
Role of Neutrophils in Cardiac Injury and Repair Following Myocardial Infarction

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
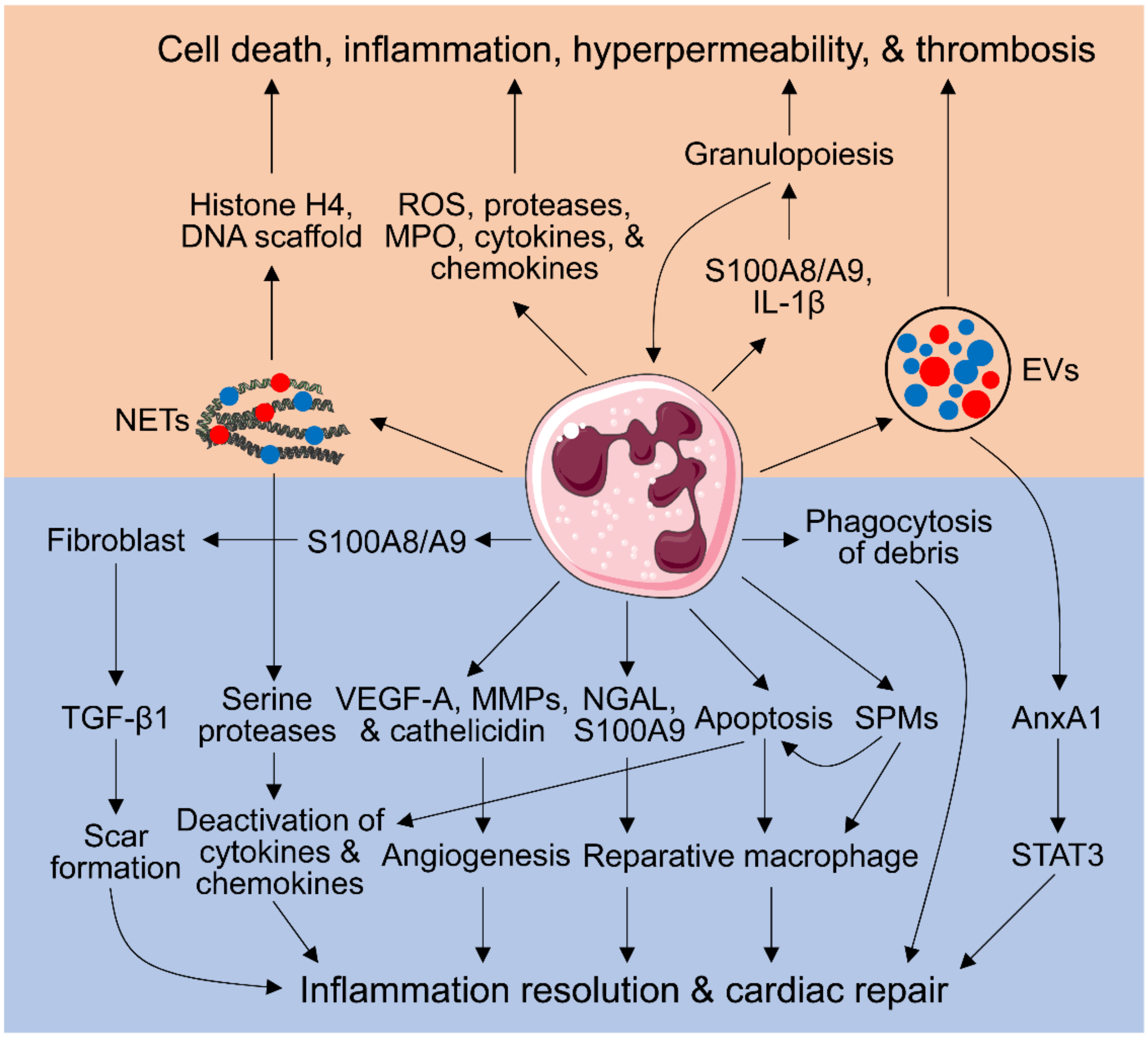
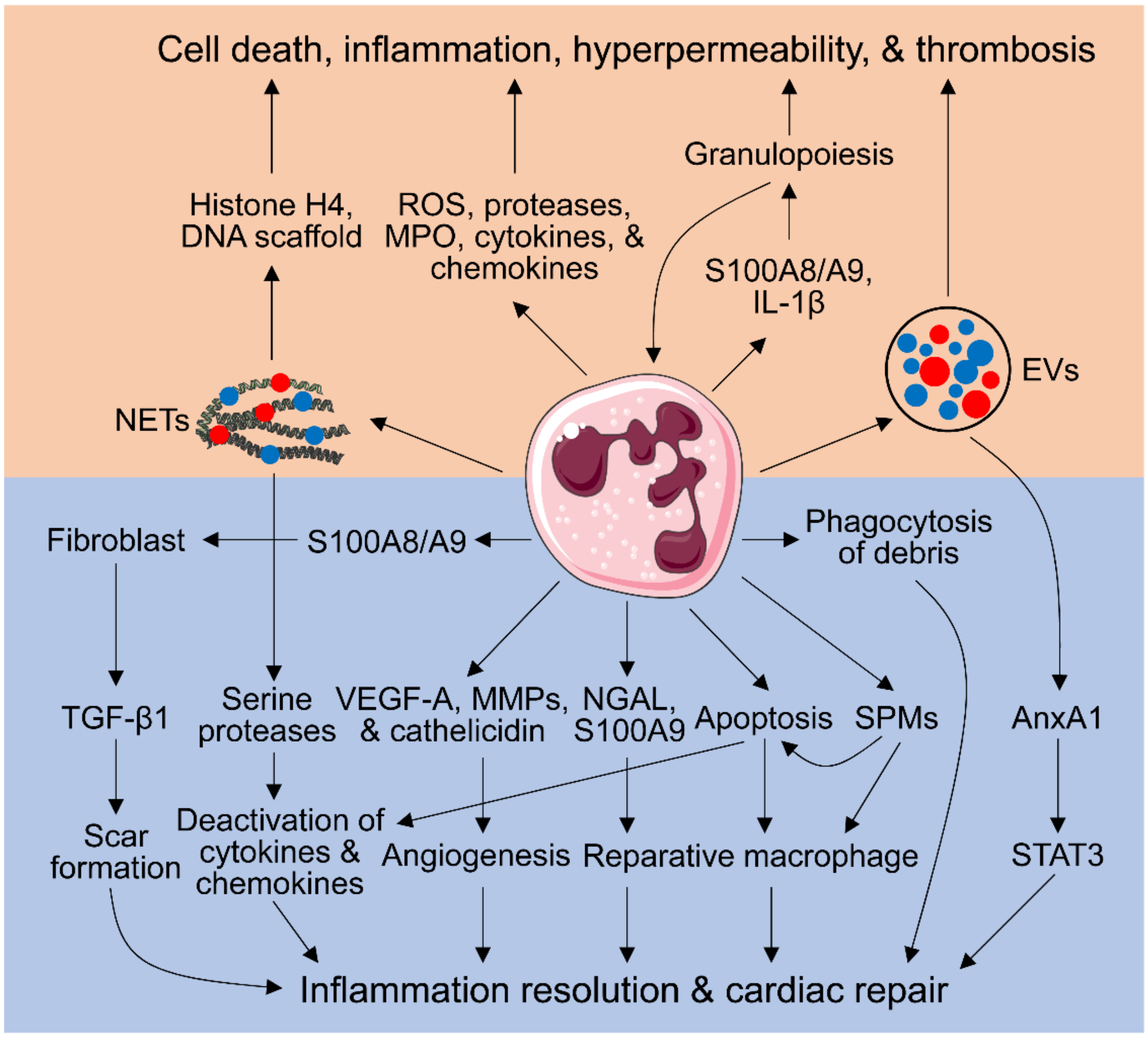
2. Neutrophil-Mediated Cardiac Injury
2.1. Neutrophil Respiratory Burst, Degranulation, Secretion of Inflammatory Mediators, and No-Reflow Induced by Neutrophils
2.2. Neutrophil Extracellular Traps (NETs)
2.3. Extracellular Vesicles (EVs)
2.4. Aggravating Granulopoiesis by Neutrophils
3. Neutrophil-Dependent Myocardial Repair
3.1. Phagocytosis of Tissue and Cellular Debris by Neutrophils
3.2. Inflammation Resolution Promoted by Apoptotic Neutrophils
3.3. Inducing a Pro-Reparative Macrophage Phenotype by Neutrophils
3.4. Pro-Angiogenic Neutrophils
3.5. Neutrophil Generation of Specialized Pro-Resolving Mediators (SPMs)
3.6. Regulation of Fibroblast Functions by Neutrophils
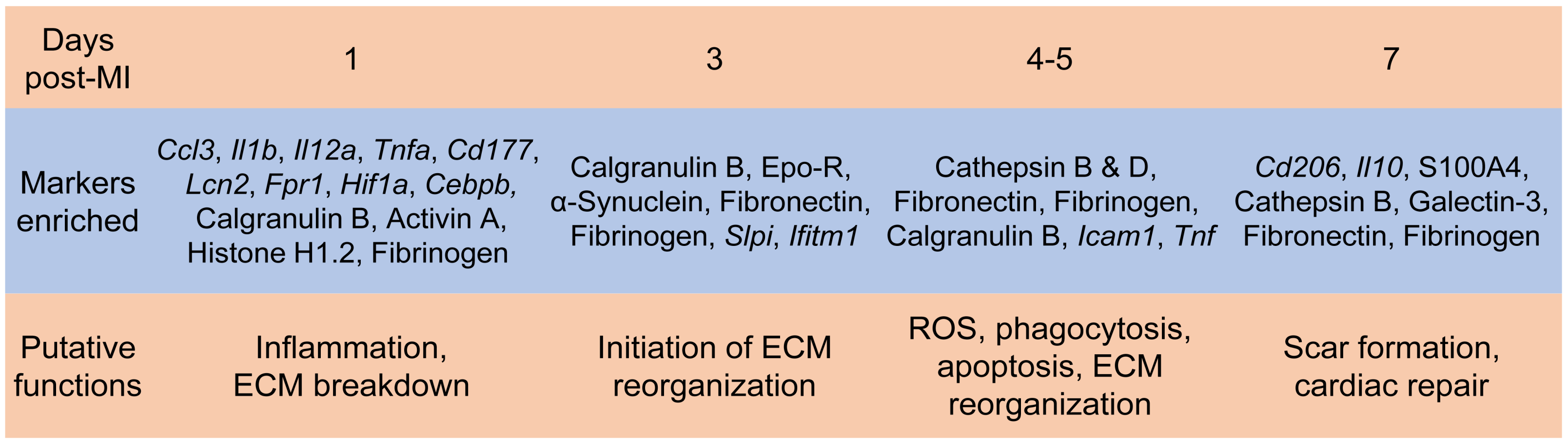
4. Neutrophil Heterogeneity and Plasticity
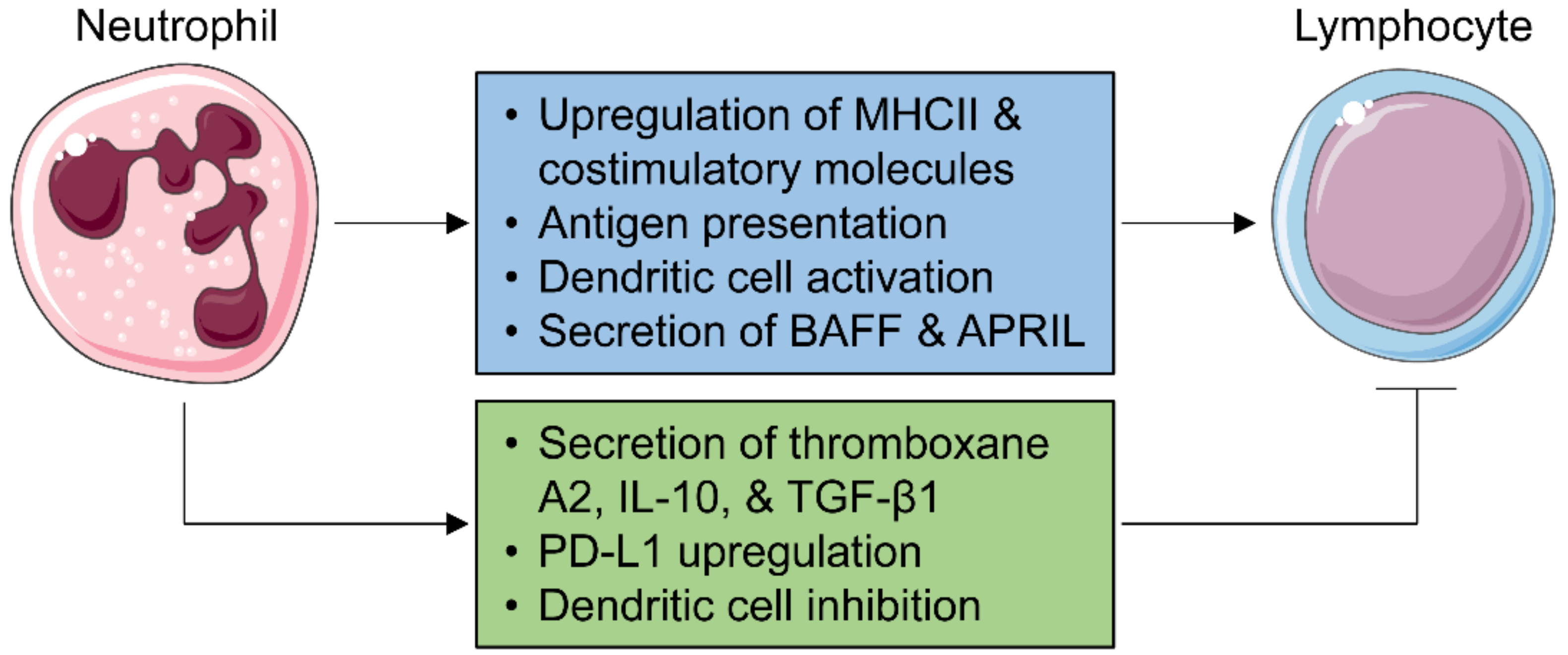
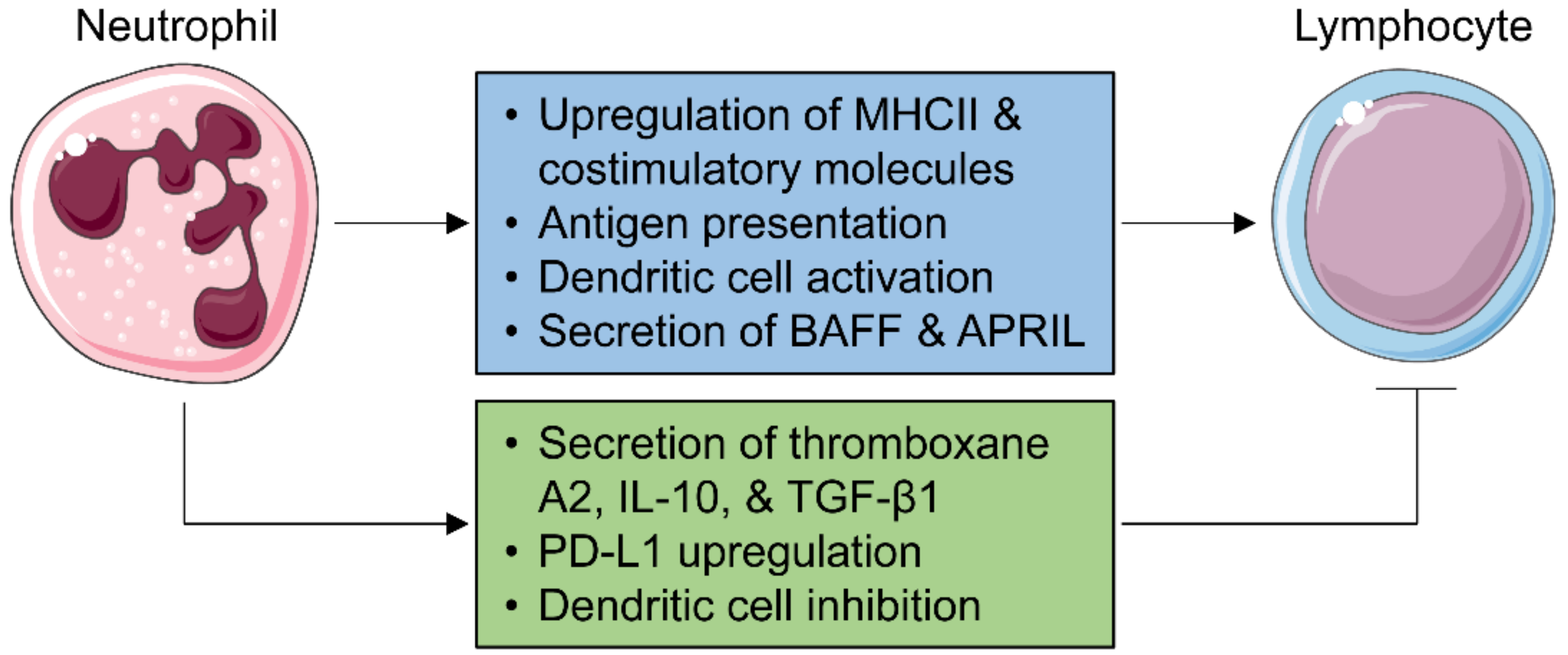
5. Neutrophils and Adaptive Immunity
6. Anti-Neutrophil Strategies and Future Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Manz, M.G.; Boettcher, S. Emergency granulopoiesis. Nat. Rev. Immunol. 2014, 14, 302–314. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.H. Homeostatic and pathogenic extramedullary hematopoiesis. J. Blood Med. 2010, 1, 13–19. [Google Scholar] [CrossRef] [Green Version]
- Eash, K.J.; Greenbaum, A.M.; Gopalan, P.K.; Link, D.C. CXCR2 and CXCR4 antagonistically regulate neutrophil trafficking from murine bone marrow. J. Clin. Investig. 2010, 120, 2423–2431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devi, S.; Wang, Y.; Chew, W.K.; Lima, R.; González, N.A.; Mattar, C.N.; Chong, S.Z.; Schlitzer, A.; Bakocevic, N.; Chew, S.; et al. Neutrophil mobilization via plerixafor-mediated CXCR4 inhibition arises from lung demargination and blockade of neutrophil homing to the bone marrow. J. Exp. Med. 2013, 210, 2321–2336. [Google Scholar] [CrossRef] [PubMed]
- Eash, K.J.; Means, J.M.; White, D.W.; Link, D.C. CXCR4 is a key regulator of neutrophil release from the bone marrow under basal and stress granulopoiesis conditions. Blood 2009, 113, 4711–4719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casanova-Acebes, M.; Nicolas-Avila, J.A.; Li, J.L.; Garcia-Silva, S.; Balachander, A.; Rubio-Ponce, A.; Weiss, L.A.; Adrover, J.M.; Burrows, K.; González, N.A.; et al. Neutrophils instruct homeostatic and pathological states in naive tissues. J. Exp. Med. 2018, 215, 2778–2795. [Google Scholar] [CrossRef]
- Ballesteros, I.; Rubio-Ponce, A.; Genua, M.; Lusito, E.; Kwok, I.; Fernandez-Calvo, G.; Khoyratty, T.E.; van Grinsven, E.; Gonzalez-Hernandez, S.; Nicolas-Avila, J.A.; et al. Co-option of Neutrophil Fates by Tissue Environments. Cell 2020, 183, 1282–1297.e1218. [Google Scholar] [CrossRef]
- Dutta, P.; Courties, G.; Wei, Y.; Leuschner, F.; Gorbatov, R.; Robbins, C.S.; Iwamoto, Y.; Thompson, B.; Carlson, A.L.; Heidt, T.; et al. Myocardial infarction accelerates atherosclerosis. Nature 2012, 487, 325–329. [Google Scholar] [CrossRef] [Green Version]
- Epelman, S.; Liu, P.P.; Mann, D.L. Role of innate and adaptive immune mechanisms in cardiac injury and repair. Nat. Rev. Immunol. 2015, 15, 117–129. [Google Scholar] [CrossRef]
- Puhl, S.L.; Steffens, S. Neutrophils in Post-myocardial Infarction Inflammation: Damage vs. Resolution? Front Cardiovasc. Med. 2019, 6, 25. [Google Scholar] [CrossRef] [Green Version]
- Peiseler, M.; Kubes, P. More friend than foe: The emerging role of neutrophils in tissue repair. J. Clin. Investig. 2019, 129, 2629–2639. [Google Scholar] [CrossRef] [Green Version]
- Futosi, K.; Fodor, S.; Mocsai, A. Reprint of Neutrophil cell surface receptors and their intracellular signal transduction pathways. Int. Immunopharmacol. 2013, 17, 1185–1197. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Yang, X.; Chatterjee, V.; Meegan, J.E.; Beard, R.S., Jr.; Yuan, S.Y. Role of Neutrophil Extracellular Traps and Vesicles in Regulating Vascular Endothelial Permeability. Front. Immunol. 2019, 10, 1037. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Yabluchanskiy, A.; Lindsey, M.L. Neutrophil roles in left ventricular remodeling following myocardial infarction. Fibrogenesis Tissue Repair 2013, 6, 11. [Google Scholar] [CrossRef] [Green Version]
- Carbone, F.; Nencioni, A.; Mach, F.; Vuilleumier, N.; Montecucco, F. Pathophysiological role of neutrophils in acute myocardial infarction. Thromb. Haemost. 2013, 110, 501–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chia, S.; Nagurney, J.T.; Brown, D.F.; Raffel, O.C.; Bamberg, F.; Senatore, F.; Wackers, F.J.; Jang, I.K. Association of leukocyte and neutrophil counts with infarct size, left ventricular function and outcomes after percutaneous coronary intervention for ST-elevation myocardial infarction. Am. J. Cardiol. 2009, 103, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Guasti, L.; Dentali, F.; Castiglioni, L.; Maroni, L.; Marino, F.; Squizzato, A.; Ageno, W.; Gianni, M.; Gaudio, G.; Grandi, A.M.; et al. Neutrophils and clinical outcomes in patients with acute coronary syndromes and/or cardiac revascularisation. A systematic review on more than 34,000 subjects. Thromb. Haemost. 2011, 106, 591–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillipson, M.; Kubes, P. The Healing Power of Neutrophils. Trends Immunol. 2019, 40, 635–647. [Google Scholar] [CrossRef]
