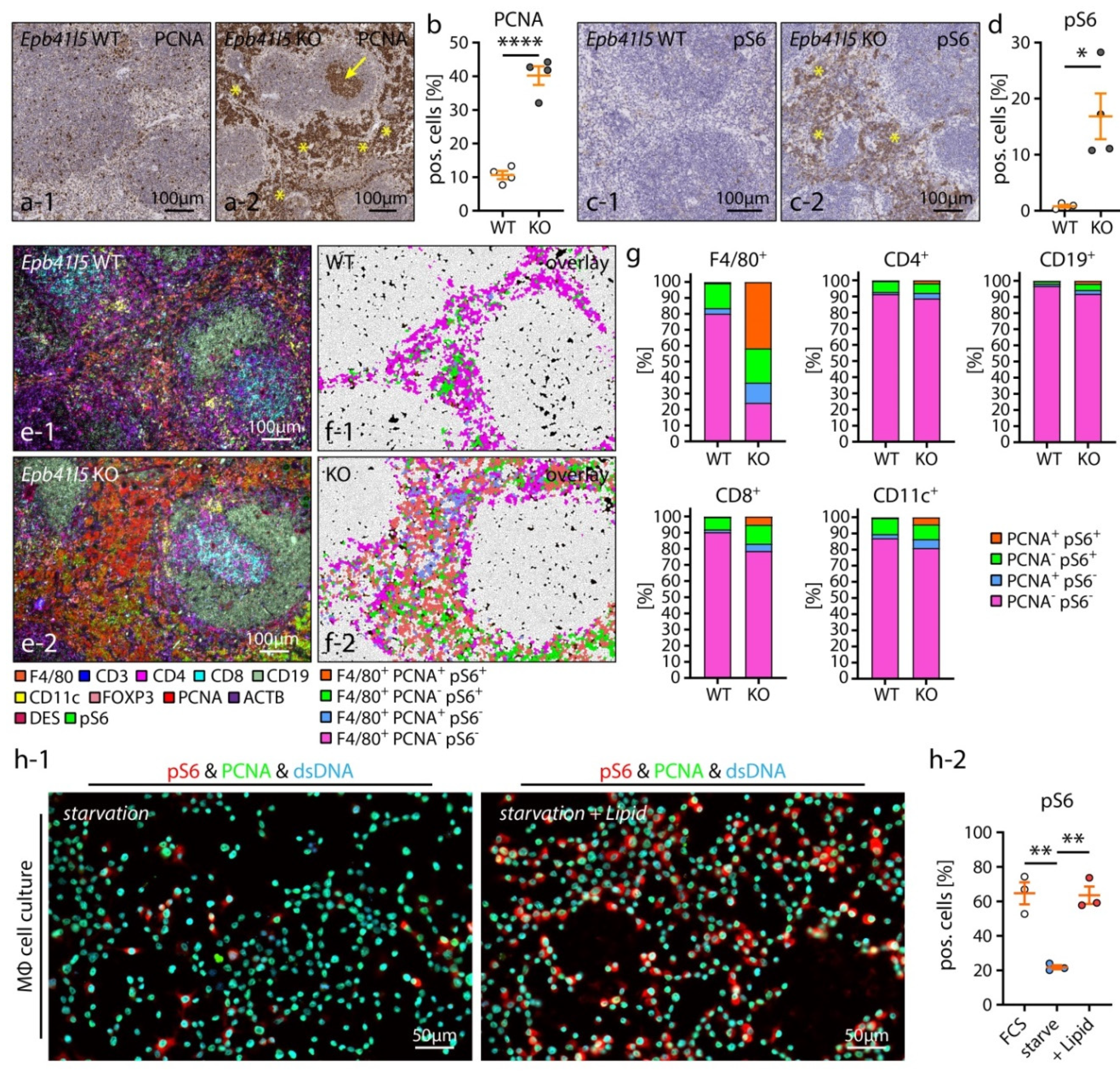
3.5. Hematopoietic Niche Expansion Correlates to RpMΦ Proliferation and mTOR Activation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fissell, W.H.; Miner, J.H. What is the glomerular ultrafiltration barrier? J. Am. Soc. Nephrol. JASN 2018, 29, 2262–2264. [Google Scholar] [CrossRef] [Green Version]
- Moeller, M.J.; Chia-Gil, A. A step forward in understanding glomerular filtration. Nat. Rev. Nephrol. 2020, 16, 431–432. [Google Scholar] [CrossRef] [PubMed]
- Greenbaum, L.A.; Benndorf, R.; Smoyer, W.E. Childhood nephrotic syndrome—Current and future therapies. Nat. Rev. Nephrol. 2012, 8, 445–458. [Google Scholar] [CrossRef] [PubMed]
- Brinkkoetter, P.T.; Ising, C.; Benzing, T. The role of the podocyte in albumin filtration. Nat. Rev. Nephrol. 2013, 9, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Grahammer, F.; Schell, C.; Huber, T.B. The podocyte slit diaphragm—From a thin grey line to a complex signalling hub. Nat. Rev. Nephrol. 2013, 9, 587–598. [Google Scholar] [CrossRef]
- Li, A.S.; Ingham, J.F.; Lennon, R. Genetic disorders of the glomerular filtration barrier. Clin. J. Am. Soc. Nephrol. CJASN 2020, 15, 1818–1828. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, S.; Zaritsky, J.J.; Fornoni, A.; Smoyer, W.E. Dyslipidaemia in nephrotic syndrome: Mechanisms and treatment. Nat. Rev. Nephrol. 2018, 14, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Mario, F.D.; Pofi, R.; Gigante, A.; Rivoli, L.; Rosato, E.; Isidori, A.M.; Cianci, R.; Barbano, B. Hypothyroidism and nephrotic syndrome: Why, when and how to treat. Curr. Vasc. Pharmacol. 2017, 15, 398–403. [Google Scholar] [CrossRef] [Green Version]
- Loscalzo, J. Venous thrombosis in the nephrotic syndrome. N. Engl. J. Med. 2013, 368, 956–958. [Google Scholar] [CrossRef] [PubMed]
- Pippin, J.W.; Brinkkoetter, P.T.; Cormack-Aboud, F.C.; Durvasula, R.V.; Hauser, P.V.; Kowalewska, J.; Krofft, R.D.; Logar, C.M.; Marshall, C.B.; Ohse, T.; et al. Inducible rodent models of acquired podocyte diseases. Am. J. Physiol. Ren. Physiol. 2009, 296, F213–F229. [Google Scholar] [CrossRef]
- Simic, I.; Tabatabaeifar, M.; Schaefer, F. Animal models of nephrotic syndrome. Pediatr. Nephrol. 2013, 28, 2079–2088. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.W.; Dettmar, A.K.; Kronbichler, A.; Gee, H.Y.; Saleem, M.; Kim, S.H.; Shin, J.I. Recent advances of animal model of focal segmental glomerulosclerosis. Clin. Exp. Nephrol. 2018, 22, 752–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moeller, M.J.; Sanden, S.K.; Soofi, A.; Wiggins, R.C.; Holzman, L.B. Podocyte-specific expression of cre recombinase in transgenic mice. Genesis 2003, 35, 39–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Widmeier, E.; Yu, S.; Nag, A.; Chung, Y.W.; Nakayama, M.; Fernandez-Del-Rio, L.; Hugo, H.; Schapiro, D.; Buerger, F.; Choi, W.I.; et al. Adck4 deficiency destabilizes the coenzyme q complex, which is rescued by 2,4-dihydroxybenzoic acid treatment. J. Am. Soc. Nephrol. JASN 2020, 31, 1191–1211. [Google Scholar] [CrossRef] [PubMed]
