
Glial Cells as Therapeutic Approaches in Brain Ischemia-Reperfusion Injury

 , , ,
, , , 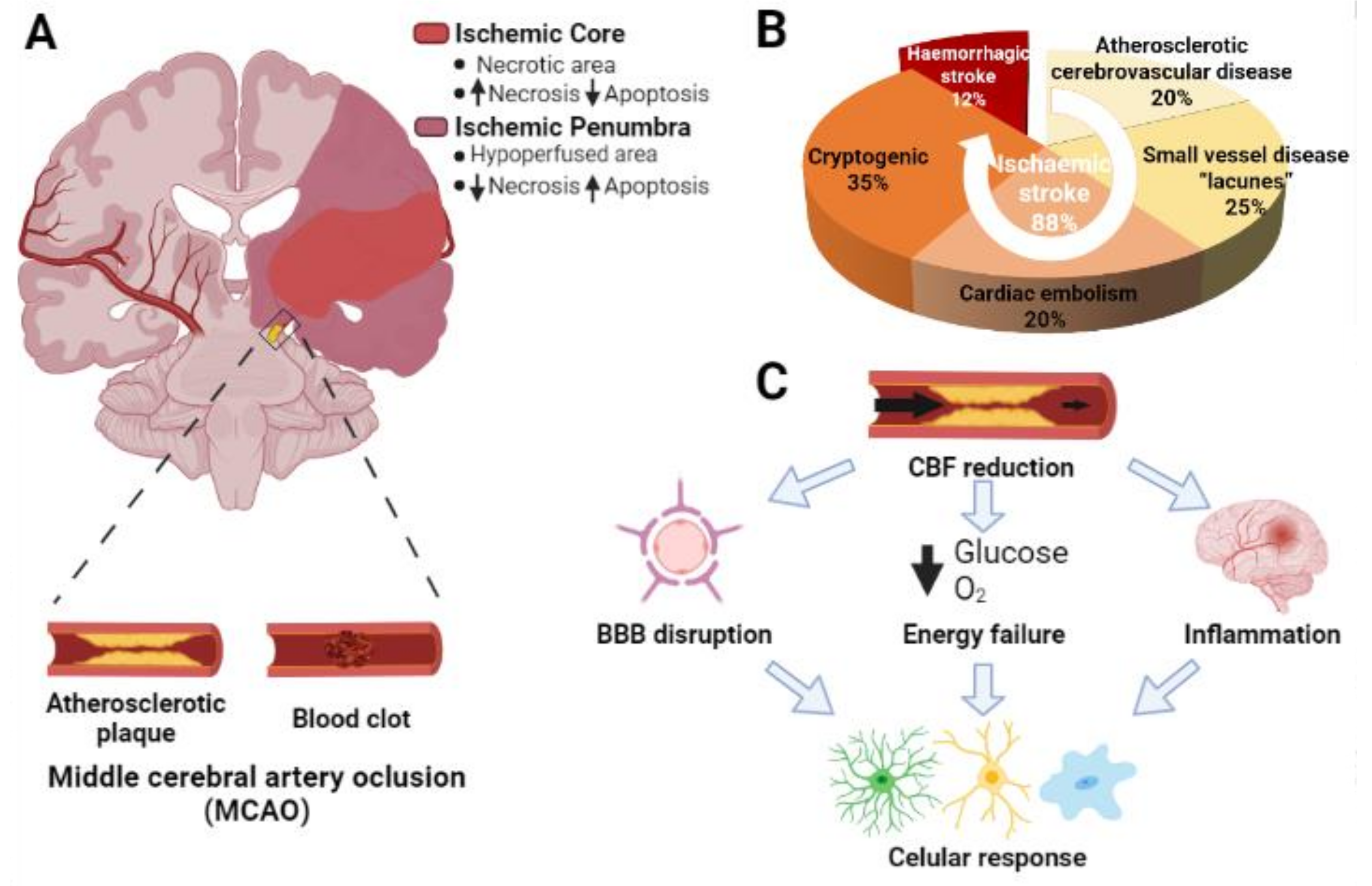{kind=link}
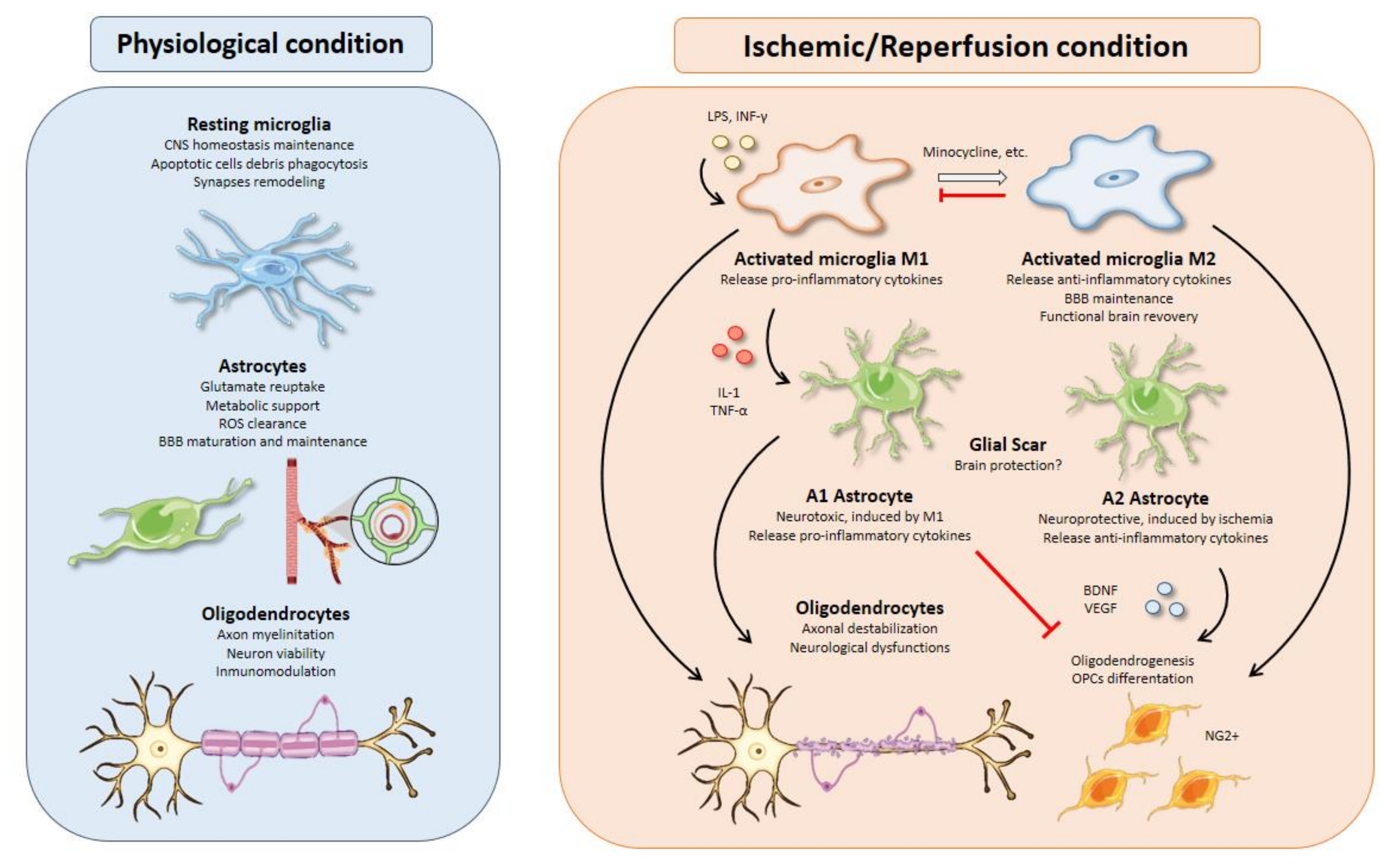{kind=link}
Abstract
:1. Introduction
2. Microglia
2.1. Microglial Activation Timing upon Brain Ischemia
2.2. M1 versus M2 Microglial Profiles
2.3. Inductors of Microglial Activation in Stroke
2.4. Therapies Based on Activated Microglia
3. Astrocytes
3.1. Excitotoxicity Modulation
3.2. Astrocyte-Neuron Metabolic Relationships in Stroke
3.3. Oxidative Stress Management
3.4. BBB Integrity and Edema
3.4.1. Negative Effectors
3.4.2. Positive Effectors
3.5. Inflammation and Glial Scar Formation
3.6. Neurogenesis and Synaptogenesis
3.7. Astrocytes as Preconditioning Vehicles
4. Oligodendrocytes
4.1. Oligodendrocyte Response to Ischemia
4.2. Oligodendrogenesis
4.3. Interaction with Other Glial Cells
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Virani, S.S.; Alonso, A.; Aparicio, H.J.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics—2021 Update: A Report From the American Heart Association. Circulation 2021, 143, e254–e743. [Google Scholar] [CrossRef]
- Neurology, T.L. A unified European action plan on stroke. Lancet Neurol. 2020, 19, 963. [Google Scholar] [CrossRef]
- Katan, M.; Luft, A. Global Burden of Stroke. Semin. Neurol. 2018, 38, 208–211. [Google Scholar] [CrossRef] [Green Version]
- Venketasubramanian, N.; Yoon, B.W.; Pandian, J.; Navarro, J.C. Stroke Epidemiology in South, East, and South-East Asia: A Review. J. Stroke 2017, 19, 286–294. [Google Scholar] [CrossRef]
- García-Moncó, J.C.; Cabrera-Muras, A.; Collía-Fernández, A.; Erburu-Iriarte, M.; Rodrigo-Armenteros, P.; Oyarzun-Irazu, I.; Martínez-Condor, D.; Bilbao-González, A.; Carmona-Abellán, M.; Caballero-Romero, I.; et al. Neurological reasons for consultation and hospitalization during the COVID-19 pandemic. Neurol. Sci. 2020, 41, 3031–3038. [Google Scholar] [CrossRef]
- Cagnazzo, F.; Piotin, M.; Escalard, S.; Maier, B.; Ribo, M.; Requena, M.; Pop, R.; Hasiu, A.; Gasparotti, R.; Mardighian, D.; et al. European Multicenter Study of ET-COVID-19. Stroke 2021, 52, 31–39. [Google Scholar] [CrossRef]
- Fifi, J.T.; Mocco, J. COVID-19 related stroke in young individuals. Lancet Neurol. 2020, 19, 713–715. [Google Scholar] [CrossRef]
- Yaghi, S.; Bernstein, R.A.; Passman, R.; Okin, P.M.; Furie, K.L. Cryptogenic Stroke. Circ. Res. 2017, 120, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Sarmah, D.; Banerjee, M.; Datta, A.; Kalia, K.; Dhar, S.; Yavagal, D.R.; Bhattacharya, P. Nanotechnology in the diagnosis and treatment of stroke. Drug Discov. Today 2020, 26, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Meschia, J.F.; Brott, T. Ischaemic stroke. Eur. J. Neurol. 2018, 25, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Campbell, B.C.V.; De Silva, D.A.; Macleod, M.R.; Coutts, S.B.; Schwamm, L.H.; Davis, S.M.; Donnan, G.A. Ischaemic stroke. Nat. Rev. Dis. Primers 2019, 5, 70. [Google Scholar] [CrossRef]
- Xing, C.; Arai, K.; Lo, E.H.; Hommel, M. Pathophysiologic cascades in ischemic stroke. Int. J. Stroke 2012, 7, 378–385. [Google Scholar] [CrossRef]
- Dirnagl, U.; Iadecola, C.; Moskowitz, M.A. Pathobiology of ischaemic stroke: An integrated view. Trends Neurosci. 1999, 22, 391–397. [Google Scholar] [CrossRef]
- Xu, S.; Lu, J.; Shao, A.; Zhang, J.H.; Zhang, J. Glial Cells: Role of the Immune Response in Ischemic Stroke. Front. Immunol. 2020, 11, 294. [Google Scholar] [CrossRef]
- Picard, K.; St-Pierre, M.K.; Vecchiarelli, H.A.; Bordeleau, M.; Tremblay, M. Neuroendocrine, neuroinflammatory and pathological outcomes of chronic stress: A story of microglial remodeling. Neurochem. Int. 2021, 145, 104987. [Google Scholar] [CrossRef] [PubMed]
- Kreutzberg, G.W. Microglia: A sensor for pathological events in the CNS. Trends Neurosci. 1996, 19, 312–318. [Google Scholar] [CrossRef]
- Jin, R.; Yang, G.; Li, G. Inflammatory mechanisms in ischemic stroke: Role of inflammatory cells. J. Leukoc. Biol. 2010, 87, 779–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakajima, K.; Kohsaka, S. Microglia: Neuroprotective and neurotrophic cells in the central nervous system. Curr. Drug Targets Cardiovasc. Haematol. Disord. 2004, 4, 65–84. [Google Scholar] [CrossRef] [PubMed]
- Morioka, T.; Kalehua, A.N.; Streit, W.J. Characterization of microglial reaction after middle cerebral artery occlusion in rat brain. J. Comp. Neurol. 1993, 327, 123–132. [Google Scholar] [CrossRef]
- Schroeter, M.; Jander, S.; Huitinga, I.; Witte, O.W.; Stoll, G. Phagocytic response in photochemically induced infarction of rat cerebral cortex. The role of resident microglia. Stroke 1997, 28, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Rupalla, K.; Allegrini, P.R.; Sauer, D.; Wiessner, C. Time course of microglia activation and apoptosis in various brain regions after permanent focal cerebral ischemia in mice. Acta Neuropathol. 1998, 96, 172–178. [Google Scholar] [CrossRef]
- Gorlamandala, N.; Parmar, J.; Craig, A.J.; Power, J.M.; Moorhouse, A.J.; Krishnan, A.V.; Housley, G.D. Focal Ischaemic Infarcts Expand Faster in Cerebellar Cortex than Cerebral Cortex in a Mouse Photothrombotic Stroke Model. Transl. Stroke Res. 2018, 9, 643–653. [Google Scholar] [CrossRef]
- Neher, J.J.; Emmrich, J.V.; Fricker, M.; Mander, P.K.; Théry, C.; Brown, G.C. Phagocytosis executes delayed neuronal death after focal brain ischemia. Proc. Natl. Acad. Sci. USA 2013, 110, E4098–E4107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perego, C.; Fumagalli, S.; De Simoni, M.G. Temporal pattern of expression and colocalization of microglia/macrophage phenotype markers following brain ischemic injury in mice. J. Neuroinflamm. 2011, 8, 174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, X.; Yamashita, T. Microglia in central nervous system repair after injury. J. Biochem. 2016, 159, 491–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davalos, D.; Grutzendler, J.; Yang, G.; Kim, J.V.; Zuo, Y.; Jung, S.; Littman, D.R.; Dustin, M.L.; Gan, W.B. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005, 8, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Prinz, M.; Mildner, A. Microglia in the CNS: Immigrants from another world. Glia 2011, 59, 177–187. [Google Scholar] [CrossRef]
- Ma, Y.M.; Li, C.D.; Zhu, Y.B.; Xu, X.; Li, J.; Cao, H. The mechanism of RvD1 alleviates type 2 diabetic neuropathic pain by influencing microglia polarization in rats. Zhongguo Ying Yong Sheng Li Xue Za Zhi 2017, 33, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Morrison, H.W.; Filosa, J.A. A quantitative spatiotemporal analysis of microglia morphology during ischemic stroke and reperfusion. J. Neuroinflamm. 2013, 10, 4. [Google Scholar] [CrossRef] [Green Version]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [Green Version]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef] [PubMed]
- Colton, C.A. Heterogeneity of microglial activation in the innate immune response in the brain. J. Neuroimmune Pharmacol. 2009, 4, 399–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef]
- Lyu, J.; Xie, D.; Bhatia, T.N.; Leak, R.K.; Hu, X.; Jiang, X. Microglial/Macrophage polarization and function in brain injury and repair after stroke. CNS Neurosci. Ther. 2021, 27, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, M.; Ninomiya, I.; Hatakeyama, M.; Takahashi, T.; Shimohata, T. Microglia and Monocytes/Macrophages Polarization Reveal Novel Therapeutic Mechanism against Stroke. Int. J. Mol. Sci. 2017, 18, 2135. [Google Scholar] [CrossRef]
- Kwon, H.S.; Koh, S.H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 42. [Google Scholar] [CrossRef] [PubMed]