- Dogan, I.; Karaman, K.; Sonmez, B.; Celik, S.; Turker, O. Relationship between serum neutrophil count and infarct size in patients with acute myocardial infarction. Nucl. Med. Commun. 2009, 30, 797–801. [Google Scholar] [CrossRef]
- Arruda-Olson, A.M.; Reeder, G.S.; Bell, M.R.; Weston, S.A.; Roger, V.L. Neutrophilia predicts death and heart failure after myocardial infarction: A community-based study. Circ. Cardiovasc. Qual. Outcomes 2009, 2, 656–662. [Google Scholar] [CrossRef] [Green Version]
- Romson, J.L.; Hook, B.G.; Kunkel, S.L.; Abrams, G.D.; Schork, M.A.; Lucchesi, B.R. Reduction of the extent of ischemic myocardial injury by neutrophil depletion in the dog. Circulation 1983, 67, 1016–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jolly, S.R.; Kane, W.J.; Hook, B.G.; Abrams, G.D.; Kunkel, S.L.; Lucchesi, B.R. Reduction of myocardial infarct size by neutrophil depletion: Effect of duration of occlusion. Am. Heart J. 1986, 112, 682–690. [Google Scholar] [CrossRef] [Green Version]
- Carbone, F.; Crowe, L.A.; Roth, A.; Burger, F.; Lenglet, S.; Braunersreuther, V.; Brandt, K.J.; Quercioli, A.; Mach, F.; Vallee, J.P.; et al. Treatment with anti-RANKL antibody reduces infarct size and attenuates dysfunction impacting on neutrophil-mediated injury. J. Mol. Cell Cardiol. 2016, 94, 82–94. [Google Scholar] [CrossRef] [Green Version]
- Hiroi, T.; Wajima, T.; Negoro, T.; Ishii, M.; Nakano, Y.; Kiuchi, Y.; Mori, Y.; Shimizu, S. Neutrophil TRPM2 channels are implicated in the exacerbation of myocardial ischaemia/reperfusion injury. Cardiovasc. Res. 2013, 97, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Amulic, B.; Cazalet, C.; Hayes, G.L.; Metzler, K.D.; Zychlinsky, A. Neutrophil function: From mechanisms to disease. Annu. Rev. Immunol. 2012, 30, 459–489. [Google Scholar] [CrossRef]
- Vinten-Johansen, J. Involvement of neutrophils in the pathogenesis of lethal myocardial reperfusion injury. Cardiovasc. Res. 2004, 61, 481–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiStasi, M.R.; Ley, K. Opening the flood-gates: How neutrophil-endothelial interactions regulate permeability. Trends Immunol. 2009, 30, 547–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prabhu, S.D. Cytokine-induced modulation of cardiac function. Circ. Res. 2004, 95, 1140–1153. [Google Scholar] [CrossRef]
- Niccoli, G.; Burzotta, F.; Galiuto, L.; Crea, F. Myocardial no-reflow in humans. J. Am. Coll. Cardiol. 2009, 54, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, R.; Charron, T.; Puley, G.; Dick, A.; Strauss, B.H. Microvascular obstruction and the no-reflow phenomenon after percutaneous coronary intervention. Circulation 2008, 117, 3152–3156. [Google Scholar] [CrossRef] [Green Version]
- Hansen, P.R. Role of neutrophils in myocardial ischemia and reperfusion. Circulation 1995, 91, 1872–1885. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Liu, Y.; Liu, Y.; Song, X.; Zhang, M.; Xu, F.; Yuan, F.; Lyu, S. Prognostic Association of Circulating Neutrophil Count with No-Reflow in Patients with ST-Segment Elevation Myocardial Infarction following Successful Primary Percutaneous Intervention. Dis. Markers 2017, 2017, 8458492. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2018, 18, 134–147. [Google Scholar] [CrossRef]
- Doring, Y.; Libby, P.; Soehnlein, O. Neutrophil Extracellular Traps Participate in Cardiovascular Diseases: Recent Experimental and Clinical Insights. Circ. Res. 2020, 126, 1228–1241. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Jorch, S.K.; Kubes, P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat. Med. 2017, 23, 279–287. [Google Scholar] [CrossRef]
- Bonaventura, A.; Vecchie, A.; Abbate, A.; Montecucco, F. Neutrophil Extracellular Traps and Cardiovascular Diseases: An Update. Cells 2020, 9, 231. [Google Scholar] [CrossRef] [Green Version]
- Stakos, D.A.; Kambas, K.; Konstantinidis, T.; Mitroulis, I.; Apostolidou, E.; Arelaki, S.; Tsironidou, V.; Giatromanolaki, A.; Skendros, P.; Konstantinides, S.; et al. Expression of functional tissue factor by neutrophil extracellular traps in culprit artery of acute myocardial infarction. Eur. Heart J. 2015, 36, 1405–1414. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D., Jr.; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 15880–15885. [Google Scholar] [CrossRef] [Green Version]
- Ge, L.; Zhou, X.; Ji, W.J.; Lu, R.Y.; Zhang, Y.; Zhang, Y.D.; Ma, Y.Q.; Zhao, J.H.; Li, Y.M. Neutrophil extracellular traps in ischemia-reperfusion injury-induced myocardial no-reflow: Therapeutic potential of DNase-based reperfusion strategy. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H500–H509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahlenberg, J.M.; Carmona-Rivera, C.; Smith, C.K.; Kaplan, M.J. Neutrophil extracellular trap-associated protein activation of the NLRP3 inflammasome is enhanced in lupus macrophages. J. Immunol. 2013, 190, 1217–1226. [Google Scholar] [CrossRef] [Green Version]