- Schell, C.; Sabass, B.; Helmstaedter, M.; Geist, F.; Abed, A.; Yasuda-Yamahara, M.; Sigle, A.; Maier, J.I.; Grahammer, F.; Siegerist, F.; et al. Arp3 controls the podocyte architecture at the kidney filtration barrier. Dev. Cell 2018, 47, 741–757.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuda, J.; Maier, M.; Aoudjit, L.; Baldwin, C.; Takano, T. Arhgef7 (beta-pix) is required for the maintenance of podocyte architecture and glomerular function. J. Am. Soc. Nephrol. JASN 2020, 31, 996–1008. [Google Scholar] [CrossRef]
- Wang, Y.; Pedigo, C.E.; Inoue, K.; Tian, X.; Cross, E.; Ebenezer, K.; Li, W.; Wang, Z.; Shin, J.W.; Schwartze, E.; et al. Murine epsins play an integral role in podocyte function. J. Am. Soc. Nephrol. JASN 2020, 31, 2870–2886. [Google Scholar] [CrossRef]
- Artelt, N.; Ludwig, T.A.; Rogge, H.; Kavvadas, P.; Siegerist, F.; Blumenthal, A.; van den Brandt, J.; Otey, C.A.; Bang, M.L.; Amann, K.; et al. The role of palladin in podocytes. J. Am. Soc. Nephrol. JASN 2018, 29, 1662–1678. [Google Scholar] [CrossRef] [Green Version]
- Koehler, S.; Brahler, S.; Braun, F.; Hagmann, H.; Rinschen, M.M.; Spath, M.R.; Hohne, M.; Wunderlich, F.T.; Schermer, B.; Benzing, T.; et al. Construction of a viral t2a-peptide based knock-in mouse model for enhanced cre recombinase activity and fluorescent labeling of podocytes. Kidney Int. 2017, 91, 1510–1517. [Google Scholar] [CrossRef]
- Lausecker, F.; Tian, X.; Inoue, K.; Wang, Z.; Pedigo, C.E.; Hassan, H.; Liu, C.; Zimmer, M.; Jinno, S.; Huckle, A.L.; et al. Vinculin is required to maintain glomerular barrier integrity. Kidney Int. 2018, 93, 643–655. [Google Scholar] [CrossRef] [Green Version]
- Schell, C.; Rogg, M.; Suhm, M.; Helmstadter, M.; Sellung, D.; Yasuda-Yamahara, M.; Kretz, O.; Kuttner, V.; Suleiman, H.; Kollipara, L.; et al. The ferm protein epb41l5 regulates actomyosin contractility and focal adhesion formation to maintain the kidney filtration barrier. Proc. Natl. Acad. Sci. USA 2017, 114, E4621–E4630. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Suh, J.H.; Go, G.; Miner, J.H. Feasibility of repairing glomerular basement membrane defects in alport syndrome. J. Am. Soc. Nephrol. JASN 2014, 25, 687–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.A.; Latz, E.; Moore, K.J.; Golenbock, D.T. The nalp3 inflammasome is involved in the innate immune response to amyloid-beta. Nat. Immunol. 2008, 9, 857–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogg, M.; Maier, J.I.; Dotzauer, R.; Artelt, N.; Kretz, O.; Helmstadter, M.; Abed, A.; Sammarco, A.; Sigle, A.; Sellung, D.; et al. Srgap1 controls small rho gtpases to regulate podocyte foot process maintenance. J. Am. Soc. Nephrol. JASN 2021, 32, 563–579. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.I.; Rogg, M.; Helmstadter, M.; Sammarco, A.; Schilling, O.; Sabass, B.; Miner, J.H.; Dengjel, J.; Walz, G.; Werner, M.; et al. Epb41l5 controls podocyte extracellular matrix assembly by adhesome-dependent force transmission. Cell Rep. 2021, 34, 108883. [Google Scholar] [CrossRef] [PubMed]
