
PFKFB3 Inhibition Impairs Erlotinib-Induced Autophagy in NSCLCs

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Culture
2.3. Antibodies and Western Blotting
2.4. Growth Rate Assay
2.5. GFP-LC3 Visualization
2.6. Acridine Orange Staining
2.7. Statistics
3. Results
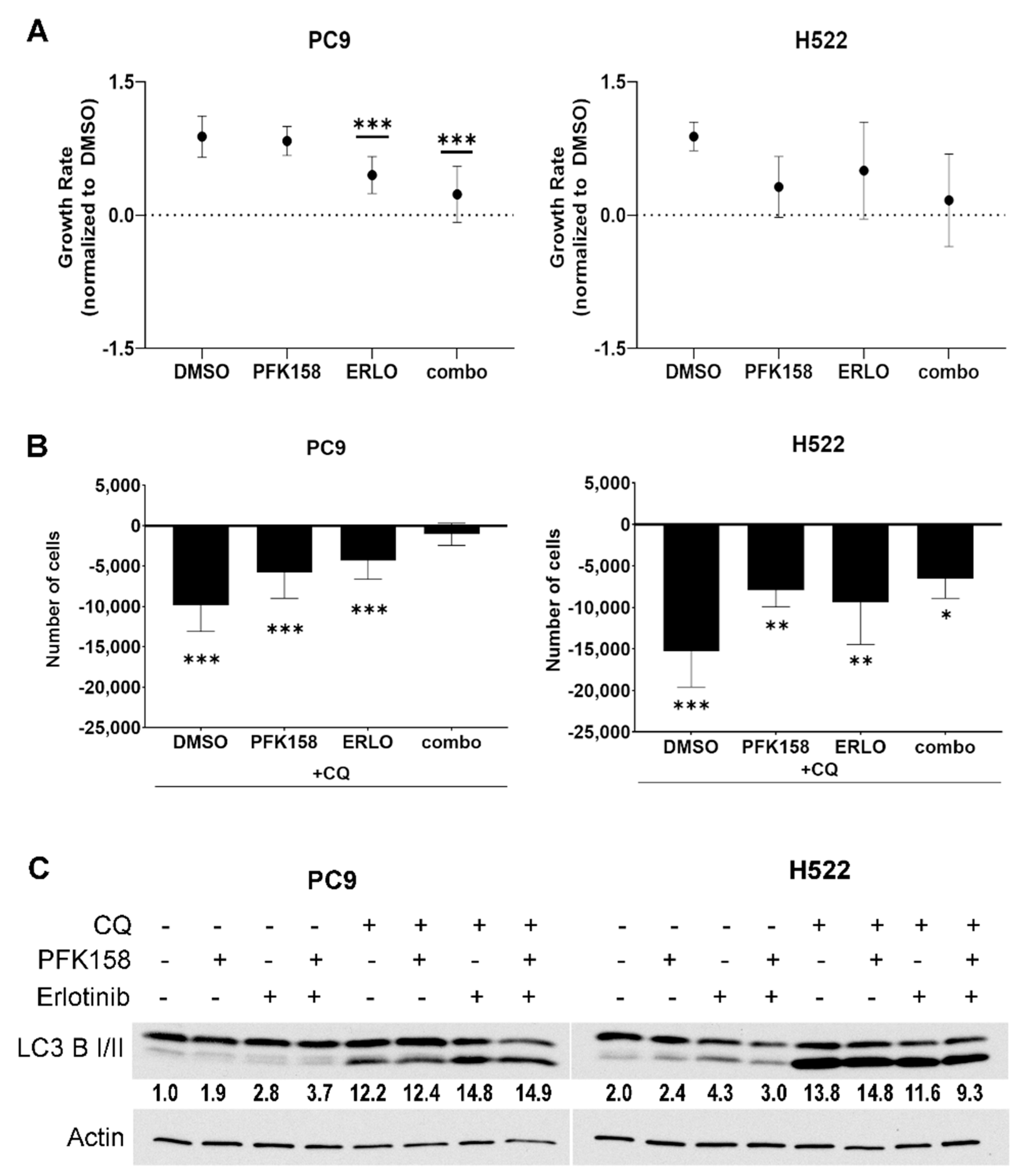
3.1. PFKFB3 Inhibition Limits the Cytotoxic Effects of Chloroquine in Erlotinib-Treated mutEGFR NSCLCs
3.2. PFKFB3 Inhibition Promotes Autophagosome Formation in Erlotinib-Treated NSCLCs
3.3. PFKFB3 Inhibition Blocks Erlotinib-Induced Turnover of p62
3.4. PFKFB3 Inhibition Promotes Accumulation of Intact Acidic Vesicular Organelles in Erlotinib-Treated NSCLC Cells
3.5. PFKFB3 Inhibition Abrogates Stress-Induced AMPK Activation in Erlotinib-Treated NSCLC Cells
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics. Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Santoni-Rugiu, E.; Melchior, L.C.; Urbanska, E.M.; Jakobsen, J.N.; Stricker, K.; Grauslund, M.; Sorensen, J.B. Intrinsic resistance to EGFR-tyrosine kinase inhibitors in EGFR-mutant non-small cell lung cancer: Differences and similarities with acquired resistance. Cancers 2019, 11, 923. [Google Scholar] [CrossRef] [Green Version]
- Mu, Y.; Hao, X.; Xing, P.; Hu, X.; Wang, Y.; Li, T.; Zhang, J.; Xu, Z.; Li, J. Acquired resistance to osimertinib in patients with non-small-cell lung cancer: Mechanisms and clinical outcomes. J. Cancer Res. Clin. Oncol. 2020, 146, 2427–2433. [Google Scholar] [CrossRef]
- Faehling, M.; Schwenk, B.; Kramberg, S.; Eckert, R.; Volckmar, A.L.; Stenzinger, A.; Strater, J. Oncogenic driver mutations, treatment, and EGFR-TKI resistance in a Caucasian population with non-small cell lung cancer: Survival in clinical practice. Oncotarget 2017, 8, 77897–77914. [Google Scholar] [CrossRef] [Green Version]
- Pao, W.; Miller, V.A.; Politi, K.A.; Riely, G.J.; Somwar, R.; Zakowski, M.F.; Kris, M.G.; Varmus, H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005, 2, e73. [Google Scholar] [CrossRef] [Green Version]
- Ward, P.S.; Thompson, C.B. Metabolic reprogramming: A cancer hallmark even warburg did not anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef] [Green Version]
- van der Steen, N.; Giovannetti, E.; Carbone, D.; Leonetti, A.; Rolfo, C.D.; Peters, G.J. Resistance to epidermal growth factor receptor inhibition in non-small cell lung cancer. Cancer Drug Resist. 2018, 1, 230–249. [Google Scholar] [CrossRef] [Green Version]
- Nakashima, N.; Liu, D.; Nakano, T.; Zhang, X.; Go, T.; Kakehi, Y.; Yokomise, H. The clinical significance of autophagy in patients with non small cell lung cancer. J. Clin. Oncol. 2018, 36, e24268. [Google Scholar] [CrossRef]
