
Cell Death in Coronavirus Infections: Uncovering Its Role during COVID-19

 ,
,  , , and
, , and
Abstract
1. Introduction
2. SARS-CoV-2: Structural Characteristics and Diffusion
3. Activation of the Immune Response Driven by SARS-CoV-2
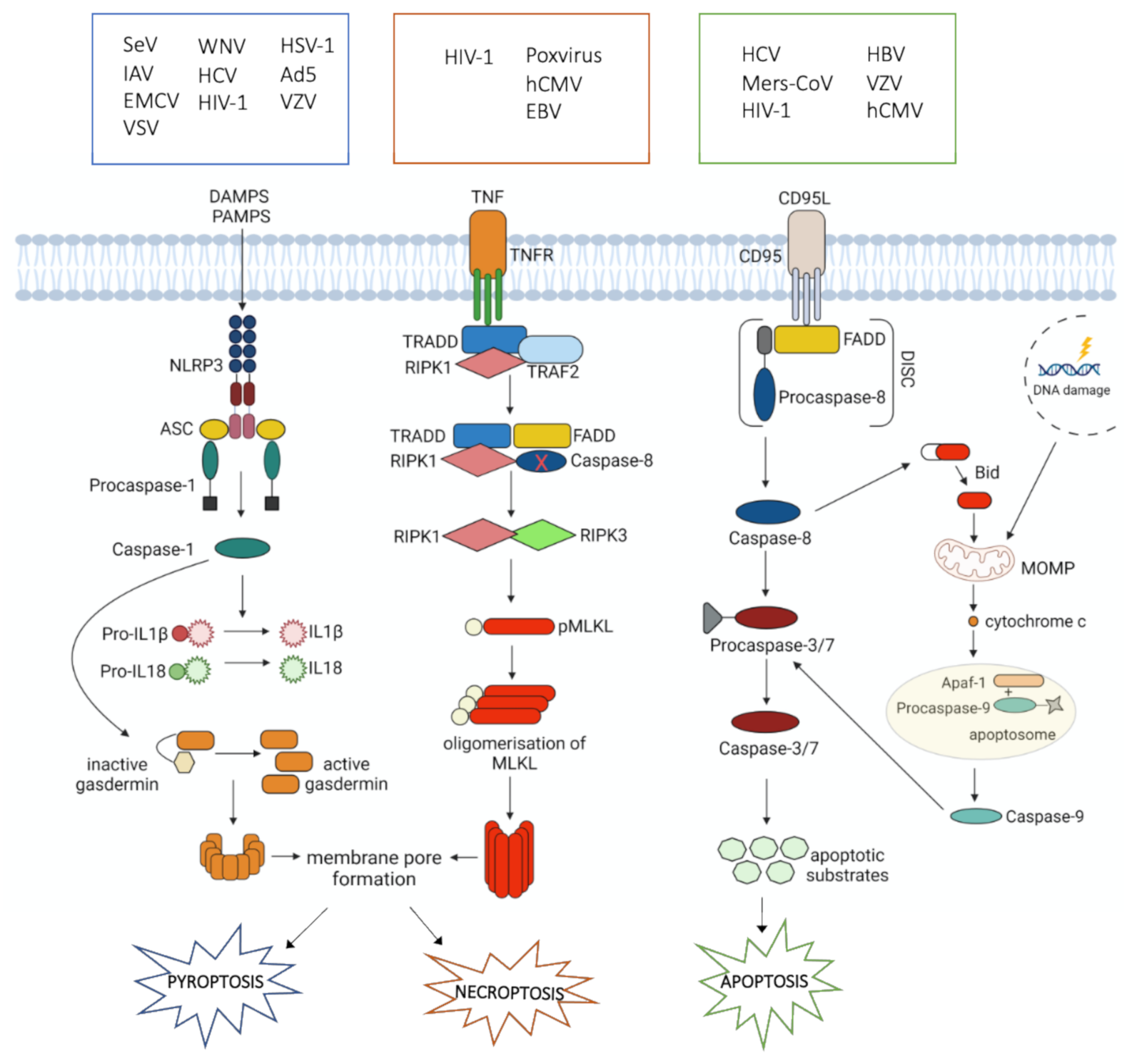
4. Cell Death during Viral Infections
4.1. Apoptosis
4.2. Necroptosis
4.3. Pyroptosis
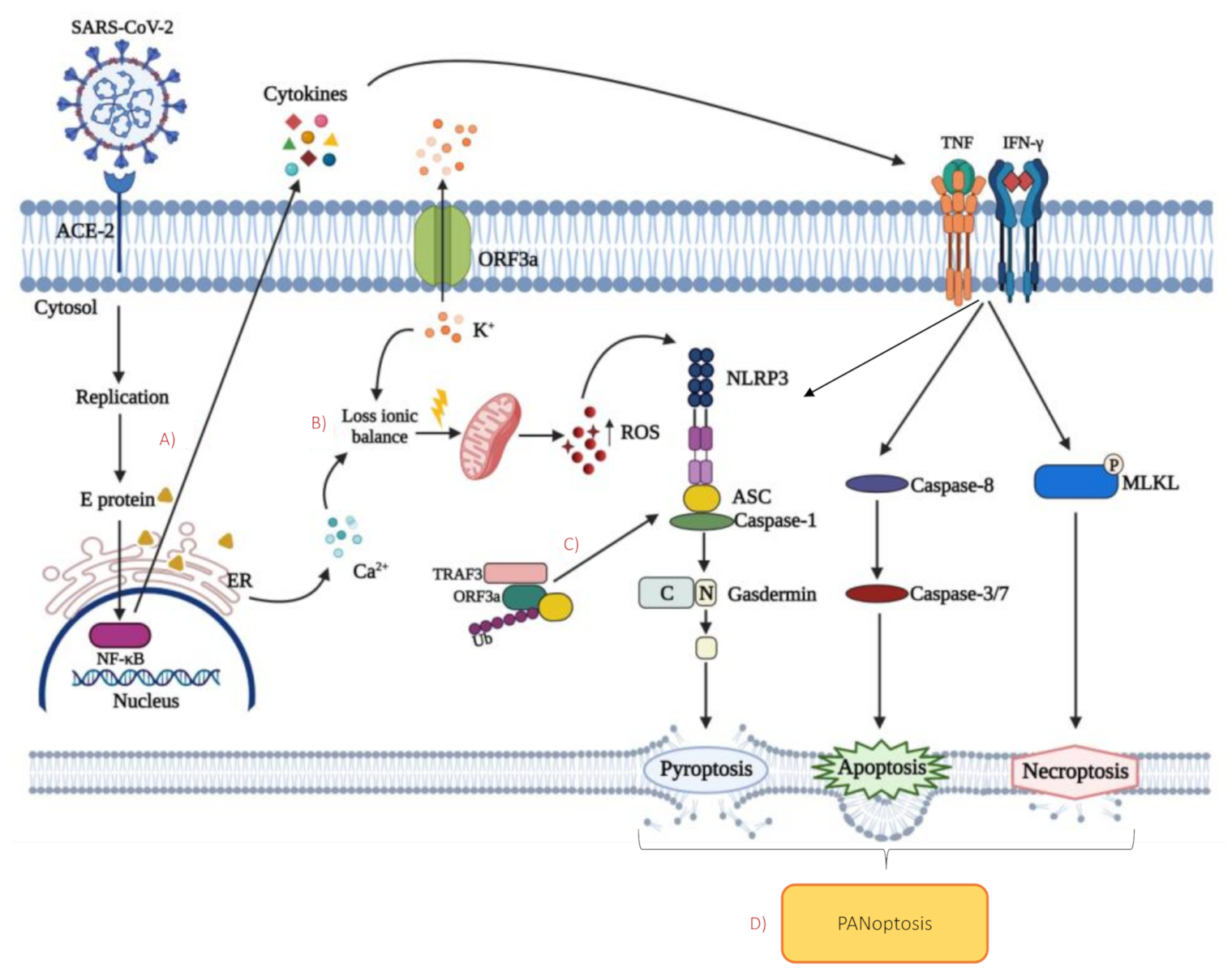
5. Cell Death during SARS-CoV-2 Infection
5.1. Apoptosis
5.2. Necroptosis
5.3. Pyroptosis
5.4. PANoptosis
6. Strategies for Apoptosis Modulation in SARS-CoV-2 Infection
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Liu, Y.C.; Kuo, R.L.; Shih, S.R. COVID-19: The first documented coronavirus pandemic in history. Biomed. J. 2020, 43, 328–333. [Google Scholar] [CrossRef]
- Coronaviridae Study Group of the International Committee on Taxonomy of V. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, W.G. Natural history of COVID-19 and current knowledge on treatment therapeutic options. Biomed. Pharmacother. 2020, 129, 110493. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.Y.; Zhao, R.; Gao, L.J.; Gao, X.F.; Wang, D.P.; Cao, J.M. SARS-CoV-2: Structure, Biology, and Structure-Based Therapeutics Development. Front. Cell Infect. Microbiol. 2020, 10, 587269. [Google Scholar] [CrossRef]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef]
- Naqvi, A.A.T.; Fatima, K.; Mohammad, T.; Fatima, U.; Singh, I.K.; Singh, A.; Atif, S.M.; Hariprasad, G.; Hasan, G.M.; Hassan, M.I. Insights into SARS-CoV-2 genome, structure, evolution, pathogenesis and therapies: Structural genomics approach. Biochim. Biophys. Acta. Mol. Basis Dis. 2020, 1866, 165878. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef] [PubMed]
- Letko, M.; Marzi, A.; Munster, V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat. Microbiol. 2020, 5, 562–569. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Salamanna, F.; Maglio, M.; Landini, M.P.; Fini, M. Body Localization of ACE-2: On the Trail of the Keyhole of SARS-CoV-2. Front. Med. 2020, 7, 594495. [Google Scholar] [CrossRef]
- Lauer, S.A.; Grantz, K.H.; Bi, Q.; Jones, F.K.; Zheng, Q.; Meredith, H.R.; Azman, A.S.; Reich, N.G.; Lessler, J. The Incubation Period of Coronavirus Disease 2019 (COVID-19) From Publicly Reported Confirmed Cases: Estimation and Application. Ann. Intern. Med. 2020, 172, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Paderno, A.; Schreiber, A.; Grammatica, A.; Raffetti, E.; Tomasoni, M.; Gualtieri, T.; Taboni, S.; Zorzi, S.; Lombardi, D.; Deganello, A.; et al. Smell and taste alterations in COVID-19: A cross-sectional analysis of different cohorts. Int. Forum Allergy Rhinol. 2020, 10, 955–962. [Google Scholar] [CrossRef] [PubMed]
- Sedaghat, A.R.; Gengler, I.; Speth, M.M. Olfactory Dysfunction: AHighly Prevalent Symptom of COVID-19 With Public Health Significance. Otolaryngol. Head Neck Surg. 2020, 163, 12–15. [Google Scholar] [CrossRef]
- Yan, C.H.; Faraji, F.; Prajapati, D.P.; Boone, C.E.; DeConde, A.S. Association of chemosensory dysfunction COVID-19 in patients presenting with influenza-like symptoms. Int. Forum Allergy Rhinol. 2020, 10, 806–813. [Google Scholar] [CrossRef] [PubMed]
- Merad, M.; Martin, J.C. Pathological inflammation in patients with COVID-19: A key role for monocytes and macrophages. Nat. Rev. Immunol. 2020, 20, 355–362. [Google Scholar] [CrossRef]
- Osuchowski, M.F.; Winkler, M.S.; Skirecki, T.; Cajander, S.; Shankar-Hari, M.; Lachmann, G.; Monneret, G.; Venet, F.; Bauer, M.; Brunkhorst, F.M.; et al. The COVID-19 puzzle: Deciphering pathophysiology and phenotypes of a new disease entity. Lancet Respir. Med. 2021, 9, 622–642. [Google Scholar] [CrossRef]
- Soy, M.; Keser, G.; Atagunduz, P.; Tabak, F.; Atagunduz, I.; Kayhan, S. Cytokine storm in COVID-19: Pathogenesis and overview of anti-inflammatory agents used in treatment. Clin. Rheumatol. 2020, 39, 2085–2094. [Google Scholar] [CrossRef]
- Gangaev, A.; Ketelaars, S.L.C.; Isaeva, O.I.; Patiwael, S.; Dopler, A.; Hoefakker, K.; De Biasi, S.; Gibellini, L.; Mussini, C.; Guaraldi, G.; et al. Identification and characterization of a SARS-CoV-2 specific CD8(+) T cell response with immunodominant features. Nat. Commun. 2021, 12, 2593. [Google Scholar] [CrossRef] [PubMed]
- Kuri-Cervantes, L.; Pampena, M.B.; Meng, W.; Rosenfeld, A.M.; Ittner, C.A.G.; Weisman, A.R.; Agyekum, R.S.; Mathew, D.; Baxter, A.E.; Vella, L.A.; et al. Comprehensive mapping of immune perturbations associated with severe COVID-19. Sci. Immunol. 2020. [Google Scholar] [CrossRef]
- Laing, A.G.; Lorenc, A.; Del Molino Del Barrio, I.; Das, A.; Fish, M.; Monin, L.; Munoz-Ruiz, M.; McKenzie, D.R.; Hayday, T.S.; Francos-Quijorna, I.; et al. A dynamic COVID-19 immune signature includes associations with poor prognosis. Nat. Med. 2020, 26, 1623–1635. [Google Scholar] [CrossRef] [PubMed]
- Wilk, A.J.; Rustagi, A.; Zhao, N.Q.; Roque, J.; Martinez-Colon, G.J.; McKechnie, J.L.; Ivison, G.T.; Ranganath, T.; Vergara, R.; Hollis, T.; et al. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat. Med. 2020, 26, 1070–1076. [Google Scholar] [CrossRef] [PubMed]
