
Optical Microscopy and the Extracellular Matrix Structure: A Review

Abstract
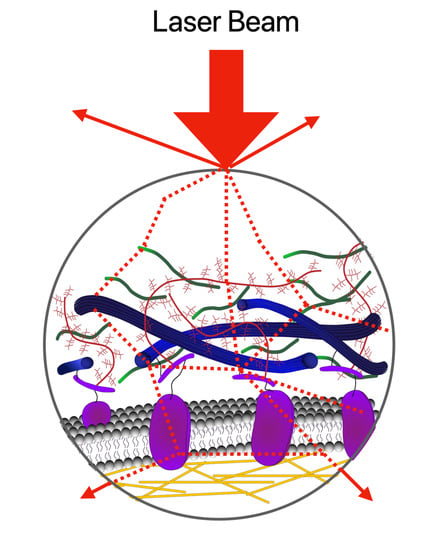
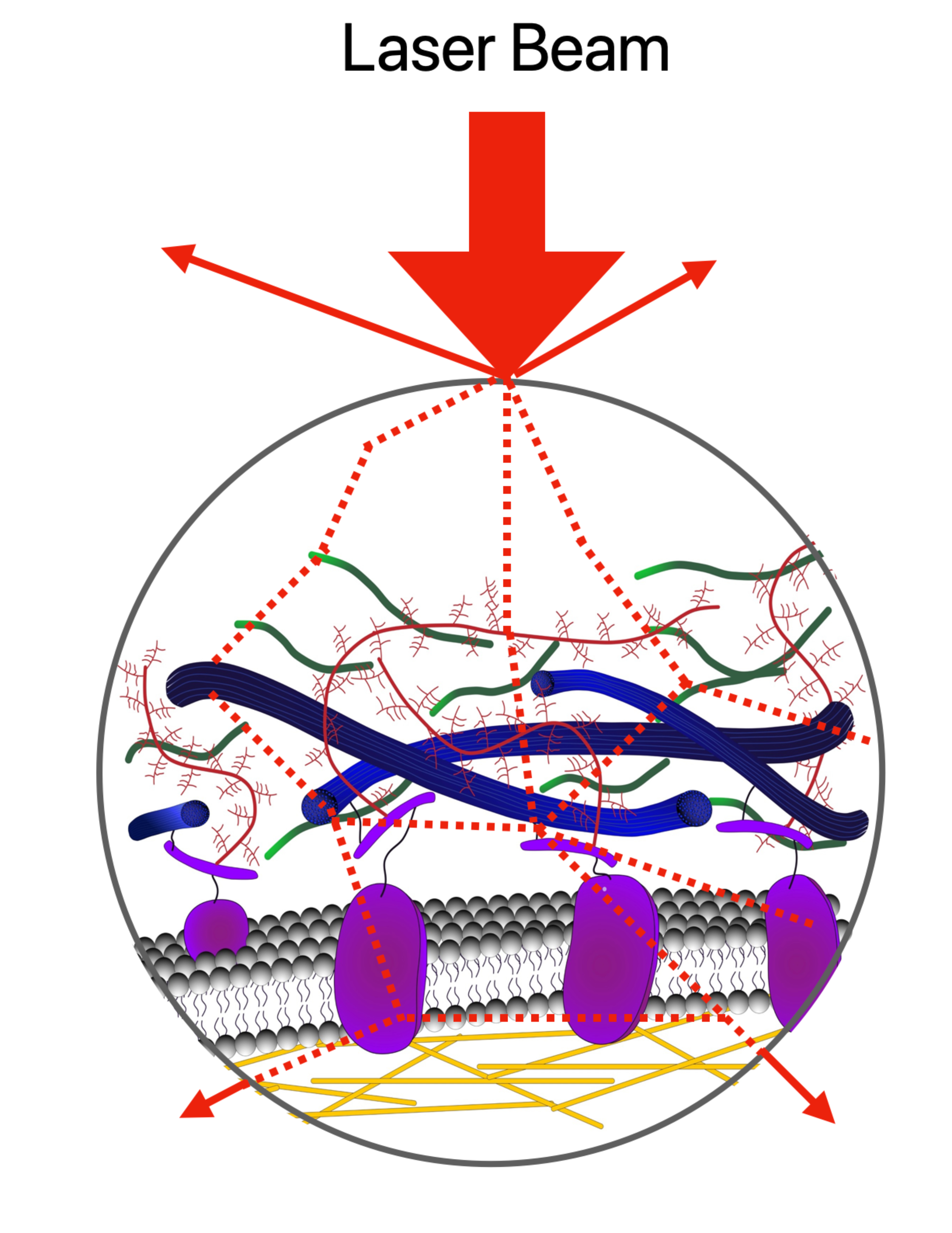
:
1. Introduction
1.1. ECM Fibrous Proteins: Collagens, Elastins, Fibronectins, and Laminins
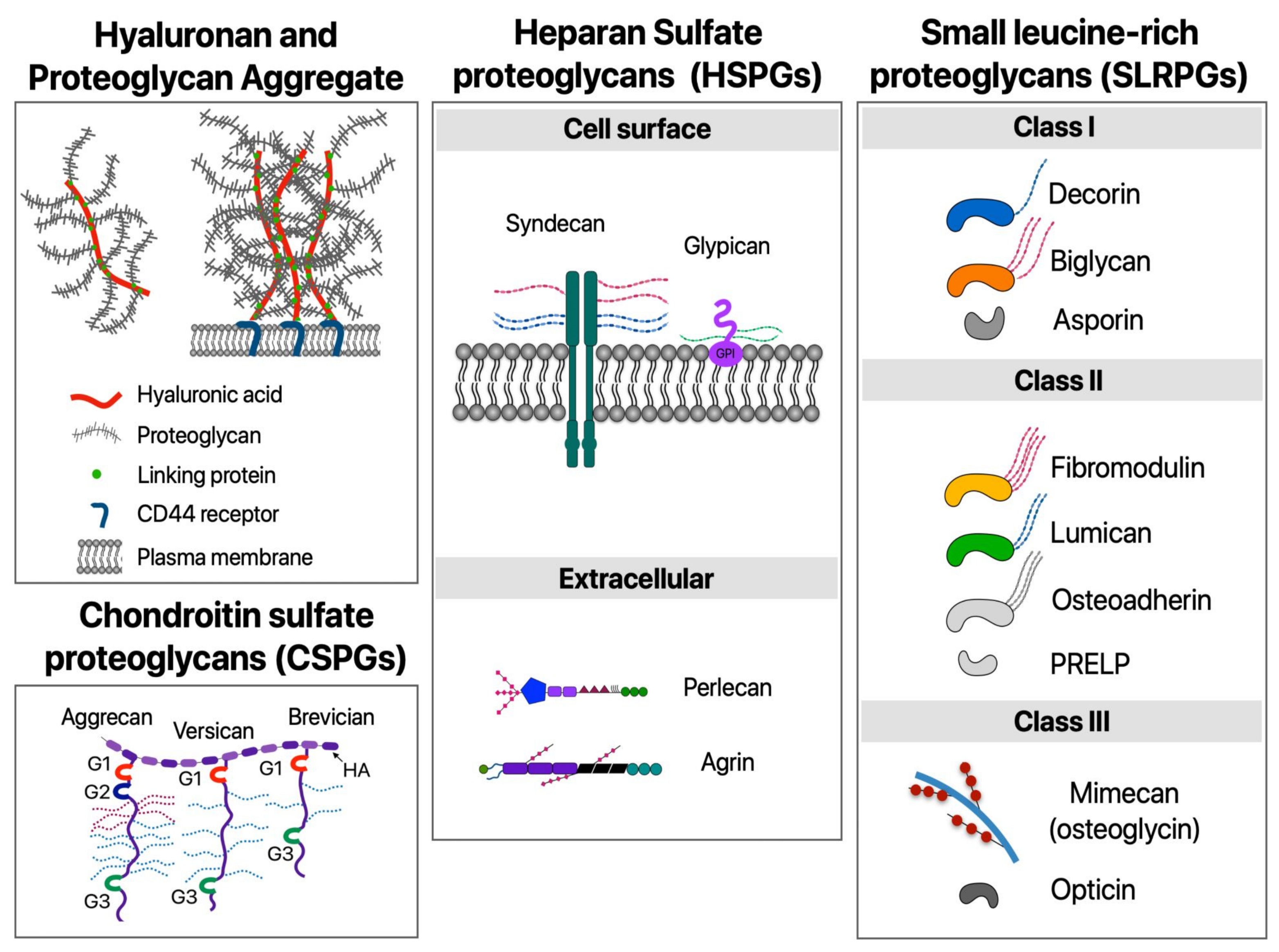
1.2. ECM Proteoglycans and Hyaluronan
1.3. The Role of the ECM in Tissue Repair and Chronic Diseases
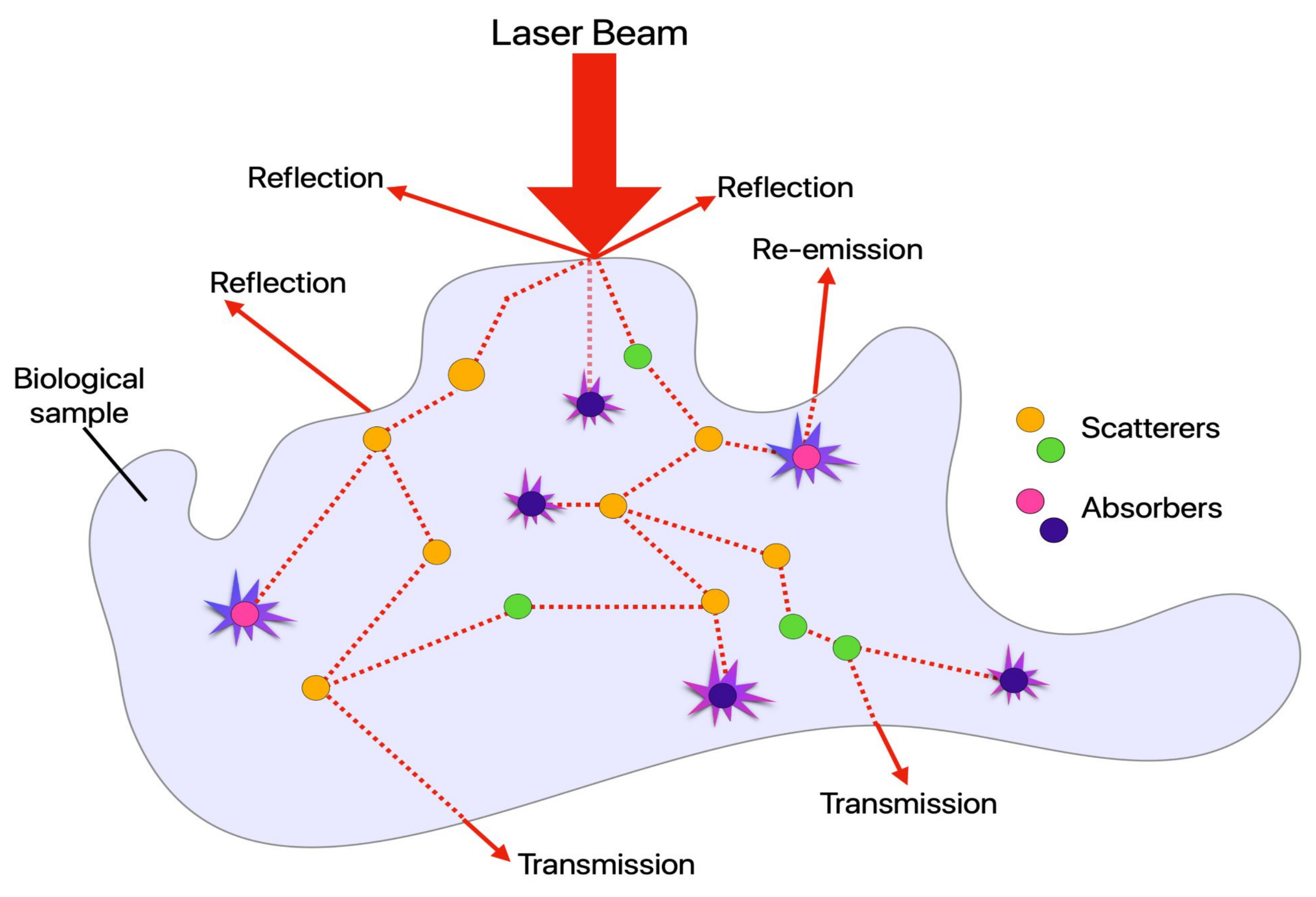
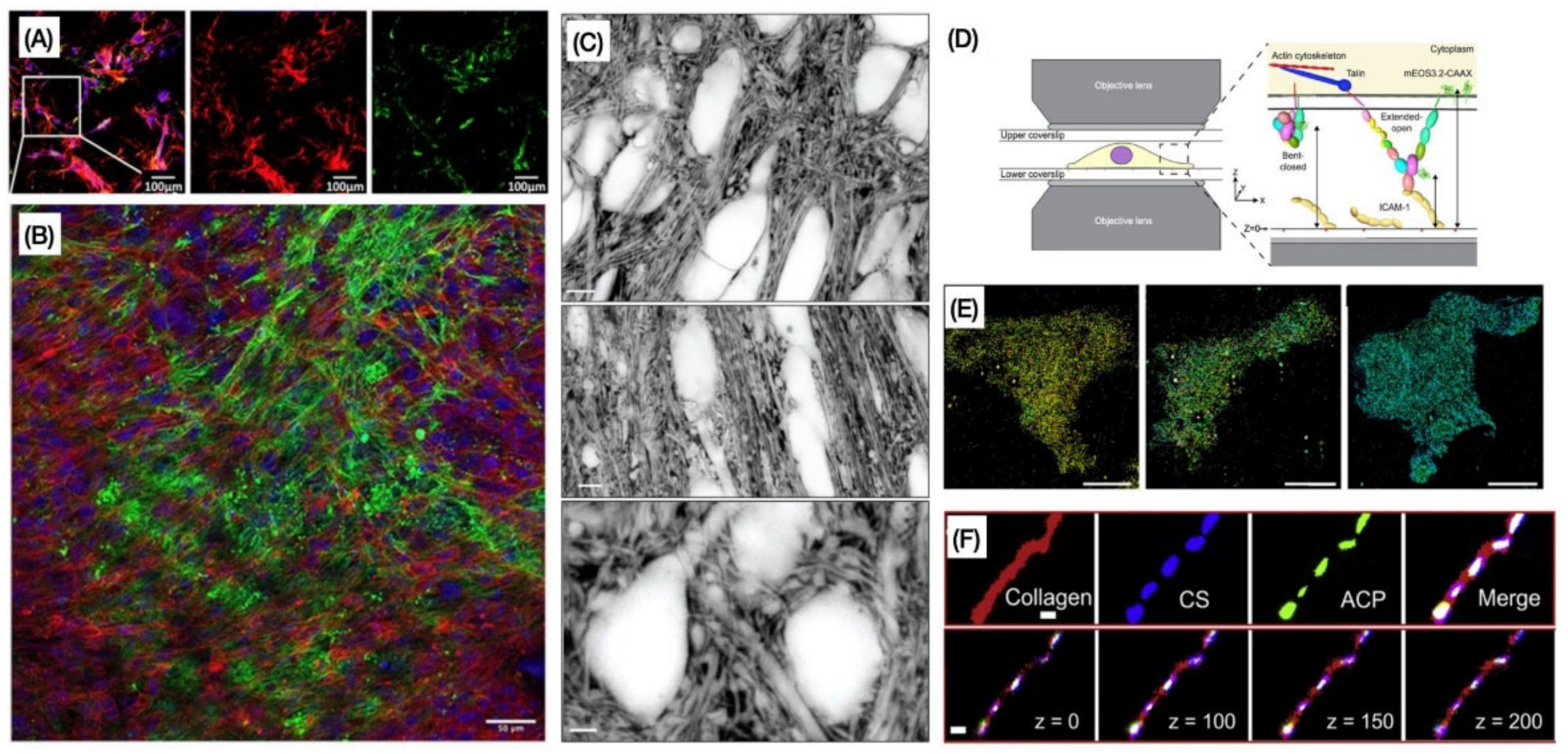
2. Imaging the ECM Components: Past, Present, and Future Challenges
2.1. Absorption, Scattering, Refraction, and Fluorescence: An Overview of Conventional (Linear) Optical Microscopy Modalities
2.1.1. Widefield Fluorescence Microscopy (WFFM)
2.1.2. Total Internal Reflection (TIRF) Microscopy
2.1.3. Laser Scanning Confocal Microscopy (LSCM)
2.1.4. Slit Scanning Confocal (SC) and Spinning Disk Confocal Microscopy (SDCM)
2.1.5. Structured Illumination Microscopy (SIM)
2.1.6. Stimulated Emission Depletion Microscopy (STED) and Ground State Depletion Microscopy (GSD)
2.1.7. Photoactivated Localization Microscopy (PALM/fPALM)
2.1.8. Stochastic Optical Reconstruction Microscopy (STORM/dSTORM)
2.1.9. Fluorescence Lifetime Imaging Microscopy (FLIM)
2.1.10. Additional Modalities: Fluctuation-Based Super-Resolution Microscopy (FSM) and Pixel Reassignment Super-Resolution Microscopy (PRSM)
2.2. Nonlinear Optical Microscopy (NLOM)
2.2.1. Two-Photon Excitation Fluorescence (TPEF)
2.2.2. Multiharmonic Imaging Microscopy (MHIM)
2.3. Raman-Based Modalities
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Olszta, M.J.; Cheng, X.; Jee, S.S.; Kumar, R.; Kim, Y.Y.; Kaufman, M.J.; Douglas, E.P.; Gower, L.B. Bone structure and formation: A new perspective. Mater. Sci. Eng. Rep. 2007, 58, 77–116. [Google Scholar] [CrossRef]
- Elfenbein, A.; Simons, M. Auxiliary and autonomous proteoglycan signaling networks. Methods Enzymol. 2010, 480, 3–31. [Google Scholar] [PubMed] [Green Version]
- Ruoslahti, E. Brain extracellular matrix. Glycobiology 1996, 6, 489–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Härtig, W.; Derouiche, A.; Welt, K.; Brauer, K.; Grosche, J.; Mäder, M.; Reichenbach, A.; Brückner, G. Cortical neurons immunoreactive for the potassium channel Kv3. 1b subunit are predominantly surrounded by perineuronal nets presumed as a buffering system for cations. Brain Res. 1999, 842, 15–29. [Google Scholar] [CrossRef]
- Kaushik, R.; Lipachev, N.; Matuszko, G.; Kochneva, A.; Dvoeglazova, A.; Becker, A.; Paveliev, M.; Dityatev, A. Fine structure analysis of perineuronal nets in the ketamine model of schizophrenia. Eur. J. Neurosci. 2020, 1–17. [Google Scholar] [CrossRef]
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The extracellular matrix at a glance. J. Cell Sci. 2010, 123, 4195–4200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iozzo, R.V.; Schaefer, L. Proteoglycan form and function: A comprehensive nomenclature of proteoglycans. Matrix Biol. 2015, 42, 11–55. [Google Scholar] [CrossRef] [PubMed]
- Theocharis, A.D.; Skandalis, S.S.; Gialeli, C.; Karamanos, N.K. Extracellular matrix structure. Adv. Drug Deliv. Rev. 2016, 97, 4–27. [Google Scholar] [CrossRef]
- Muiznieks, L.D.; Keeley, F.W. Molecular assembly and mechanical properties of the extracellular matrix: A fibrous protein perspective. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 866–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, M.K.; Hahn, R.A. Collagens. Cell Tissue Res. 2010, 339, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Gosline, J.; Lillie, M.; Carrington, E.; Guerette, P.; Ortlepp, C.; Savage, K. Elastic proteins: Biological roles and mechanical properties. Philosophical Transactions of the Royal Society of London. Ser. B Biol. Sci. 2002, 357, 121–132. [Google Scholar]
- Zollinger, A.J.; Smith, M.L. Fibronectin, the extracellular glue. Matrix Biol. 2017, 60, 27–37. [Google Scholar] [CrossRef]
- Aumailley, M. The laminin family. Cell Adhes. Migr. 2013, 7, 48–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fratzl, P. Collagen: Structure and mechanics, an introduction. In Collagen; Springer: Boston, MA, USA, 2008; pp. 1–13. [Google Scholar]
- Green, E.M.; Mansfield, J.C.; Bell, J.S.; Winlove, C.P. The structure and micromechanics of elastic tissue. Interface Focus 2014, 4, 20130058. [Google Scholar] [CrossRef] [Green Version]
- Parisi, L.; Toffoli, A.; Ghezzi, B.; Mozzoni, B.; Lumetti, S.; Macaluso, G.M. A glance on the role of fibronectin in controlling cell response at biomaterial interface. Jpn. Dent. Sci. Rev. 2020, 56, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Lenselink, E.A. Role of fibronectin in normal wound healing. Int. Wound J. 2015, 12, 313–316. [Google Scholar] [CrossRef]
- Islam, S.; Watanabe, H. Versican: A Dynamic Regulator of the Extracellular Matrix. J. Histochem. Cytochem. 2020, 68, 763–775. [Google Scholar] [CrossRef]
- Koch, C.D.; Lee, C.M.; Apte, S.S. Aggrecan in cardiovascular development and disease. J. Histochem. Cytochem. 2020, 68, 777–795. [Google Scholar] [CrossRef]
- Hayes, A.J.; Melrose, J. Aggrecan, the Primary Weight-Bearing Cartilage Proteoglycan, Has Context-Dependent, Cell-Directive Properties in Embryonic Development and Neurogenesis: Aggrecan Glycan Side Chain Modifications Convey Interactive Biodiversity. Biomolecules 2020, 10, 1244. [Google Scholar] [CrossRef]
- Abatangelo, G.; Vindigni, V.; Avruscio, G.; Pandis, L.; Brun, P. Hyaluronic acid: Redefining its role. Cells 2020, 9, 1743. [Google Scholar] [CrossRef] [PubMed]
- Robins, S. Biochemistry and functional significance of collagen cross-linking. Biochem. Soc. Trans. 2007, 35, 849–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callaghan, T.; Wilhelm, K.P. A review of ageing and an examination of clinical methods in the assessment of ageing skin. Part I: Cellular and molecular perspectives of skin ageing. Int. J. Cosmet. Sci. 2008, 30, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Calleja-Agius, J.; Muscat-Baron, Y.; Brincat, M.P. Skin ageing. Menopause Int. 2007, 13, 60–64. [Google Scholar] [CrossRef] [Green Version]