- Hu, Q.; Shi, H.; Zeng, T.; Liu, H.; Su, Y.; Cheng, X.; Ye, J.; Yin, Y.; Liu, M.; Zheng, H.; et al. Increased neutrophil extracellular traps activate NLRP3 and inflammatory macrophages in adult-onset Still’s disease. Arthritis Res. 2019, 21, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Yang, D.; Wang, X.; Zhu, Z.; Wang, T.; Ma, A.; Liu, P. Neutrophil extracellular traps and dsDNA predict outcomes among patients with ST-elevation myocardial infarction. Sci. Rep. 2019, 9, 11599. [Google Scholar] [CrossRef] [Green Version]
- Hofbauer, T.M.; Mangold, A.; Scherz, T.; Seidl, V.; Panzenbock, A.; Ondracek, A.S.; Muller, J.; Schneider, M.; Binder, T.; Hell, L.; et al. Neutrophil extracellular traps and fibrocytes in ST-segment elevation myocardial infarction. Basic Res. Cardiol. 2019, 114, 33. [Google Scholar] [CrossRef] [Green Version]
- Helseth, R.; Shetelig, C.; Andersen, G.O.; Langseth, M.S.; Limalanathan, S.; Opstad, T.B.; Arnesen, H.; Hoffmann, P.; Eritsland, J.; Seljeflot, I. Neutrophil Extracellular Trap Components Associate with Infarct Size, Ventricular Function, and Clinical Outcome in STEMI. Mediat. Inflamm. 2019, 2019, 7816491. [Google Scholar] [CrossRef]
- Mangold, A.; Alias, S.; Scherz, T.; Hofbauer, M.; Jakowitsch, J.; Panzenbock, A.; Simon, D.; Laimer, D.; Bangert, C.; Kammerlander, A.; et al. Coronary neutrophil extracellular trap burden and deoxyribonuclease activity in ST-elevation acute coronary syndrome are predictors of ST-segment resolution and infarct size. Circ. Res. 2015, 116, 1182–1192. [Google Scholar] [CrossRef] [Green Version]
- Savchenko, A.S.; Borissoff, J.I.; Martinod, K.; De Meyer, S.F.; Gallant, M.; Erpenbeck, L.; Brill, A.; Wang, Y.; Wagner, D.D. VWF-mediated leukocyte recruitment with chromatin decondensation by PAD4 increases myocardial ischemia/reperfusion injury in mice. Blood 2014, 123, 141–148. [Google Scholar] [CrossRef] [Green Version]
- Du, M.; Yang, W.; Schmull, S.; Gu, J.; Xue, S. Inhibition of peptidyl arginine deiminase-4 protects against myocardial infarction induced cardiac dysfunction. Int. Immunopharmacol. 2020, 78, 106055. [Google Scholar] [CrossRef]
- Silvestre-Roig, C.; Braster, Q.; Wichapong, K.; Lee, E.Y.; Teulon, J.M.; Berrebeh, N.; Winter, J.; Adrover, J.M.; Santos, G.S.; Froese, A.; et al. Externalized histone H4 orchestrates chronic inflammation by inducing lytic cell death. Nature 2019, 569, 236–240. [Google Scholar] [CrossRef]
- Schauer, C.; Janko, C.; Munoz, L.E.; Zhao, Y.; Kienhofer, D.; Frey, B.; Lell, M.; Manger, B.; Rech, J.; Naschberger, E.; et al. Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nat. Med. 2014, 20, 511–517. [Google Scholar] [CrossRef]
- Hahn, J.; Schauer, C.; Czegley, C.; Kling, L.; Petru, L.; Schmid, B.; Weidner, D.; Reinwald, C.; Biermann, M.H.C.; Blunder, S.; et al. Aggregated neutrophil extracellular traps resolve inflammation by proteolysis of cytokines and chemokines and protection from antiproteases. FASEB J. 2019, 33, 1401–1414. [Google Scholar] [CrossRef]
- Eghbalzadeh, K.; Georgi, L.; Louis, T.; Zhao, H.; Keser, U.; Weber, C.; Mollenhauer, M.; Conforti, A.; Wahlers, T.; Paunel-Gorgulu, A. Compromised Anti-inflammatory Action of Neutrophil Extracellular Traps in PAD4-Deficient Mice Contributes to Aggravated Acute Inflammation After Myocardial Infarction. Front Immunol. 2019, 10, 2313. [Google Scholar] [CrossRef]
- Mangold, A.; Hofbauer, T.M.; Ondracek, A.S.; Artner, T.; Scherz, T.; Speidl, W.S.; Krychtiuk, K.A.; Sadushi-Kolici, R.; Jakowitsch, J.; Lang, I.M. Neutrophil extracellular traps and monocyte subsets at the culprit lesion site of myocardial infarction patients. Sci. Rep. 2019, 9, 16304. [Google Scholar] [CrossRef]
- Chatterjee, V.; Yang, X.; Ma, Y.; Wu, M.H.; Yuan, S.Y. Extracellular vesicles: New players in regulating vascular barrier function. Am. J. Physiol. Heart Circ. Physiol. 2020, 319, H1181–H1196. [Google Scholar] [CrossRef] [PubMed]
- Ridger, V.C.; Boulanger, C.M.; Angelillo-Scherrer, A.; Badimon, L.; Blanc-Brude, O.; Bochaton-Piallat, M.L.; Boilard, E.; Buzas, E.I.; Caporali, A.; Dignat-George, F.; et al. Microvesicles in vascular homeostasis and diseases. Position Paper of the European Society of Cardiology (ESC) Working Group on Atherosclerosis and Vascular Biology. Thromb. Haemost. 2017, 117, 1296–1316. [Google Scholar] [CrossRef]
- Timar, C.I.; Lorincz, A.M.; Csepanyi-Komi, R.; Valyi-Nagy, A.; Nagy, G.; Buzas, E.I.; Ivanyi, Z.; Kittel, A.; Powell, D.W.; McLeish, K.R.; et al. Antibacterial effect of microvesicles released from human neutrophilic granulocytes. Blood 2013, 121, 510–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalli, J.; Montero-Melendez, T.; Norling, L.V.; Yin, X.; Hinds, C.; Haskard, D.; Mayr, M.; Perretti, M. Heterogeneity in neutrophil microparticles reveals distinct proteome and functional properties. Mol. Cell Proteom. 2013, 12, 2205–2219. [Google Scholar] [CrossRef] [Green Version]
- Mesri, M.; Altieri, D.C. Endothelial cell activation by leukocyte microparticles. J. Immunol. 1998, 161, 4382–4387. [Google Scholar] [PubMed]
- Mesri, M.; Altieri, D.C. Leukocyte microparticles stimulate endothelial cell cytokine release and tissue factor induction in a JNK1 signaling pathway. J. Biol. Chem. 1999, 274, 23111–23118. [Google Scholar] [CrossRef] [Green Version]
- Ajikumar, A.; Long, M.B.; Heath, P.R.; Wharton, S.B.; Ince, P.G.; Ridger, V.C.; Simpson, J.E. Neutrophil-Derived Microvesicle Induced Dysfunction of Brain Microvascular Endothelial Cells In Vitro. Int. J. Mol. Sci. 2019, 20, 5227. [Google Scholar] [CrossRef] [Green Version]