- Zhang, Z.; Lian, X.; Xie, W.; Quan, J.; Liao, M.; Wu, Y.; Yang, Z.Z.; Wang, G. Role of PARP1-mediated autophagy in EGFR-TKI resistance in non-small cell lung cancer. Sci. Rep. 2020, 10, 20924. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Lu, Y.; Pan, T.; Fan, Z. Roles of autophagy in cetuximab-mediated cancer therapy against EGFR. Autophagy 2010, 6, 1066–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fung, C.; Chen, X.; Grandis, J.R.; Duvvuri, U. EGFR tyrosine kinase inhibition induces autophagy in cancer cells. Cancer Biol. Ther. 2012, 13, 1417–1424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.Y.; Lam, S.K.; Mak, J.C.; Zheng, C.Y.; Ho, J.C. Erlotinib-induced autophagy in epidermal growth factor receptor mutated non-small cell lung cancer. Lung Cancer 2013, 81, 354–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Gao, S.; Wang, D.; Song, D.; Feng, Y. Colorectal cancer cells are resistant to anti-EGFR monoclonal antibody through adapted autophagy. Am. J. Transl. Res. 2016, 8, 1190–1196. [Google Scholar] [PubMed]
- Li, L.; Wang, Y.; Jiao, L.; Lin, C.; Lu, C.; Zhang, K.; Hu, C.; Ye, J.; Zhang, D.; Wu, H.; et al. Protective autophagy decreases osimertinib cytotoxicity through regulation of stem cell-like properties in lung cancer. Cancer Lett 2019, 452, 191–202. [Google Scholar] [CrossRef]
- Cao, Q.; You, X.; Xu, L.; Wang, L.; Chen, Y. PAQR3 suppresses the growth of non-small cell lung cancer cells via modulation of EGFR-mediated autophagy. Autophagy 2020, 16, 1236–1247. [Google Scholar] [CrossRef] [PubMed]
- Hirschey, M.D.; DeBerardinis, R.J.; Diehl, A.M.E.; Drew, J.E.; Frezza, C.; Green, M.F.; Jones, L.W.; Ko, Y.H.; Le, A.; Lea, M.A.; et al. Dysregulated metabolism contributes to oncogenesis. Semin Cancer Biol. 2015, 35, S129–S150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Schaftingen, E.; Hue, L.; Hers, H.G. Fructose 2,6-bisphosphate, the probably structure of the glucose- and glucagon-sensitive stimulator of phosphofructokinase. Biochem. J. 1980, 192, 897–901. [Google Scholar] [CrossRef] [Green Version]
- Van Schaftingen, E.; Hue, L.; Hers, H.G. Control of the fructose-6-phosphate/fructose 1,6-bisphosphate cycle in isolated hepatocytes by glucose and glucagon. Role of a low-molecular-weight stimulator of phosphofructokinase. Biochem. J. 1980, 192, 887–895. [Google Scholar] [CrossRef] [Green Version]
- Atsumi, T.; Chesney, J.; Metz, C.; Leng, L.; Donnelly, S.; Makita, Z.; Mitchell, R.; Bucala, R. High expression of inducible 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (iPFK-2; PFKFB3) in human cancers. Cancer Res. 2002, 62, 5881–5887. [Google Scholar] [PubMed]
- Vivanco, I. Targeting molecular addictions in cancer. Br. J. Cancer 2014, 111, 2033–2038. [Google Scholar] [CrossRef] [Green Version]
- Minchenko, O.; Opentanova, I.; Minchenko, D.; Ogura, T.; Esumi, H. Hypoxia induces transcription of 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase-4 gene via hypoxia-inducible factor-1alpha activation. FEBS Lett 2004, 576, 14–20. [Google Scholar] [CrossRef] [Green Version]
- Telang, S.; Yalcin, A.; Clem, A.L.; Bucala, R.; Lane, A.N.; Eaton, J.W.; Chesney, J. Ras transformation requires metabolic control by 6-phosphofructo-2-kinase. Oncogene 2006, 25, 7225–7234. [Google Scholar] [CrossRef] [Green Version]