- Mehlem, A.; Hagberg, C.E.; Muhl, L.; Eriksson, U.; Falkevall, A. Imaging of neutral lipids by oil red o for analyzing the metabolic status in health and disease. Nat. Protoc. 2013, 8, 1149–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gut, G.; Herrmann, M.D.; Pelkmans, L. Multiplexed protein maps link subcellular organization to cellular states. Science 2018, 361, eaar7042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bankhead, P.; Loughrey, M.B.; Fernandez, J.A.; Dombrowski, Y.; McArt, D.G.; Dunne, P.D.; McQuaid, S.; Gray, R.T.; Murray, L.J.; Coleman, H.G.; et al. Qupath: Open source software for digital pathology image analysis. Sci. Rep. 2017, 7, 16878. [Google Scholar] [CrossRef] [Green Version]
- El Nahas, A.M.; Bassett, A.H.; Cope, G.H.; Le Carpentier, J.E. Role of growth hormone in the development of experimental renal scarring. Kidney Int. 1991, 40, 29–34. [Google Scholar] [CrossRef] [Green Version]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahe, F. Vsearch: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Eren, A.M.; Morrison, H.G.; Lescault, P.J.; Reveillaud, J.; Vineis, J.H.; Sogin, M.L. Minimum entropy decomposition: Unsupervised oligotyping for sensitive partitioning of high-throughput marker gene sequences. ISME J. 2015, 9, 968–979. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. Qiime allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Angly, F.E.; Dennis, P.G.; Skarshewski, A.; Vanwonterghem, I.; Hugenholtz, P.; Tyson, G.W. Copyrighter: A rapid tool for improving the accuracy of microbial community profiles through lineage-specific gene copy number correction. Microbiome 2014, 2, 11. [Google Scholar] [CrossRef] [PubMed]
- Metsalu, T.; Vilo, J. Clustvis: A web tool for visualizing clustering of multivariate data using principal component analysis and heatmap. Nucleic Acids Res. 2015, 43, W566–W570. [Google Scholar] [CrossRef]
- Zakrzewski, M.; Proietti, C.; Ellis, J.J.; Hasan, S.; Brion, M.J.; Berger, B.; Krause, L. Calypso: A user-friendly web-server for mining and visualizing microbiome-environment interactions. Bioinformatics 2017, 33, 782–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, Z.; Fan, Y.; Li, A.; Shen, Q.; Wu, J.; Ren, L.; Lu, H.; Ding, S.; Ren, H.; Liu, C.; et al. Alterations of the human gut microbiome in chronic kidney disease. Adv. Sci. 2020, 7, 2001936. [Google Scholar] [CrossRef]