- Dalli, J.; Norling, L.V.; Renshaw, D.; Cooper, D.; Leung, K.Y.; Perretti, M. Annexin 1 mediates the rapid anti-inflammatory effects of neutrophil-derived microparticles. Blood 2008, 112, 2512–2519. [Google Scholar] [CrossRef] [Green Version]
- Headland, S.E.; Jones, H.R.; Norling, L.V.; Kim, A.; Souza, P.R.; Corsiero, E.; Gil, C.D.; Nerviani, A.; Dell’Accio, F.; Pitzalis, C.; et al. Neutrophil-derived microvesicles enter cartilage and protect the joint in inflammatory arthritis. Sci. Transl. Med. 2015, 7, 315ra190. [Google Scholar] [CrossRef]
- Zhao, C.; Zhang, B.; Jiang, J.; Wang, Y.; Wu, Y. Up-regulation of ANXA1 suppresses polymorphonuclear neutrophil infiltration and myeloperoxidase activity by activating STAT3 signaling pathway in rat models of myocardial ischemia-reperfusion injury. Cell Signal 2019, 62, 109325. [Google Scholar] [CrossRef]
- Rhys, H.I.; Dell’Accio, F.; Pitzalis, C.; Moore, A.; Norling, L.V.; Perretti, M. Neutrophil Microvesicles from Healthy Control and Rheumatoid Arthritis Patients Prevent the Inflammatory Activation of Macrophages. EBioMedicine 2018, 29, 60–69. [Google Scholar] [CrossRef] [Green Version]
- Johnson, B.L., III; Kuethe, J.W.; Caldwell, C.C. Neutrophil derived microvesicles: Emerging role of a key mediator to the immune response. Endocr. Metab. Immune Disord. Drug Targets 2014, 14, 210–217. [Google Scholar] [CrossRef] [Green Version]
- Bonaventura, A.; Montecucco, F.; Dallegri, F.; Carbone, F.; Luscher, T.F.; Camici, G.G.; Liberale, L. Novel findings in neutrophil biology and their impact on cardiovascular disease. Cardiovasc. Res. 2019, 115, 1266–1285. [Google Scholar] [CrossRef]
- Semerad, C.L.; Liu, F.; Gregory, A.D.; Stumpf, K.; Link, D.C. G-CSF is an essential regulator of neutrophil trafficking from the bone marrow to the blood. Immunity 2002, 17, 413–423. [Google Scholar] [CrossRef] [Green Version]
- Poller, W.C.; Nahrendorf, M.; Swirski, F.K. Hematopoiesis and Cardiovascular Disease. Circ. Res 2020, 126, 1061–1085. [Google Scholar] [CrossRef]
- Sreejit, G.; Abdel Latif, A.; Murphy, A.J.; Nagareddy, P.R. Emerging roles of neutrophil-borne S100A8/A9 in cardiovascular inflammation. Pharm. Res. 2020, 161, 105212. [Google Scholar] [CrossRef]
- Sreejit, G.; Abdel-Latif, A.; Athmanathan, B.; Annabathula, R.; Dhyani, A.; Noothi, S.K.; Quaife-Ryan, G.A.; Al-Sharea, A.; Pernes, G.; Dragoljevic, D.; et al. Neutrophil-Derived S100A8/A9 Amplify Granulopoiesis After Myocardial Infarction. Circulation 2020, 141, 1080–1094. [Google Scholar] [CrossRef]
- Marinkovic, G.; Grauen Larsen, H.; Yndigegn, T.; Szabo, I.A.; Mares, R.G.; de Camp, L.; Weiland, M.; Tomas, L.; Goncalves, I.; Nilsson, J.; et al. Inhibition of pro-inflammatory myeloid cell responses by short-term S100A9 blockade improves cardiac function after myocardial infarction. Eur. Heart J. 2019, 40, 2713–2723. [Google Scholar] [CrossRef]
- Li, Y.; Chen, B.; Yang, X.; Zhang, C.; Jiao, Y.; Li, P.; Liu, Y.; Li, Z.; Qiao, B.; Bond Lau, W.; et al. S100a8/a9 Signaling Causes Mitochondrial Dysfunction and Cardiomyocyte Death in Response to Ischemic/Reperfusion Injury. Circulation 2019, 140, 751–764. [Google Scholar] [CrossRef]
- Kolaczkowska, E.; Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. [Google Scholar] [CrossRef]
- Daseke, M.J., II; Chalise, U.; Becirovic-Agic, M.; Salomon, J.D.; Cook, L.M.; Case, A.J.; Lindsey, M.L. Neutrophil signaling during myocardial infarction wound repair. Cell Signal 2021, 77, 109816. [Google Scholar] [CrossRef]
- Dewitt, S.; Tian, W.; Hallett, M.B. Localised PtdIns(3,4,5)P3 or PtdIns(3,4)P2 at the phagocytic cup is required for both phagosome closure and Ca2+ signalling in HL60 neutrophils. J. Cell Sci. 2006, 119, 443–451. [Google Scholar] [CrossRef] [Green Version]
- Hsu, M.J.; Lee, S.S.; Lee, S.T.; Lin, W.W. Signaling mechanisms of enhanced neutrophil phagocytosis and chemotaxis by the polysaccharide purified from Ganoderma lucidum. Br. J. Pharm. 2003, 139, 289–298. [Google Scholar] [CrossRef] [Green Version]
- El Kebir, D.; Filep, J.G. Modulation of Neutrophil Apoptosis and the Resolution of Inflammation through beta2 Integrins. Front Immunol. 2013, 4, 60. [Google Scholar] [CrossRef] [Green Version]
- Garlichs, C.D.; Eskafi, S.; Cicha, I.; Schmeisser, A.; Walzog, B.; Raaz, D.; Stumpf, C.; Yilmaz, A.; Bremer, J.; Ludwig, J.; et al. Delay of neutrophil apoptosis in acute coronary syndromes. J. Leukoc. Biol. 2004, 75, 828–835. [Google Scholar] [CrossRef]
- Bratton, D.L.; Henson, P.M. Neutrophil clearance: When the party is over, clean-up begins. Trends Immunol. 2011, 32, 350–357. [Google Scholar] [CrossRef] [Green Version]
- Colom, B.; Bodkin, J.V.; Beyrau, M.; Woodfin, A.; Ody, C.; Rourke, C.; Chavakis, T.; Brohi, K.; Imhof, B.A.; Nourshargh, S. Leukotriene B4-Neutrophil Elastase Axis Drives Neutrophil Reverse Transendothelial Cell Migration In Vivo. Immunity 2015, 42, 1075–1086. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Hossain, M.; Thanabalasuriar, A.; Gunzer, M.; Meininger, C.; Kubes, P. Visualizing the function and fate of neutrophils in sterile injury and repair. Science 2017, 358, 111–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fullerton, J.N.; Gilroy, D.W. Resolution of inflammation: A new therapeutic frontier. Nat. Rev. Drug Discov. 2016, 15, 551–567. [Google Scholar] [CrossRef]