- Cordero-Espinoza, L.; Hagen, T. Increased concentrations of fructose 2,6-bisphosphate contribute to the Warburg effect in phosphatase and tensin homolog (PTEN)-deficient cells. J. Biol. Chem. 2013, 288, 36020–36028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bando, H.; Atsumi, T.; Nishio, T.; Niwa, H.; Mishima, S.; Shimizu, C.; Yoshioka, N.; Bucala, R.; Koike, T. Phosphorylation of the 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatase/PFKFB3 family of glycolytic regulators in human cancer. Clin. Cancer Res. 2005, 11, 5784–5792. [Google Scholar] [CrossRef] [Green Version]
- O’Neal, J.; Clem, A.; Reynolds, L.; Dougherty, S.; Imbert-Fernandez, Y.; Telang, S.; Chesney, J.; Clem, B.F. Inhibition of 6-phosphofructo-2-kinase (PFKFB3) suppresses glucose metabolism and the growth of HER2+ breast cancer. Breast Cancer Res. Treat. 2016, 160, 29–40. [Google Scholar] [CrossRef]
- Klarer, A.C.; O’Neal, J.; Imbert-Fernandez, Y.; Clem, A.; Ellis, S.R.; Clark, J.; Clem, B.; Chesney, J.; Telang, S. Inhibition of 6-phosphofructo-2-kinase (PFKFB3) induces autophagy as a survival mechanism. Cancer Metab. 2014, 2, 2. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.; Jin, L.; Deng, C.; Guan, Y.; Kalogera, E.; Ray, U.; Thirusangu, P.; Staub, J.; Sarkar Bhattacharya, S.; Xu, H.; et al. Inhibition of PFKFB3 induces cell death and synergistically enhances chemosensitivity in endometrial cancer. Oncogene 2021, 40, 1409–1424. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Zhou, N.; Zhang, D.; Zhang, K.; Zheng, W.; Bao, Y.; Yang, W. PFKFB3 inhibition attenuates oxaliplatin-induced autophagy and enhances its cytotoxicity in colon cancer cells. Int. J. Mol. Sci. 2019, 20, 5415. [Google Scholar] [CrossRef] [Green Version]
- Ramirez-Peinado, S.; Leon-Annicchiarico, C.L.; Galindo-Moreno, J.; Iurlaro, R.; Caro-Maldonado, A.; Prehn, J.H.; Ryan, K.M.; Munoz-Pinedo, C. Glucose-starved cells do not engage in prosurvival autophagy. J. Biol. Chem. 2013, 288, 30387–30398. [Google Scholar] [CrossRef] [Green Version]
- Mondal, S.; Roy, D.; Sarkar Bhattacharya, S.; Jin, L.; Jung, D.; Zhang, S.; Kalogera, E.; Staub, J.; Wang, Y.; Xuyang, W.; et al. Therapeutic targeting of PFKFB3 with a novel glycolytic inhibitor PFK158 promotes lipophagy and chemosensitivity in gynecologic cancers. Int. J. Cancer 2019, 144, 178–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, S.; Wei, X.; Xu, S.; Sun, H.; Wang, W.; Liu, L.; Jiang, X.; Zhang, Y.; Che, Y. 6-Phosphofructo-2-kinase/fructose-2,6-bisphosphatase isoform 3 spatially mediates autophagy through the AMPK signaling pathway. Oncotarget 2017, 8, 80909–80922. [Google Scholar] [CrossRef] [Green Version]
- La Belle Flynn, A.; Calhoun, B.C.; Sharma, A.; Chang, J.C.; Almasan, A.; Schiemann, W.P. Autophagy inhibition elicits emergence from metastatic dormancy by inducing and stabilizing Pfkfb3 expression. Nat. Commun. 2019, 10, 3668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lypova, N.; Telang, S.; Chesney, J.; Imbert-Fernandez, Y. Increased 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3 activity in response to EGFR signaling contributes to non-small cell lung cancer cell survival. J. Biol. Chem. 2019, 294, 10530–10543. [Google Scholar] [CrossRef] [PubMed]