- Schultz, G.S.; Wysocki, A. Interactions between extracellular matrix and growth factors in wound healing. Wound Repair Regen. 2009, 17, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Velnar, T.; Bailey, T.; Smrkolj, V. The wound healing process: An overview of the cellular and molecular mechanisms. J. Int. Med Res. 2009, 37, 1528–1542. [Google Scholar] [CrossRef] [PubMed]
- Szauter, K.M.; Cao, T.; Boyd, C.D.; Csiszar, K. Lysyl oxidase in development, aging and pathologies of the skin. Pathologie Biologie 2005, 53, 448–456. [Google Scholar] [CrossRef]
- Schäfer, M.; Werner, S. Cancer as an overhealing wound: An old hypothesis revisited. Nat. Rev. Mol. Cell Biol. 2008, 9, 628–638. [Google Scholar] [CrossRef]
- Lin, T.C.; Yang, C.H.; Cheng, L.H.; Chang, W.T.; Lin, Y.R.; Cheng, H.C. Fibronectin in cancer: Friend or foe. Cells 2020, 9, 27. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y. Cellular and molecular mechanisms of renal fibrosis. Nat. Rev. Nephrology 2011, 7, 684. [Google Scholar] [CrossRef]
- King Jr, T.E.; Pardo, A.; Selman, M. Idiopathic pulmonary fibrosis. The Lancet 2011, 378, 1949–1961. [Google Scholar] [CrossRef]
- Ekanayaka, R.P.; Tilakaratne, W.M. Oral submucous fibrosis: Review on mechanisms of malignant transformation. Oral Surgery, Oral Med. Oral Pathol. Oral Radiol. 2016, 122, 192–199. [Google Scholar] [CrossRef]
- Hinderer, S.; Schenke-Layland, K. Cardiac fibrosis—A short review of causes and therapeutic strategies. Adv. Drug Deliv. Rev. 2019, 146, 77–82. [Google Scholar] [CrossRef]
- Boor, P.; Floege, J. Chronic kidney disease growth factors in renal fibrosis. Clin. Exp. Pharmacol. Physiol. 2011, 38, 441–450. [Google Scholar] [CrossRef]
- Hegele, R.G. The pathology of asthma: Brief review. Immunopharmacology 2000, 48, 257–262. [Google Scholar] [CrossRef]
- Horn, M.A.; Trafford, A.W. Aging and the cardiac collagen matrix: Novel mediators of fibrotic remodelling. J. Mol. Cell. Cardiol. 2016, 93, 175–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zipfel, W.R.; Williams, R.M.; Christie, R.; Nikitin, A.Y.; Hyman, B.T.; Webb, W.W. Live tissue intrinsic emission microscopy using multiphoton-excited native fluorescence and second harmonic generation. Proc. Natl. Acad. Sci. USA 2003, 100, 7075–7080. [Google Scholar] [CrossRef] [Green Version]
- Mostaço-Guidolin, L.B.; Ko, A.C.T.; Wang, F.; Xiang, B.; Hewko, M.; Tian, G.; Major, A.; Shiomi, M.; Sowa, M.G. Collagen morphology and texture analysis: From statistics to classification. Sci. Rep. 2013, 3, 1–10. [Google Scholar] [CrossRef]
- Mostaço-Guidolin, L.B.; Ko, A.C.; Popescu, D.P.; Smith, M.S.; Kohlenberg, E.K.; Shiomi, M.; Major, A.; Sowa, M.G. Evaluation of texture parameters for the quantitative description of multimodal nonlinear optical images from atherosclerotic rabbit arteries. Phy. Med. Biol. 2011, 56, 5319. [Google Scholar] [CrossRef] [Green Version]
- Helmchen, F.; Denk, W. Deep tissue two-photon microscopy. Nat. Methods 2005, 2, 932–940. [Google Scholar] [CrossRef]
- Campagnola, P.J.; Loew, L.M. Second-harmonic imaging microscopy for visualizing biomolecular arrays in cells, tissues and organisms. Nat. Biotechnol. 2003, 21, 1356–1360. [Google Scholar] [CrossRef] [PubMed]
- Masters, B. Encyclopedia of Life Sciences (ELS); John Wiley & Sons, Ltd.: Chichester, UK, 2010; pp. 1134–1139. [Google Scholar]
- McGucken, W. Nineteenth-Century Spectroscopy; Johns Hopkins Press: Baltimore, MD, USA, 1969. [Google Scholar]
- Kasten, F.H. The origins of modern fluorescence microscopy and fluorescent probes. In Cell Structure and Function by Microspectrofluorometry; Elsevier: Amsterdam, The Netherlands, 1989; pp. 3–50. [Google Scholar]
- Combs, C.A.; Shroff, H. Fluorescence microscopy: A concise guide to current imaging methods. Curr. Protoc. Neurosci. 2017, 79, 2.1.1–2.1.14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galbraith, C.G.; Galbraith, J.A. Super-resolution microscopy at a glance. J. Cell Sci. 2011, 124, 1607–1611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacquemet, G.; Carisey, A.F.; Hamidi, H.; Henriques, R.; Leterrier, C. The cell biologist’s guide to super-resolution microscopy. J. Cell Sci. 2020, 133, jcs240713. [Google Scholar] [CrossRef]
- Graham, J.; Raghunath, M.; Vogel, V. Fibrillar fibronectin plays a key role as nucleator of collagen I polymerization during macromolecular crowding-enhanced matrix assembly. Biom. Sci. 2019, 7, 4519–4535. [Google Scholar] [CrossRef] [Green Version]
- Malakpour-Permlid, A.; Buzzi, I.; Hegardt, C.; Johansson, F.; Oredsson, S. Identification of extracellular matrix proteins secreted by human dermal fibroblasts cultured in 3D electrospun scaffolds. Sci. Rep. 2021, 11, 6655. [Google Scholar] [CrossRef]
- Tønnesen, J.; Inavalli, V.K.; Nägerl, U.V. Super-resolution imaging of the extracellular space in living brain tissue. Cell 2018, 172, 1108–1121. [Google Scholar] [CrossRef] [Green Version]
- Moore, T.I.; Aaron, J.; Chew, T.L.; Springer, T.A. Measuring integrin conformational change on the cell surface with superresolution microscopy. Cell Rep. 2018, 22, 1903–1912. [Google Scholar] [CrossRef] [Green Version]
- He, H.; Shao, C.; Mu, Z.; Mao, C.; Sun, J.; Chen, C.; Tang, R.; Gu, X. Promotion effect of immobilized chondroitin sulfate on intrafibrillar mineralization of collagen. Carbohydr. Polym. 2020, 229, 115547. [Google Scholar] [CrossRef] [PubMed]
- Denk, W.; Strickler, J.H.; Webb, W.W. Two-photon laser scanning fluorescence microscopy. Science 1990, 248, 73–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaspro, A. New World Microscopy; IEEE: New York, NY, USA, 1996. [Google Scholar]