- Huynh, M.L.; Fadok, V.A.; Henson, P.M. Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-beta1 secretion and the resolution of inflammation. J. Clin. Investig. 2002, 109, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kim, H.J.; Yamamoto, S.; Kang, X.; Ma, X. Regulation of interleukin-10 gene expression in macrophages engulfing apoptotic cells. J. Interferon Cytokine Res. 2010, 30, 113–122. [Google Scholar] [CrossRef] [Green Version]
- Elliott, M.R.; Ravichandran, K.S. Clearance of apoptotic cells: Implications in health and disease. J. Cell Biol. 2010, 189, 1059–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, S.; Leitch, A.E.; Duffin, R.; Haslett, C.; Rossi, A.G. Neutrophil apoptosis: Relevance to the innate immune response and inflammatory disease. J. Innate Immun. 2010, 2, 216–227. [Google Scholar] [CrossRef] [Green Version]
- El Kebir, D.; Filep, J.G. Targeting neutrophil apoptosis for enhancing the resolution of inflammation. Cells 2013, 2, 330–348. [Google Scholar] [CrossRef]
- Miles, K.; Clarke, D.J.; Lu, W.; Sibinska, Z.; Beaumont, P.E.; Davidson, D.J.; Barr, T.A.; Campopiano, D.J.; Gray, M. Dying and necrotic neutrophils are anti-inflammatory secondary to the release of alpha-defensins. J. Immunol. 2009, 183, 2122–2132. [Google Scholar] [CrossRef]
- Iyer, R.P.; Patterson, N.L.; Zouein, F.A.; Ma, Y.; Dive, V.; de Castro Bras, L.E.; Lindsey, M.L. Early matrix metalloproteinase-12 inhibition worsens post-myocardial infarction cardiac dysfunction by delaying inflammation resolution. Int. J. Cardiol. 2015, 185, 198–208. [Google Scholar] [CrossRef] [Green Version]
- Lindsey, M.L.; Jung, M.; Yabluchanskiy, A.; Cannon, P.L.; Iyer, R.P.; Flynn, E.R.; DeLeon-Pennell, K.Y.; Valerio, F.M.; Harrison, C.L.; Ripplinger, C.M.; et al. Exogenous CXCL4 infusion inhibits macrophage phagocytosis by limiting CD36 signalling to enhance post-myocardial infarction cardiac dilation and mortality. Cardiovasc. Res. 2019, 115, 395–408. [Google Scholar] [CrossRef]
- Jones, H.R.; Robb, C.T.; Perretti, M.; Rossi, A.G. The role of neutrophils in inflammation resolution. Semin. Immunol. 2016, 28, 137–145. [Google Scholar] [CrossRef]
- Ariel, A.; Fredman, G.; Sun, Y.P.; Kantarci, A.; Van Dyke, T.E.; Luster, A.D.; Serhan, C.N. Apoptotic neutrophils and T cells sequester chemokines during immune response resolution through modulation of CCR5 expression. Nat. Immunol. 2006, 7, 1209–1216. [Google Scholar] [CrossRef] [Green Version]
- Farrera, C.; Fadeel, B. Macrophage clearance of neutrophil extracellular traps is a silent process. J. Immunol. 2013, 191, 2647–2656. [Google Scholar] [CrossRef] [Green Version]
- Lazzaretto, B.; Fadeel, B. Intra- and Extracellular Degradation of Neutrophil Extracellular Traps by Macrophages and Dendritic Cells. J. Immunol. 2019, 203, 2276–2290. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Mouton, A.J.; Lindsey, M.L. Cardiac macrophage biology in the steady-state heart, the aging heart, and following myocardial infarction. Transl. Res. 2018, 191, 15–28. [Google Scholar] [CrossRef] [Green Version]
- Marwick, J.A.; Mills, R.; Kay, O.; Michail, K.; Stephen, J.; Rossi, A.G.; Dransfield, I.; Hirani, N. Neutrophils induce macrophage anti-inflammatory reprogramming by suppressing NF-kappaB activation. Cell Death Dis. 2018, 9, 665. [Google Scholar] [CrossRef]
- Horckmans, M.; Ring, L.; Duchene, J.; Santovito, D.; Schloss, M.J.; Drechsler, M.; Weber, C.; Soehnlein, O.; Steffens, S. Neutrophils orchestrate post-myocardial infarction healing by polarizing macrophages towards a reparative phenotype. Eur. Heart J. 2017, 38, 187–197. [Google Scholar] [CrossRef] [Green Version]
- Wan, E.; Yeap, X.Y.; Dehn, S.; Terry, R.; Novak, M.; Zhang, S.; Iwata, S.; Han, X.; Homma, S.; Drosatos, K.; et al. Enhanced efferocytosis of apoptotic cardiomyocytes through myeloid-epithelial-reproductive tyrosine kinase links acute inflammation resolution to cardiac repair after infarction. Circ. Res. 2013, 113, 1004–1012. [Google Scholar] [CrossRef]
- Marinkovic, G.; Koenis, D.S.; de Camp, L.; Jablonowski, R.; Graber, N.; de Waard, V.; de Vries, C.J.; Goncalves, I.; Nilsson, J.; Jovinge, S.; et al. S100A9 Links Inflammation and Repair in Myocardial Infarction. Circ. Res. 2020, 127, 664–676. [Google Scholar] [CrossRef]
- Otsuka, K.; Terasaki, F.; Ikemoto, M.; Fujita, S.; Tsukada, B.; Katashima, T.; Kanzaki, Y.; Sohmiya, K.; Kono, T.; Toko, H.; et al. Suppression of inflammation in rat autoimmune myocarditis by S100A8/A9 through modulation of the proinflammatory cytokine network. Eur. J. Heart Fail 2009, 11, 229–237. [Google Scholar] [CrossRef]
- Besnier, M.; Galaup, A.; Nicol, L.; Henry, J.P.; Coquerel, D.; Gueret, A.; Mulder, P.; Brakenhielm, E.; Thuillez, C.; Germain, S.; et al. Enhanced angiogenesis and increased cardiac perfusion after myocardial infarction in protein tyrosine phosphatase 1B-deficient mice. FASEB J. 2014, 28, 3351–3361. [Google Scholar] [CrossRef]
- Zhang, X.; Zhu, D.; Wei, L.; Zhao, Z.; Qi, X.; Li, Z.; Sun, D. OSM Enhances Angiogenesis and Improves Cardiac Function after Myocardial Infarction. Biomed. Res. Int. 2015, 2015, 317905. [Google Scholar] [CrossRef]
- Taichman, N.S.; Young, S.; Cruchley, A.T.; Taylor, P.; Paleolog, E. Human neutrophils secrete vascular endothelial growth factor. J. Leukoc. Biol. 1997, 62, 397–400. [Google Scholar] [CrossRef] [PubMed]