- Gibellini, L.; De Biasi, S.; Paolini, A.; Borella, R.; Boraldi, F.; Mattioli, M.; Lo Tartaro, D.; Fidanza, L.; Caro-Maldonado, A.; Meschiari, M.; et al. Altered bioenergetics and mitochondrial dysfunction of monocytes in patients with COVID-19 pneumonia. EMBO Mol. Med. 2020, 12, e13001. [Google Scholar] [CrossRef]
- Sica, A.; Guarneri, V.; Gennari, A. Myelopoiesis, metabolism and therapy: A crucial crossroads in cancer progression. Cell Stress 2019, 3, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Silvin, A.; Chapuis, N.; Dunsmore, G.; Goubet, A.G.; Dubuisson, A.; Derosa, L.; Almire, C.; Henon, C.; Kosmider, O.; Droin, N.; et al. Elevated Calprotectin and Abnormal Myeloid Cell Subsets Discriminate Severe from Mild COVID-19. Cell 2020, 182, 1401–1418.e18. [Google Scholar] [CrossRef] [PubMed]
- De Biasi, S.; Meschiari, M.; Gibellini, L.; Bellinazzi, C.; Borella, R.; Fidanza, L.; Gozzi, L.; Iannone, A.; Lo Tartaro, D.; Mattioli, M.; et al. Marked T cell activation, senescence, exhaustion and skewing towards TH17 in patients with COVID-19 pneumonia. Nat. Commun. 2020, 11, 3434. [Google Scholar] [CrossRef]
- De Biasi, S.; Gibellini, L.; Lo Tartaro, D.; Puccio, S.; Rabacchi, C.; Mazza, E.M.C.; Brummelman, J.; Williams, B.; Kaihara, K.; Forcato, M.; et al. Circulating mucosal-associated invariant T cells identify patients responding to anti-PD-1 therapy. Nat. Commun. 2021, 12, 1669. [Google Scholar] [CrossRef]
- Rydyznsk, M.C.; Ramirez, S.I.; Dan, J.M.; Grifoni, A.; Hastie, K.M.; Weiskopf, D.; Belanger, S.; Abbott, R.K.; Kim, C.; Choi, J.; et al. Antigen-Specific Adaptive Immunity to SARS-CoV-2 in Acute COVID-19 and Associations with Age and Disease Severity. Cell 2020, 183, 996–1012.e19. [Google Scholar] [CrossRef] [PubMed]
- Grifoni, A.; Weiskopf, D.; Ramirez, S.I.; Mateus, J.; Dan, J.M.; Moderbacher, C.R.; Rawlings, S.A.; Sutherland, A.; Premkumar, L.; Jadi, R.S.; et al. Targets of T Cell Responses to SARS-CoV-2 Coronavirus in Humans with COVID-19 Disease and Unexposed Individuals. Cell 2020, 181, 1489–1501.e15. [Google Scholar] [CrossRef]
- De Biasi, S.; Lo Tartaro, D.; Meschiari, M.; Gibellini, L.; Bellinazzi, C.; Borella, R.; Fidanza, L.; Mattioli, M.; Paolini, A.; Gozzi, L.; et al. Expansion of plasmablasts and loss of memory B cells in peripheral blood from COVID-19 patients with pneumonia. Eur. J. Immunol. 2020, 50, 1283–1294. [Google Scholar] [CrossRef] [PubMed]
- Diao, B.; Wang, C.; Tan, Y.; Chen, X.; Liu, Y.; Ning, L.; Chen, L.; Li, M.; Liu, Y.; Wang, G.; et al. Reduction and Functional Exhaustion of T Cells in Patients With Coronavirus Disease 2019 (COVID-19). Front. Immunol. 2020, 11, 827. [Google Scholar] [CrossRef] [PubMed]
- Cupps, T.R.; Fauci, A.S. Corticosteroid-mediated immunoregulation in man. Immunol. Rev. 1982, 65, 133–155. [Google Scholar] [CrossRef]
- Liu, J.; Li, H.; Luo, M.; Liu, J.; Wu, L.; Lin, X.; Li, R.; Wang, Z.; Zhong, H.; Zheng, W.; et al. Lymphopenia predicted illness severity and recovery in patients with COVID-19: A single-center, retrospective study. PLoS ONE 2020, 15, e0241659. [Google Scholar] [CrossRef] [PubMed]
- Peruzzi, B.; Bencini, S.; Capone, M.; Mazzoni, A.; Maggi, L.; Salvati, L.; Vanni, A.; Orazzini, C.; Nozzoli, C.; Morettini, A.; et al. Quantitative and qualitative alterations of circulating myeloid cells and plasmacytoid DC in SARS-CoV-2 infection. Immunology 2020, 161, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Giamarellos-Bourboulis, E.J.; Netea, M.G.; Rovina, N.; Akinosoglou, K.; Antoniadou, A.; Antonakos, N.; Damoraki, G.; Gkavogianni, T.; Adami, M.E.; Katsaounou, P.; et al. Complex Immune Dysregulation in COVID-19 Patients with Severe Respiratory Failure. Cell Host Microbe. 2020, 27, 992–1000.e3. [Google Scholar] [CrossRef]
- Kratofil, R.M.; Kubes, P.; Deniset, J.F. Monocyte Conversion During Inflammation and Injury. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 35–42. [Google Scholar] [CrossRef]
- Sanchez-Cerrillo, I.; Landete, P.; Aldave, B.; Sanchez-Alonso, S.; Sanchez-Azofra, A.; Marcos-Jimenez, A.; Avalos, E.; Alcaraz-Serna, A.; de Los Santos, I.; Mateu-Albero, T.; et al. COVID-19 severity associates with pulmonary redistribution of CD1c+ DCs and inflammatory transitional and nonclassical monocytes. J. Clin. Investig. 2020, 130, 6290–6300. [Google Scholar] [CrossRef]
- Alzaid, F.; Julla, J.B.; Diedisheim, M.; Potier, C.; Potier, L.; Velho, G.; Gaborit, B.; Manivet, P.; Germain, S.; Vidal-Trecan, T.; et al. Monocytopenia, monocyte morphological anomalies and hyperinflammation characterise severe COVID-19 in type 2 diabetes. EMBO Mol. Med. 2020, 12, e13038. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Kennedy, P.G. Viruses, apoptosis, and neuroinflammation—A double-edged sword. J. Neurovirol. 2015, 21, 1–7. [Google Scholar] [CrossRef]
- Hengartner, M.O. The biochemistry of apoptosis. Nature 2000, 407, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazi, A.; Dixit, V.M. Death receptors: Signaling and modulation. Science 1998, 281, 1305–1308. [Google Scholar] [CrossRef] [PubMed]
- Pereira, W.O.; Amarante-Mendes, G.P. Apoptosis: A programme of cell death or cell disposal? Scand. J. Immunol. 2011, 73, 401–407. [Google Scholar] [CrossRef]