- Alkmin, S.; Brodziski, R.; Simon, H.; Hinton, D.; Goldsmith, R.H.; Patankar, M.; Campagnola, P.J. Role of Collagen Fiber Morphology on Ovarian Cancer Cell Migration Using Image-Based Models of the Extracellular Matrix. Cancers 2020, 12, 1390. [Google Scholar] [CrossRef]
- Mostaço-Guidolin, L.B.; Smith, M.S.; Hewko, M.; Schattka, B.; Sowa, M.G.; Major, A.; Ko, A.C.T. Fractal dimension and di-rectional analysis of elastic and collagen fiber arrangement in unsectioned arterial tissues affected by atherosclerosis and aging. J. Applied Physiol. 2019, 126, 638–646. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Fu, Y.; Zickmund, P.; Shi, R.; Cheng, J.-X. Coherent Anti-Stokes Raman Scattering Imaging of Axonal Myelin in Live Spinal Tissues. Biophys. J. 2005, 89, 581–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozel, B.A.; Rongish, B.; Czirok, A.; Zach, J.; Little, C.D.; Davis, E.C.; Knutsen, R.H.; Wagenseil, J.; Levy, M.A.; Mecham, R.P. Elastic fiber formation: A dynamic view of extracellular matrix assembly using timer reporters. J. Cell. Physiol. 2006, 207, 87–96. [Google Scholar] [CrossRef]
- Linder, S. The matrix corroded: Podosomes and invadopodia in extracellular matrix degradation. Trends Cell Biol. 2007, 17, 107–117. [Google Scholar] [CrossRef]
- Hayn, A.; Fischer, T.; Mierke, C.T. Inhomogeneities in 3D Collagen Matrices Impact Matrix Mechanics and Cancer Cell Migration. Front. Cell Dev. Biol. 2020, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Utsunomiya, N.; Utsunomiya, A.; Chino, T.; Hasegawa, M.; Oyama, N. Gene silencing of extracellular matrix protein 1 (ECM1) results in phenotypic alterations of dermal fibroblasts reminiscent of clinical features of lichen sclerosus. J. Dermatol. Sci. 2020, 100, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Cottle, L.; Loudovaris, T.; Xiao, D.; Yang, P.; Thomas, H.E.; Kebede, M.A.; Thorn, P. Enhanced structure and function of human pluripotent stem cell-derived beta-cells cultured on extracellular matrix. Stem Cells Transl. Med. 2021, 10, 492–505. [Google Scholar] [CrossRef]
- Doyle, A.D.; Sykora, D.J.; Pacheco, G.G.; Kutys, M.L.; Yamada, K.M. 3D mesenchymal cell migration is driven by anterior cellular contraction that generates an extracellular matrix prestrain. Dev. Cell 2021, 56, 826–841.e4. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Shklover, J.; Parvizi, M.; Sherlock, B.; Alfonso-Garcia, A.; Haudenschild, A.K.; Griffiths, L.G.; Marcu, L. Label-Free Assessment of Collagenase Digestion on Bovine Pericardium Properties by Fluorescence Lifetime Imaging. Ann. Biomed. Eng. 2018, 46, 1870–1881. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Portalatin, N.; Alfonso-Garcia, A.; Liu, J.C.; Marcu, L.; Panitch, A. Physical, Biomechanical, and Optical Charac-terization of Collagen and Elastin Blend Hydrogels. Annals Biomed. Eng. 2020, 48, 2924–2935. [Google Scholar] [CrossRef]
- Haudenschild, A.K.; Sherlock, B.; Zhou, X.; Hu, J.C.; Leach, J.K.; Marcu, L.; Athanasiou, K.A. Non-destructive detection of matrix stabilization correlates with enhanced mechanical properties of self-assembled articular cartilage. J. Tissue Eng. Regen. Med. 2019, 13, 637–648. [Google Scholar] [CrossRef]
- Barlow, A.M.; Mostaço-Guidolin, L.; Osei, E.; Booth, S.; Hackett, T.-L. Super resolution measurement of collagen fibers in biological samples: Validation of a commercial solution for multiphoton microscopy. PLoS ONE 2020, 15, e0229278. [Google Scholar] [CrossRef] [Green Version]
- Cavinato, C.; Murtada, S.I.; Rojas, A.; Humphrey, J.D. Evolving structure-function relations during aortic maturation and aging revealed by multiphoton microscopy. Mec. Ageing Dev. 2021, 196, 111471. [Google Scholar] [CrossRef] [PubMed]
- Jadidi, M.; Sherifova, S.; Sommer, G.; Kamenskiy, A.; Holzapfel, G.A. Constitutive modeling using structural information on collagen fiber direction and dispersion in human superficial femoral artery specimens of different ages. Acta Biomater. 2020, 121, 461–474. [Google Scholar] [CrossRef] [PubMed]
- Xi, G.; Guo, W.; Kang, D.; Ma, J.; Fu, F.; Qiu, L.; Zheng, L.; He, J.; Fang, N.; Chen, J.; et al. Large-scale tumor-associated collagen signatures identify high-risk breast cancer patients. Theranostics 2021, 11, 3229–3243. [Google Scholar] [CrossRef]
- Hsiao, C.Y.; Teng, X.; Su, T.H.; Lee, P.H.; Kao, J.H.; Huang, K.W. Improved second harmonic generation and two-photon excitation fluorescence microscopy-based quantitative assessments of liver fibrosis through auto-correction and optimal sam-pling. Quantitative Imag. Med. Surg. 2021, 11, 351. [Google Scholar] [CrossRef]
- Atkuru, S.; Muniraj, G.; Sudhaharan, T.; Chiam, K.; Wright, G.D.; Sriram, G. Cellular ageing of oral fibroblasts differentially modulates extracellular matrix organization. J. Periodontal Res. 2021, 56, 108–120. [Google Scholar] [CrossRef]
- Pendleton, E.G.; Tehrani, K.F.; Barrow, R.P.; Mortensen, L.J. Second harmonic generation characterization of collagen in whole bone. Biom. Optics Express 2020, 11, 4379–4396. [Google Scholar] [CrossRef] [PubMed]
- Xydias, D.; Ziakas, G.; Psilodimitrakopoulos, S.; Lemonis, A.; Bagli, E.; Fotsis, T.; Gravanis, A.; Tzeranis, D.S.; Stratakis, E. Three-dimensional characterization of collagen remodeling in cell-seeded collagen scaffolds via polarization second harmonic generation. Biom. Optics Express 2021, 12, 1136–1153. [Google Scholar] [CrossRef]
- Voytik-Harbin, S.L.; Rajwa, B.; Robinson, J. Chapter 27 Three-dimensional imaging of extracellular matrix and extracellular matrix-cell interactions. Mitosis Meiosis Part A 2001, 63, 583–597. [Google Scholar] [CrossRef]
- Gubarkova, E.V.; Elagin, V.V.; Dudenkova, V.V.; Kuznetsov, S.S.; Karabut, M.M.; Potapov, A.L.; Vorontsov, D.A.; Vorontsov, A.Y.; Sirotkina, M.A.; Zagaynova, E.V.; et al. Multiphoton tomography in differentiation of morphological and molecular subtypes of breast cancer: A quantitative analysis. J. Biophotonics 2021, 14, e202000471. [Google Scholar] [CrossRef]