- Hafner, M.; Niepel, M.; Sorger, P.K. Alternative drug sensitivity metrics improve preclinical cancer pharmacogenomics. Nat. Biotechnol. 2017, 35, 500–502. [Google Scholar] [CrossRef] [PubMed]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef] [PubMed]
- Pierzynska-Mach, A.; Janowski, P.A.; Dobrucki, J.W. Evaluation of acridine orange, LysoTracker Red, and quinacrine as fluorescent probes for long-term tracking of acidic vesicles. Cytom. A 2014, 85, 729–737. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Ling, Y.H.; Sironi, J.; Schwartz, E.L.; Perez-Soler, R.; Piperdi, B. The autophagy inhibitor chloroquine overcomes the innate resistance of wild-type EGFR non-small-cell lung cancer cells to erlotinib. J. Thorac. Oncol. 2013, 8, 693–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdalla, F.C.; Abeliovich, H.; Abraham, R.T.; Acevedo-Arozena, A.; Adeli, K.; Agholme, L.; Agnello, M.; Agostinis, P.; Aguirre-Ghiso, J.A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012, 8, 445–544. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef]
- Fader, C.M.; Colombo, M.I. Autophagy and multivesicular bodies: Two closely related partners. Cell Death Differ. 2009, 16, 70–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- du Toit, A.; Hofmeyr, J.S.; Gniadek, T.J.; Loos, B. Measuring autophagosome flux. Autophagy 2018, 14, 1060–1071. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, E.E.; Pocceschi, M.G.; Kong, X.; Leeper, D.B.; Caro, J.; Limesand, K.H.; Burd, R. Control of glycolytic flux by AMP-activated protein kinase in tumor cells adapted to low pH. Transl. Oncol. 2012, 5, 208–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Mosquera, L.; Yambire, K.F.; Couto, R.; Pereyra, L.; Pabis, K.; Ponsford, A.H.; Diogo, C.V.; Stagi, M.; Milosevic, I.; Raimundo, N. Mitochondrial respiratory chain deficiency inhibits lysosomal hydrolysis. Autophagy 2019, 15, 1572–1591. [Google Scholar] [CrossRef] [Green Version]
- Jia, J.; Bissa, B.; Brecht, L.; Allers, L.; Choi, S.W.; Gu, Y.; Zbinden, M.; Burge, M.R.; Timmins, G.; Hallows, K.; et al. AMPK, a regulator of metabolism and autophagy, is activated by lysosomal damage via a novel galectin-directed ubiquitin signal transduction system. Mol. Cell 2020, 77, 951–969.e9. [Google Scholar] [CrossRef]
- Inpanathan, S.; Botelho, R.J. The Lysosome signaling platform: Adapting with the times. Front. Cell Dev. Biol. 2019, 7, 113. [Google Scholar] [CrossRef] [Green Version]
- Jang, M.; Park, R.; Kim, H.; Namkoong, S.; Jo, D.; Huh, Y.H.; Jang, I.S.; Lee, J.I.; Park, J. AMPK contributes to autophagosome maturation and lysosomal fusion. Sci. Rep. 2018, 8, 12637. [Google Scholar] [CrossRef] [Green Version]