- Troiano, L.; Ferraresi, R.; Lugli, E.; Nemes, E.; Roat, E.; Nasi, M.; Pinti, M.; Cossarizza, A. Multiparametric analysis of cells with different mitochondrial membrane potential during apoptosis by polychromatic flow cytometry. Nat. Protoc. 2007, 2, 2719–2727. [Google Scholar] [CrossRef]
- Fleisher, T.A. Apoptosis. Ann. Allergy Asthma Immunol. 1997, 78, 245–250. [Google Scholar] [CrossRef]
- Baloul, L.; Camelo, S.; Lafon, M. Up-regulation of Fas ligand (FasL) in the central nervous system: A mechanism of immune evasion by rabies virus. J. Neurovirol. 2004, 10, 372–382. [Google Scholar] [CrossRef]
- Zhou, X.; Jiang, W.; Liu, Z.; Liu, S.; Liang, X. Virus Infection and Death Receptor-Mediated Apoptosis. Viruses 2017, 9, 316. [Google Scholar] [CrossRef]
- Cossarizza, A.; Stent, G.; Mussini, C.; Paganelli, R.; Borghi, V.; Nuzzo, C.; Pinti, M.; Pedrazzi, J.; Benatti, F.; Esposito, R.; et al. Deregulation of the CD95/CD95L system in lymphocytes from patients with primary acute HIV infection. AIDS 2000, 14, 345–355. [Google Scholar] [CrossRef]
- Nasi, M.; Pinti, M.; Bugarini, R.; Troiano, L.; Lugli, E.; Bellodi, C.; Mussini, C.; Borghi, V.; Trenti, T.; Balli, F.; et al. Genetic polymorphisms of Fas (CD95) and Fas ligand (CD178) influence the rise in CD4+ T cell count after antiretroviral therapy in drug-naive HIV-positive patients. Immunogenetics 2005, 57, 628–635. [Google Scholar] [CrossRef]
- Pinti, M.; Pedrazzi, J.; Benatti, F.; Sorrentino, V.; Nuzzo, C.; Cavazzuti, V.; Biswas, P.; Petrusca, D.N.; Mussini, C.; De Rienzo, B.; et al. Differential down-regulation of CD95 or CD95L in chronically HIV-infected cells of monocytic or lymphocytic origin: Cellular studies and molecular analysis by quantitative competitive RT-PCR. FEBS Lett. 1999, 458, 209–214. [Google Scholar] [CrossRef]
- Herbeuval, J.P.; Grivel, J.C.; Boasso, A.; Hardy, A.W.; Chougnet, C.; Dolan, M.J.; Yagita, H.; Lifson, J.D.; Shearer, G.M. CD4+ T-cell death induced by infectious and noninfectious HIV-1: Role of type 1 interferon-dependent, TRAIL/DR5-mediated apoptosis. Blood 2005, 106, 3524–3531. [Google Scholar] [CrossRef]
- Herbeuval, J.P.; Hardy, A.W.; Boasso, A.; Anderson, S.A.; Dolan, M.J.; Dy, M.; Shearer, G.M. Regulation of TNF-related apoptosis-inducing ligand on primary CD4+ T cells by HIV-1: Role of type I IFN-producing plasmacytoid dendritic cells. Proc. Natl. Acad. Sci. USA 2005, 102, 13974–13979. [Google Scholar] [CrossRef]
- Van Grevenynghe, J.; Cubas, R.A.; Noto, A.; DaFonseca, S.; He, Z.; Peretz, Y.; Filali-Mouhim, A.; Dupuy, F.P.; Procopio, F.A.; Chomont, N.; et al. Loss of memory B cells during chronic HIV infection is driven by Foxo3a- and TRAIL-mediated apoptosis. J. Clin. Investig. 2011, 121, 3877–3888. [Google Scholar] [CrossRef]
- Taya, N.; Torimoto, Y.; Shindo, M.; Hirai, K.; Hasebe, C.; Kohgo, Y. Fas-mediated apoptosis of peripheral blood mononuclear cells in patients with hepatitis C. Br. J. Haematol. 2000, 110, 89–97. [Google Scholar] [CrossRef]
- Mundt, B.; Kuhnel, F.; Zender, L.; Paul, Y.; Tillmann, H.; Trautwein, C.; Manns, M.P.; Kubicka, S. Involvement of TRAIL and its receptors in viral hepatitis. FASEB J. 2003, 17, 94–96. [Google Scholar] [CrossRef] [PubMed]
- Silberstein, E.; Ulitzky, L.; Lima, L.A.; Cehan, N.; Teixeira-Carvalho, A.; Roingeard, P.; Taylor, D.R. HCV-Mediated Apoptosis of Hepatocytes in Culture and Viral Pathogenesis. PLoS ONE 2016, 11, e0155708. [Google Scholar] [CrossRef] [PubMed]
- Janssen, H.L.; Higuchi, H.; Abdulkarim, A.; Gores, G.J. Hepatitis B virus enhances tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) cytotoxicity by increasing TRAIL-R1/death receptor 4 expression. J. Hepatol. 2003, 39, 414–420. [Google Scholar] [CrossRef]
- Brazeau, E.; Mahalingam, R.; Gilden, D.; Wellish, M.; Kaufer, B.B.; Osterrieder, N.; Pugazhenthi, S. Varicella-zoster virus-induced apoptosis in MeWo cells is accompanied by down-regulation of Bcl-2 expression. J. Neurovirol. 2010, 16, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, I.; Takizawa, T.; Ohba, Y.; Nakanishi, Y. Co-expression of Fas and Fas-ligand on the surface of influenza virus-infected cells. Cell Death Differ. 1998, 5, 426–431. [Google Scholar] [CrossRef]
- Hocke, A.C.; Becher, A.; Knepper, J.; Peter, A.; Holland, G.; Tonnies, M.; Bauer, T.T.; Schneider, P.; Neudecker, J.; Muth, D.; et al. Emerging human middle East respiratory syndrome coronavirus causes widespread infection and alveolar damage in human lungs. Am. J. Respir. Crit. Care. Med. 2013, 188, 882–886. [Google Scholar] [CrossRef]
- Shokri, S.; Mahmoudvand, S.; Taherkhani, R.; Farshadpour, F. Modulation of the immune response by Middle East respiratory syndrome coronavirus. J. Cell Physiol. 2019, 234, 2143–2151. [Google Scholar] [CrossRef]
- Poole, E.; Lau, J.C.H.; Sinclair, J. Latent infection of myeloid progenitors by human cytomegalovirus protects cells from FAS-mediated apoptosis through the cellular IL-10/PEA-15 pathway. J. Gen. Virol. 2015, 96, 2355–2359. [Google Scholar] [CrossRef] [PubMed]
- Seirafian, S.; Prod’homme, V.; Sugrue, D.; Davies, J.; Fielding, C.; Tomasec, P.; Wilkinson, G.W.G. Human cytomegalovirus suppresses Fas expression and function. J. Gen. Virol. 2014, 95, 933–939. [Google Scholar] [CrossRef] [PubMed]