- Leibovich, S.J.; Chen, J.F.; Pinhal-Enfield, G.; Belem, P.C.; Elson, G.; Rosania, A.; Ramanathan, M.; Montesinos, C.; Jacobson, M.; Schwarzschild, M.A.; et al. Synergistic up-regulation of vascular endothelial growth factor expression in murine macrophages by adenosine A(2A) receptor agonists and endotoxin. Am. J. Pathol. 2002, 160, 2231–2244. [Google Scholar] [CrossRef] [Green Version]
- Christoffersson, G.; Vagesjo, E.; Vandooren, J.; Liden, M.; Massena, S.; Reinert, R.B.; Brissova, M.; Powers, A.C.; Opdenakker, G.; Phillipson, M. VEGF-A recruits a proangiogenic MMP-9-delivering neutrophil subset that induces angiogenesis in transplanted hypoxic tissue. Blood 2012, 120, 4653–4662. [Google Scholar] [CrossRef]
- Yabluchanskiy, A.; Ma, Y.; Iyer, R.P.; Hall, M.E.; Lindsey, M.L. Matrix metalloproteinase-9: Many shades of function in cardiovascular disease. Physiology 2013, 28, 391–403. [Google Scholar] [CrossRef] [Green Version]
- Lindsey, M.L.; Escobar, G.P.; Dobrucki, L.W.; Goshorn, D.K.; Bouges, S.; Mingoia, J.T.; McClister, D.M., Jr.; Su, H.; Gannon, J.; MacGillivray, C.; et al. Matrix metalloproteinase-9 gene deletion facilitates angiogenesis after myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H232–H239. [Google Scholar] [CrossRef] [Green Version]
- Massena, S.; Christoffersson, G.; Vagesjo, E.; Seignez, C.; Gustafsson, K.; Binet, F.; Herrera Hidalgo, C.; Giraud, A.; Lomei, J.; Westrom, S.; et al. Identification and characterization of VEGF-A-responsive neutrophils expressing CD49d, VEGFR1, and CXCR4 in mice and humans. Blood 2015, 126, 2016–2026. [Google Scholar] [CrossRef] [PubMed]
- Christoffersson, G.; Lomei, J.; O’Callaghan, P.; Kreuger, J.; Engblom, S.; Phillipson, M. Vascular sprouts induce local attraction of proangiogenic neutrophils. J. Leukoc. Biol. 2017, 102, 741–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soehnlein, O.; Wantha, S.; Simsekyilmaz, S.; Doring, Y.; Megens, R.T.; Mause, S.F.; Drechsler, M.; Smeets, R.; Weinandy, S.; Schreiber, F.; et al. Neutrophil-derived cathelicidin protects from neointimal hyperplasia. Sci. Transl. Med. 2011, 3, 103ra198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferraro, B.; Leoni, G.; Hinkel, R.; Ormanns, S.; Paulin, N.; Ortega-Gomez, A.; Viola, J.R.; de Jong, R.; Bongiovanni, D.; Bozoglu, T.; et al. Pro-Angiogenic Macrophage Phenotype to Promote Myocardial Repair. J. Am. Coll. Cardiol. 2019, 73, 2990–3002. [Google Scholar] [CrossRef] [PubMed]
- Basil, M.C.; Levy, B.D. Specialized pro-resolving mediators: Endogenous regulators of infection and inflammation. Nat. Rev. Immunol. 2016, 16, 51–67. [Google Scholar] [CrossRef]
- Arita, M.; Bianchini, F.; Aliberti, J.; Sher, A.; Chiang, N.; Hong, S.; Yang, R.; Petasis, N.A.; Serhan, C.N. Stereochemical assignment, antiinflammatory properties, and receptor for the omega-3 lipid mediator resolvin E1. J. Exp. Med. 2005, 201, 713–722. [Google Scholar] [CrossRef]
- Krishnamoorthy, S.; Recchiuti, A.; Chiang, N.; Yacoubian, S.; Lee, C.H.; Yang, R.; Petasis, N.A.; Serhan, C.N. Resolvin D1 binds human phagocytes with evidence for proresolving receptors. Proc. Natl. Acad. Sci. USA 2010, 107, 1660–1665. [Google Scholar] [CrossRef] [Green Version]
- Chiang, N.; Dalli, J.; Colas, R.A.; Serhan, C.N. Identification of resolvin D2 receptor mediating resolution of infections and organ protection. J. Exp. Med. 2015, 212, 1203–1217. [Google Scholar] [CrossRef] [Green Version]
- Sekheri, M.; El Kebir, D.; Edner, N.; Filep, J.G. 15-Epi-LXA4 and 17-epi-RvD1 restore TLR9-mediated impaired neutrophil phagocytosis and accelerate resolution of lung inflammation. Proc. Natl. Acad. Sci. USA 2020, 117, 7971–7980. [Google Scholar] [CrossRef] [Green Version]
- Kain, V.; Halade, G.V. Role of neutrophils in ischemic heart failure. Pharmacol. Ther. 2020, 205, 107424. [Google Scholar] [CrossRef]
- Dalli, J.; Chiang, N.; Serhan, C.N. Elucidation of novel 13-series resolvins that increase with atorvastatin and clear infections. Nat. Med. 2015, 21, 1071–1075. [Google Scholar] [CrossRef]
- Kain, V.; Jadapalli, J.K.; Tourki, B.; Halade, G.V. Inhibition of FPR2 impaired leukocytes recruitment and elicited non-resolving inflammation in acute heart failure. Pharm. Res. 2019, 146, 104295. [Google Scholar] [CrossRef]
- Ma, Y.; Iyer, R.P.; Jung, M.; Czubryt, M.P.; Lindsey, M.L. Cardiac Fibroblast Activation Post-Myocardial Infarction: Current Knowledge Gaps. Trends Pharm. Sci. 2017, 38, 448–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Halade, G.V.; Zhang, J.; Ramirez, T.A.; Levin, D.; Voorhees, A.; Jin, Y.F.; Han, H.C.; Manicone, A.M.; Lindsey, M.L. Matrix metalloproteinase-28 deletion exacerbates cardiac dysfunction and rupture after myocardial infarction in mice by inhibiting M2 macrophage activation. Circ. Res. 2013, 112, 675–688. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. The role of transforming growth factor (TGF)-beta in the infarcted myocardium. J. Thorac. Dis. 2017, 9, S52–S63. [Google Scholar] [CrossRef] [Green Version]
- Curaj, A.; Schumacher, D.; Rusu, M.; Staudt, M.; Li, X.; Simsekyilmaz, S.; Jankowski, V.; Jankowski, J.; Dumitrascu, A.R.; Hausenloy, D.J.; et al. Neutrophils Modulate Fibroblast Function and Promote Healing and Scar Formation after Murine Myocardial Infarction. Int. J. Mol. Sci. 2020, 21, 3685. [Google Scholar] [CrossRef] [PubMed]
