Abstract
Recent studies on liver disease burden worldwide estimated that cirrhosis is the 11th most common cause of death globally, and there is a great need for new therapies to limit the progression of liver injuries in the early stages. Cholestasis is caused by accumulation of hydrophobic bile acids (BA) in the liver due to dysfunctional BA efflux or bile flow into the gall bladder. Therefore, strategies to increase detoxification of hydrophobic BA and downregulate genes involved in BA production are largely investigated. Farnesoid X receptor (FXR) has a central role in BA homeostasis and recent publications revealed that changes in autophagy due to BA-induced reactive oxygen species and increased anti-oxidant response via nuclear factor E2-related factor 2 (NRF2), result in dysregulation of FXR signaling. Several mechanistic studies have identified new dysfunctions of the cholestatic liver at cellular and molecular level, opening new venues for developing more performant therapies.
1. Introduction
According to studies on the burden of liver diseases worldwide, it is estimated that approximately 1 million people die each year due to complications of cirrhosis, and 1 million due to viral hepatitis and hepatocellular carcinoma [1]. Cirrhosis is currently the 11th most common cause of death globally, and liver cancer is the 16th leading cause of death. For patients with end-stage liver disease, the only available therapy is liver transplantation, and at the present rate, less than 10% of transplantation needs are met [1]. A common denominator of a large spectrum of hepatic diseases is liver fibrosis, and therefore a robust number of research studies aim to develop new therapies to mitigate this condition.
Cholestasis is a clinical syndrome with intra- or extra-hepatic etiology, resulting from the obstruction of bile secretion and flow from the liver into the gall bladder and duodenum [2]. Cholestasis can be caused by mutations of genes encoding for proteins with roles in bile transport from hepatocytes into cholangiocytes and bile ducts, resulting in the retention of BA in the liver. The bile flow can also be obstructed by gall stones due to metabolic dysfunctions (cholelithiasis), by tumors (hepatocellular carcinoma, cholangiocarcinoma), or by parasitic infections [2]. Other causes of cholestasis include immune-mediated conditions such as primary sclerosing cholangitis (PSC), primary biliary cirrhosis (PBC), and also exposure to certain medications which negatively affect the liver (non-steroidal anti-inflammatory drugs, anti-diabetic medications). Numerous cholestatic disorders are chronic and may lead to liver fibrosis and cirrhosis if left untreated.
Cholestatic liver injury is a complex disease with a multitude of dysfunctions including not only BA metabolism and transport, but also excessive cell proliferation especially of cholangiocytes and hepatic stellate cells (HSC) which become activated and initiate signaling pathways involving anti-oxidant and immune responses. Therefore, the mechanistic studies on cholestasis encompass a very large area starting with dysregulation of BA metabolism, transport, and signaling on parenchymal and non-parenchymal cells and expanding to inflammation, fibrosis, and eventually carcinogenesis.
BA have critical roles in the regulation of a multitude of physiological processes related to nutrition and digestion, mediating the transport and metabolism of lipids, influencing glucose and insulin sensitivity and modulating the overall energy expenditure in the body [3,4]. BA are signaling molecules acting on receptors that have been defined as BA sensors, and are from two different classes of receptors: (i) nuclear receptors such as farnesoid X receptor (FXR; also known as NR1H4), pregnane X receptor (PXR), constitutive androstane receptor (CAR), and vitamin D receptor (VDR), that become activated by BA and bind to specific response elements on target genes influencing the rate of their transcription; and (ii) membrane-bound G-protein coupled receptors such as Takeda G-protein receptor 5 (TGR5) or G protein-coupled BA receptor 1 (GPBAR1) and spingosine-1-phosphate receptor 2 (S1PR2). In this review we focus on FXR since it is the main player in dysregulation of BA homeostasis in the context of cholestatic injury of the liver. Known therapies as well as new potential drug targets from mechanistic studies on cholestasis-induced liver fibrosis, with focus on dysfunctions in metabolism and transport of hydrophobic BA, are outlined. Several molecular and cellular signaling pathways initiated by FXR in the liver are described. Special attention is also given to a possible role of FXR in autophagy in the context of hepatic cholestasis, bringing light on new potential drug targets for more efficient therapies.