- Nailwal, H.; Chan, F.K. Necroptosis in anti-viral inflammation. Cell Death Differ. 2019, 26, 4–13. [Google Scholar] [CrossRef]
- Khoury, M.K.; Gupta, K.; Franco, S.R.; Liu, B. Necroptosis in the Pathophysiology of Disease. Am. J. Pathol. 2020, 190, 272–285. [Google Scholar] [CrossRef]
- Dhuriya, Y.K.; Sharma, D. Necroptosis: A regulated inflammatory mode of cell death. J. Neuroinflamm. 2018, 15, 199. [Google Scholar] [CrossRef]
- Kang, T.B.; Yang, S.H.; Toth, B.; Kovalenko, A.; Wallach, D. Caspase-8 blocks kinase RIPK3-mediated activation of the NLRP3 inflammasome. Immunity 2013, 38, 27–40. [Google Scholar] [CrossRef]
- Lawlor, K.E.; Khan, N.; Mildenhall, A.; Gerlic, M.; Croker, B.A.; D’Cruz, A.A.; Hall, C.; Kaur Spall, S.; Anderton, H.; Masters, S.L.; et al. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat. Commun. 2015, 6, 6282. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.S.; Park, H.L. Exploitation of necroptosis for treatment of caspase-compromised cancers. Oncol. Lett. 2017, 14, 1207–1214. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhou, Q.; Snipas, S.; Orth, K.; Muzio, M.; Dixit, V.M.; Salvesen, G.S. Target protease specificity of the viral serpin CrmA. Analysis of five caspases. J. Biol. Chem. 1997, 272, 7797–7800. [Google Scholar] [CrossRef]
- Omoto, S.; Guo, H.; Talekar, G.R.; Roback, L.; Kaiser, W.J.; Mocarski, E.S. Suppression of RIP3-dependent necroptosis by human cytomegalovirus. J. Biol. Chem. 2015, 290, 11635–11648. [Google Scholar] [CrossRef]
- Liu, X.; Li, Y.; Peng, S.; Yu, X.; Li, W.; Shi, F.; Luo, X.; Tang, M.; Tan, Z.; Bode, A.M.; et al. Epstein-Barr virus encoded latent membrane protein 1 suppresses necroptosis through targeting RIPK1/3 ubiquitination. Cell Death Dis. 2018, 9, 53. [Google Scholar] [CrossRef]
- Terahara, K.; Iwabuchi, R.; Iwaki, R.; Takahashi, Y.; Tsunetsugu-Yokota, Y. Substantial induction of non-apoptotic CD4 T-cell death during the early phase of HIV-1 infection in a humanized mouse model. Microbes. Infect. 2021, 23, 104767. [Google Scholar] [CrossRef]
- Hughes, M.A.; Powley, I.R.; Jukes-Jones, R.; Horn, S.; Feoktistova, M.; Fairall, L.; Schwabe, J.W.; Leverkus, M.; Cain, K.; MacFarlane, M. Co-operative and Hierarchical Binding of c-FLIP and Caspase-8: A Unified Model Defines How c-FLIP Isoforms Differentially Control Cell Fate. Mol. Cell 2016, 61, 834–849. [Google Scholar] [CrossRef] [PubMed]
- Bertin, J.; Armstrong, R.C.; Ottilie, S.; Martin, D.A.; Wang, Y.; Banks, S.; Wang, G.H.; Senkevich, T.G.; Alnemri, E.S.; Moss, B.; et al. Death effector domain-containing herpesvirus and poxvirus proteins inhibit both Fas- and TNFR1-induced apoptosis. Proc. Natl. Acad. Sci. USA 1997, 94, 1172–1176. [Google Scholar] [CrossRef] [PubMed]
- Thome, M.; Schneider, P.; Hofmann, K.; Fickenscher, H.; Meinl, E.; Neipel, F.; Mattmann, C.; Burns, K.; Bodmer, J.L.; Schroter, M.; et al. Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature 1997, 386, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Garvey, T.; Bertin, J.; Siegel, R.; Lenardo, M.; Cohen, J. The death effector domains (DEDs) of the molluscum contagiosum virus MC159 v-FLIP protein are not functionally interchangeable with each other or with the DEDs of caspase-8. Virology 2002, 300, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Garvey, T.L.; Bertin, J.; Siegel, R.M.; Wang, G.H.; Lenardo, M.J.; Cohen, J.I. Binding of FADD and caspase-8 to molluscum contagiosum virus MC159 v-FLIP is not sufficient for its antiapoptotic function. J. Virol. 2002, 76, 697–706. [Google Scholar] [CrossRef]
- Chaudhary, P.M.; Jasmin, A.; Eby, M.T.; Hood, L. Modulation of the NF-kappa B pathway by virally encoded death effector domains-containing proteins. Oncogene 1999, 18, 5738–5746. [Google Scholar] [CrossRef]
- Sun, Q.; Matta, H.; Chaudhary, P.M. The human herpes virus 8-encoded viral FLICE inhibitory protein protects against growth factor withdrawal-induced apoptosis via NF-kappa B activation. Blood 2003, 101, 1956–1961. [Google Scholar] [CrossRef][Green Version]
- Frank, D.; Vince, J.E. Pyroptosis versus necroptosis: Similarities, differences, and crosstalk. Cell Death Differ. 2019, 26, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Hilbi, H.; Moss, J.E.; Hersh, D.; Chen, Y.; Arondel, J.; Banerjee, S.; Flavell, R.A.; Yuan, J.; Sansonetti, P.J.; Zychlinsky, A. Shigella-induced apoptosis is dependent on caspase-1 which binds to IpaB. J. Biol. Chem. 1998, 273, 32895–32900. [Google Scholar] [CrossRef]
- Monack, D.M.; Raupach, B.; Hromockyj, A.E.; Falkow, S. Salmonella typhimurium invasion induces apoptosis in infected macrophages. Proc. Natl. Acad. Sci. USA 1996, 93, 9833–9838. [Google Scholar] [CrossRef] [PubMed]
- Mencacci, A.; Bacci, A.; Cenci, E.; Montagnoli, C.; Fiorucci, S.; Casagrande, A.; Flavell, R.A.; Bistoni, F.; Romani, L. Interleukin 18 restores defective Th1 immunity to Candida albicans in caspase 1-deficient mice. Infect. Immun. 2000, 68, 5126–5131. [Google Scholar] [CrossRef]
- Kanneganti, T.D.; Body-Malapel, M.; Amer, A.; Park, J.H.; Whitfield, J.; Franchi, L.; Taraporewala, Z.F.; Miller, D.; Patton, J.T.; Inohara, N.; et al. Critical role for Cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and double-stranded RNA. J. Biol. Chem. 2006, 281, 36560–36568. [Google Scholar] [CrossRef] [PubMed]