- Qin, C.; Zhou, L.Q.; Ma, X.T.; Hu, Z.W.; Yang, S.; Chen, M.; Bosco, D.B.; Wu, L.J.; Tian, D.S. Dual Functions of Microglia in Ischemic Stroke. Neurosci. Bull. 2019, 35, 921–933. [Google Scholar] [CrossRef]
- Gaojian, T.; Dingfei, Q.; Linwei, L.; Xiaowei, W.; Zheng, Z.; Wei, L.; Tong, Z.; Benxiang, N.; Yanning, Q.; Wei, Z.; et al. Parthenolide promotes the repair of spinal cord injury by modulating M1/M2 polarization via the NF-κB and STAT 1/3 signaling pathway. Cell Death Discov. 2020, 6, 97. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Li, W.B.; Qu, Y.W.; Gao, J.X.; Tang, Y.S.; Wang, D.J.; Pan, Y.J. Bone marrow mesenchymal stem cells induce M2 microglia polarization through PDGF-AA/MANF signaling. World J. Stem Cells 2020, 12, 633–658. [Google Scholar] [CrossRef]
- Parisi, C.; Napoli, G.; Pelegrin, P.; Volonté, C. M1 and M2 Functional Imprinting of Primary Microglia: Role of P2X7 Activation and miR-125b. Mediat. Inflamm. 2016, 2016, 2989548. [Google Scholar] [CrossRef] [Green Version]
- Tu, H.; Chu, H.; Guan, S.; Hao, F.; Xu, N.; Zhao, Z.; Liang, Y. The role of the M1/M2 microglia in the process from cancer pain to morphine tolerance. Tissue Cell 2021, 68, 101438. [Google Scholar] [CrossRef] [PubMed]
- Lana, D.; Ugolini, F.; Nosi, D.; Wenk, G.L.; Giovannini, M.G. The Emerging Role of the Interplay Among Astrocytes, Microglia, and Neurons in the Hippocampus in Health and Disease. Front. Aging Neurosci. 2021, 13, 651973. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef] [PubMed]
- Dabrowska, S.; Andrzejewska, A.; Lukomska, B.; Janowski, M. Neuroinflammation as a target for treatment of stroke using mesenchymal stem cells and extracellular vesicles. J. Neuroinflamm. 2019, 16, 178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilchrist, S.E.; Goudarzi, S.; Hafizi, S. Gas6 Inhibits Toll-Like Receptor-Mediated Inflammatory Pathways in Mouse Microglia via Axl and Mer. Front. Cell Neurosci. 2020, 14, 576650. [Google Scholar] [CrossRef]
- Lu, Y.; Li, X.; Liu, S.; Zhang, Y.; Zhang, D. Toll-like Receptors and Inflammatory Bowel Disease. Front. Immunol. 2018, 9, 72. [Google Scholar] [CrossRef] [Green Version]
- Pan, W.; Kastin, A.J. Tumor necrosis factor and stroke: Role of the blood-brain barrier. Prog. Neurobiol. 2007, 83, 363–374. [Google Scholar] [CrossRef] [Green Version]
- Welser, J.V.; Li, L.; Milner, R. Microglial activation state exerts a biphasic influence on brain endothelial cell proliferation by regulating the balance of TNF and TGF-beta1. J. Neuroinflamm. 2010, 7, 89. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, T.G.; Alves-Silva, J.; de Souza, J.M.; Real, A.; Doria, J.G.; Vieira, E.L.M.; Gomes, G.F.; de Oliveira, A.C.; Miranda, A.S.; Ribeiro, F.M. Metabotropic glutamate receptor 5 ablation accelerates age-related neurodegeneration and neuroinflammation. Neurochem. Int. 2019, 126, 218–228. [Google Scholar] [CrossRef]
- Rahman, M.S.; Yang, J.; Luan, Y.; Qiu, Z.; Zhang, J.; Lu, H.; Chen, X.; Liu, Y. Attenuation of Acute Intracerebral Hemorrhage-Induced Microglial Activation and Neuronal Death Mediated by the Blockade of Metabotropic Glutamate Receptor 5 In Vivo. Neurochem. Res. 2020, 45, 1230–1243. [Google Scholar] [CrossRef]
- Kaiser, M.; Penk, A.; Franke, H.; Krügel, U.; Nörenberg, W.; Huster, D.; Schaefer, M. Lack of functional P2X7 receptor aggravates brain edema development after middle cerebral artery occlusion. Purinergic Signal. 2016, 12, 453–463. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, H.M.; di Lucente, J.; Chen, Y.J.; Cui, Y.; Ibrahim, R.H.; Pennington, M.W.; Jin, L.W.; Maezawa, I.; Wulff, H. Biophysical basis for Kv1.3 regulation of membrane potential changes induced by P2X4-mediated calcium entry in microglia. Glia 2020, 68, 2377–2394. [Google Scholar] [CrossRef]
- Hou, K.; Li, G.; Yu, J.; Xu, K.; Wu, W. Receptors, Channel Proteins, and Enzymes Involved in Microglia-mediated Neuroinflammation and Treatments by Targeting Microglia in Ischemic Stroke. Neuroscience 2021, 460, 167–180. [Google Scholar] [CrossRef]
- Chen, Y.; Yu, S.; Wu, Q.; Tang, Y.; Jiang, C.; Zhuang, Z.; Zhao, N.; Huang, B.; Xie, L. Effect of bone marrow mesenchymal stem cells conditioned medium on microglia and its secretion of arginase 1 in rats. Zhongguo Xiu Fu Chong Jian Wai Ke Za Zhi 2017, 31, 1500–1505. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zhou, M.; Li, Y.; Li, Y.; Hua, Y.; Fan, Y. Minocycline promotes functional recovery in ischemic stroke by modulating microglia polarization through STAT1/STAT6 pathways. Biochem. Pharmacol. 2021, 186, 114464. [Google Scholar] [CrossRef] [PubMed]
- Serhan, A.; Aerts, J.L.; Boddeke, E.; Kooijman, R. Neuroprotection by Insulin-like Growth Factor-1 in Rats with Ischemic Stroke is Associated with Microglial Changes and a Reduction in Neuroinflammation. Neuroscience 2020, 426, 101–114. [Google Scholar] [CrossRef]
- Scholz, R.; Sobotka, M.; Caramoy, A.; Stempfl, T.; Moehle, C.; Langmann, T. Minocycline counter-regulates pro-inflammatory microglia responses in the retina and protects from degeneration. J. Neuroinflamm. 2015, 12, 209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomás-Camardiel, M.; Rite, I.; Herrera, A.J.; de Pablos, R.M.; Cano, J.; Machado, A.; Venero, J.L. Minocycline reduces the lipopolysaccharide-induced inflammatory reaction, peroxynitrite-mediated nitration of proteins, disruption of the blood-brain barrier, and damage in the nigral dopaminergic system. Neurobiol. Dis. 2004, 16, 190–201. [Google Scholar] [CrossRef]
- Jolivel, V.; Bicker, F.; Binamé, F.; Ploen, R.; Keller, S.; Gollan, R.; Jurek, B.; Birkenstock, J.; Poisa-Beiro, L.; Bruttger, J.; et al. Perivascular microglia promote blood vessel disintegration in the ischemic penumbra. Acta Neuropathol. 2015, 129, 279–295. [Google Scholar] [CrossRef]
- Lampl, Y.; Boaz, M.; Gilad, R.; Lorberboym, M.; Dabby, R.; Rapoport, A.; Anca-Hershkowitz, M.; Sadeh, M. Minocycline treatment in acute stroke: An open-label, evaluator-blinded study. Neurology 2007, 69, 1404–1410. [Google Scholar] [CrossRef]
- Kikuchi, K.; Tanaka, E.; Murai, Y.; Tancharoen, S. Clinical trials in acute ischemic stroke. CNS Drugs 2014, 28, 929–938. [Google Scholar] [CrossRef]
- Liu, Z.J.; Ran, Y.Y.; Qie, S.Y.; Gong, W.J.; Gao, F.H.; Ding, Z.T.; Xi, J.N. Melatonin protects against ischemic stroke by modulating microglia/macrophage polarization toward anti-inflammatory phenotype through STAT3 pathway. CNS Neurosci. Ther. 2019, 25, 1353–1362. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.M.; Gu, S.S.; Mei, W.H.; Zhou, J.; Wang, Z.Z.; Xiao, W. Ginkgolides and bilobalide protect BV2 microglia cells against OGD/reoxygenation injury by inhibiting TLR2/4 signaling pathways. Cell Stress Chaperones 2016, 21, 1037–1053. [Google Scholar] [CrossRef]
- Xi, Z.; Xu, C.; Chen, X.; Wang, B.; Zhong, Z.; Sun, Q.; Sun, Y.; Bian, L. Protocatechuic Acid Suppresses Microglia Activation and Facilitates M1 to M2 Phenotype Switching in Intracerebral Hemorrhage Mice. J. Stroke Cereb. Dis. 2021, 30, 105765. [Google Scholar] [CrossRef]
- Li, D.; Wang, C.; Yao, Y.; Chen, L.; Liu, G.; Zhang, R.; Liu, Q.; Shi, F.D.; Hao, J. mTORC1 pathway disruption ameliorates brain inflammation following stroke via a shift in microglia phenotype from M1 type to M2 type. FASEB J. 2016, 30, 3388–3399. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Lu, W.; Wang, Y.; Li, J.; Luo, Y. A20-Binding Inhibitor of NF-κB 1 Ameliorates Neuroinflammation and Mediates Antineuroinflammatory Effect of Electroacupuncture in Cerebral Ischemia/Reperfusion Rats. Evid. Based Complement. Altern. Med. 2020, 2020, 6980398. [Google Scholar] [CrossRef]