- Benboujja, F.; Hartnick, C. Quantitative evaluation of the human vocal fold extracellular matrix using multiphoton microscopy and optical coherence tomography. Sci. Rep. 2021, 11, 2440. [Google Scholar] [CrossRef]
- Pintér, A.; Hevesi, Z.; Zahola, P.; Alpár, A.; Hanics, J. Chondroitin sulfate proteoglycan-5 forms perisynaptic matrix assemblies in the adult rat cortex. Cell. Signalling 2020, 74, 109710. [Google Scholar] [CrossRef]
- Hayes, A.; Melrose, J. 3D distribution of perlecan within intervertebral disc chondrons suggests novel regulatory roles for this multifunctional modular heparan sulphate proteoglycan. Eur. Cells Mater. 2021, 41, 73–89. [Google Scholar] [CrossRef]
- Rácz, É.; Gaál, B.; Matesz, C. Heterogeneous expression of extracellular matrix molecules in the red nucleus of the rat. Neurosci. 2016, 322, 1–17. [Google Scholar] [CrossRef]
- Dzyubenko, E.; Manrique-Castano, D.; Kleinschnitz, C.; Faissner, A.; Hermann, D.M. Topological remodeling of cortical per-ineuronal nets in focal cerebral ischemia and mild hypoperfusion. Matrix Biol. 2018, 74, 121–132. [Google Scholar] [CrossRef]
- Soria, F.N.; Paviolo, C.; Doudnikoff, E.; Arotcarena, M.L.; Lee, A.; Danné, N.; Mandal, A.K.; Gosset, P.; Dehay, B.; Groc, L.; et al. Synucleinopathy alters nanoscale organization and diffusion in the brain extracellular space through hyaluronan re-modeling. Nat. Comm. 2020, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- De Angelis, J.E.; Lagendijk, A.K.; Chen, H.; Tromp, A.; Bower, N.; Tunny, K.A.; Brooks, A.; Bakkers, J.; Francois, M.; Yap, A.S.; et al. Tmem2 Regulates Embryonic Vegf Signaling by Controlling Hyaluronic Acid Turnover. Dev. Cell 2017, 40, 123–136. [Google Scholar] [CrossRef] [Green Version]
- Hu, S.; Tee, Y.H.; Kabla, A.; Zaidel-Bar, R.; Bershadsky, A.; Hersen, P. Structured illumination microscopy reveals focal ad-hesions are composed of linear subunits. Cytoskeleton 2015, 72, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Spiess, M.; Hernandez-Varas, P.; Oddone, A.; Olofsson, H.; Blom, H.; Waithe, D.; Lock, J.; Lakadamyali, M.; Strömblad, S. Active and inactive β1 integrins segregate into distinct nanoclusters in focal adhesions. J. Cell Biol. 2018, 217, 1929–1940. [Google Scholar] [CrossRef] [Green Version]
- Tobin, S.J.; Cacao, E.E.; Hong, D.W.W.; Terenius, L.; Vukojevic, V.; Jovanović-Talisman, T. Nanoscale Effects of Ethanol and Naltrexone on Protein Organization in the Plasma Membrane Studied by Photoactivated Localization Microscopy (PALM). PLoS ONE 2014, 9, e87225. [Google Scholar] [CrossRef]
- Changede, R.; Cai, H.; Wind, S.J.; Sheetz, M.P. Integrin nanoclusters can bridge thin matrix fibres to form cell–matrix adhesions. Nat. Mater. 2019, 18, 1366–1375. [Google Scholar] [CrossRef]
- Fan, Z.; Kiosses, W.B.; Sun, H.; Orecchioni, M.; Ghosheh, Y.; Zajonc, D.M.; Arnaout, M.A.; Gutierrez, E.; Groisman, A.; Ginsberg, M.H.; et al. High-Affinity Bent β2-Integrin Molecules in Arresting Neutrophils Face Each Other through Binding to ICAMs In cis. Cell Rep. 2019, 26, 119–130.e5. [Google Scholar] [CrossRef] [Green Version]
- Deschout, H.; Lukes, T.; Sharipov, A.; Szlag, D.; Feletti, L.; Vandenberg, W.; Dedecker, P.; Hofkens, J.; Leutenegger, M.; Lasser, T.; et al. Complementarity of PALM and SOFI for super-resolution live-cell imaging of focal adhesions. Nat. Commun. 2016, 7, 13693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vega, M.E.; Kastberger, B.; Wehrle-Haller, B.; Schwarzbauer, J.E. Stimulation of Fibronectin Matrix Assembly by Lysine Acet-ylation. Cells 2020, 9, 655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coito, A.; Binder, J.; de Sousa, M.; Kupiec-Weglinski, J.W. The expression of extracellular matrix proteins during accelerated rejection of cardiac allografts in sensitized rats. Transplantation 1994, 57, 599–605. [Google Scholar] [CrossRef]
- Codron, P.; Letournel, F.; Marty, S.; Renaud, L.; Bodin, A.; Duchesne, M.; Verny, C.; Lenaers, G.; Duyckaerts, C.; Julien, J.P.; et al. STochastic Optical Reconstruction Microscopy (STORM) reveals the nanoscale organization of pathological aggregates in human brain. Neuropath. Appl. Neurobiol. 2021, 47, 127. [Google Scholar] [CrossRef]
- Zhou, Z.; Zhang, L.; Li, J.; Shi, Y.; Wu, Z.; Zheng, H.; Wang, Z.; Zhao, W.; Pan, H.; Wang, Q.; et al. Polyelectrolyte–calcium complexes as a pre-precursor induce biomimetic mineralization of collagen. Nanoscale 2021, 13, 953–967. [Google Scholar] [CrossRef] [PubMed]
- Yao, S.; Xu, Y.; Shao, C.; Nudelman, F.; Sommerdijk, N.A.J.M.; Tang, R. A Biomimetic Model for Mineralization of Type-I Collagen Fibrils. In Breast Cancer; Springer Science and Business Media LLC: Berlin, Gernmany, 2019; Volume 1944, pp. 39–54. [Google Scholar]
- Okkelman, I.A.; McGarrigle, R.; O’Carroll, S.; Berrio, D.C.; Schenke-Layland, K.; Hynes, J.; Dmitriev, R.I. Extracellular Ca2+-Sensing Fluorescent Protein Biosensor Based on a Collagen-Binding Domain. ACS Appl. Bio Mater. 2020, 3, 5310–5321. [Google Scholar] [CrossRef]
- Szmacinski, H.; Hegde, K.; Zeng, H.-H.; Eslami, K.; Puche, A.C.; Lengyel, I.; Thompson, R.B. Imaging hydroxyapatite in sub-retinal pigment epithelial deposits by fluorescence lifetime imaging microscopy with tetracycline staining. J. Biomed. Opt. 2020, 25, 047001. [Google Scholar] [CrossRef] [Green Version]
- Lima, M.A.; Cavalheiro, R.P.; Viana, G.M.; Meneghetti, M.C.; Rudd, T.R.; Skidmore, M.; Powell, A.K.; Yates, E.A. 19F labelled glycosaminoglycan probes for solution NMR and non-linear (CARS) microscopy. Glycoconj. J. 2016, 34, 405–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sehm, T.; Uckermann, O.; Galli, R.; Meinhardt, M.; Rickelt, E.; Krex, D.; Schackert, G.; Kirsch, M. Label-free multiphoton mi-croscopy as a tool to investigate alterations of cerebral aneurysms. Sci. Rep. 2020, 10, 12359. [Google Scholar] [CrossRef]
- Jacques, S.L. Fractal Nature of Light Scattering in Tissues. J. Innov. Opt. Heal. Sci. 2011, 4, 1–7. [Google Scholar] [CrossRef]