2. FXR
Numerous studies on FXR−/− mice revealed essential roles of FXR in liver functions including the regulation of BA, lipid, and glucose homeostasis [5]. FXR activators have been successfully applied for treatment of dyslipidemia, insulin resistance, and steatosis in obesity, diabetes, alcohol-induced liver injury, and non-alcohol fatty liver or steatohepatitis [5,6,7]. However, the role of FXR in cholestasis in animal models or clinical studies is still unclear and controversial. Cholestatic liver disorders have a large range of different etiologies resulting in reduced bile flow and interruption of enterohepatic circulation, leading to accumulation of toxic bile, rich in hydrophobic BA in the liver. Excessive BA in the liver cause hepatocyte damage, activation of Kupffer cells, HSC, and cholangiocytes, followed by hepatic inflammation and liver fibrosis.
FXR is the main sensor and regulator of BA metabolism in the liver and intestine, with a role in maintaining BA homeostasis during the daily cycles of feeding [8,9]. In Figure 1, a schematic representation describes the most important signaling pathways with impact in cholestasis, that are induced by changes in BA concentrations within the liver versus ileum. A more comprehensive illustration of the overall effects of FXR in liver diseases, including modulation of inflammation, portal hypertension, and carcinogenesis, was published before by Fuchs et al. [10]. In the liver, FXR represses genes involved in BA synthesis via small heterodimer partner (SHP) which is a co-repressor that competes with peroxisome proliferator activated receptor gamma coactivator 1 alpha (PGC1α) which activates genes that stimulate BA synthesis [11]. Overall, FXR in the liver and ileum control BA homeostasis modulating genes of BA synthesis and transport. Therefore, several cholestatic diseases are associated with dysfunctions in signaling pathways regulated by FXR.
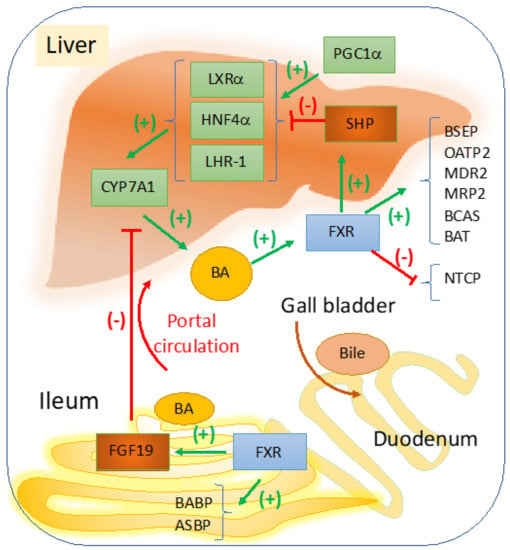
Figure 1.
FXR regulation of bile acid synthesis, transport and hepato-enteric circulation. Cyp7a1 is the rate limiting enzyme in BA synthesis, and it is transactivated by LXRα, HNF4α, and LHR-1 which in turn are stimulated by various ligands derived from lipid nutrients including cholesterol (for LXRα), fatty acyl-CoA (for HNF4α), dietary phospholipids for LRH-1 or liver receptor homologue-1 [18,19]. When BA becomes abundant in the liver, a negative feedback loop starts to function via FXR to maintain balance and to control the elimination of excess BA. BA-induced activation of FXR results in upregulation of SHP which is a co-repressor of LXRα, HNF4α, and LHR-1. FXR upregulates genes with role in BA conjugation, i.e., BA CoA synthase (BCAS) and BA CoA: amino acid N-acetyltransferase (BAT). FXR mediates the bile formation and flow from the liver to the gallbladder by upregulating bile salt export pump (BSEP, ABCB11) which exports BA from hepatocytes; multidrug resistance associated 2 (MRP2, ABCC2) for the transport of BA amongst many other components of the bile into the gallbladder. Other proteins such as multidrug resistance protein 2 (MDR2), a transporter of phospholipids, and ABCG5, ABCG8 that transport cholesterol to the gallbladder are also upregulated by FXR. In the intestine, FXR transactivates ileum BA binding protein (IBABP) and mediates BA reabsorption by apical sodium-dependent bile salt transporter (ASBT). From portal circulation BA are reabsorbed into hepatocytes via sodium-dependent taurocholate cotransport peptide (NTCP), and organic anion transport protein 2 (OATP2). In the intestine, BA-activated FXR upregulates FGF19, a growth factor that is secreted into the portal circulation and inhibits Cyp7a1-controled BA synthesis in hepatocytes. BA synthesis is upregulated when BA concentration in the liver is low and SHP is replaced by PGC1α co-activator of genes that stimulate Cyp7a1 synthesis [11].