- Li, F.; Ma, Q.; Zhao, H.; Wang, R.; Tao, Z.; Fan, Z.; Zhang, S.; Li, G.; Luo, Y. L-3-n-Butylphthalide reduces ischemic stroke injury and increases M2 microglial polarization. Metab. Brain Dis. 2018, 33, 1995–2003. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Wang, X.; Yang, S.; Huang, J.; Xue, X.; Zheng, Y.; Shang, G.; Tao, J.; Chen, L. Electroacupunctre improves motor impairment via inhibition of microglia-mediated neuroinflammation in the sensorimotor cortex after ischemic stroke. Life Sci. 2016, 151, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Zhang, Z.; Bai, F.; Jiang, T.; Yan, C.; Wang, Q. Electroacupuncture Pretreatment Alleviates Cerebral Ischemic Injury Through α7 Nicotinic Acetylcholine Receptor-Mediated Phenotypic Conversion of Microglia. Front. Cell Neurosci. 2019, 13, 537. [Google Scholar] [CrossRef] [PubMed]
- Mason, S. Lactate Shuttles in Neuroenergetics-Homeostasis, Allostasis and Beyond. Front. Neurosci. 2017, 11, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Chopp, M. Astrocytes, therapeutic targets for neuroprotection and neurorestoration in ischemic stroke. Prog. Neurobiol. 2016, 144, 103–120. [Google Scholar] [CrossRef] [Green Version]
- Chisholm, N.C.; Henderson, M.L.; Selvamani, A.; Park, M.J.; Dindot, S.; Miranda, R.C.; Sohrabji, F. Histone methylation patterns in astrocytes are influenced by age following ischemia. Epigenetics 2015, 10, 142–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, T.; Kawahara, K.; Kosugi, T.; Tanaka, M. Nitric oxide produced during sublethal ischemia is crucial for the preconditioning-induced down-regulation of glutamate transporter GLT-1 in neuron/astrocyte co-cultures. Neurochem. Res. 2006, 31, 49–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeevalk, G.D.; Davis, N.; Hyndman, A.G.; Nicklas, W.J. Origins of the extracellular glutamate released during total metabolic blockade in the immature retina. J. Neurochem. 1998, 71, 2373–2381. [Google Scholar] [CrossRef] [Green Version]
- Ouyang, Y.B.; Voloboueva, L.A.; Xu, L.J.; Giffard, R.G. Selective dysfunction of hippocampal CA1 astrocytes contributes to delayed neuronal damage after transient forebrain ischemia. J. Neurosci. 2007, 27, 4253–4260. [Google Scholar] [CrossRef] [Green Version]
- Harvey, B.K.; Airavaara, M.; Hinzman, J.; Wires, E.M.; Chiocco, M.J.; Howard, D.B.; Shen, H.; Gerhardt, G.; Hoffer, B.J.; Wang, Y. Targeted over-expression of glutamate transporter 1 (GLT-1) reduces ischemic brain injury in a rat model of stroke. PLoS ONE 2011, 6, e22135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weller, M.L.; Stone, I.M.; Goss, A.; Rau, T.; Rova, C.; Poulsen, D.J. Selective overexpression of excitatory amino acid transporter 2 (EAAT2) in astrocytes enhances neuroprotection from moderate but not severe hypoxia-ischemia. Neuroscience 2008, 155, 1204–1211. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; He, P.; Fan, Y.Y.; Zhang, J.X.; Yan, H.J.; Hu, W.W.; Ohtsu, H.; Chen, Z. Carnosine protects against permanent cerebral ischemia in histidine decarboxylase knockout mice by reducing glutamate excitotoxicity. Free Radic. Biol. Med. 2010, 48, 727–735. [Google Scholar] [CrossRef]
- Cui, X.; Li, L.; Hu, Y.Y.; Ren, S.; Zhang, M.; Li, W.B. Sulbactam Plays Neuronal Protective Effect Against Brain Ischemia via Upregulating GLT1 in Rats. Mol. Neurobiol. 2015, 51, 1322–1333. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.L.; Dogan, A.; Todd, K.G.; Bowen, K.K.; Kim, B.T.; Rothstein, J.D.; Dempsey, R.J. Antisense knockdown of the glial glutamate transporter GLT-1, but not the neuronal glutamate transporter EAAC1, exacerbates transient focal cerebral ischemia-induced neuronal damage in rat brain. J. Neurosci. 2001, 21, 1876–1883. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Vitery, M.D.C.; Chen, J.; Osei-Owusu, J.; Chu, J.; Qiu, Z. Glutamate-Releasing SWELL1 Channel in Astrocytes Modulates Synaptic Transmission and Promotes Brain Damage in Stroke. Neuron 2019, 102, 813–827.e6. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Jin, Y.; Behr, M.J.; Feustel, P.J.; Morrison, J.P.; Kimelberg, H.K. Behavioral and histological neuroprotection by tamoxifen after reversible focal cerebral ischemia. Exp. Neurol. 2005, 196, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Duan, S.; Anderson, C.M.; Keung, E.C.; Chen, Y.; Chen, Y.; Swanson, R.A. P2X7 receptor-mediated release of excitatory amino acids from astrocytes. J. Neurosci. 2003, 23, 1320–1328. [Google Scholar] [CrossRef] [Green Version]
- Liang, Z.; Wang, X.; Hao, Y.; Qiu, L.; Lou, Y.; Zhang, Y.; Ma, D.; Feng, J. The Multifaceted Role of Astrocyte Connexin 43 in Ischemic Stroke Through Forming Hemichannels and Gap Junctions. Front. Neurol. 2020, 11, 703. [Google Scholar] [CrossRef]
- Deng, Z.H.; Liao, J.; Zhang, J.Y.; Liang, C.; Song, C.H.; Han, M.; Wang, L.H.; Xue, H.; Zhang, K.; Zabeau, L.; et al. Inhibition of the connexin 43 elevation may be involved in the neuroprotective activity of leptin against brain ischemic injury. Cell Mol. Neurobiol. 2014, 34, 871–879. [Google Scholar] [CrossRef]
- Turner, D.A.; Adamson, D.C. Neuronal-astrocyte metabolic interactions: Understanding the transition into abnormal astrocytoma metabolism. J. Neuropathol. Exp. Neurol. 2011, 70, 167–176. [Google Scholar] [CrossRef]
- MacVicar, B.A.; Newman, E.A. Astrocyte regulation of blood flow in the brain. Cold Spring Harb Perspect. Biol. 2015, 7. [Google Scholar] [CrossRef]
- Pellerin, L.; Magistretti, P.J. Glutamate uptake into astrocytes stimulates aerobic glycolysis: A mechanism coupling neuronal activity to glucose utilization. Proc. Natl. Acad. Sci. USA 1994, 91, 10625–10629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dienel, G.A. Lack of appropriate stoichiometry: Strong evidence against an energetically important astrocyte-neuron lactate shuttle in brain. J. Neurosci. Res. 2017, 95, 2103–2125. [Google Scholar] [CrossRef] [Green Version]
- Hertz, L.; Chen, Y. Glycogenolysis, an Astrocyte-Specific Reaction, is Essential for Both Astrocytic and Neuronal Activities Involved in Learning. Neuroscience 2018, 370, 27–36. [Google Scholar] [CrossRef]
- Takahashi, S.; Iizumi, T.; Mashima, K.; Abe, T.; Suzuki, N. Roles and regulation of ketogenesis in cultured astroglia and neurons under hypoxia and hypoglycemia. ASN Neuro 2014, 6. [Google Scholar] [CrossRef]
- Guzman, M.; Blazquez, C. Is there an astrocyte-neuron ketone body shuttle? Trends Endocrinol. Metab. 2001, 12, 169–173. [Google Scholar] [CrossRef]
- Iglesias, J.; Morales, L.; Barreto, G.E. Metabolic and Inflammatory Adaptation of Reactive Astrocytes: Role of PPARs. Mol. Neurobiol. 2017, 54, 2518–2538. [Google Scholar] [CrossRef]
- Bazzigaluppi, P.; Lake, E.M.; Beckett, T.L.; Koletar, M.M.; Weisspapir, I.; Heinen, S.; Mester, J.; Lai, A.; Janik, R.; Dorr, A.; et al. Imaging the Effects of beta-Hydroxybutyrate on Peri-Infarct Neurovascular Function and Metabolism. Stroke 2018, 49, 2173–2181. [Google Scholar] [CrossRef] [PubMed]
- Gibson, C.L.; Murphy, A.N.; Murphy, S.P. Stroke outcome in the ketogenic state--a systematic review of the animal data. J. Neurochem. 2012, 123 (Suppl. 2), 52–57. [Google Scholar] [CrossRef] [Green Version]
- Yin, J.; Han, P.; Tang, Z.; Liu, Q.; Shi, J. Sirtuin 3 mediates neuroprotection of ketones against ischemic stroke. J. Cereb. Blood Flow Metab. 2015, 35, 1783–1789. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Deepa, S.S.; Etzler, J.C.; Ryu, J.; Mao, X.; Fang, Q.; Liu, D.D.; Torres, J.M.; Jia, W.; Lechleiter, J.D.; et al. Adiponectin activates AMP-activated protein kinase in muscle cells via APPL1/LKB1-dependent and phospholipase C/Ca2+/Ca2+/calmodulin-dependent protein kinase kinase-dependent pathways. J. Biol. Chem. 2009, 284, 22426–22435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018, 15, 490–503. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Qin, C.; Huang, J.; Tang, X.; Liu, C.; Huang, K.; Xu, J.; Guo, G.; Tong, A.; Zhou, L. The role of astrocytes in oxidative stress of central nervous system: A mixed blessing. Cell Prolif. 2020, 53, e12781. [Google Scholar] [CrossRef] [Green Version]