- Ramezani, A.; Raj, D.S. The gut microbiome, kidney disease, and targeted interventions. J. Am. Soc. Nephrol. JASN 2014, 25, 657–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lobel, L.; Cao, Y.G.; Fenn, K.; Glickman, J.N.; Garrett, W.S. Diet posttranslationally modifies the mouse gut microbial proteome to modulate renal function. Science 2020, 369, 1518–1524. [Google Scholar] [CrossRef]
- Meijers, B.; Evenepoel, P.; Anders, H.J. Intestinal microbiome and fitness in kidney disease. Nat. Rev. Nephrol. 2019, 15, 531–545. [Google Scholar] [CrossRef] [PubMed]
- Andersen, K.; Kesper, M.S.; Marschner, J.A.; Konrad, L.; Ryu, M.; Kumar Vr, S.; Kulkarni, O.P.; Mulay, S.R.; Romoli, S.; Demleitner, J.; et al. Intestinal dysbiosis, barrier dysfunction, and bacterial translocation account for ckd-related systemic inflammation. J. Am. Soc. Nephrol. JASN 2017, 28, 76–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emal, D.; Rampanelli, E.; Stroo, I.; Butter, L.M.; Teske, G.J.; Claessen, N.; Stokman, G.; Florquin, S.; Leemans, J.C.; Dessing, M.C. Depletion of gut microbiota protects against renal ischemia-reperfusion injury. J. Am. Soc. Nephrol. JASN 2017, 28, 1450–1461. [Google Scholar] [CrossRef] [Green Version]
- Mishima, E.; Fukuda, S.; Shima, H.; Hirayama, A.; Akiyama, Y.; Takeuchi, Y.; Fukuda, N.N.; Suzuki, T.; Suzuki, C.; Yuri, A.; et al. Alteration of the intestinal environment by lubiprostone is associated with amelioration of adenine-induced ckd. J. Am. Soc. Nephrol. JASN 2015, 26, 1787–1794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, Y.; Feng, D.; Law, H.K.; Qu, W.; Wu, Y.; Zhu, G.H.; Huang, W.Y. Compositional alterations of gut microbiota in children with primary nephrotic syndrome after initial therapy. BMC Nephrol. 2019, 20, 434. [Google Scholar] [CrossRef]
- Tsuji, S.; Akagawa, S.; Akagawa, Y.; Yamaguchi, T.; Kino, J.; Yamanouchi, S.; Kimata, T.; Hashiyada, M.; Akane, A.; Kaneko, K. Idiopathic nephrotic syndrome in children: Role of regulatory t cells and gut microbiota. Pediatr. Res. 2020, 89, 1185–1191. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.H.; Mitch, W.E. Mechanisms of muscle wasting in chronic kidney disease. Nat. Rev. Nephrol. 2014, 10, 504–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matyjek, A.; Literacki, S.; Niemczyk, S.; Rymarz, A. Protein energy-wasting associated with nephrotic syndrome—The comparison of metabolic pattern in severe nephrosis to different stages of chronic kidney disease. BMC Nephrol. 2020, 21, 346. [Google Scholar] [CrossRef]
- Elias, A.N.; Carreon, G.; Vaziri, N.D.; Pandian, M.R.; Oveisi, F. The pituitary-gonadal axis in experimental nephrotic syndrome in male rats. J. Lab. Clin. Med. 1992, 120, 949–954. [Google Scholar]
- Glass, A.R.; Beach, J.; Vigersky, R.A. Hypogonadotropic hypogonadism in nephrotic rats: Increased sensitivity to negative feedback effects of testosterone. Metab. Clin. Exp. 1985, 34, 574–579. [Google Scholar] [CrossRef]