- Rajan, J.V.; Rodriguez, D.; Miao, E.A.; Aderem, A. The NLRP3 inflammasome detects encephalomyocarditis virus and vesicular stomatitis virus infection. J. Virol. 2011, 85, 4167–4172. [Google Scholar] [CrossRef] [PubMed]
- Kanneganti, T.D. Central roles of NLRs and inflammasomes in viral infection. Nat. Rev. Immunol. 2010, 10, 688–698. [Google Scholar] [CrossRef] [PubMed]
- Ramos, H.J.; Lanteri, M.C.; Blahnik, G.; Negash, A.; Suthar, M.S.; Brassil, M.M.; Sodhi, K.; Treuting, P.M.; Busch, M.P.; Norris, P.J.; et al. IL-1beta signaling promotes CNS-intrinsic immune control of West Nile virus infection. PLoS Pathog. 2012, 8, e1003039. [Google Scholar] [CrossRef]
- Kofahi, H.M.; Taylor, N.G.; Hirasawa, K.; Grant, M.D.; Russell, R.S. Hepatitis C Virus Infection of Cultured Human Hepatoma Cells Causes Apoptosis and Pyroptosis in Both Infected and Bystander Cells. Sci. Rep. 2016, 6, 37433. [Google Scholar] [CrossRef]
- Doitsh, G.; Galloway, N.L.; Geng, X.; Yang, Z.; Monroe, K.M.; Zepeda, O.; Hunt, P.W.; Hatano, H.; Sowinski, S.; Munoz-Arias, I.; et al. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature 2014, 505, 509–514. [Google Scholar] [CrossRef]
- Muruve, D.A.; Petrilli, V.; Zaiss, A.K.; White, L.R.; Clark, S.A.; Ross, P.J.; Parks, R.J.; Tschopp, J. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature 2008, 452, 103–107. [Google Scholar] [CrossRef]
- Barlan, A.U.; Griffin, T.M.; McGuire, K.A.; Wiethoff, C.M. Adenovirus membrane penetration activates the NLRP3 inflammasome. J. Virol. 2011, 85, 146–155. [Google Scholar] [CrossRef]
- Nour, A.M.; Reichelt, M.; Ku, C.C.; Ho, M.Y.; Heineman, T.C.; Arvin, A.M. Varicella-zoster virus infection triggers formation of an interleukin-1beta (IL-1beta)-processing inflammasome complex. J. Biol. Chem. 2011, 286, 17921–17933. [Google Scholar] [CrossRef]
- Johnston, J.B.; Barrett, J.W.; Nazarian, S.H.; Goodwin, M.; Ricciuto, D.; Wang, G.; McFadden, G. A poxvirus-encoded pyrin domain protein interacts with ASC-1 to inhibit host inflammatory and apoptotic responses to infection. Immunity 2005, 23, 587–598. [Google Scholar] [CrossRef]
- Stasakova, J.; Ferko, B.; Kittel, C.; Sereinig, S.; Romanova, J.; Katinger, H.; Egorov, A. Influenza A mutant viruses with altered NS1 protein function provoke caspase-1 activation in primary human macrophages, resulting in fast apoptosis and release of high levels of interleukins 1beta and 18. J. Gen. Virol. 2005, 86, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Bergsbaken, T.; Fink, S.L.; Cookson, B.T. Pyroptosis: Host cell death and inflammation. Nat. Rev. Microbiol. 2009, 7, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Jose, R.J.; Manuel, A. COVID-19 cytokine storm: The interplay between inflammation and coagulation. Lancet Respir. Med. 2020, 8, e46–e47. [Google Scholar] [CrossRef]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J. Hlh Across Speciality Collaboration, U.K., COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034. [Google Scholar] [CrossRef]
- Liu, Y.; Garron, T.M.; Chang, Q.; Su, Z.; Zhou, C.; Qiu, Y.; Gong, E.C.; Zheng, J.; Yin, Y.W.; Ksiazek, T.; et al. Cell-Type Apoptosis in Lung during SARS-CoV-2 Infection. Pathogens 2021, 10, 509. [Google Scholar] [CrossRef]
- Bianchi, M.; Borsetti, A.; Ciccozzi, M.; Pascarella, S. SARS-Cov-2 ORF3a: Mutability and function. Int. J. Biol. Macromol. 2021, 170, 820–826. [Google Scholar] [CrossRef]
- Ren, Y.; Shu, T.; Wu, D.; Mu, J.; Wang, C.; Huang, M.; Han, Y.; Zhang, X.Y.; Zhou, W.; Qiu, Y.; et al. The ORF3a protein of SARS-CoV-2 induces apoptosis in cells. Cell Mol. Immunol. 2020, 17, 881–883. [Google Scholar] [CrossRef]
- Issa, E.; Merhi, G.; Panossian, B.; Salloum, T.; Tokajian, S. SARS-CoV-2 and ORF3a: Nonsynonymous Mutations, Functional Domains, and Viral Pathogenesis. mSystems 2020, 170, 820–826. [Google Scholar] [CrossRef]
- Lu, W.; Zheng, B.J.; Xu, K.; Schwarz, W.; Du, L.; Wong, C.K.; Chen, J.; Duan, S.; Deubel, V.; Sun, B. Severe acute respiratory syndrome-associated coronavirus 3a protein forms an ion channel and modulates virus release. Proc. Natl. Acad. Sci. USA 2006, 103, 12540–12545. [Google Scholar] [CrossRef] [PubMed]
- Scott, C.; Griffin, S. Viroporins: Structure, function and potential as antiviral targets. J. Gen. Virol. 2015, 96, 2000–2027. [Google Scholar] [CrossRef] [PubMed]
- Castano-Rodriguez, C.; Honrubia, J.M.; Gutierrez-Alvarez, J.; DeDiego, M.L.; Nieto-Torres, J.L.; Jimenez-Guardeno, J.M.; Regla-Nava, J.A.; Fernandez-Delgado, R.; Verdia-Baguena, C.; Queralt-Martin, M.; et al. Role of Severe Acute Respiratory Syndrome Coronavirus Viroporins E, 3a, and 8a in Replication and Pathogenesis. mBio 2018. [Google Scholar] [CrossRef]
- Wang, F.; Nie, J.; Wang, H.; Zhao, Q.; Xiong, Y.; Deng, L.; Song, S.; Ma, Z.; Mo, P.; Zhang, Y. Characteristics of Peripheral Lymphocyte Subset Alteration in COVID-19 Pneumonia. J. Infect. Dis. 2020, 221, 1762–1769. [Google Scholar] [CrossRef] [PubMed]