- Jimenez-Blasco, D.; Santofimia-Castano, P.; Gonzalez, A.; Almeida, A.; Bolanos, J.P. Astrocyte NMDA receptors’ activity sustains neuronal survival through a Cdk5-Nrf2 pathway. Cell Death Differ. 2015, 22, 1877–1889. [Google Scholar] [CrossRef] [Green Version]
- Takagi, T.; Kitashoji, A.; Iwawaki, T.; Tsuruma, K.; Shimazawa, M.; Yoshimura, S.; Iwama, T.; Hara, H. Temporal activation of Nrf2 in the penumbra and Nrf2 activator-mediated neuroprotection in ischemia-reperfusion injury. Free Radic. Biol. Med. 2014, 72, 124–133. [Google Scholar] [CrossRef]
- Skowronska, K.; Obara-Michlewska, M.; Zielinska, M.; Albrecht, J. NMDA Receptors in Astrocytes: In Search for Roles in Neurotransmission and Astrocytic Homeostasis. Int. J. Mol. Sci. 2019, 20, 309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dzamba, D.; Honsa, P.; Valny, M.; Kriska, J.; Valihrach, L.; Novosadova, V.; Kubista, M.; Anderova, M. Quantitative Analysis of Glutamate Receptors in Glial Cells from the Cortex of GFAP/EGFP Mice Following Ischemic Injury: Focus on NMDA Receptors. Cell Mol. Neurobiol. 2015, 35, 1187–1202. [Google Scholar] [CrossRef]
- Wang, H.; Yan, H.; Zhang, S.; Wei, X.; Zheng, J.; Li, J. The GluN3A subunit exerts a neuroprotective effect in brain ischemia and the hypoxia process. ASN Neuro 2013, 5, 231–242. [Google Scholar] [CrossRef]
- Tschopp, J.; Schroder, K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat. Rev. Immunol. 2010, 10, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Juliana, C.; Fernandes-Alnemri, T.; Kang, S.; Farias, A.; Qin, F.; Alnemri, E.S. Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J. Biol. Chem. 2012, 287, 36617–36622. [Google Scholar] [CrossRef] [Green Version]
- Abais, J.M.; Xia, M.; Zhang, Y.; Boini, K.M.; Li, P.L. Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector? Antioxid. Redox Signal. 2015, 22, 1111–1129. [Google Scholar] [CrossRef] [Green Version]
- Xu, N.; Zhang, Y.; Doycheva, D.M.; Ding, Y.; Zhang, Y.; Tang, J.; Guo, H.; Zhang, J.H. Adiponectin attenuates neuronal apoptosis induced by hypoxia-ischemia via the activation of AdipoR1/APPL1/LKB1/AMPK pathway in neonatal rats. Neuropharmacology 2018, 133, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Yin, Y.; Cui, J.; Yan, J.; Zhu, Y.; Guan, Y.; Wei, G.; Weng, Y.; Wu, X.; Guo, C.; et al. Chikusetsu Saponin IVa Ameliorates Cerebral Ischemia Reperfusion Injury in Diabetic Mice via Adiponectin-Mediated AMPK/GSK-3beta Pathway In Vivo and In Vitro. Mol. Neurobiol. 2016, 53, 728–743. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wu, X.; Luo, J.; Zhao, L.; Li, X.; Guo, H.; Bai, H.; Cui, W.; Guo, W.; Feng, D.; et al. Adiponectin peptide alleviates oxidative stress and NLRP3 inflammasome activation after cerebral ischemia-reperfusion injury by regulating AMPK/GSK-3beta. Exp. Neurol. 2020, 329, 113302. [Google Scholar] [CrossRef]
- Allaman, I.; Belanger, M.; Magistretti, P.J. Astrocyte-neuron metabolic relationships: For better and for worse. Trends Neurosci. 2011, 34, 76–87. [Google Scholar] [CrossRef]
- Sarkar, S.; Mukherjee, A.; Swarnakar, S.; Das, N. Nanocapsulated Ascorbic Acid in Combating Cerebral Ischemia Reperfusion- Induced Oxidative Injury in Rat Brain. Curr. Alzheimer Res. 2016, 13, 1363–1373. [Google Scholar] [CrossRef]
- Peng, L.; Zhao, Y.; Li, Y.; Zhou, Y.; Li, L.; Lei, S.; Yu, S.; Zhao, Y. Effect of DJ-1 on the neuroprotection of astrocytes subjected to cerebral ischemia/reperfusion injury. J. Mol. Med. 2019, 97, 189–199. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Jiang, Q.; Tuccitto, A.; Chan, D.; Alqawlaq, S.; Won, G.J.; Sivak, J.M. The AMPK-PGC-1alpha signaling axis regulates the astrocyte glutathione system to protect against oxidative and metabolic injury. Neurobiol. Dis. 2018, 113, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Trendelenburg, G.; Prass, K.; Priller, J.; Kapinya, K.; Polley, A.; Muselmann, C.; Ruscher, K.; Kannbley, U.; Schmitt, A.O.; Castell, S.; et al. Serial analysis of gene expression identifies metallothionein-II as major neuroprotective gene in mouse focal cerebral ischemia. J. Neurosci. 2002, 22, 5879–5888. [Google Scholar] [CrossRef] [Green Version]
- Manley, G.T.; Fujimura, M.; Ma, T.; Noshita, N.; Filiz, F.; Bollen, A.W.; Chan, P.; Verkman, A.S. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat. Med. 2000, 6, 159–163. [Google Scholar] [CrossRef]
- Luo, J.; Chen, H.; Kintner, D.B.; Shull, G.E.; Sun, D. Decreased neuronal death in Na+/H+ exchanger isoform 1-null mice after in vitro and in vivo ischemia. J. Neurosci. 2005, 25, 11256–11268. [Google Scholar] [CrossRef] [Green Version]
- Begum, G.; Song, S.; Wang, S.; Zhao, H.; Bhuiyan, M.I.H.; Li, E.; Nepomuceno, R.; Ye, Q.; Sun, M.; Calderon, M.J.; et al. Selective knockout of astrocytic Na(+) /H(+) exchanger isoform 1 reduces astrogliosis, BBB damage, infarction, and improves neurological function after ischemic stroke. Glia 2018, 66, 126–144. [Google Scholar] [CrossRef]
- Zhang, H.T.; Zhang, P.; Gao, Y.; Li, C.L.; Wang, H.J.; Chen, L.C.; Feng, Y.; Li, R.Y.; Li, Y.L.; Jiang, C.L. Early VEGF inhibition attenuates blood-brain barrier disruption in ischemic rat brains by regulating the expression of MMPs. Mol. Med. Rep. 2017, 15, 57–64. [Google Scholar] [CrossRef] [Green Version]
- Reeson, P.; Tennant, K.A.; Gerrow, K.; Wang, J.; Weiser Novak, S.; Thompson, K.; Lockhart, K.L.; Holmes, A.; Nahirney, P.C.; Brown, C.E. Delayed inhibition of VEGF signaling after stroke attenuates blood-brain barrier breakdown and improves functional recovery in a comorbidity-dependent manner. J. Neurosci. 2015, 35, 5128–5143. [Google Scholar] [CrossRef]
- Jiang, L.; Pan, C.L.; Wang, C.Y.; Liu, B.Q.; Han, Y.; Hu, L.; Liu, L.; Yang, Y.; Qu, J.W.; Liu, W.T. Selective suppression of the JNK-MMP2/9 signal pathway by tetramethylpyrazine attenuates neuropathic pain in rats. J. Neuroinflamm. 2017, 14, 174. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; An, Q.; Wang, T.; Gao, S.; Zhou, G. Corrigendum to “Autophagy- and MMP-2/9-mediated Reduction and Redistribution of ZO-1 Contribute to Hyperglycemia-increased Blood-Brain Barrier Permeability During Early Reperfusion in Stroke” [Neuroscience 377 (2018) 126-137]. Neuroscience 2018, 386, 351. [Google Scholar] [CrossRef]
- Yang, Y.; Estrada, E.Y.; Thompson, J.F.; Liu, W.; Rosenberg, G.A. Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J. Cereb. Blood Flow Metab. 2007, 27, 697–709. [Google Scholar] [CrossRef] [PubMed]
- Asahi, M.; Wang, X.; Mori, T.; Sumii, T.; Jung, J.C.; Moskowitz, M.A.; Fini, M.E.; Lo, E.H. Effects of matrix metalloproteinase-9 gene knock-out on the proteolysis of blood-brain barrier and white matter components after cerebral ischemia. J. Neurosci. 2001, 21, 7724–7732. [Google Scholar] [CrossRef] [Green Version]
- Saha, R.N.; Pahan, K. Signals for the induction of nitric oxide synthase in astrocytes. Neurochem. Int. 2006, 49, 154–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michinaga, S.; Koyama, Y. Dual Roles of Astrocyte-Derived Factors in Regulation of Blood-Brain Barrier Function after Brain Damage. Int. J. Mol. Sci. 2019, 20, 571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, Y.; Zheng, G.; Xu, M.; Li, Y.; Chen, X.; Zhu, W.; Tong, Y.; Chung, S.K.; Liu, K.J.; Shen, J. Caveolin-1 regulates nitric oxide-mediated matrix metalloproteinases activity and blood-brain barrier permeability in focal cerebral ischemia and reperfusion injury. J. Neurochem. 2012, 120, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Hogan-Cann, A.D.; Globa, A.K.; Lu, P.; Nagy, J.I.; Bamji, S.X.; Anderson, C.M. Astrocytes drive cortical vasodilatory signaling by activating endothelial NMDA receptors. J. Cereb. Blood Flow Metab. 2019, 39, 481–496. [Google Scholar] [CrossRef]