- Suttie, A.W. Histopathology of the spleen. Toxicol. Pathol. 2006, 34, 466–503. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.; Prabhu, K.S.; Paulson, R.F. Monocyte-derived macrophages expand the murine stress erythropoietic niche during the recovery from anemia. Blood 2018, 132, 2580–2593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobsen, R.N.; Perkins, A.C.; Levesque, J.P. Macrophages and regulation of erythropoiesis. Curr. Opin. Hematol. 2015, 22, 212–219. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, M.M.; Kopsombut, P.; Bondurant, M.C.; Price, J.O.; Koury, M.J. Adherence to macrophages in erythroblastic islands enhances erythroblast proliferation and increases erythrocyte production by a different mechanism than erythropoietin. Blood 2008, 111, 1700–1708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasan, S.; Johnson, M.C.; Kini, A.R.; Baldea, A.J.; Muthumalaiappan, K. A shift in myeloid cell phenotype via down regulation of siglec-1 in island macrophages of bone marrow is associated with decreased late erythroblasts seen in anemia of critical illness. Front. Med. 2019, 6, 260. [Google Scholar] [CrossRef]
- Ohyagi, H.; Onai, N.; Sato, T.; Yotsumoto, S.; Liu, J.; Akiba, H.; Yagita, H.; Atarashi, K.; Honda, K.; Roers, A.; et al. Monocyte-derived dendritic cells perform hemophagocytosis to fine-tune excessive immune responses. Immunity 2013, 39, 584–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxton, R.A.; Sabatini, D.M. Mtor signaling in growth, metabolism, and disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knight, Z.A.; Schmidt, S.F.; Birsoy, K.; Tan, K.; Friedman, J.M. A critical role for mtorc1 in erythropoiesis and anemia. eLife 2014, 3, e01913. [Google Scholar] [CrossRef] [PubMed]
- Weichhart, T.; Hengstschlager, M.; Linke, M. Regulation of innate immune cell function by mtor. Nat. Rev. Immunol. 2015, 15, 599–614. [Google Scholar] [CrossRef]
- Castellano, B.M.; Thelen, A.M.; Moldavski, O.; Feltes, M.; van der Welle, R.E.; Mydock-McGrane, L.; Jiang, X.; van Eijkeren, R.J.; Davis, O.B.; Louie, S.M.; et al. Lysosomal cholesterol activates mtorc1 via an slc38a9-niemann-pick c1 signaling complex. Science 2017, 355, 1306–1311. [Google Scholar] [CrossRef] [Green Version]
- Menon, D.; Salloum, D.; Bernfeld, E.; Gorodetsky, E.; Akselrod, A.; Frias, M.A.; Sudderth, J.; Chen, P.H.; DeBerardinis, R.; Foster, D.A. Lipid sensing by mtor complexes via de novo synthesis of phosphatidic acid. J. Biol. Chem. 2017, 292, 6303–6311. [Google Scholar] [CrossRef] [Green Version]
- Um, S.H.; Frigerio, F.; Watanabe, M.; Picard, F.; Joaquin, M.; Sticker, M.; Fumagalli, S.; Allegrini, P.R.; Kozma, S.C.; Auwerx, J.; et al. Absence of s6k1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature 2004, 431, 200–205. [Google Scholar] [CrossRef]