- Tam, S.Y.; Wu, V.W.; Law, H.K. Influence of autophagy on the efficacy of radiotherapy. Radiat. Oncol. 2017, 12, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.G.; Shin, J.H.; Shim, H.S.; Lee, C.Y.; Kim, D.J.; Kim, Y.S.; Chung, K.Y. Autophagy contributes to the chemo-resistance of non-small cell lung cancer in hypoxic conditions. Respir. Res. 2015, 16, 138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, Y.; Kim, M.; Jung, H.S.; Kim, Y.; Jeoung, D. Targeting Autophagy for Overcoming Resistance to Anti-EGFR Treatments. Cancers 2019, 11, 1374. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Hu, Y.F.; Feng, X.M.; Tian, T.; Guo, Y.H.; Ma, J.W.; Nan, K.J.; Zhang, H.Y. EGFR inhibitors and autophagy in cancer treatment. Tumor Biol. 2014, 35, 11701–11709. [Google Scholar] [CrossRef]
- Henson, E.; Chen, Y.; Gibson, S. EGFR Family members’ regulation of autophagy is at a crossroads of cell survival and death in cancer. Cancers 2017, 9, 27. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Zhang, M.; Liu, H.; Yin, W. AZD9291 promotes autophagy and inhibits PI3K/Akt pathway in NSCLC cancer cells. J. Cell Biochem. 2019, 120, 756–767. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Nam, B.; Choi, Y.J.; Kim, S.Y.; Lee, J.E.; Sung, K.J.; Kim, W.S.; Choi, C.M.; Chang, E.J.; Koh, J.S.; et al. Enhanced glycolysis supports cell survival in EGFR-mutant lung adenocarcinoma by inhibiting autophagy-mediated EGFR degradation. Cancer Res. 2018, 78, 4482–4496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weihua, Z.; Tsan, R.; Huang, W.C.; Wu, Q.; Chiu, C.H.; Fidler, I.J.; Hung, M.C. Survival of cancer cells is maintained by EGFR independent of its kinase activity. Cancer Cell 2008, 13, 385–393. [Google Scholar] [CrossRef] [Green Version]
- Sakuma, Y.; Matsukuma, S.; Nakamura, Y.; Yoshihara, M.; Koizume, S.; Sekiguchi, H.; Saito, H.; Nakayama, H.; Kameda, Y.; Yokose, T.; et al. Enhanced autophagy is required for survival in EGFR-independent EGFR-mutant lung adenocarcinoma cells. Lab. Investig. 2013, 93, 1137–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, X.; Thapa, N.; Sun, Y.; Anderson, R.A. A kinase-independent role for EGF receptor in autophagy initiation. Cell 2015, 160, 145–160. [Google Scholar] [CrossRef] [Green Version]
- Almacellas, E.; Pelletier, J.; Manzano, A.; Gentilella, A.; Ambrosio, S.; Mauvezin, C.; Tauler, A. Phosphofructokinases axis controls glucose-dependent mTORC1 activation driven by E2F1. iScience 2019, 20, 434–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar Bhattacharya, S.; Thirusangu, P.; Jin, L.; Roy, D.; Jung, D.; Xiao, Y.; Staub, J.; Roy, B.; Molina, J.R.; Shridhar, V. PFKFB3 inhibition reprograms malignant pleural mesothelioma to nutrient stress-induced macropinocytosis and ER stress as independent binary adaptive responses. Cell Death Dis. 2019, 10, 725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jager, S.; Bucci, C.; Tanida, I.; Ueno, T.; Kominami, E.; Saftig, P.; Eskelinen, E.L. Role for Rab7 in maturation of late autophagic vacuoles. J. Cell Sci. 2004, 117, 4837–4848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hulsmann, H.J.; Rolff, J.; Bender, C.; Jarahian, M.; Korf, U.; Herwig, R.; Frohlich, H.; Thomas, M.; Merk, J.; Fichtner, I.; et al. Activation of AMP-activated protein kinase sensitizes lung cancer cells and H1299 xenografts to erlotinib. Lung Cancer 2014, 86, 151–157. