
Live Fluorescence Imaging of F-Actin Organization in Chick Whole Embryo Cultures Using SiR-Actin


Abstract
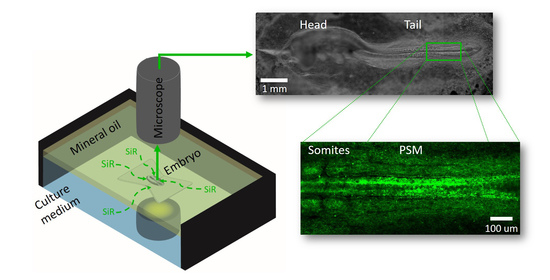
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Culture Medium
2.2. Submerged Filter Paper Sandwich (Figure 1)
2.3. SiR-Actin Live Fluorescence Microscopy (Figure 2)
2.4. SiR-Actin and Phalloidin Co-Staining in Fixated Samples (Figure 3A)
3. Results
3.1. Proof of Principle—Increasing Fluorescence Signal While Development Progresses
3.2. Identification of a Safe and Effective Sir-Actin Concentration
3.3. Confocal Imaging of Whole Embryos in Fixated Samples and In Vivo
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Heisenberg, C.-P.; Bellaïche, Y. Forces in Tissue Morphogenesis and Patterning. Cell 2013, 153, 948–962. [Google Scholar] [CrossRef] [Green Version]
- Paluch, E.K.; Nelson, C.M.; Biais, N.; Fabry, B.; Moeller, J.; Pruitt, B.L.; Wollnik, C.; Kudryasheva, G.; Rehfeldt, F.; Federle, W. Mechanotransduction: Use the force(s). BMC Biol. 2015, 13, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Chugh, P.; Clark, A.G.; Smith, M.B.; Cassani, D.A.D.; Dierkes, K.; Ragab, A.; Roux, P.P.; Charras, G.; Salbreux, G.; Paluch, E. Actin cortex architecture regulates cell surface tension. Nat. Cell Biol. 2017, 19, 689–697. [Google Scholar] [CrossRef]
- Rozbicki, E.; Chuai, M.; Karjalainen, A.I.; Song, F.; Sang, H.M.; Martin, R.; MacDonald, M.P.; Weijer, C.J. Myosin II-mediated cell shape changes and cell intercalation contribute to primitive streak formation. Nat. Cell Biol. 2015, 17, 397–408. [Google Scholar] [CrossRef] [Green Version]
- Wulf, E.; Deboben, A.; Bautz, F.A.; Faulstich, H.; Wieland, T. Fluorescent phallotoxin, a tool for the visualization of cellular actin. Proc. Natl. Acad. Sci. USA 1979, 76, 4498–4502. [Google Scholar] [CrossRef] [Green Version]
- Vandekerckhove, J.; Deboben, A.; Nassal, M.; Wieland, T. The phalloidin binding site of F-actin. EMBO J. 1985, 4, 2815–2818. [Google Scholar] [CrossRef]
- Belin, B.J.; Goins, L.M.; Mullins, R.D. Comparative analysis of tools for live cell imaging of actin network architecture. BioArchitecture 2014, 4, 189–202. [Google Scholar] [CrossRef]
- Lazarides, E.; Weber, K. Actin Antibody: The Specific Visualization of Actin Filaments in Non-Muscle Cells. Proc. Natl. Acad. Sci. USA 1974, 71, 2268–2272. [Google Scholar] [CrossRef] [Green Version]
- Melak, M.; Plessner, M.; Grosse, R. Actin visualization at a glance. J. Cell Sci. 2017, 130, 525–530. [Google Scholar] [CrossRef] [Green Version]
- Waterman-Storer, C.M.; Desai, A.; Bulinski, J.C.; Salmon, E. Fluorescent speckle microscopy, a method to visualize the dynamics of protein assemblies in living cells. Curr. Biol. 1998, 8, 1227–1230. [Google Scholar] [CrossRef] [Green Version]
- Schmit, A.C.; Lambert, A.M. Microinjected fluorescent phalloidin in vivo reveals the F-actin dynamics and assembly in higher plant mitotic cells. Plant Cell 1990, 2, 129–138. [Google Scholar]
- McNeil, P.L.; Warder, E. Glass beads load macromolecules into living cells. J. Cell Sci. 1987, 88, 669–678. [Google Scholar] [CrossRef] [PubMed]
- Sliogeryte, K.; Thorpe, S.D.; Wang, Z.; Thompson, C.L.; Gavara, N.; Knight, M.M. Differential effects of LifeAct-GFP and actin-GFP on cell mechanics assessed using micropipette aspiration. J. Biomech. 2016, 49, 310–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukinavicius, G.; Reymond, L.; D’Este, E.; Masharina, A.; Göttfert, F.; Ta, H.; Güther, A.; Fournier, M.; Rizzo, S.; Waldmann, H.; et al. Fluorogenic probes for live-cell imaging of the cytoskeleton. Nat. Methods 2014, 11, 731–733. [Google Scholar] [CrossRef]
- Lukinavičius, G.; Umezawa, K.; Olivier, N.; Honigmann, A.; Yang, G.; Plass, T.; Mueller, V.; Reymond, L.; Corrêa, I.R., Jr.; Luo, Z.-G.; et al. A near-infrared fluorophore for live-cell super-resolution microscopy of cellular proteins. Nat. Chem. 2013, 5, 132–139. [Google Scholar] [CrossRef]
- Milroy, L.-G.; Rizzo, S.; Calderon, A.; Ellinger, B.; Erdmann, S.; Mondry, J.; Verveer, P.; Bastiaens, P.; Waldmann, H.; Dehmelt, L.; et al. Selective Chemical Imaging of Static Actin in Live Cells. J. Am. Chem. Soc. 2012, 134, 8480–8486. [Google Scholar] [CrossRef]