- Yan, J.; Horng, T. Lipid metabolism in regulation of macrophage functions. Trends Cell Biol. 2020, 30, 979–989. [Google Scholar] [CrossRef]
- Jiang, Q.; He, X.; Zou, Y.; Ding, Y.; Li, H.; Chen, H. Altered gut microbiome promotes proteinuria in mice induced by adriamycin. AMB Express 2018, 8, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, B.J.; Wearsch, P.A.; Veloo, A.C.M.; Rodriguez-Palacios, A. The genus alistipes: Gut bacteria with emerging implications to inflammation, cancer, and mental health. Front. Immunol. 2020, 11, 906. [Google Scholar] [CrossRef] [PubMed]
- Kaur, H.; Das, C.; Mande, S.S. In silico analysis of putrefaction pathways in bacteria and its implication in colorectal cancer. Front. Microbiol. 2017, 8, 2166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leong, S.C.; Sirich, T.L. Indoxyl sulfate-review of toxicity and therapeutic strategies. Toxins 2016, 8, 358. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, T.; Ise, M.; Seo, H.; Niwa, T. Indoxyl sulfate increases the gene expressions of tgf-beta 1, timp-1 and pro-alpha 1(i) collagen in uremic rat kidneys. Kidney Int. Suppl. 1997, 62, S15–S22. [Google Scholar] [PubMed]
- Park, S.J.; Shin, J.I. Complications of nephrotic syndrome. Korean J. Pediatr. 2011, 54, 322–328. [Google Scholar] [CrossRef]
- Wong, E.K.; Brady, M.; Sheerin, N.S. Severe intestinal oedema due to nephrotic syndrome. QJM Mon. J. Assoc. Physicians 2013, 106, 191–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, W.H.W.; Li, D.Y.; Hazen, S.L. Dietary metabolism, the gut microbiome, and heart failure. Nat. Rev. Cardiol. 2019, 16, 137–154. [Google Scholar] [CrossRef] [PubMed]
- Chiu, S.C.; Liu, H.H.; Chen, C.L.; Chen, P.R.; Liu, M.C.; Lin, S.Z.; Chang, K.T. Extramedullary hematopoiesis (emh) in laboratory animals: Offering an insight into stem cell research. Cell Transpl. 2015, 24, 349–366. [Google Scholar] [CrossRef]
- Iorember, F.; Aviles, D. Anemia in nephrotic syndrome: Approach to evaluation and treatment. Pediatr. Nephrol. 2017, 32, 1323–1330. [Google Scholar] [CrossRef]
- Prinsen, B.H.; de Sain-van der Velden, M.G.; Kaysen, G.A.; Straver, H.W.; van Rijn, H.J.; Stellaard, F.; Berger, R.; Rabelink, T.J. Transferrin synthesis is increased in nephrotic patients insufficiently to replace urinary losses. J. Am. Soc. Nephrol. JASN 2001, 12, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Feinstein, S.; Becker-Cohen, R.; Algur, N.; Raveh, D.; Shalev, H.; Shvil, Y.; Frishberg, Y. Erythropoietin deficiency causes anemia in nephrotic children with normal kidney function. Am. J. Kidney Dis. 2001, 37, 736–742. [Google Scholar] [CrossRef]