- Mayinger, F. Optical Measurements: Techniques and Applications; Springer Science & Business Media: Berlin, Germany, 2013. [Google Scholar]
- Tuchin, V.V. Light scattering study of tissues. Physics-Uspekhi 1997, 40, 495–515. [Google Scholar] [CrossRef]
- Clarke, R.J.; Oprysa, A. Fluorescence and Light Scattering. J. Chem. Educ. 2004, 81, 705. [Google Scholar] [CrossRef]
- Xu, Z.; Reilley, M.; Li, R.; Xu, M. Mapping absolute tissue endogenous fluorophore concentrations with chemometric wide-field fluorescence microscopy. J. Biomed. Opt. 2017, 22, 066009. [Google Scholar] [CrossRef]
- Riis, S.; Hansen, A.C.; Johansen, L.; Lund, K.; Hoeeg, C.; Pitsa, A.; Hyldig, K.; Zachar, V.; Fink, T.; Pennisi, C.P. Fabrication and characterization of extracellular matrix scaffolds obtained from adipose-derived stem cells. Methods 2020, 171, 68–76. [Google Scholar] [CrossRef]
- Neill, T.; Schaefer, L.; Iozzo, R.V. Decorin: A guardian from the matrix. Am. J. Pathol. 2012, 181, 380–387. [Google Scholar] [CrossRef] [Green Version]
- Axelrod, D. Total Internal Reflection Fluorescence Microscopy in Cell Biology. Traffic 2001, 2, 764–774. [Google Scholar] [CrossRef]
- Truskey, G.A.; Burmeister, J.S.; Grapa, E.; Reichert, W.M. Total internal reflection fluorescence microscopy (TIRFM). II. Topo-graphical mapping of relative cell/substratum separation distances. J. Cell Sci. 1992, 103, 491–499. [Google Scholar] [CrossRef]
- Steffen, A.; Le Dez, G.; Poincloux, R.; Recchi, C.; Nassoy, P.; Rottner, K.; Galli, T.; Chavrier, P. MT1-MMP-Dependent Invasion Is Regulated by TI-VAMP/VAMP7. Curr. Biol. 2008, 18, 926–931. [Google Scholar] [CrossRef]
- Chery, D.R.; Han, B.; Zhou, Y.; Wang, C.; Adams, S.M.; Chandrasekaran, P.; Kwok, B.; Heo, S.-J.; Enomoto-Iwamoto, M.; Lu, X.L.; et al. Decorin regulates cartilage pericellular matrix micromechanobiology. Matrix Biol. 2021, 96, 1–17. [Google Scholar] [CrossRef]
- Umana-Diaz, C.; Pichol-Thievend, C.; Marchand, M.F.; Atlas, Y.; Salza, R.; Malbouyres, M.; Barret, A.; Teillon, J.; ArdidieRobouant, C.; Ruggiero, F.; et al. Scavenger Receptor Cysteine-Rich domains of Lysyl Oxidase-Like2 regulate endo-thelial ECM and angiogenesis through non-catalytic scaffolding mechanisms. Matrix Biol. 2020, 88, 33–52. [Google Scholar] [CrossRef]
- Claxton, N.S.; Fellers, T.J.; Davidson, M.W. Laser scanning confocal microscopy. Encyclop. Med. Devices Instrum. 2006, 21, 1–37. [Google Scholar]
- Xu, C.; Inai, R.; Kotaki, M.; Ramakrishna, S. Electrospun nanofiber fabrication as synthetic extracellular matrix and its potential for vascular tissue engineering. Tissue Eng. 2004, 10, 1160–1168. [Google Scholar] [CrossRef]
- Elliott, A.D. Confocal Microscopy: Principles and Modern Practices. Curr. Protoc. Cytom. 2020, 92, e68. [Google Scholar] [CrossRef]
- Gräf, R.; Zimmermann, T. Live Cell Spinning Disk Microscopy. Blue Biotech. 2005, 95, 57–75. [Google Scholar] [CrossRef]
- Sheppard, C.; Mao, X. Confocal Microscopes with Slit Apertures. J. Mod. Opt. 1988, 35, 1169–1185. [Google Scholar] [CrossRef]
- Gustafsson, M.G.L. Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy. SHORT COMMUNICATION. J. Microsc. 2000, 198, 82–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schermelleh, L.; Carlton, P.; Haase, S.; Shao, L.; Winoto, L.; Kner, P.; Burke, B.; Cardoso, M.C.; Agard, D.A.; Gustafsson, M.G.L.; et al. Subdiffraction Multicolor Imaging of the Nuclear Periphery with 3D Structured Illumination Microscopy. Science 2008, 320, 1332–1336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kner, P.; Chhun, B.; Griffis, E.; Winoto, L.; Gustafsson, M.G.L. Super-resolution video microscopy of live cells by structured illumination. Nat. Methods 2009, 6, 339–342. [Google Scholar] [CrossRef] [Green Version]
- Fiolka, R.; Shao, L.; Rego, E.H.; Davidson, M.W.; Gustafsson, M.G.L. Time-lapse two-color 3D imaging of live cells with doubled resolution using structured illumination. Proc. Natl. Acad. Sci. USA 2012, 109, 5311–5315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.; Li, D.; Zhang, S.; Yang, Y.; Liu, J.J.; Wang, X.; Liu, C.; Milkie, D.E.; Moore, R.P.; Tulu, U.S.; et al. Visualizing intra-cellular organelle and cytoskeletal interactions at nanoscale resolution on millisecond timescales. Cell 2018, 175, 1430–1442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engel, U. Structured illumination superresolution imaging of the cytoskeleton. Mitosis Meiosis Part A 2014, 123, 315–333. [Google Scholar] [CrossRef]
- Heine, J.; Reuss, M.; Harke, B.; D’Este, E.; Sahl, S.J.; Hell, S.W. Adaptive-illumination STED nanoscopy. Proc. Nat. Ac. Sci. USA 2017, 114, 9797–9802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bottanelli, F.; Kromann, E.B.; Allgeyer, E.; Erdmann, R.S.; Baguley, S.W.; Sirinakis, G.; Schepartz, A.; Baddeley, D.; Toomre, D.K.; E Rothman, J.; et al. Two-colour live-cell nanoscale imaging of intracellular targets. Nature Comm. 2021, 7, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Göttfert, F.; Wurm, C.A.; Mueller, V.; Berning, S.; Cordes, V.C.; Honigmann, A.; Hell, S.W. Coaligned Dual-Channel STED Nanoscopy and Molecular Diffusion Analysis at 20 nm Resolution. Biophys. J. 2013, 105, L01–L03. [Google Scholar] [CrossRef] [Green Version]
- Fölling, J.; Bossi, M.; Bock, H.; Medda, R.; A Wurm, C.; Hein, B.; Jakobs, S.; Eggeling, C.; Hell, S.W. Fluorescence nanoscopy by ground-state depletion and single-molecule return. Nat. Methods 2008, 5, 943–945. [Google Scholar] [CrossRef]
- Stockhammer, A.; Bottanelli, F. Appreciating the small things in life: STED microscopy in living cells. J. Phys. D Appl. Phys. 2021, 54, 033001. [Google Scholar] [CrossRef]
- Wu, Z.; Xu, X.; Xi, P. Stimulated emission depletion microscopy for biological imaging in four dimensions: A review. Microsc. Res. Tech. 2021, 1–12. [Google Scholar] [CrossRef]