- Hammond, T.R.; Gadea, A.; Dupree, J.; Kerninon, C.; Nait-Oumesmar, B.; Aguirre, A.; Gallo, V. Astrocyte-derived endothelin-1 inhibits remyelination through notch activation. Neuron 2014, 81, 588–602. [Google Scholar] [CrossRef] [Green Version]
- Lo, A.C.; Chen, A.Y.; Hung, V.K.; Yaw, L.P.; Fung, M.K.; Ho, M.C.; Tsang, M.C.; Chung, S.S.; Chung, S.K. Endothelin-1 overexpression leads to further water accumulation and brain edema after middle cerebral artery occlusion via aquaporin 4 expression in astrocytic end-feet. J. Cereb. Blood Flow Metab. 2005, 25, 998–1011. [Google Scholar] [CrossRef] [Green Version]
- Matsuo, Y.; Mihara, S.; Ninomiya, M.; Fujimoto, M. Protective effect of endothelin type A receptor antagonist on brain edema and injury after transient middle cerebral artery occlusion in rats. Stroke 2001, 32, 2143–2148. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, J.I.; Dodelet-Devillers, A.; Kebir, H.; Ifergan, I.; Fabre, P.J.; Terouz, S.; Sabbagh, M.; Wosik, K.; Bourbonniere, L.; Bernard, M.; et al. The Hedgehog pathway promotes blood-brain barrier integrity and CNS immune quiescence. Science 2011, 334, 1727–1731. [Google Scholar] [CrossRef] [Green Version]
- Xia, Y.P.; He, Q.W.; Li, Y.N.; Chen, S.C.; Huang, M.; Wang, Y.; Gao, Y.; Huang, Y.; Wang, M.D.; Mao, L.; et al. Recombinant human sonic hedgehog protein regulates the expression of ZO-1 and occludin by activating angiopoietin-1 in stroke damage. PLoS ONE 2013, 8, e68891. [Google Scholar] [CrossRef] [Green Version]
- Zhu, S.L.; Luo, M.Q.; Peng, W.X.; Li, Q.X.; Feng, Z.Y.; Li, Z.X.; Wang, M.X.; Feng, X.X.; Liu, F.; Huang, J.L. Sonic hedgehog signalling pathway regulates apoptosis through Smo protein in human umbilical vein endothelial cells. Rheumatology 2015, 54, 1093–1102. [Google Scholar] [CrossRef] [Green Version]
- Mizee, M.R.; Nijland, P.G.; van der Pol, S.M.; Drexhage, J.A.; van Het Hof, B.; Mebius, R.; van der Valk, P.; van Horssen, J.; Reijerkerk, A.; de Vries, H.E. Astrocyte-derived retinoic acid: A novel regulator of blood-brain barrier function in multiple sclerosis. Acta Neuropathol. 2014, 128, 691–703. [Google Scholar] [CrossRef]
- Madathil, S.K.; Carlson, S.W.; Brelsfoard, J.M.; Ye, P.; D’Ercole, A.J.; Saatman, K.E. Astrocyte-Specific Overexpression of Insulin-Like Growth Factor-1 Protects Hippocampal Neurons and Reduces Behavioral Deficits following Traumatic Brain Injury in Mice. PLoS ONE 2013, 8, e67204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okoreeh, A.K.; Bake, S.; Sohrabji, F. Astrocyte-specific insulin-like growth factor-1 gene transfer in aging female rats improves stroke outcomes. Glia 2017, 65, 1043–1058. [Google Scholar] [CrossRef] [PubMed]
- Bake, S.; Okoreeh, A.K.; Alaniz, R.C.; Sohrabji, F. Insulin-Like Growth Factor (IGF)-I Modulates Endothelial Blood-Brain Barrier Function in Ischemic Middle-Aged Female Rats. Endocrinology 2016, 157, 61–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pekny, M.; Wilhelmsson, U.; Tatlisumak, T.; Pekna, M. Astrocyte activation and reactive gliosis-A new target in stroke? Neurosci. Lett. 2019, 689, 45–55. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Barres, B.A. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef] [Green Version]
- Perez-Alvarez, M.J.; Wandosell, F. Stroke and Neuroinflamation: Role of Sexual Hormones. Curr. Pharm. Des. 2016, 22, 1334–1349. [Google Scholar] [CrossRef]
- Nayak, A.R.; Kashyap, R.S.; Kabra, D.; Purohit, H.J.; Taori, G.M.; Daginawala, H.F. Time course of inflammatory cytokines in acute ischemic stroke patients and their relation to inter-alfa trypsin inhibitor heavy chain 4 and outcome. Ann. Indian Acad. Neurol. 2012, 15, 181–185. [Google Scholar] [CrossRef]
- Kuboyama, K.; Harada, H.; Tozaki-Saitoh, H.; Tsuda, M.; Ushijima, K.; Inoue, K. Astrocytic P2Y(1) receptor is involved in the regulation of cytokine/chemokine transcription and cerebral damage in a rat model of cerebral ischemia. J. Cereb. Blood Flow Metab. 2011, 31, 1930–1941. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Liu, Z.Q.; Zhou, H.; Wang, Z.L.; Tao, Y.H.; Tong, Y. P2Y1 receptor antagonists mitigate oxygen and glucose deprivationinduced astrocyte injury. Mol. Med. Rep. 2018, 17, 1819–1824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Li, Z.; Yao, Y.; Jin, W.N.; Wood, K.; Liu, Q.; Shi, F.D.; Hao, J. Astrocyte-derived interleukin-15 exacerbates ischemic brain injury via propagation of cellular immunity. Proc. Natl. Acad. Sci. USA 2017, 114, E396–E405. [Google Scholar] [CrossRef] [Green Version]
- Lee, G.A.; Lin, T.N.; Chen, C.Y.; Mau, S.Y.; Huang, W.Z.; Kao, Y.C.; Ma, R.Y.; Liao, N.S. Interleukin 15 blockade protects the brain from cerebral ischemia-reperfusion injury. Brain Behav. Immun. 2018, 73, 562–570. [Google Scholar] [CrossRef] [PubMed]
- Cekanaviciute, E.; Fathali, N.; Doyle, K.P.; Williams, A.M.; Han, J.; Buckwalter, M.S. Astrocytic transforming growth factor-beta signaling reduces subacute neuroinflammation after stroke in mice. Glia 2014, 62, 1227–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zamanian, J.L.; Xu, L.; Foo, L.C.; Nouri, N.; Zhou, L.; Giffard, R.G.; Barres, B.A. Genomic analysis of reactive astrogliosis. J. Neurosci. 2012, 32, 6391–6410. [Google Scholar] [CrossRef] [Green Version]
- Rakers, C.; Schleif, M.; Blank, N.; Matušková, H.; Ulas, T.; Händler, K.; Torres, S.V.; Schumacher, T.; Tai, K.; Schultze, J.L.; et al. Stroke target identification guided by astrocyte transcriptome analysis. Glia 2019, 67, 619–633. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009, 32, 638–647. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, N.; Nandi, S.; Garg, S.; Ghosh, S.; Ghosh, S.; Samat, R.; Ghosh, S. Targeting Chondroitin Sulfate Proteoglycans: An Emerging Therapeutic Strategy to Treat CNS Injury. ACS Chem. Neurosci. 2020, 11, 231–232. [Google Scholar] [CrossRef]
- Pekny, M.; Johansson, C.B.; Eliasson, C.; Stakeberg, J.; Wallen, A.; Perlmann, T.; Lendahl, U.; Betsholtz, C.; Berthold, C.H.; Frisen, J. Abnormal reaction to central nervous system injury in mice lacking glial fibrillary acidic protein and vimentin. J. Cell Biol. 1999, 145, 503–514. [Google Scholar] [CrossRef]
- Li, L.; Lundkvist, A.; Andersson, D.; Wilhelmsson, U.; Nagai, N.; Pardo, A.C.; Nodin, C.; Stahlberg, A.; Aprico, K.; Larsson, K.; et al. Protective role of reactive astrocytes in brain ischemia. J. Cereb. Blood Flow Metab. 2008, 28, 468–481. [Google Scholar] [CrossRef] [Green Version]
- Cho, S.; Park, E.M.; Febbraio, M.; Anrather, J.; Park, L.; Racchumi, G.; Silverstein, R.L.; Iadecola, C. The class B scavenger receptor CD36 mediates free radical production and tissue injury in cerebral ischemia. J. Neurosci. 2005, 25, 2504–2512. [Google Scholar] [CrossRef]
- Bao, Y.; Qin, L.; Kim, E.; Bhosle, S.; Guo, H.; Febbraio, M.; Haskew-Layton, R.E.; Ratan, R.; Cho, S. CD36 is involved in astrocyte activation and astroglial scar formation. J. Cereb. Blood Flow Metab. 2012, 32, 1567–1577. [Google Scholar] [CrossRef] [PubMed]
- Morizawa, Y.M.; Hirayama, Y.; Ohno, N.; Shibata, S.; Shigetomi, E.; Sui, Y.; Nabekura, J.; Sato, K.; Okajima, F.; Takebayashi, H.; et al. Author Correction: Reactive astrocytes function as phagocytes after brain ischemia via ABCA1-mediated pathway. Nature Commun. 2017, 8, 1598. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.Y.; Chung, W.S. The roles of astrocytic phagocytosis in maintaining homeostasis of brains. J. Pharmacol. Sci. 2021, 145, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.J.; Jin, K.; Mao, X.O.; Xie, L.; Greenberg, D.A. Intracerebral chondroitinase ABC and heparan sulfate proteoglycan glypican improve outcome from chronic stroke in rats. Proc. Natl. Acad. Sci. USA 2012, 109, 9155–9160. [Google Scholar] [CrossRef] [Green Version]