- Artinger, K.; Kirsch, A.H.; Aringer, I.; Schabhuttl, C.; Rosenkranz, A.R.; Eller, P.; Rho, E.; Eller, K. The spleen plays no role in nephrotoxic serum nephritis, but constitutes a place of compensatory haematopoiesis. PLoS ONE 2015, 10, e0135087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, H.; Han, S.; Xu, Y.; Li, Y.; Wang, H.; Yang, L.J.; Reeves, W.H. Toll-like receptor 7-stimulated tumor necrosis factor alpha causes bone marrow damage in systemic lupus erythematosus. Arthritis Rheumatol. 2014, 66, 140–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chasis, J.A.; Mohandas, N. Erythroblastic islands: Niches for erythropoiesis. Blood 2008, 112, 470–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neuhaus, T.J.; Wadhwa, M.; Callard, R.; Barratt, T.M. Increased il-2, il-4 and interferon-gamma (ifn-gamma) in steroid-sensitive nephrotic syndrome. Clin. Exp. Immunol. 1995, 100, 475–479. [Google Scholar] [CrossRef] [PubMed]
- Schachter, A.D. The pediatric nephrotic syndrome spectrum: Clinical homogeneity and molecular heterogeneity. Pediatr. Transpl. 2004, 8, 344–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ai, X.M.; Ho, L.C.; Han, L.L.; Lu, J.J.; Yue, X.; Yang, N.Y. The role of splenectomy in lipid metabolism and atherosclerosis (as). Lipids Health Dis. 2018, 17, 186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westerterp, M.; Gourion-Arsiquaud, S.; Murphy, A.J.; Shih, A.; Cremers, S.; Levine, R.L.; Tall, A.R.; Yvan-Charvet, L. Regulation of hematopoietic stem and progenitor cell mobilization by cholesterol efflux pathways. Cell Stem Cell 2012, 11, 195–206. [Google Scholar] [CrossRef] [Green Version]
- Landau, D.; Gurevich, E.; Kapelushnik, J.; Tamary, H.; Shelef, I.; Lazar, I. Association between childhood nephrotic syndrome and hemophagocytic lymphohistiocytosis. Pediatr. Nephrol. 2013, 28, 2389–2392. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Garcia, V.; Gonzalez-Ramos, S.; Martin-Sanz, P.; Castrillo, A.; Bosca, L. Contribution of extramedullary hematopoiesis to atherosclerosis. The spleen as a neglected hub of inflammatory cells. Front. Immunol. 2020, 11, 586527. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maier, J.I.; Rogg, M.; Helmstädter, M.; Sammarco, A.; Walz, G.; Werner, M.; Schell, C. A Novel Model for Nephrotic Syndrome Reveals Associated Dysbiosis of the Gut Microbiome and Extramedullary Hematopoiesis. Cells 2021, 10, 1509. https://doi.org/10.3390/cells10061509
Maier JI, Rogg M, Helmstädter M, Sammarco A, Walz G, Werner M, Schell C. A Novel Model for Nephrotic Syndrome Reveals Associated Dysbiosis of the Gut Microbiome and Extramedullary Hematopoiesis. Cells. 2021; 10(6):1509. https://doi.org/10.3390/cells10061509
Chicago/Turabian StyleMaier, Jasmin I., Manuel Rogg, Martin Helmstädter, Alena Sammarco, Gerd Walz, Martin Werner, and Christoph Schell. 2021. "A Novel Model for Nephrotic Syndrome Reveals Associated Dysbiosis of the Gut Microbiome and Extramedullary Hematopoiesis" Cells 10, no. 6: 1509. https://doi.org/10.3390/cells10061509