- Chrysanthopoulou, A.; Mitroulis, I.; Apostolidou, E.; Arelaki, S.; Mikroulis, D.; Konstantinidis, T.; Sivridis, E.; Koffa, M.; Giatromanolaki, A.; Boumpas, D.T.; et al. Neutrophil extracellular traps promote differentiation and function of fibroblasts. J. Pathol. 2014, 233, 294–307. [Google Scholar] [CrossRef]
- Wu, Y.; Li, Y.; Zhang, C.; Wang, Y.; Cui, W.; Li, H.; Du, J. S100a8/a9 released by CD11b+Gr1+ neutrophils activates cardiac fibroblasts to initiate angiotensin II-Induced cardiac inflammation and injury. Hypertension 2014, 63, 1241–1250. [Google Scholar] [CrossRef] [Green Version]
- Silvestre-Roig, C.; Hidalgo, A.; Soehnlein, O. Neutrophil heterogeneity: Implications for homeostasis and pathogenesis. Blood 2016, 127, 2173–2181. [Google Scholar] [CrossRef] [Green Version]
- Filep, J.G.; Ariel, A. Neutrophil heterogeneity and fate in inflamed tissues: Implications for the resolution of inflammation. Am. J. Physiol. Cell Physiol. 2020, 319, C510–C532. [Google Scholar] [CrossRef]
- Liew, P.X.; Kubes, P. The Neutrophil’s Role During Health and Disease. Physiol. Rev. 2019, 99, 1223–1248. [Google Scholar] [CrossRef]
- Ma, Y.; Yabluchanskiy, A.; Iyer, R.P.; Cannon, P.L.; Flynn, E.R.; Jung, M.; Henry, J.; Cates, C.A.; Deleon-Pennell, K.Y.; Lindsey, M.L. Temporal neutrophil polarization following myocardial infarction. Cardiovasc. Res. 2016, 110, 51–61. [Google Scholar] [CrossRef] [Green Version]
- Daseke, M.J., II; Tenkorang-Impraim, M.A.A.; Ma, Y.; Chalise, U.; Konfrst, S.R.; Garrett, M.R.; DeLeon-Pennell, K.Y.; Lindsey, M.L. Exogenous IL-4 shuts off pro-inflammation in neutrophils while stimulating anti-inflammation in macrophages to induce neutrophil phagocytosis following myocardial infarction. J. Mol. Cell Cardiol. 2020, 145, 112–121. [Google Scholar] [CrossRef]
- Daseke, M.J., II; Valerio, F.M.; Kalusche, W.J.; Ma, Y.; DeLeon-Pennell, K.Y.; Lindsey, M.L. Neutrophil proteome shifts over the myocardial infarction time continuum. Basic Res. Cardiol. 2019, 114, 37. [Google Scholar] [CrossRef] [Green Version]
- Vafadarnejad, E.; Rizzo, G.; Krampert, L.; Arampatzi, P.; Arias-Loza, A.P.; Nazzal, Y.; Rizakou, A.; Knochenhauer, T.; Bandi, S.R.; Nugroho, V.A.; et al. Dynamics of Cardiac Neutrophil Diversity in Murine Myocardial Infarction. Circ. Res. 2020, 127, e232–e249. [Google Scholar] [CrossRef]
- Calcagno, D.M.; Zhang, C.; Toomu, A.; Huang, K.; Ninh, V.K.; Miyamoto, S.; Aguirre, A.D.; Fu, Z.; Heller Brown, J.; King, K.R. SiglecF(HI) Marks Late-Stage Neutrophils of the Infarcted Heart: A Single-Cell Transcriptomic Analysis of Neutrophil Diversification. J. Am. Heart Assoc. 2021, 10, e019019. [Google Scholar] [CrossRef]
- Mao, H.; Kano, G.; Hudson, S.A.; Brummet, M.; Zimmermann, N.; Zhu, Z.; Bochner, B.S. Mechanisms of Siglec-F-induced eosinophil apoptosis: A role for caspases but not for SHP-1, Src kinases, NADPH oxidase or reactive oxygen. PLoS ONE 2013, 8, e68143. [Google Scholar] [CrossRef]
- Lok, L.S.C.; Dennison, T.W.; Mahbubani, K.M.; Saeb-Parsy, K.; Chilvers, E.R.; Clatworthy, M.R. Phenotypically distinct neutrophils patrol uninfected human and mouse lymph nodes. Proc. Natl. Acad. Sci. USA 2019, 116, 19083–19089. [Google Scholar] [CrossRef] [Green Version]
- Bogoslowski, A.; Butcher, E.C.; Kubes, P. Neutrophils recruited through high endothelial venules of the lymph nodes via PNAd intercept disseminating Staphylococcus aureus. Proc. Natl. Acad. Sci. USA 2018, 115, 2449–2454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorlino, C.V.; Ranocchia, R.P.; Harman, M.F.; Garcia, I.A.; Crespo, M.I.; Moron, G.; Maletto, B.A.; Pistoresi-Palencia, M.C. Neutrophils exhibit differential requirements for homing molecules in their lymphatic and blood trafficking into draining lymph nodes. J. Immunol. 2014, 193, 1966–1974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hampton, H.R.; Bailey, J.; Tomura, M.; Brink, R.; Chtanova, T. Microbe-dependent lymphatic migration of neutrophils modulates lymphocyte proliferation in lymph nodes. Nat. Commun. 2015, 6, 7139. [Google Scholar] [CrossRef] [PubMed]
- Rigby, D.A.; Ferguson, D.J.; Johnson, L.A.; Jackson, D.G. Neutrophils rapidly transit inflamed lymphatic vessel endothelium via integrin-dependent proteolysis and lipoxin-induced junctional retraction. J. Leukoc. Biol. 2015, 98, 897–912. [Google Scholar] [CrossRef]
- Hampton, H.R.; Chtanova, T. The lymph node neutrophil. Semin. Immunol. 2016, 28, 129–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogoslowski, A.; Wijeyesinghe, S.; Lee, W.Y.; Chen, C.S.; Alanani, S.; Jenne, C.; Steeber, D.A.; Scheiermann, C.; Butcher, E.C.; Masopust, D.; et al. Neutrophils Recirculate through Lymph Nodes to Survey Tissues for Pathogens. J. Immunol. 2020, 204, 2552–2561. [Google Scholar] [CrossRef] [PubMed]
- Abi Abdallah, D.S.; Egan, C.E.; Butcher, B.A.; Denkers, E.Y. Mouse neutrophils are professional antigen-presenting cells programmed to instruct Th1 and Th17 T-cell differentiation. Int. Immunol. 2011, 23, 317–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vono, M.; Lin, A.; Norrby-Teglund, A.; Koup, R.A.; Liang, F.; Lore, K. Neutrophils acquire the capacity for antigen presentation to memory CD4(+) T cells in vitro and ex vivo. Blood 2017, 129, 1991–2001. [Google Scholar] [CrossRef] [Green Version]