- Bellesi, S.; Metafuni, E.; Hohaus, S.; Maiolo, E.; Marchionni, F.; D’Innocenzo, S.; La Sorda, M.; Ferraironi, M.; Ramundo, F.; Fantoni, M.; et al. Increased CD95 (Fas) and PD-1 expression in peripheral blood T lymphocytes in COVID-19 patients. Br. J. Haematol. 2020, 191, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Liu, Y.; Cao, L.; Wang, D.; Guo, M.; Jiang, A.; Guo, D.; Hu, W.; Yang, J.; Tang, Z.; et al. Transcriptomic characteristics of bronchoalveolar lavage fluid and peripheral blood mononuclear cells in COVID-19 patients. Emerg. Microbes. Infect. 2020, 9, 761–770. [Google Scholar] [CrossRef]
- Thompson, E.A.; Cascino, K.; Ordonez, A.A.; Zhou, W.; Vaghasia, A.; Hamacher-Brady, A.; Brady, N.R.; Sun, I.H.; Wang, R.; Rosenberg, A.Z.; et al. Metabolic programs define dysfunctional immune responses in severe COVID-19 patients. Cell Rep. 2021, 34, 108863. [Google Scholar] [CrossRef]
- Chu, H.; Zhou, J.; Wong, B.H.; Li, C.; Chan, J.F.; Cheng, Z.S.; Yang, D.; Wang, D.; Lee, A.C.; Li, C.; et al. Middle East Respiratory Syndrome Coronavirus Efficiently Infects Human Primary T Lymphocytes and Activates the Extrinsic and Intrinsic Apoptosis Pathways. J. Infect. Dis. 2016, 213, 904–914. [Google Scholar] [CrossRef]
- Helal, M.A.; Shouman, S.; Abdelwaly, A.; Elmehrath, A.O.; Essawy, M.; Sayed, S.M.; Saleh, A.H.; El-Badri, N. Molecular basis of the potential interaction of SARS-CoV-2 spike protein to CD147 in COVID-19 associated-lymphopenia. J. Biomol. Struct. Dyn. 2020, 16, 1–11. [Google Scholar] [CrossRef]
- Kang, M.J.; Kim, H.P.; Lee, K.S.; Yoo, Y.D.; Kwon, Y.T.; Kim, K.M.; Kim, T.Y.; Yi, E.C. Proteomic analysis reveals that CD147/EMMPRIN confers chemoresistance in cancer stem cell-like cells. Proteomics 2013, 13, 1714–1725. [Google Scholar] [CrossRef]
- Wang, K.; Chen, W.; Zhang, Z.; Deng, Y.; Lian, J.Q.; Du, P.; Wei, D.; Zhang, Y.; Sun, X.X.; Gong, L.; et al. CD147-spike protein is a novel route for SARS-CoV-2 infection to host cells. Signal Transduct Target Ther. 2020, 5, 283. [Google Scholar] [CrossRef] [PubMed]
- Arunachalam, P.S.; Wimmers, F.; Mok, C.K.P.; Perera, R.; Scott, M.; Hagan, T.; Sigal, N.; Feng, Y.; Bristow, L.; Tak-Yin Tsang, O.; et al. Systems biological assessment of immunity to mild versus severe COVID-19 infection in humans. Science 2020, 369, 1210–1220. [Google Scholar] [CrossRef] [PubMed]
- Chua, R.L.; Lukassen, S.; Trump, S.; Hennig, B.P.; Wendisch, D.; Pott, F.; Debnath, O.; Thurmann, L.; Kurth, F.; Volker, M.T.; et al. COVID-19 severity correlates with airway epithelium-immune cell interactions identified by single-cell analysis. Nat. Biotechnol. 2020, 38, 970–979. [Google Scholar] [CrossRef]
- Liao, M.; Liu, Y.; Yuan, J.; Wen, Y.; Xu, G.; Zhao, J.; Cheng, L.; Li, J.; Wang, X.; Wang, F.; et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. 2020, 26, 842–844. [Google Scholar] [CrossRef]
- Nienhold, R.; Ciani, Y.; Koelzer, V.H.; Tzankov, A.; Haslbauer, J.D.; Menter, T.; Schwab, N.; Henkel, M.; Frank, A.; Zsikla, V.; et al. Two distinct immunopathological profiles in autopsy lungs of COVID-19. Nat. Commun. 2020, 11, 5086. [Google Scholar] [CrossRef]
- Wauters, E.; Van Mol, P.; Garg, A.D.; Jansen, S.; Van Herck, Y.; Vanderbeke, L.; Bassez, A.; Boeckx, B.; Malengier-Devlies, B.; Timmerman, A.; et al. Discriminating mild from critical COVID-19 by innate and adaptive immune single-cell profiling of bronchoalveolar lavages. Cell Res. 2021, 31, 272–290. [Google Scholar] [CrossRef]
- Swiecki, M.; Colonna, M. The multifaceted biology of plasmacytoid dendritic cells. Nat. Rev. Immunol. 2015, 15, 471–485. [Google Scholar] [CrossRef]
- Liu, C.; Martins, A.J.; Lau, W.W.; Rachmaninoff, N.; Chen, J.; Imberti, L.; Mostaghimi, D.; Fink, D.L.; Burbelo, P.D.; Dobbs, K.; et al. Time-resolved systems immunology reveals a late juncture linked to fatal COVID-19. Cell 2021, 184, 1836–1857.e22. [Google Scholar] [CrossRef]
- Li, S.; Zhang, Y.; Guan, Z.; Li, H.; Ye, M.; Chen, X.; Shen, J.; Zhou, Y.; Shi, Z.L.; Zhou, P.; et al. SARS-CoV-2 triggers inflammatory responses and cell death through caspase-8 activation. Signal Transduct. Target. Ther. 2020, 5, 235. [Google Scholar] [CrossRef]
- Cao, L.; Mu, W. Necrostatin-1 and necroptosis inhibition: Pathophysiology and therapeutic implications. Pharmacol. Res. 2021, 163, 105297. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Jiang, W.; Yan, Y.; Gong, T.; Han, J.; Tian, Z.; Zhou, R. RNA viruses promote activation of the NLRP3 inflammasome through a RIP1-RIP3-DRP1 signaling pathway. Nat. Immunol. 2014, 15, 1126–1133. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Yin, C.; Boyd, D.F.; Quarato, G.; Ingram, J.P.; Shubina, M.; Ragan, K.B.; Ishizuka, T.; Crawford, J.C.; Tummers, B.; et al. Influenza Virus Z-RNAs Induce ZBP1-Mediated Necroptosis. Cell 2020, 180, 1115–1129.e13. [Google Scholar] [CrossRef]
- Shashaty, M.G.S.; Reilly, J.P.; Faust, H.E.; Forker, C.M.; Ittner, C.A.G.; Zhang, P.X.; Hotz, M.J.; Fitzgerald, D.; Yang, W.; Anderson, B.J.; et al. Plasma receptor interacting protein kinase-3 levels are associated with acute respiratory distress syndrome in sepsis and trauma: A cohort study. Crit. Care 2019, 23, 235. [Google Scholar] [CrossRef]
- Nakamura, H.; Kinjo, T.; Arakaki, W.; Miyagi, K.; Tateyama, M.; Fujita, J. Serum levels of receptor-interacting protein kinase-3 in patients with COVID-19. Crit. Care 2020, 24, 484. [Google Scholar] [CrossRef]