- Belov, V.N.; Stoldt, S.; Rüttger, F.; John, M.; Seikowski, J.; Schimpfhauser, J.; Hell, S.W. Synthesis of Fluorescent Jasplakinolide Analogues for Live-Cell STED Microscopy of Actin. J. Org. Chem. 2020, 85, 7267–7275. [Google Scholar] [CrossRef]
- Dumbović, G.; Sanjuan, X.; Perucho, M.; Forcales, S.-V. Stimulated emission depletion (STED) super resolution imaging of RNA- and protein-containing domains in fixed cells. Methods 2021, 187, 68–76. [Google Scholar] [CrossRef]
- Damenti, M.; Coceano, G.; Pennacchietti, F.; Bodén, A.; Testa, I. STED and parallelized RESOLFT optical nanoscopy of the tubular endoplasmic reticulum and its mitochondrial contacts in neuronal cells. Neurobiol. Dis. 2021, 155, 105361. [Google Scholar] [CrossRef]
- Moerner, W.E.W.E. Single-Molecule Spectroscopy, Imaging, and Photocontrol: Foundations for Super-Resolution Microscopy (Nobel Lecture). Angew. Chem. Int. Ed. 2015, 54, 8067–8093. [Google Scholar] [CrossRef] [PubMed]
- Von Diezmann, L.; Shechtman, Y.; Moerner, W.E. Three-Dimensional Localization of Single Molecules for Super-Resolution Imaging and Single-Particle Tracking. Chem. Rev. 2017, 117, 7244–7275. [Google Scholar] [CrossRef]
- Brown, T.A.; Fetter, R.D.; Tkachuk, A.N.; Clayton, D.A. Approaches toward super-resolution fluorescence imaging of mito-chondrial proteins using PALM. Methods 2010, 51, 458–463. [Google Scholar] [CrossRef] [Green Version]
- Scarselli, M.; Annibale, P.; McCormick, P.J.; Kolachalam, S.; Aringhieri, S.; Radenovic, A.; Corsini, G.U.; Maggio, R. Revealing G-protein-coupled receptor oligomerization at the single-molecule level through a nanoscopic lens: Methods, dynamics and biological function. FEBS J. 2016, 283, 1197–1217. [Google Scholar] [CrossRef] [PubMed]
- Bartle, E.I.; Rao, T.C.; Urner, T.M.; Mattheyses, A.L. Bridging the gap: Super-resolution microscopy of epithelial cell junctions. Tissue Barriers 2017, 6, e1404189. [Google Scholar] [CrossRef]
- Shroff, H.; White, H.; Betzig, E. Photoactivated Localization Microscopy (PALM) of Adhesion Complexes. Curr. Protoc. Cell Biol. 2008, 41, 4.21.1–4.21.27. [Google Scholar] [CrossRef] [Green Version]
- Deschout, H.; Platzman, I.; Sage, D.; Feletti, L.; Spatz, J.P.; Radenovic, A. Investigating focal adhesion substructures by local-ization microscopy. Biophy. J. 2017, 113, 2508–2518. [Google Scholar] [CrossRef] [Green Version]
- Tam, J.; Merino, D. Stochastic optical reconstruction microscopy (STORM) in comparison with stimulated emission depletion (STED) and other imaging methods. J. Neurochem. 2015, 135, 643–658. [Google Scholar] [CrossRef]
- Heilemann, M.; Van De Linde, S.; Schüttpelz, M.; Kasper, R.; Seefeldt, B.; Mukherjee, A.; Tinnefeld, P.; Sauer, M. Subdiffrac-tionresolution fluorescence imaging with conventional fluorescent probes. Angewandte Chemie Int. Ed. 2008, 47, 6172–6176. [Google Scholar] [CrossRef]
- Chen, B.; Gong, W.; Yang, Z.; Pan, W.; Verwilst, P.; Shin, J.; Yan, W.; Liu, L.; Qu, J.; Kim, J.S. STORM imaging of mitochondrial dynamics using a vicinal-dithiol-proteins-targeted probe. Biomaterials 2020, 243, 119938. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Liu, Y. Imaging Higher-order Chromatin Structures in Single Cells Using Stochastic Optical Reconstruction Microscopy. BIO-Protocol. 2019, 9, e3160. [Google Scholar] [CrossRef]
- Beliveau, B.J.; Boettiger, A.N.; Nir, G.; Bintu, B.; Yin, P.; Zhuang, X.; Wu, C.-T. In situ super-resolution imaging of genomic DNA with OligoSTORM and OligoDNA-PAINT. In Super-Resolution Microscopy; Springer: Berin, Germany, 2017; pp. 231–252. [Google Scholar]
- Huang, B.; Wang, W.; Bates, M.; Zhuang, X. Three-Dimensional Super-Resolution. Science 2008, 319, 810–813. [Google Scholar] [CrossRef] [Green Version]
- Van Munster, E.; Gadella, T.; Rietdorf, J. Microscopy techniques, Advances in Biochemical Engineering/Biotechnology; Springer: Berlin, Germany, 2005; pp. 143–175. [Google Scholar]
- König, K. Review: Clinical in vivo multiphoton FLIM tomography. Methods Appl. Fluoresc. 2020, 8, 034002. [Google Scholar] [CrossRef]
- Datta, R.; Heaster, T.M.; Sharick, J.T.; Gillette, A.A.; Skala, M.C. Fluorescence lifetime imaging microscopy: Fundamentals and advances in instrumentation, analysis, and applications. J. Biomed. Opt. 2020, 25, 071203. [Google Scholar] [CrossRef]
- Padilla-Parra, S.; Audugé, N.; Coppey-Moisan, M.; Tramier, M. Non fitting based FRET–FLIM analysis approaches applied to quantify protein–protein interactions in live cells. Biophys. Rev. 2011, 3, 63–70. [Google Scholar] [CrossRef] [Green Version]
- Osterlund, E.J.; Liu, Q.; Andrews, D.W. The Use of FLIM-FRET for the Detection of Mitochondria-Associated Protein Interactions. In Advanced Structural Safety Studies; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2015; Volume 1264, pp. 395–419. [Google Scholar]
- Rajoria, S.; Zhao, L.; Intes, X.; Barroso, M. FLIM-FRET for Cancer Applications. Curr. Mol. Imaging 2015, 3, 144–161. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Wang, H.; Wang, X.; Aoki, Y.; Wang, X.; Yang, Y.; Cheng, X.; Wang, Z.; Wang, X. Aurora-A/SOX8/FOXK1 signaling axis promotes chemoresistance via suppression of cell senescence and induction of glucose metabolism in ovarian cancer or-ganoids and cells. Theranostics 2020, 10, 6928. [Google Scholar] [CrossRef] [PubMed]
- Phipps, J.E.; Sun, Y.; Fishbein, M.C.; Marcu, L. A fluorescence lifetime imaging classification method to investigate the collagen to lipid ratio in fibrous caps of atherosclerotic plaque. Lasers Surg. Med. 2012, 44, 564–571. [Google Scholar] [CrossRef] [Green Version]
- Elson, D.; A Jo, J.; Marcu, L. Miniaturized side-viewing imaging probe for fluorescence lifetime imaging (FLIM): Validation with fluorescence dyes, tissue structural proteins and tissue specimens. New J. Phys. 2007, 9, 127. [Google Scholar] [CrossRef]