- Tuinstra, H.M.; Ducommun, M.M.; Briley, W.E.; Shea, L.D. Gene delivery to overcome astrocyte inhibition of axonal growth: An in vitro model of the glial scar. Biotechnol. Bioeng. 2013, 110, 947–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Overman, J.J.; Clarkson, A.N.; Wanner, I.B.; Overman, W.T.; Eckstein, I.; Maguire, J.L.; Dinov, I.D.; Toga, A.W.; Carmichael, S.T. A role for ephrin-A5 in axonal sprouting, recovery, and activity-dependent plasticity after stroke. Proc. Natl. Acad. Sci. USA 2012, 109, E2230–E2239. [Google Scholar] [CrossRef] [Green Version]
- Liauw, J.; Hoang, S.; Choi, M.; Eroglu, C.; Choi, M.; Sun, G.H.; Percy, M.; Wildman-Tobriner, B.; Bliss, T.; Guzman, R.G.; et al. Thrombospondins 1 and 2 are necessary for synaptic plasticity and functional recovery after stroke. J. Cereb. Blood Flow Metab. 2008, 28, 1722–1732. [Google Scholar] [CrossRef] [Green Version]
- Thored, P.; Arvidsson, A.; Cacci, E.; Ahlenius, H.; Kallur, T.; Darsalia, V.; Ekdahl, C.T.; Kokaia, Z.; Lindvall, O. Persistent production of neurons from adult brain stem cells during recovery after stroke. Stem Cells 2006, 24, 739–747. [Google Scholar] [CrossRef]
- Li, M.; Sun, L.; Li, Y.; Xie, C.; Wan, D.; Luo, Y. Oxygen glucose deprivation/reperfusion astrocytes promotes primary neural stem/progenitor cell proliferation by releasing high-mobility group box 1. Neurochem. Res. 2014, 39, 1440–1450. [Google Scholar] [CrossRef]
- Kinoshita, A.; Yamada, K.; Kohmura, E.; Hayakawa, T. Effect of astrocyte-derived factors on ischemic brain edema induced by rat MCA occlusion. APMIS 1990, 98, 851–857. [Google Scholar] [CrossRef]
- Zhang, J.; Yu, Z.; Yu, Z.; Yang, Z.; Zhao, H.; Liu, L.; Zhao, J. rAAV-mediated delivery of brain-derived neurotrophic factor promotes neurite outgrowth and protects neurodegeneration in focal ischemic model. Int. J. Clin. Exp. Pathol. 2011, 4, 496–504. [Google Scholar]
- Reitmeir, R.; Kilic, E.; Kilic, U.; Bacigaluppi, M.; ElAli, A.; Salani, G.; Pluchino, S.; Gassmann, M.; Hermann, D.M. Post-acute delivery of erythropoietin induces stroke recovery by promoting perilesional tissue remodelling and contralesional pyramidal tract plasticity. Brain 2011, 134, 84–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gozes, I.; Divinsky, I.; Pilzer, I.; Fridkin, M.; Brenneman, D.E.; Spier, A.D. From vasoactive intestinal peptide (VIP) through activity-dependent neuroprotective protein (ADNP) to NAP: A view of neuroprotection and cell division. J. Mol. Neurosci. 2003, 20, 315–322. [Google Scholar] [CrossRef]
- Sragovich, S.; Merenlender-Wagner, A.; Gozes, I. ADNP Plays a Key Role in Autophagy: From Autism to Schizophrenia and Alzheimer’s Disease. Bioessays 2017, 39. [Google Scholar] [CrossRef] [PubMed]
- Greggio, S.; de Paula, S.; de Oliveira, I.M.; Trindade, C.; Rosa, R.M.; Henriques, J.A.; DaCosta, J.C. NAP prevents acute cerebral oxidative stress and protects against long-term brain injury and cognitive impairment in a model of neonatal hypoxia-ischemia. Neurobiol. Dis. 2011, 44, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Leker, R.R.; Teichner, A.; Grigoriadis, N.; Ovadia, H.; Brenneman, D.E.; Fridkin, M.; Giladi, E.; Romano, J.; Gozes, I. NAP, a femtomolar-acting peptide, protects the brain against ischemic injury by reducing apoptotic death. Stroke 2002, 33, 1085–1092. [Google Scholar] [CrossRef] [Green Version]
- Magnusson, J.P.; Goritz, C.; Tatarishvili, J.; Dias, D.O.; Smith, E.M.; Lindvall, O.; Kokaia, Z.; Frisen, J. A latent neurogenic program in astrocytes regulated by Notch signaling in the mouse. Science 2014, 346, 237–241. [Google Scholar] [CrossRef]
- Guo, Z.; Zhang, L.; Wu, Z.; Chen, Y.; Wang, F.; Chen, G. In vivo direct reprogramming of reactive glial cells into functional neurons after brain injury and in an Alzheimer’s disease model. Cell Stem Cell 2014, 14, 188–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitagawa, K.; Matsumoto, M.; Tagaya, M.; Hata, R.; Ueda, H.; Niinobe, M.; Handa, N.; Fukunaga, R.; Kimura, K.; Mikoshiba, K.; et al. ‘Ischemic tolerance’ phenomenon found in the brain. Brain Res. 1990, 528, 21–24. [Google Scholar] [CrossRef] [Green Version]
- Hirayama, Y.; Ikeda-Matsuo, Y.; Notomi, S.; Enaida, H.; Kinouchi, H.; Koizumi, S. Astrocyte-mediated ischemic tolerance. J. Neurosci. 2015, 35, 3794–3805. [Google Scholar] [CrossRef]
- Baranova, O.; Miranda, L.F.; Pichiule, P.; Dragatsis, I.; Johnson, R.S.; Chavez, J.C. Neuron-specific inactivation of the hypoxia inducible factor 1 alpha increases brain injury in a mouse model of transient focal cerebral ischemia. J. Neurosci. 2007, 27, 6320–6332. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, S.; Sakakima, H.; Terashi, T.; Takada, S.; Nakanishi, K.; Kikuchi, K. Preconditioning exercise reduces brain damage and neuronal apoptosis through enhanced endogenous 14-3-3gamma after focal brain ischemia in rats. Brain Struct. Funct. 2019, 224, 727–738. [Google Scholar] [CrossRef]
- Narayanan, S.V.; Dave, K.R.; Perez-Pinzon, M.A. Ischemic Preconditioning Protects Astrocytes against Oxygen Glucose Deprivation Via the Nuclear Erythroid 2-Related Factor 2 Pathway. Transl. Stroke Res. 2018, 9, 99–109. [Google Scholar] [CrossRef]
- Narayanan, S.V.; Perez-Pinzon, M.A. Ischemic preconditioning treatment of astrocytes transfers ischemic tolerance to neurons. Cond. Med. 2017, 1, 2–8. [Google Scholar]
- Gao, C.; Li, Z.; Bai, J.; Wang, R.; Gao, L. Involvement of monocarboxylate transporters in the cross-tolerance between epilepsy and cerebral infarction: A promising choice towards new treatments. Neurosci. Lett. 2019, 707, 134305. [Google Scholar] [CrossRef]
- Xu, L.; Cao, H.; Xie, Y.; Zhang, Y.; Du, M.; Xu, X.; Ye, R.; Liu, X. Exosome-shuttled miR-92b-3p from ischemic preconditioned astrocytes protects neurons against oxygen and glucose deprivation. Brain Res. 2019, 1717, 66–73. [Google Scholar] [CrossRef]
- Carter, H.H.; Maxwell, J.D.; Hellsten, Y.; Thompson, A.; Thijssen, D.H.J.; Jones, H. The impact of acute remote ischaemic preconditioning on cerebrovascular function. Eur. J. Appl. Physiol. 2020, 120, 603–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connolly, M.; Bilgin-Freiert, A.; Ellingson, B.; Dusick, J.R.; Liebeskind, D.; Saver, J.; Gonzalez, N.R. Peripheral vascular disease as remote ischemic preconditioning, for acute stroke. Clin. Neurol. Neurosurg. 2013, 115, 2124–2129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, J.M.; McLaughlin, M.; Yool, D.; Zhang, S.-C.; Fowler, J.H.; Montague, P.; Barrie, J.A.; McCulloch, M.C.; Duncan, I.D.; Garbern, J.; et al. Oligodendroglial modulation of fast axonal transport in a mouse model of hereditary spastic paraplegia. J. Cell Biol. 2004, 166, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, S.; Gritti, L.; Crooks, D.; Dombrowski, Y. Oligodendrocytes in Development, Myelin Generation and Beyond. Cells 2019, 8, 1424. [Google Scholar] [CrossRef] [Green Version]
- Kirby, B.B.; Takada, N.; Latimer, A.J.; Shin, J.; Carney, T.J.; Kelsh, R.N.; Appel, B. In vivo time-lapse imaging shows dynamic oligodendrocyte progenitor behavior during zebrafish development. Nat. Neurosci. 2006, 9, 1506–1511. [Google Scholar] [CrossRef]
- Nishiyama, A.; Komitova, M.; Suzuki, R.; Zhu, X. Polydendrocytes (NG2 cells): Multifunctional cells with lineage plasticity. Nat. Rev. Neurosci. 2009, 10, 9–22. [Google Scholar] [CrossRef]