- Faust, H.; Mangalmurti, N.S. Collateral damage: Necroptosis in the development of lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 2020, 318, L215–L225. [Google Scholar] [CrossRef]
- Chen, I.Y.; Moriyama, M.; Chang, M.F.; Ichinohe, T. Severe Acute Respiratory Syndrome Coronavirus Viroporin 3a Activates the NLRP3 Inflammasome. Front. Microbiol. 2019, 10, 50. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Ockinger, J.; Yu, J.; Byles, V.; McColl, A.; Hofer, A.M.; Horng, T. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc. Natl. Acad. Sci. USA 2012, 109, 11282–11287. [Google Scholar] [CrossRef]
- De Diego, M.L.; Nieto-Torres, J.L.; Regla-Nava, J.A.; Jimenez-Guardeno, J.M.; Fernandez-Delgado, R.; Fett, C.; Castano-Rodriguez, C.; Perlman, S.; Enjuanes, L. Inhibition of NF-kappaB-mediated inflammation in severe acute respiratory syndrome coronavirus-infected mice increases survival. J. Virol. 2014, 88, 913–924. [Google Scholar] [CrossRef] [PubMed]
- Kanzawa, N.; Nishigaki, K.; Hayashi, T.; Ishii, Y.; Furukawa, S.; Niiro, A.; Yasui, F.; Kohara, M.; Morita, K.; Matsushima, K.; et al. Augmentation of chemokine production by severe acute respiratory syndrome coronavirus 3a/X1 and 7a/X4 proteins through NF-kappaB activation. FEBS Lett. 2006, 580, 6807–6812. [Google Scholar] [CrossRef] [PubMed]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [PubMed]
- Siu, K.L.; Yuen, K.S.; Castano-Rodriguez, C.; Ye, Z.W.; Yeung, M.L.; Fung, S.Y.; Yuan, S.; Chan, C.P.; Yuen, K.Y.; Enjuanes, L.; et al. Severe acute respiratory syndrome coronavirus ORF3a protein activates the NLRP3 inflammasome by promoting TRAF3-dependent ubiquitination of ASC. FASEB J. 2019, 33, 8865–8877. [Google Scholar] [CrossRef]
- Yap, J.K.Y.; Moriyama, M.; Iwasaki, A. Inflammasomes and Pyroptosis as Therapeutic Targets for COVID-19. J. Immunol. 2020, 205, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Place, D.E.; Kanneganti, T.D. Recent advances in inflammasome biology. Curr. Opin. Immunol. 2018, 50, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, K.; Xiong, S.; DiZerega, G.S. Effect of angiotensin II and angiotensin(1-7) on hematopoietic recovery after intravenous chemotherapy. Cancer Chemother Pharmacol. 2003, 51, 97–106. [Google Scholar] [CrossRef]
- Sun, N.N.; Yu, C.H.; Pan, M.X.; Zhang, Y.; Zheng, B.J.; Yang, Q.J.; Zheng, Z.M.; Meng, Y. Mir-21 Mediates the Inhibitory Effect of Ang (1-7) on AngII-induced NLRP3 Inflammasome Activation by Targeting Spry1 in lung fibroblasts. Sci. Rep. 2017, 7, 14369. [Google Scholar] [CrossRef]
- Zhao, M.; Bai, M.; Ding, G.; Zhang, Y.; Huang, S.; Jia, Z.; Zhang, A. Angiotensin II Stimulates the NLRP3 Inflammasome to Induce Podocyte Injury and Mitochondrial Dysfunction. Kidney Dis. 2018, 4, 83–94. [Google Scholar] [CrossRef]
- Chappell, M.C.; Pirro, N.T.; Sykes, A.; Ferrario, C.M. Metabolism of angiotensin-(1-7) by angiotensin-converting enzyme. Hypertension 1998, 31, 362–367. [Google Scholar] [CrossRef]
- Fleming, I. Signaling by the angiotensin-converting enzyme. Circ. Res. 2006, 98, 887–896. [Google Scholar] [CrossRef]
- Schwacke, J.H.; Spainhour, J.C.; Ierardi, J.L.; Chaves, J.M.; Arthur, J.M.; Janech, M.G.; Velez, J.C. Network modeling reveals steps in angiotensin peptide processing. Hypertension 2013, 61, 690–700. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Singh, K.D.; Karnik, S.S. Angiotensin Receptors: Structure, Function, Signaling and Clinical Applications. J. Cell Signal 2016. [Google Scholar] [CrossRef]
- Mori, J.; Oudit, G.Y.; Lopaschuk, G.D. SARS-CoV-2 perturbs the renin-angiotensin system and energy metabolism. Am. J. Physiol. Endocrinol. Metab. 2020, 319, E43–E47. [Google Scholar] [CrossRef] [PubMed]
- Ratajczak, M.Z.; Bujko, K.; Ciechanowicz, A.; Sielatycka, K.; Cymer, M.; Marlicz, W.; Kucia, M. SARS-CoV-2 Entry Receptor ACE2 Is Expressed on Very Small CD45(-) Precursors of Hematopoietic and Endothelial Cells and in Response to Virus Spike Protein Activates the Nlrp3 Inflammasome. Stem. Cell Rev. Rep. 2021, 17, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Karki, R.; Sharma, B.R.; Tuladhar, S.; Williams, E.P.; Zalduondo, L.; Samir, P.; Zheng, M.; Sundaram, B.; Banoth, B.; Malireddi, R.K.S.; et al. Synergism of TNF-alpha and IFN-gamma Triggers Inflammatory Cell Death, Tissue Damage, and Mortality in SARS-CoV-2 Infection and Cytokine Shock Syndromes. Cell 2021, 184, 149–168.e17. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gao, W.; Shi, X.; Ding, J.; Liu, W.; He, H.; Wang, K.; Shao, F. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 2017, 547, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Moulian, N.; Truffault, F.; Gaudry-Talarmain, Y.M.; Serraf, A.; Berrih-Aknin, S. In vivo and in vitro apoptosis of human thymocytes are associated with nitrotyrosine formation. Blood 2001, 97, 3521–3530. [Google Scholar] [CrossRef]
- Vig, M.; Srivastava, S.; Kandpal, U.; Sade, H.; Lewis, V.; Sarin, A.; George, A.; Bal, V.; Durdik, J.M.; Rath, S. Inducible nitric oxide synthase in T cells regulates T cell death and immune memory. J. Clin. Investig. 2004, 113, 1734–1742. [Google Scholar] [CrossRef]