- Hachem, J.-P.; Roelandt, T.; Schürer, N.; Pu, X.; Fluhr, J.; Giddelo, C.; Man, M.-Q.; Crumrine, D.; Roseeuw, D.; Feingold, K.R.; et al. Acute Acidification of Stratum Corneum Membrane Domains Using Polyhydroxyl Acids Improves Lipid Processing and Inhibits Degradation of Corneodesmosomes. J. Investig. Dermatol. 2010, 130, 500–510. [Google Scholar] [CrossRef] [Green Version]
- Dertinger, T.; Colyer, R.; Iyer, G.; Weiss, S.; Enderlein, J. Fast, background-free, 3D super-resolution optical fluctuation imaging (SOFI). Proc. Natl. Acad. Sci. USA 2009, 106, 22287–22292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Culley, S.; Tosheva, K.L.; Pereira, P.; Henriques, R. SRRF: Universal live-cell super-resolution microscopy. Int. J. Biochem. Cell Biol. 2018, 101, 74–79. [Google Scholar] [CrossRef]
- Wu, Y.; Shroff, H. Faster, sharper, and deeper: Structured illumination microscopy for biological imaging. Nat. Methods 2018, 15, 1011–1019. [Google Scholar] [CrossRef]
- Vangindertael, J.; Camacho, R.; Sempels, W.; Mizuno, H.; Dedecker, P.; Janssen, K.P.F.P. An introduction to optical super-resolution microscopy for the adventurous biologist. Methods Appl. Fluoresc. 2018, 6, 022003. [Google Scholar] [CrossRef]
- Huff, J. The Airyscan detector from ZEISS: Confocal imaging with improved signal-to-noise ratio and super-resolution. Nat. Methods 2015, 12, i–ii. [Google Scholar] [CrossRef]
- Schenke-Layland, K. Non-invasive multiphoton imaging of extracellular matrix structures. J. Biophotonics 2008, 1, 451–462. [Google Scholar] [CrossRef] [Green Version]
- Provenzano, P.P.; Eliceiri, K.W.; Campbell, J.M.; Inman, D.R.; White, J.G.; Keely, P.J. Collagen reorganization at the tu-mor-stromal interface facilitates local invasion. BMC Med. 2006, 4, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rueden, C.T.; Conklin, M.W.; Provenzano, P.; Keely, P.J.; Eliceiri, K.W. Nonlinear optical microscopy and computational analysis of intrinsic signatures in breast cancer. In Proceedings of the 2009 Annual International Conference of the IEEE Engineering in Medicine and Biology Society, Minneapolis, MN, USA, 3–6 September 2009; Volume 2009, pp. 4077–4080. [Google Scholar]
- Wong, S.; Nathanson, M.H.; Chen, J.; Jain, D. Evaluation of Barrett Esophagus by Multiphoton Microscopy. Arch. Pathol. Lab. Med. 2014, 138, 204–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.-J.; Jee, S.-H.; Dong, C.-Y. Multiphoton microscopy: A new paradigm in dermatological imaging. Eur. J. Dermatol. 2007, 17, 361–366. [Google Scholar] [CrossRef]
- Bacskai, B.J.; Hyman, B.T. Alzheimer’s disease: What multiphoton microscopy teaches us. The neuroscientist 2002, 8, 386–390. [Google Scholar] [CrossRef]
- Chen, X.; Nadiarynkh, O.; Plotnikov, S.; Campagnola, P.J. Second harmonic generation microscopy for quantitative analysis of collagen fibrillar structure. Nat. Protocols 2012, 7, 654–669. [Google Scholar] [CrossRef] [PubMed]
- Rubart, M. Two-Photon Microscopy of Cells and Tissue. Circ. Res. 2004, 95, 1154–1166. [Google Scholar] [CrossRef] [Green Version]
- Shih, C.C.; Oakley, D.M.; Joens, M.S.; Roth, R.A.; Fitzpatrick, J.A. Nonlinear optical imaging of extracellular matrix proteins. Echinoderms Part B 2018, 143, 57–78. [Google Scholar] [CrossRef]
- Mostaço-Guidolin, L.; Rosin, N.L.; Hackett, T.-L. Imaging Collagen in Scar Tissue: Developments in Second Harmonic Generation Microscopy for Biomedical Applications. Int. J. Mol. Sci. 2017, 18, 1772. [Google Scholar] [CrossRef]
- Parodi, V.; Jacchetti, E.; Osellame, R.; Cerullo, G.; Polli, D.; Raimondi, M.T. Nonlinear Optical Microscopy: From Fundamentals to Applications in Live Bioimaging. Front. Bioeng. Biotechnol. 2020, 8, 585363. [Google Scholar] [CrossRef]
- Débarre, D.; Supatto, W.; Pena, A.-M.; Fabre, A.; Tordjmann, T.; Combettes, L.; Schanne-Klein, M.-C.; Beaurepaire, E. Imaging lipid bodies in cells and tissues using third-harmonic generation microscopy. Nat. Methods 2005, 3, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Zoumi, A.; Yeh, A.; Tromberg, B.J. Imaging cells and extracellular matrix in vivo by using second-harmonic generation and two-photon excited fluorescence. Proc. Natl. Acad. Sci. USA 2002, 99, 11014–11019. [Google Scholar] [CrossRef] [Green Version]
- Nadiarnykh, O.; LaComb, R.B.; A Brewer, M.; Campagnola, P.J. Alterations of the extracellular matrix in ovarian cancer studied by Second Harmonic Generation imaging microscopy. BMC Cancer 2010, 10, 94. [Google Scholar] [CrossRef] [Green Version]
- Burke, K.; Brown, E. The use of second harmonic generation to image the extracellular matrix during tumor progression. Intra Vital 2014, 3, e984509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tilbury, K.; Hocker, J.; Wen, B.L.; Sandbo, N.; Singh, V.; Campagnola, P.J. Second harmonic generation microscopy analysis of extracellular matrix changes in human idiopathic pulmonary fibrosis. J. Biomed. Opt. 2014, 19, 086014. [Google Scholar] [CrossRef]
- Sun, W.; Chang, S.; Tai, D.C.S.; Tan, N.; Xiao, G.; Tang, H.; Yu, H. Nonlinear optical microscopy: Use of second harmonic generation and two-photon microscopy for automated quantitative liver fibrosis studies. J. Biomed. Opt. 2008, 13, 064010. [Google Scholar] [CrossRef] [PubMed]
- Deka, G.; Wu, W.-W.; Kao, F.-J. In vivo wound healing diagnosis with second harmonic and fluorescence lifetime imaging. J. Biomed. Opt. 2013, 18, 061222. [Google Scholar] [CrossRef] [Green Version]
- Israel, J.S.; Esquibel, C.R.; Dingle, A.M.; Liu, Y.; Keikhosravi, A.; Pisaniello, J.A.; Hesse, M.A.; Brodnick, S.K.; Novello, J.; Krugner-Higby, L.; et al. Quantification of collagen organization after nerve repair. Plastic Reconstruc. Surg/Global Open 2017, 5, e1586. [Google Scholar] [CrossRef]
- Aptel, F.; Olivier, N.; Deniset-Besseau, A.; Legeais, J.M.; Plamann, K.; Schanne-Klein, M.C.; Beaurepaire, E. Multimodal non-linear imaging of the human cornea. Investigative Ophthalm. Vis. Sci. 2010, 51, 2459–2465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masihzadeh, O.; Lei, T.C.; Domingue, S.R.; Kahook, M.Y.; Bartels, R.; Ammar, D.A. Third harmonic generation microscopy of a mouse retina. Mol. Vis. 2015, 21, 538–547. [Google Scholar] [PubMed]