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.M.; Yun, M.R.; Hong, Y.K.; Solca, F.; Kim, J.H.; Kim, H.J.; Cho, B.C. Glycolysis inhibition sensitizes non-small cell lung cancer with T790M mutation to irreversible EGFR inhibitors via translational suppression of Mcl-1 by AMPK activation. Mol. Cancer 2013, 12, 2145–2156. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Itahana, Y.; Ong, C.C.; Itahana, K. A redox-dependent mechanism for AMPK dysregulation interrupts metabolic adaptation of cancer under glucose deprivation. bioRxiv 2021. [Google Scholar] [CrossRef]
- Ren, Y.; Chen, J.; Chen, P.; Hao, Q.; Cheong, L.K.; Tang, M.; Hong, L.L.; Hu, X.Y.; Celestial, T.Y.; Bay, B.H.; et al. Oxidative stress-mediated AMPK inactivation determines the high susceptibility of LKB1-mutant NSCLC cells to glucose starvation. Free Radic. Biol. Med. 2021, 166, 128–139. [Google Scholar] [CrossRef]
- Kaminskyy, V.O.; Piskunova, T.; Zborovskaya, I.B.; Tchevkina, E.M.; Zhivotovsky, B. Suppression of basal autophagy reduces lung cancer cell proliferation and enhances caspase-dependent and -independent apoptosis by stimulating ROS formation. Autophagy 2012, 8, 1032–1044. [Google Scholar] [CrossRef]
- Santana-Codina, N.; Mancias, J.D.; Kimmelman, A.C. The role of autophagy in cancer. Annu. Rev. Cancer Biol. 2017, 1, 19–39. [Google Scholar] [CrossRef]
- Marinkovic, M.; Sprung, M.; Buljubasic, M.; Novak, I. Autophagy modulation in cancer: Current knowledge on action and therapy. Oxid. Med. Cell Longev. 2018, 2018, 8023821. [Google Scholar] [CrossRef] [Green Version]
- Poillet-Perez, L.; White, E. Role of tumor and host autophagy in cancer metabolism. Genes Dev. 2019, 33, 610–619. [Google Scholar] [CrossRef] [Green Version]
- Amaravadi, R.K.; Kimmelman, A.C.; Debnath, J. Targeting autophagy in cancer: Recent advances and future directions. Cancer Discov. 2019, 9, 1167–1181. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lypova, N.; Dougherty, S.M.; Lanceta, L.; Chesney, J.; Imbert-Fernandez, Y. PFKFB3 Inhibition Impairs Erlotinib-Induced Autophagy in NSCLCs. Cells 2021, 10, 1679. https://doi.org/10.3390/cells10071679
Lypova N, Dougherty SM, Lanceta L, Chesney J, Imbert-Fernandez Y. PFKFB3 Inhibition Impairs Erlotinib-Induced Autophagy in NSCLCs. Cells. 2021; 10(7):1679. https://doi.org/10.3390/cells10071679
Chicago/Turabian StyleLypova, Nadiia, Susan M. Dougherty, Lilibeth Lanceta, Jason Chesney, and Yoannis Imbert-Fernandez. 2021. "PFKFB3 Inhibition Impairs Erlotinib-Induced Autophagy in NSCLCs" Cells 10, no. 7: 1679. https://doi.org/10.3390/cells10071679