- Bennouna, S.; Bliss, S.K.; Curiel, T.J.; Denkers, E.Y. Cross-talk in the innate immune system: Neutrophils instruct recruitment and activation of dendritic cells during microbial infection. J. Immunol. 2003, 171, 6052–6058. [Google Scholar] [CrossRef] [Green Version]
- Van Gisbergen, K.P.; Sanchez-Hernandez, M.; Geijtenbeek, T.B.; van Kooyk, Y. Neutrophils mediate immune modulation of dendritic cells through glycosylation-dependent interactions between Mac-1 and DC-SIGN. J. Exp. Med. 2005, 201, 1281–1292. [Google Scholar] [CrossRef]
- Castell, S.D.; Harman, M.F.; Moron, G.; Maletto, B.A.; Pistoresi-Palencia, M.C. Neutrophils Which Migrate to Lymph Nodes Modulate CD4(+) T Cell Response by a PD-L1 Dependent Mechanism. Front. Immunol. 2019, 10, 105. [Google Scholar] [CrossRef]
- Scapini, P.; Bazzoni, F.; Cassatella, M.A. Regulation of B-cell-activating factor (BAFF)/B lymphocyte stimulator (BLyS) expression in human neutrophils. Immunol. Lett. 2008, 116, 1–6. [Google Scholar] [CrossRef]
- Huard, B.; McKee, T.; Bosshard, C.; Durual, S.; Matthes, T.; Myit, S.; Donze, O.; Frossard, C.; Chizzolini, C.; Favre, C.; et al. APRIL secreted by neutrophils binds to heparan sulfate proteoglycans to create plasma cell niches in human mucosa. J. Clin. Investig. 2008, 118, 2887–2895. [Google Scholar] [CrossRef]
- Yang, C.W.; Unanue, E.R. Neutrophils control the magnitude and spread of the immune response in a thromboxane A2-mediated process. J. Exp. Med. 2013, 210, 375–387. [Google Scholar] [CrossRef] [Green Version]
- Doz, E.; Lombard, R.; Carreras, F.; Buzoni-Gatel, D.; Winter, N. Mycobacteria-infected dendritic cells attract neutrophils that produce IL-10 and specifically shut down Th17 CD4 T cells through their IL-10 receptor. J. Immunol. 2013, 191, 3818–3826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Odobasic, D.; Kitching, A.R.; Yang, Y.; O’Sullivan, K.M.; Muljadi, R.C.; Edgtton, K.L.; Tan, D.S.; Summers, S.A.; Morand, E.F.; Holdsworth, S.R. Neutrophil myeloperoxidase regulates T-cell-driven tissue inflammation in mice by inhibiting dendritic cell function. Blood 2013, 121, 4195–4204. [Google Scholar] [CrossRef]
- De Kleijn, S.; Langereis, J.D.; Leentjens, J.; Kox, M.; Netea, M.G.; Koenderman, L.; Ferwerda, G.; Pickkers, P.; Hermans, P.W. IFN-gamma-stimulated neutrophils suppress lymphocyte proliferation through expression of PD-L1. PLoS ONE 2013, 8, e72249. [Google Scholar] [CrossRef] [Green Version]
- Kamenyeva, O.; Boularan, C.; Kabat, J.; Cheung, G.Y.; Cicala, C.; Yeh, A.J.; Chan, J.L.; Periasamy, S.; Otto, M.; Kehrl, J.H. Neutrophil recruitment to lymph nodes limits local humoral response to Staphylococcus aureus. PLoS Pathog. 2015, 11, e1004827. [Google Scholar] [CrossRef]
- Silvestre-Roig, C.; Braster, Q.; Ortega-Gomez, A.; Soehnlein, O. Neutrophils as regulators of cardiovascular inflammation. Nat. Rev. Cardiol. 2020, 17, 327–340. [Google Scholar] [CrossRef] [PubMed]
- Soehnlein, O.; Steffens, S.; Hidalgo, A.; Weber, C. Neutrophils as protagonists and targets in chronic inflammation. Nat. Rev. Immunol. 2017, 17, 248–261. [Google Scholar] [CrossRef] [PubMed]
- Stackowicz, J.; Jonsson, F.; Reber, L.L. Mouse Models and Tools for the in vivo Study of Neutrophils. Front. Immunol. 2019, 10, 3130. [Google Scholar] [CrossRef] [Green Version]
- Boivin, G.; Faget, J.; Ancey, P.B.; Gkasti, A.; Mussard, J.; Engblom, C.; Pfirschke, C.; Contat, C.; Pascual, J.; Vazquez, J.; et al. Durable and controlled depletion of neutrophils in mice. Nat. Commun. 2020, 11, 2762. [Google Scholar] [CrossRef]
- Reber, L.L.; Gillis, C.M.; Starkl, P.; Jonsson, F.; Sibilano, R.; Marichal, T.; Gaudenzio, N.; Berard, M.; Rogalla, S.; Contag, C.H.; et al. Neutrophil myeloperoxidase diminishes the toxic effects and mortality induced by lipopolysaccharide. J. Exp. Med. 2017, 214, 1249–1258. [Google Scholar] [CrossRef]
- Litt, M.R.; Jeremy, R.W.; Weisman, H.F.; Winkelstein, J.A.; Becker, L.C. Neutrophil depletion limited to reperfusion reduces myocardial infarct size after 90 min of ischemia. Evidence for neutrophil-mediated reperfusion injury. Circulation 1989, 80, 1816–1827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Prieto, J.; Villena-Gutierrez, R.; Gomez, M.; Bernardo, E.; Pun-Garcia, A.; Garcia-Lunar, I.; Crainiciuc, G.; Fernandez-Jimenez, R.; Sreeramkumar, V.; Bourio-Martinez, R.; et al. Neutrophil stunning by metoprolol reduces infarct size. Nat. Commun. 2017, 8, 14780. [Google Scholar] [CrossRef] [PubMed]
- Andreadou, I.; Cabrera-Fuentes, H.A.; Devaux, Y.; Frangogiannis, N.G.; Frantz, S.; Guzik, T.; Liehn, E.A.; Gomes, C.P.C.; Schulz, R.; Hausenloy, D.J. Immune cells as targets for cardioprotection: New players and novel therapeutic opportunities. Cardiovasc. Res. 2019, 115, 1117–1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, Y. Role of Neutrophils in Cardiac Injury and Repair Following Myocardial Infarction. Cells 2021, 10, 1676. https://doi.org/10.3390/cells10071676
Ma Y. Role of Neutrophils in Cardiac Injury and Repair Following Myocardial Infarction. Cells. 2021; 10(7):1676. https://doi.org/10.3390/cells10071676
Chicago/Turabian StyleMa, Yonggang. 2021. "Role of Neutrophils in Cardiac Injury and Repair Following Myocardial Infarction" Cells 10, no. 7: 1676. https://doi.org/10.3390/cells10071676
APA StyleMa, Y. (2021). Role of Neutrophils in Cardiac Injury and Repair Following Myocardial Infarction. Cells, 10(7), 1676. https://doi.org/10.3390/cells10071676