- Bubb, M.R.; Senderowicz, A.M.; Sausville, E.A.; Duncan, K.L.; Korn, E.D. Jasplakinolide, a cytotoxic natural product, induces actin polymerization and competitively inhibits the binding of phalloidin to F-actin. J. Biol. Chem. 1994, 269, 14869–14871. [Google Scholar] [CrossRef]
- D’Este, E.; Kamin, D.; Göttfert, F.; El-Hady, A.; Hell, S.W. STED Nanoscopy Reveals the Ubiquity of Subcortical Cytoskeleton Periodicity in Living Neurons. Cell Rep. 2015, 10, 1246–1251. [Google Scholar] [CrossRef] [Green Version]
- D’Este, E.; Kamin, D.; Velte, C.; Göttfert, F.; Simons, M.; Hell, S.W. Subcortical cytoskeleton periodicity throughout the nervous system. Sci. Rep. 2016, 6, 22741. [Google Scholar] [CrossRef] [Green Version]
- Gokhin, D.S.; Nowak, R.B.; Khoory, J.A.; De La Piedra, A.; Ghiran, I.C.; Fowler, V.M. Dynamic actin filaments control the mechanical behavior of the human red blood cell membrane. Mol. Biol. Cell 2015, 26, 1699–1710. [Google Scholar] [CrossRef]
- Tyszka, J.; Bickmeyer, U.; Raitzsch, M.; Bijma, J.; Kaczmarek, K.; Mewes, A.; Topa, P.; Janse, M. Form and function of F-actin during biomineralization revealed from live experiments on foraminifera. Proc. Natl. Acad. Sci. USA 2019, 116, 4111–4116. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, M.; Nelemans, B.K.A.; Smit, T.H. A Submerged Filter Paper Sandwich for Long-term Ex Ovo Time-lapse Imaging of Early Chick Embryos. J. Vis. Exp. 2016, 28, e54636. [Google Scholar] [CrossRef] [Green Version]
- Chapman, S.C.; Collignon, J.; Schoenwolf, G.C.; Lumsden, A. Improved method for chick whole-embryo culture using a filter paper carrier. Dev. Dyn. 2001, 220, 284–289. [Google Scholar] [CrossRef]
- Tam, P.P. The control of somitogenesis in mouse embryos. J. Embryol. Exp. Morphol. 1981, 65, 103–128. [Google Scholar]
- Rowton, M.; Ramos, P.; Anderson, D.M.; Rhee, J.M.; Cunliffe, H.E.; Rawls, A. Regulation of mesenchymal-to-epithelial transition by PARAXIS during somitogenesis. Dev. Dyn. 2013, 242, 1332–1344. [Google Scholar] [CrossRef] [PubMed]
- Martins, G.G.; Rifes, P.; Amândio, R.; Rodrigues, G.; Palmeirim, I.; Thorsteinsdóttir, S. Dynamic 3D Cell Rearrangements Guided by a Fibronectin Matrix Underlie Somitogenesis. PLoS ONE 2009, 4, e7429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, R.; Fujii, K.; Suzuki, M.; Yamaoka, I. Organization of Actin Filaments and Zonula Adherens during Somitogenesis in the Chick Embryo. (somitogenesis/actin/zonula adherens/chick embryo). Dev. Growth Differ. 1989, 31, 123–130. [Google Scholar] [CrossRef]
- Pannett, C.; Compton, A. The cultivation of tissues in saline embryonic juice. Lancet 1924, 203, 381–384. [Google Scholar] [CrossRef]
- Voiculescu, O.; Papanayotou, C.; Stern, C.D. Spatially and temporally controlled electroporation of early chick embryos. Nat. Protoc. 2008, 3, 419–426. [Google Scholar] [CrossRef]
- Burley, R.W.; Vadehra, D.V. The Avian Egg: Chemistry and Biology; Wiley: New York, NY, USA, 1989; p. 472. [Google Scholar]
- Hamburger, V.; Hamilton, H.L. A series of normal stages in the development of the chick embryo. J. Morphol. 1951, 88, 49–92. [Google Scholar] [CrossRef]
- Lee, H.; Nagele, R.G. Toxic and teratologic effects of verapamil on early chick embryos: Evidence for the involvement of calcium in neural tube closure. Teratology 1986, 33, 203–211. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schmitz-Elbers, M.; Lukinavičius, G.; Smit, T.H. Live Fluorescence Imaging of F-Actin Organization in Chick Whole Embryo Cultures Using SiR-Actin. Cells 2021, 10, 1578. https://doi.org/10.3390/cells10071578
Schmitz-Elbers M, Lukinavičius G, Smit TH. Live Fluorescence Imaging of F-Actin Organization in Chick Whole Embryo Cultures Using SiR-Actin. Cells. 2021; 10(7):1578. https://doi.org/10.3390/cells10071578
Chicago/Turabian StyleSchmitz-Elbers, Manuel, Gražvydas Lukinavičius, and Theodoor H. Smit. 2021. "Live Fluorescence Imaging of F-Actin Organization in Chick Whole Embryo Cultures Using SiR-Actin" Cells 10, no. 7: 1578. https://doi.org/10.3390/cells10071578