- Refaeli, Y.; Van Parijs, L.; Alexander, S.I.; Abbas, A.K. Interferon gamma is required for activation-induced death of T lymphocytes. J. Exp. Med. 2002, 196, 999–1005. [Google Scholar] [CrossRef]
- Malireddi, R.K.S.; Kesavardhana, S.; Kanneganti, T.D. ZBP1 and TAK1: Master Regulators of NLRP3 Inflammasome/Pyroptosis, Apoptosis, and Necroptosis (PAN-optosis). Front. Cell. Infect. Microbiol. 2019, 9, 406. [Google Scholar] [CrossRef] [PubMed]
- Christgen, S.; Zheng, M.; Kesavardhana, S.; Karki, R.; Malireddi, R.K.S.; Banoth, B.; Place, D.E.; Briard, B.; Sharma, B.R.; Tuladhar, S.; et al. Identification of the PANoptosome: A Molecular Platform Triggering Pyroptosis, Apoptosis, and Necroptosis (PANoptosis). Front. Cell. Infect. Microbiol. 2020, 10, 237. [Google Scholar] [CrossRef]
- Lee, S.; Channappanavar, R.; Kanneganti, T.D. Coronaviruses: Innate Immunity, Inflammasome Activation, Inflammatory Cell Death, and Cytokines. Trends Immunol. 2020, 41, 1083–1099. [Google Scholar] [CrossRef]
- Chang, D.W.; Xing, Z.; Pan, Y.; Algeciras-Schimnich, A.; Barnhart, B.C.; Yaish-Ohad, S.; Peter, M.E.; Yang, X. c-FLIP(L) is a dual function regulator for caspase-8 activation and CD95-mediated apoptosis. EMBO J. 2002, 21, 3704–3714. [Google Scholar] [CrossRef] [PubMed]
- Fricker, N.; Beaudouin, J.; Richter, P.; Eils, R.; Krammer, P.H.; Lavrik, I.N. Model-based dissection of CD95 signaling dynamics reveals both a pro- and antiapoptotic role of c-FLIPL. J. Cell. Biol. 2010, 190, 377–389. [Google Scholar] [CrossRef]
- Ivanisenko, N.V.; Seyrek, K.; Kolchanov, N.A.; Ivanisenko, V.A.; Lavrik, I.N. The role of death domain proteins in host response upon SARS-CoV-2 infection: Modulation of programmed cell death and translational applications. Cell Death Discov. 2020, 6, 101. [Google Scholar] [CrossRef] [PubMed]
- Hillert, L.K.; Ivanisenko, N.V.; Busse, D.; Espe, J.; Konig, C.; Peltek, S.E.; Kolchanov, N.A.; Ivanisenko, V.A.; Lavrik, I.N. Dissecting DISC regulation via pharmacological targeting of caspase-8/c-FLIPL heterodimer. Cell Death Differ. 2020, 27, 2117–2130. [Google Scholar] [CrossRef]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.C.; Uhl, S.; Hoagland, D.; Moller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045.e9. [Google Scholar] [CrossRef]
- Sun, J.; Ye, F.; Wu, A.; Yang, R.; Pan, M.; Sheng, J.; Zhu, W.; Mao, L.; Wang, M.; Xia, Z.; et al. Comparative Transcriptome Analysis Reveals the Intensive Early Stage Responses of Host Cells to SARS-CoV-2 Infection. Front. Microbiol. 2020, 11, 593857. [Google Scholar] [CrossRef]
- Miller, S.C.; Huang, R.; Sakamuru, S.; Shukla, S.J.; Attene-Ramos, M.S.; Shinn, P.; Van Leer, D.; Leister, W.; Austin, C.P.; Xia, M. Identification of known drugs that act as inhibitors of NF-kappaB signaling and their mechanism of action. Biochem. Pharmacol. 2010, 79, 1272–1280. [Google Scholar] [CrossRef] [PubMed]
- Abu-Hamad, S.; Arbel, N.; Calo, D.; Arzoine, L.; Israelson, A.; Keinan, N.; Ben-Romano, R.; Friedman, O.; Shoshan-Barmatz, V. The VDAC1 N-terminus is essential both for apoptosis and the protective effect of anti-apoptotic proteins. J. Cell. Sci. 2009, 122, 1906–1916. [Google Scholar] [CrossRef]
- Shimizu, S.; Ide, T.; Yanagida, T.; Tsujimoto, Y. Electrophysiological study of a novel large pore formed by Bax and the voltage-dependent anion channel that is permeable to cytochrome c. J. Biol. Chem. 2000, 275, 12321–12325. [Google Scholar] [CrossRef] [PubMed]
- Guaraldi, G.; Meschiari, M.; Cozzi-Lepri, A.; Milic, J.; Tonelli, R.; Menozzi, M.; Franceschini, E.; Cuomo, G.; Orlando, G.; Borghi, V.; et al. Tocilizumab in patients with severe COVID-19: A retrospective cohort study. Lancet Rheumatol. 2020, 2, e474–e484. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Death Pathways Involved | PANoptosome Component |
|---|---|
| Pyroptosis | Caspase-1 NLRP3 ASC |
| Necroptosis | RIPK1 RIPK3 |
| Apoptosis | Caspase-8 FADD |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paolini, A.; Borella, R.; De Biasi, S.; Neroni, A.; Mattioli, M.; Lo Tartaro, D.; Simonini, C.; Franceschini, L.; Cicco, G.; Piparo, A.M.; et al. Cell Death in Coronavirus Infections: Uncovering Its Role during COVID-19. Cells 2021, 10, 1585. https://doi.org/10.3390/cells10071585
Paolini A, Borella R, De Biasi S, Neroni A, Mattioli M, Lo Tartaro D, Simonini C, Franceschini L, Cicco G, Piparo AM, et al. Cell Death in Coronavirus Infections: Uncovering Its Role during COVID-19. Cells. 2021; 10(7):1585. https://doi.org/10.3390/cells10071585
Chicago/Turabian StylePaolini, Annamaria, Rebecca Borella, Sara De Biasi, Anita Neroni, Marco Mattioli, Domenico Lo Tartaro, Cecilia Simonini, Laura Franceschini, Gerolamo Cicco, Anna Maria Piparo, and et al. 2021. "Cell Death in Coronavirus Infections: Uncovering Its Role during COVID-19" Cells 10, no. 7: 1585. https://doi.org/10.3390/cells10071585
APA StylePaolini, A., Borella, R., De Biasi, S., Neroni, A., Mattioli, M., Lo Tartaro, D., Simonini, C., Franceschini, L., Cicco, G., Piparo, A. M., Cossarizza, A., & Gibellini, L. (2021). Cell Death in Coronavirus Infections: Uncovering Its Role during COVID-19. Cells, 10(7), 1585. https://doi.org/10.3390/cells10071585