- Sommer, I.; Schachner, M. Monoclonal antibodies (O1 to O4) to oligodendrocyte cell surfaces: An immunocytological study in the central nervous system. Dev. Biol. 1981, 83, 311–327. [Google Scholar] [CrossRef]
- Braun, P.; Sandillon, F.; Edwards, A.; Matthieu, J.; Privat, A. Immunocytochemical localization by electron microscopy of 2′3′-cyclic nucleotide 3′-phosphodiesterase in developing oligodendrocytes of normal and mutant brain. J. Neurosci. 1988, 8, 3057–3066. [Google Scholar] [CrossRef] [Green Version]
- Brunner, C.; Lassmann, H.; Waehneldt, T.V.; Matthieu, J.-M.; Linington, C. Differential Ultrastructural Localization of Myelin Basic Protein, Myelin/Oligodendroglial Glycoprotein, and 2′,3′-Cyclic Nucleotide 3′-Phosphodiesterase in the CNS of Adult Rats. J. Neurochem. 1989, 52, 296–304. [Google Scholar] [CrossRef]
- Michalski, J.-P.; Anderson, C.; Beauvais, A.; De Repentigny, Y.; Kothary, R. The Proteolipid Protein Promoter Drives Expression outside of the Oligodendrocyte Lineage during Embryonic and Early Postnatal Development. PLoS ONE 2011, 6, e19772. [Google Scholar] [CrossRef] [Green Version]
- Trapp, B.D. Myelin-Associated Glycoprotein Location and Potential Functionsa. Ann. N. Y. Acad. Sci. 1990, 605, 29–43. [Google Scholar] [CrossRef]
- Raff, M.C.; Mirsky, R.; Fields, K.L.; Lisak, R.P.; Dorfman, S.H.; Silberberg, D.H.; Gregson, N.A.; Leibowitz, S.; Kennedy, M.C. Galactocerebroside is a specific cell-surface antigenic marker for oligodendrocytes in culture. Nature 1978, 274, 813–816. [Google Scholar] [CrossRef] [PubMed]
- Kang, Z.; Wang, C.; Zepp, J.; Wu, L.; Sun, K.; Zhao, J.; Chandrasekharan, U.; DiCorleto, P.E.; Trapp, B.D.; Ransohoff, R.M.; et al. Act1 mediates IL-17–induced EAE pathogenesis selectively in NG2+ glial cells. Nat. Neurosci. 2013, 16, 1401–1408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirby, L.; Jin, J.; Cardona, J.G.; Smith, M.D.; Martin, K.A.; Wang, J.; Strasburger, H.; Herbst, L.; Alexis, M.; Karnell, J.; et al. Oligodendrocyte precursor cells present antigen and are cytotoxic targets in inflammatory demyelination. Nat. Commun. 2019, 10, 3887. [Google Scholar] [CrossRef] [Green Version]
- Chew, L.-J.; King, W.C.; Kennedy, A.; Gallo, V. Interferon-γ inhibits cell cycle exit in differentiating oligodendrocyte progenitor cells. Glia 2005, 52, 127–143. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Kemper, A.; Dupree, J.L.; Harding, H.P.; Ron, D.; Popko, B. Interferon-γ inhibits central nervous system remyelination through a process modulated by endoplasmic reticulum stress. Brain 2006, 129, 1306–1318. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, G.; Hong, D.; Chen, F.; Ji, X.; Cao, G. White matter injury in ischemic stroke. Prog. Neurobiol. 2016, 141, 45–60. [Google Scholar] [CrossRef] [Green Version]
- Pantoni, L.; Garcia, J.H.; Gutierrez, J.A. Cerebral white matter is highly vulnerable to ischemia. Stroke 1996, 27, 1641–1646. [Google Scholar] [CrossRef]
- Chida, Y.; Kokubo, Y.; Sato, S.; Kuge, A.; Takemura, S.; Kondo, R.; Kayama, T. The alterations of oligodendrocyte, myelin in corpus callosum, and cognitive dysfunction following chronic cerebral ischemia in rats. Brain Res. 2011, 1414, 22–31. [Google Scholar] [CrossRef]
- Li, F.; Liu, W.C.; Wang, Q.; Sun, Y.; Wang, H.; Jin, X. NG2-glia cell proliferation and differentiation by glial growth factor 2 (GGF2), a strategy to promote functional recovery after ischemic stroke. Biochem. Pharmacol. 2020, 171, 113720. [Google Scholar] [CrossRef]
- Wu, M.N.; Fang, P.T.; Hung, C.H.; Hsu, C.Y.; Chou, P.S.; Yang, Y.H. The association between white matter changes and development of malignant middle cerebral artery infarction: A case-control study. Medicine 2021, 100, e25751. [Google Scholar] [CrossRef]
- Regenhardt, R.W.; Etherton, M.R.; Das, A.S.; Schirmer, M.D.; Hirsch, J.A.; Stapleton, C.J.; Patel, A.B.; Leslie-Mazwi, T.M.; Rost, N.S. White Matter Acute Infarct Volume After Thrombectomy for Anterior Circulation Large Vessel Occlusion Stroke is Associated with Long Term Outcomes. J. Stroke Cereb. Dis. 2021, 30, 105567. [Google Scholar] [CrossRef] [PubMed]
- Ahrendsen, J.T.; Grewal, H.S.; Hickey, S.P.; Culp, C.M.; Gould, E.A.; Shimizu, T.; Strnad, F.A.; Traystman, R.J.; Herson, P.S.; Macklin, W.B. Juvenile striatal white matter is resistant to ischemia-induced damage. Glia 2016, 64, 1972–1986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baltan, S.; Besancon, E.F.; Mbow, B.; Ye, Z.; Hamner, M.A.; Ransom, B.R. White Matter Vulnerability to Ischemic Injury Increases with Age Because of Enhanced Excitotoxicity. J. Neurosci. 2008, 28, 1479–1489. [Google Scholar] [CrossRef] [Green Version]
- Dai, X.; Chen, J.; Xu, F.; Zhao, J.; Cai, W.; Sun, Z.; Hitchens, T.K.; Foley, L.M.; Leak, R.K.; Chen, J.; et al. TGFα preserves oligodendrocyte lineage cells and improves white matter integrity after cerebral ischemia. J. Cereb. Blood Flow Metab. 2020, 40, 639–655. [Google Scholar] [CrossRef]
- Liu, S.; Jin, R.; Xiao, A.Y.; Zhong, W.; Li, G. Inhibition of CD147 improves oligodendrogenesis and promotes white matter integrity and functional recovery in mice after ischemic stroke. Brain Behav. Immun. 2019, 82, 13–24. [Google Scholar] [CrossRef]
- Juurlink, B.H.J. Response of Glial Cells to Ischemia: Roles of Reactive Oxygen Species and Glutathione. Neurosci. Biobehav. Rev. 1997, 21, 151–166. [Google Scholar] [CrossRef]
- Dewar, D.; Underhill, S.M.; Goldberg, M.P. Oligodendrocytes and ischemic brain injury. J. Cereb. Blood Flow Metab.: Off. J. Int. Soc. Cereb. Blood Flow Metab. 2003, 23, 263–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, H.; Hu, X.; Leak, R.K.; Shi, Y.; An, C.; Suenaga, J.; Chen, J.; Gao, Y. Demyelination as a rational therapeutic target for ischemic or traumatic brain injury. Exp. Neurol. 2015, 272, 17–25. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.R.; Helps, S.C.; Gibbins, I.L.; Nilsson, M.; Sims, N.R. Losses of NG2 and NeuN immunoreactivity but not astrocytic markers during early reperfusion following severe focal cerebral ischemia. Brain Res. 2003, 989, 221–230. [Google Scholar] [CrossRef]
- Shereen, A.; Nemkul, N.; Yang, D.; Adhami, F.; Dunn, R.S.; Hazen, M.L.; Nakafuku, M.; Ning, G.; Lindquist, D.M.; Kuan, C.-Y. Ex Vivo Diffusion Tensor Imaging and Neuropathological Correlation in a Murine Model of Hypoxia–Ischemia-Induced Thrombotic Stroke. J. Cereb. Blood Flow Metab. 2011, 31, 1155–1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imai, H.; Masayasu, H.; Dewar, D.; Graham, D.I.; Macrae, I.M. Ebselen protects both gray and white matter in a rodent model of focal cerebral ischemia. Stroke 2001, 32, 2149–2154. [Google Scholar] [CrossRef] [Green Version]
- Matute, C.; Domercq, M.; Pérez-Samartín, A.; Ransom, B.R. Protecting White Matter From Stroke Injury. Stroke 2013, 44, 1204–1211. [Google Scholar] [CrossRef]
- Bakiri, Y.; Burzomato, V.; Frugier, G.; Hamilton, N.B.; Káradóttir, R.; Attwell, D. Glutamatergic signaling in the brain’s white matter. Neuroscience 2009, 158, 266–274. [Google Scholar] [CrossRef]
- Káradóttir, R.; Cavelier, P.; Bergersen, L.H.; Attwell, D. NMDA receptors are expressed in oligodendrocytes and activated in ischaemia. Nature 2005, 438, 1162–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manning, S.M.; Talos, D.M.; Zhou, C.; Selip, D.B.; Park, H.K.; Park, C.J.; Volpe, J.J.; Jensen, F.E. NMDA receptor blockade with memantine attenuates white matter injury in a rat model of periventricular leukomalacia. J. Neurosci. 2008, 28, 6670–6678. [Google Scholar] [CrossRef] [Green Version]
- Matute, C.; Torre, I.; Pérez-Cerdá, F.; Pérez-Samartín, A.; Alberdi, E.; Etxebarria, E.; Arranz, A.M.; Ravid, R.; Rodríguez-Antigüedad, A.; Sánchez-Gómez, M.; et al. P2X(7) receptor blockade prevents ATP excitotoxicity in oligodendrocytes and ameliorates experimental autoimmune encephalomyelitis. J. Neurosci. 2007, 27, 9525–9533. [Google Scholar] [CrossRef] [PubMed]