- Gavgiotaki, E.; Filippidis, G.; Kalognomou, M.; Tsouko, A.; Skordos, I.; Fotakis, C.; Athanassakis, I. Third Harmonic Generation microscopy as a reliable diagnostic tool for evaluating lipid body modification during cell activation: The example of BV-2 microglia cells. J. Struct. Biol. 2015, 189, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-Y.; Hsu, C.-Y.; Sun, C.-K. Epi-third and second harmonic generation microscopic imaging of abnormal enamel. Opt. Express 2008, 16, 11670–11679. [Google Scholar] [CrossRef]
- Stoller, P.; Kim, B.-M.; Rubenchik, A.M.; Reiser, K.M.; Da Silva, L.B. Polarization-dependent optical second-harmonic imaging of a rat-tail tendon. J. Biomed. Opt. 2002, 7, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Potma, E.O.; Xie, X.S. CARS Microscopy for Biology and Medicine. Opt. Photon. News 2004, 15, 40–45. [Google Scholar] [CrossRef]
- Yu, Y.; Ramachandran, P.V.; Wang, M.C. Shedding new light on lipid functions with CARS and SRS microscopy. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2014, 1841, 1120–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pezacki, J.P.; A Blake, J.; Danielson, D.C.; Kennedy, D.C.; Lyn, R.K.; Singaravelu, R. Chemical contrast for imaging living systems: Molecular vibrations drive CARS microscopy. Nat. Chem. Biol. 2011, 7, 137–145. [Google Scholar] [CrossRef]
- Pope, I.; Langbein, W.; Borri, P.; Watson, P. Live Cell Imaging with Chemical Specificity Using Dual Frequency CARS Microscopy. Met. Enzymology 2012, 504, 273–291. [Google Scholar] [CrossRef]
- Vukosavljevic, B.; Hittinger, M.; Hachmeister, H.; Pilger, C.; Murgia, X.; Gepp, M.M.; Gentile, L.; Huwer, H.; Schneider-Daum, N.; Huser, T.; et al. Vibrational spectroscopic imaging and live cell video microscopy for studying differentiation of primary human alveolar epithelial cells. J. Biophotonics 2019, 12, e201800052. [Google Scholar] [CrossRef] [Green Version]
- Brackmann, C.; Zaborowska, M.; Sundberg, J.; Gatenholm, P.; Enejder, A. In situ imaging of collagen synthesis by osteopro-genitor cells in microporous bacterial cellulose scaffolds. Tissue Eng. Part C Methods 2012, 18, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Brackmann, C.; Dahlberg, J.-O.; Vrana, N.E.; Lally, C.; Gatenholm, P.; Enejder, A. Non-linear microscopy of smooth muscle cells in artificial extracellular matrices made of cellulose. J. Biophotonics 2012, 5, 404–414. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.-S.; Park, J.H.; Lee, S.-W.; Hahn, J.; Lee, H.; Chae, S.-W.; Lee, T.G.; Moon, D.W.; Kim, S.-H. Lipid crystals mechanically stimulate adjacent extracellular matrix in advanced atherosclerotic plaques. Atherosclerosis 2014, 237, 769–776. [Google Scholar] [CrossRef]
- Wang, H.W.; Le, T.T.; Cheng, J.X. Label-free imaging of arterial cells and extracellular matrix using a multimodal CARS mi-croscope. Opt. Commun. 2008, 281, 1813–1822. [Google Scholar] [CrossRef] [Green Version]
- Moen, E.; Bannon, D.; Kudo, T.; Graf, W.; Covert, M.; Van Valen, D. Deep learning for cellular image analysis. Nat. Methods 2019, 16, 1233–1246. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Jin, L.; Chen, J.; Fang, Q.; Ablameyko, S.; Yin, Z.; Xu, Y. A survey on applications of deep learning in microscopy image analysis. Comput. Biol. Med. 2021, 134, 104523. [Google Scholar] [CrossRef] [PubMed]
- Wershof, E.; Park, D.; Barry, D.J.; Jenkins, R.P.; Rullan, A.; Wilkins, A.; Schlegelmilch, K.; Roxanis, I.; I Anderson, K.; A Bates, P.; et al. A FIJI macro for quantifying pattern in extracellular matrix. Life Sci. Alliance 2021, 4, e202000880. [Google Scholar] [CrossRef] [PubMed]
- Hotaling, N.A.; Bharti, K.; Kriel, H.; Simon, C.G., Jr. DiameterJ: A validated open source nanofiber diameter measurement tool. Biomaterials 2015, 61, 327–338. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Technique | Main Advantages | Main Limitations | ECM and Non-ECM Components Commonly Imaged |
|---|---|---|---|
| WFFM |
|
|
|
| TIRFM |
|
|
|
| LSCM |
|
|
|
| SC/SDCM |
|
|
|
| Technique | Main Advantages | Main Limitations | ECM and Non-ECM Components Commonly Imaged |
|---|---|---|---|
| SIM |
|
|
|
| STED |
|
|
|
| STORM |
|
|
|
| PALM |
|
|
|
| FLIM |
|
|
|
| Technique | Main Advantages | Main Limitations | ECM and Non-ECM Components Commonly Imaged |
|---|---|---|---|
| TPEF |
|
|
|
| SHG (SFG) |
|
|
|
| THG (SFG) |
|
|
|
| CARS |
|
|
|
| ECM Component | Imaging Modality | Representative References |
|---|---|---|
| Collagen | WFFM, LCSM, SHG | [49,55,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74] |
| Elastin | WFFM, LCSM, TPEF | [68,69,70,71,75,76,77] |
| Fibronectin | LCSM | [49,58,62,72] |
| Laminins | LCSM | [49,61,62,72] |
| Proteoglycans | LCSM, SIM, CARS, DSCM | [74,78,79,80,81] |
| Hyaluronan | LSCM, DSCM, | [80,82,83] |
| Cell-ECM interactions | STED, PALM | [50,84,85,86,87,88,89] |
| Other EC components | PALM, STORM, TIRF, CARS, THG | [52,90,91,92,93,94,95,96,97,98] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poole, J.J.A.; Mostaço-Guidolin, L.B. Optical Microscopy and the Extracellular Matrix Structure: A Review. Cells 2021, 10, 1760. https://doi.org/10.3390/cells10071760
Poole JJA, Mostaço-Guidolin LB. Optical Microscopy and the Extracellular Matrix Structure: A Review. Cells. 2021; 10(7):1760. https://doi.org/10.3390/cells10071760
Chicago/Turabian StylePoole, Joshua J. A., and Leila B. Mostaço-Guidolin. 2021. "Optical Microscopy and the Extracellular Matrix Structure: A Review" Cells 10, no. 7: 1760. https://doi.org/10.3390/cells10071760
APA StylePoole, J. J. A., & Mostaço-Guidolin, L. B. (2021). Optical Microscopy and the Extracellular Matrix Structure: A Review. Cells, 10(7), 1760. https://doi.org/10.3390/cells10071760