- Domercq, M.; Perez-Samartin, A.; Aparicio, D.; Alberdi, E.; Pampliega, O.; Matute, C. P2X7 receptors mediate ischemic damage to oligodendrocytes. Glia 2010, 58, 730–740. [Google Scholar] [CrossRef] [PubMed]
- Del Puerto, A.; Fronzaroli-Molinieres, L.; Perez-Alvarez, M.J.; Giraud, P.; Carlier, E.; Wandosell, F.; Debanne, D.; Garrido, J.J. ATP-P2X7 Receptor Modulates Axon Initial Segment Composition and Function in Physiological Conditions and Brain Injury. Cereb. Cortex 2015, 25, 2282–2294. [Google Scholar] [CrossRef] [Green Version]
- Villa González, M.; Vallés-Saiz, L.; Hernández, I.H.; Avila, J.; Hernández, F.; Pérez-Alvarez, M.J. Focal cerebral ischemia induces changes in oligodendrocytic tau isoforms in the damaged area. Glia 2020, 68, 2471–2485. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Chopp, M.; Zhang, Z.G. Oligodendrogenesis after cerebral ischemia. Front. Cell Neurosci. 2013, 7, 201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mabuchi, T.; Kitagawa, K.; Ohtsuki, T.; Kuwabara, K.; Yagita, Y.; Yanagihara, T.; Hori, M.; Matsumoto, M. Contribution of microglia/macrophages to expansion of infarction and response of oligodendrocytes after focal cerebral ischemia in rats. Stroke 2000, 31, 1735–1743. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Nogawa, S.; Ito, D.; Suzuki, S.; Dembo, T.; Kosakai, A.; Fukuuchi, Y. Activation of NG2-positive oligodendrocyte progenitor cells during post-ischemic reperfusion in the rat brain. Neuroreport 2001, 12, 2169–2174. [Google Scholar] [CrossRef]
- Goldman, S.A.; Osorio, J. So many progenitors, so little myelin. Nat. Neurosci. 2014, 17, 483–485. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Hua, X.; Niu, H.; Sun, Z.; Zhang, L.; Li, Y.; Zhang, L.; Li, L. Cornel Iridoid Glycoside Protects Against White Matter Lesions Induced by Cerebral Ischemia in Rats via Activation of the Brain-Derived Neurotrophic Factor/Neuregulin-1 Pathway. Neuropsychiatr. Dis. Treat. 2019, 15, 3327–3340. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.S.; Chopp, M.; Kassis, H.; Jia, L.F.; Hozeska-Solgot, A.; Zhang, R.L.; Chen, C.; Cui, Y.S.; Zhang, Z.G. Valproic acid increases white matter repair and neurogenesis after stroke. Neuroscience 2012, 220, 313–321. [Google Scholar] [CrossRef] [Green Version]
- Kishida, N.; Maki, T.; Takagi, Y.; Yasuda, K.; Kinoshita, H.; Ayaki, T.; Noro, T.; Kinoshita, Y.; Ono, Y.; Kataoka, H.; et al. Role of Perivascular Oligodendrocyte Precursor Cells in Angiogenesis After Brain Ischemia. J. Am. Heart Assoc. 2019, 8, e011824. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Geng, J.; Qu, M.; Yuan, F.; Wang, Y.; Pan, J.; Li, Y.; Ma, Y.; Zhou, P.; Zhang, Z.; et al. Oligodendrocyte precursor cells transplantation protects blood-brain barrier in a mouse model of brain ischemia via Wnt/β-catenin signaling. Cell Death Dis. 2020, 11, 9. [Google Scholar] [CrossRef]
- Zhang, J.; Li, Y.; Zhang, Z.G.; Lu, M.; Borneman, J.; Buller, B.; Savant-Bhonsale, S.; Elias, S.B.; Chopp, M. Bone marrow stromal cells increase oligodendrogenesis after stroke. J. Cereb. Blood Flow Metab. 2009, 29, 1166–1174. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Wu, H.; Zhao, Y.; Guo, Y.; Chen, Y.; Dong, P.; Mu, Q.; Wang, X.; Wang, X. Bone marrow mesenchymal stromal cells alleviate brain white matter injury via the enhanced proliferation of oligodendrocyte progenitor cells in focal cerebral ischemic rats. Brain Res. 2018, 1680, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, Y.; Zheng, X.; Gao, Q.; Liu, Z.; Qu, R.; Borneman, J.; Elias, S.B.; Chopp, M. Bone marrow stromal cells protect oligodendrocytes from oxygen-glucose deprivation injury. J. Neurosci. Res. 2008, 86, 1501–1510. [Google Scholar] [CrossRef] [Green Version]
- Hughes, E.G.; Orthmann-Murphy, J.L.; Langseth, A.J.; Bergles, D.E. Myelin remodeling through experience-dependent oligodendrogenesis in the adult somatosensory cortex. Nat. Neurosci. 2018, 21, 696–706. [Google Scholar] [CrossRef] [PubMed]
- Xin, W.; Chan, J.R. Myelin plasticity: Sculpting circuits in learning and memory. Nat. Rev. Neurosci. 2020, 21, 682–694. [Google Scholar] [CrossRef] [PubMed]
- Pak, M.E.; Jung, D.H.; Lee, H.J.; Shin, M.J.; Kim, S.-Y.; Shin, Y.B.; Yun, Y.J.; Shin, H.K.; Choi, B.T. Combined therapy involving electroacupuncture and treadmill exercise attenuates demyelination in the corpus callosum by stimulating oligodendrogenesis in a rat model of neonatal hypoxia-ischemia. Exp. Neurol. 2018, 300, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Schabitz, W.R.; Berger, C.; Kollmar, R.; Seitz, M.; Tanay, E.; Kiessling, M.; Schwab, S.; Sommer, C. Effect of brain-derived neurotrophic factor treatment and forced arm use on functional motor recovery after small cortical ischemia. Stroke 2004, 35, 992–997. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, N.; Maki, T.; Shindo, A.; Liang, A.C.; Maeda, M.; Egawa, N.; Itoh, K.; Lo, E.K.; Lok, J.; Ihara, M.; et al. Astrocytes Promote Oligodendrogenesis after White Matter Damage via Brain-Derived Neurotrophic Factor. J. Neurosci. 2015, 35, 14002–14008. [Google Scholar] [CrossRef] [Green Version]
- Magami, S.; Miyamoto, N.; Ueno, Y.; Hira, K.; Tanaka, R.; Yamashiro, K.; Oishi, H.; Arai, H.; Urabe, T.; Hattori, N. The Effects of Astrocyte and Oligodendrocyte Lineage Cell Interaction on White Matter Injury under Chronic Cerebral Hypoperfusion. Neuroscience 2019, 406, 167–175. [Google Scholar] [CrossRef]
- Sozmen, E.G.; DiTullio, D.J.; Rosenzweig, S.; Hinman, J.D.; Bridges, S.P.; Marin, M.A.; Kawaguchi, R.; Coppola, G.; Carmichael, S.T. White Matter Stroke Induces a Unique Oligo-Astrocyte Niche That Inhibits Recovery. J. Neurosci. 2019, 39, 9343–9359. [Google Scholar] [CrossRef]
- Miron, V.E.; Boyd, A.; Zhao, J.W.; Yuen, T.J.; Ruckh, J.M.; Shadrach, J.L.; van Wijngaarden, P.; Wagers, A.J.; Williams, A.; Franklin, R.J.M.; et al. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat. Neurosci. 2013, 16, 1211–1218. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Li, J.; Duan, Y.; Tao, Z.; Zhao, H.; Luo, Y. Effects of Erythropoietin on Gliogenesis during Cerebral Ischemic/Reperfusion Recovery in Adult Mice. Aging Dis. 2017, 8, 410–419. [Google Scholar] [CrossRef] [Green Version]
- Cai, Z.; Lin, S.; Fan, L.W.; Pang, Y.; Rhodes, P.G. Minocycline alleviates hypoxic-ischemic injury to developing oligodendrocytes in the neonatal rat brain. Neuroscience 2006, 137, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Stinear, C.M.; Lang, C.E.; Zeiler, S.; Byblow, W.D. Advances and challenges in stroke rehabilitation. Lancet Neurol. 2020, 19, 348–360. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hernández, I.H.; Villa-González, M.; Martín, G.; Soto, M.; Pérez-Álvarez, M.J. Glial Cells as Therapeutic Approaches in Brain Ischemia-Reperfusion Injury. Cells 2021, 10, 1639. https://doi.org/10.3390/cells10071639
Hernández IH, Villa-González M, Martín G, Soto M, Pérez-Álvarez MJ. Glial Cells as Therapeutic Approaches in Brain Ischemia-Reperfusion Injury. Cells. 2021; 10(7):1639. https://doi.org/10.3390/cells10071639
Chicago/Turabian StyleHernández, Ivó H., Mario Villa-González, Gerardo Martín, Manuel Soto, and María José Pérez-Álvarez. 2021. "Glial Cells as Therapeutic Approaches in Brain Ischemia-Reperfusion Injury" Cells 10, no. 7: 1639. https://doi.org/10.3390/cells10071639
APA StyleHernández, I. H., Villa-González, M., Martín, G., Soto, M., & Pérez-Álvarez, M. J. (2021). Glial Cells as Therapeutic Approaches in Brain Ischemia-Reperfusion Injury. Cells, 10(7), 1639. https://doi.org/10.3390/cells10071639