Genetic studies revealed several defects of genes involved in FXR signaling pathway in cases of pediatric and adult cholestasis [12]. Thus, in intrahepatic cholestasis of pregnancy (ICP), a disease that occurs in 1/200 pregnancies in Caucasian women and can result in intrauterine fetal death, four heterozygous variants of FXR were found in or near the transcribed sequence of FXR being associated with downregulation of FXR target genes encoding for SHP and organic anion transporting polypeptide 1B3, upon BA stimulation [13,14]. Two different forms of inherited cholestasis, benign recurrent intrahepatic cholestasis (BRIC) and progressive familial intrahepatic cholestasis (PFIC1) were correlated with mutations in ATP8B1 (FIC1), in chromosome 18 [15], a membrane protein that activates FXR via protein kinase C-zeta, and mutations in FIC1 are associated with downstream effects of FXR on BA homeostasis [16]. Specifically, gain of function experiments in cells in vitro demonstrated that FIC1 overexpression enhanced FXR phosphorylation and nuclear localization as well as upregulation of Bsep which is transactivated by FXR. FIC1 effect was dependent on protein kinase C-zeta and also on CDCA presence in culture medium of the cells [16]. BRIC was related to a partially functional FIC1, and it has been suggested that it is caused by a deficient FIC1/PKC-zeta/FXR signaling pathway [16]. Progressive familial intrahepatic cholestasis (PFIC) is a liver injury characterized by early onset of BA accumulation in the liver, accompanied by symptoms of pruritus and malabsorption. In PFIC patients, genetic analysis identified inborn errors in CYP7A1 gene [17], suggesting formation of toxic bile due to predominant alternative biosynthesis of BA.
Table 1 summarizes the newest targets for drugs against cholestasis based on mechanistic studies on FXR and its role in BA homeostasis.

Table 1.
New drug targets related to BA homeostasis and FXR for treating cholestasis-induced hepatic fibrosis.
2.1. Targeting FXR for BA Regulation in Cholestasis
The role of FXR in the regulation of hepatic triglyceride and glucose homeostasis [6,55,56] has been well described and several FXR agonist drugs have been developed for treating dyslipidemia, nonalcoholic fatty liver disease (NAFLD), nonalcoholic steatohepatitis (NASH), and to improve liver functions [6]. However, the beneficial effects of FXR agonists in the treatment of liver fibrosis caused by biliary cholestasis, have been controversial. In cholestasis, the activation of FXR by excessive amounts of BA accumulated in the liver, is at a maximum, and it was demonstrated that administration of CDCA or DCA in their natural form, does not improve liver fibrosis in animal models of hepatic cholestasis [6]. Semi-synthetic BA and non-steroidal agonists of FXR have been developed and tested for therapies of liver and pancreas-related diseases [6]. Thus, INT747 or obeticholic acid (OCA), a 6-α-ethyl derivative of CDCA were proposed to have hepatoprotective effects on certain types of cholestasis based on animal model experiments [21,25]. Non-steroidal agonists of FXR such as GW4064 and WAY-362450, which may modulate multiple G protein-coupled receptors besides directly activating FXR, have been proposed to be used to reduce hepatic inflammation in the context of cholestasis [22,26,57].
Searches for FXR ligands on the database clinicaltrials.gov show numerous studies aiming to apply FXR ligands for many types of diseases including cholestasis but also alcohol-, obesity-, metabolic syndrome-, and diabetes-related liver injuries (e.g., alcoholic hepatitis, NASH, and NAFLD). Out of the total number of clinical trials listed, 12 studies used FXR agonist alone or in combination with either a fibrate such as benzafibrate or with ursodeoxycholic acid (UDCA), to treat biliary and cholestatic related conditions, and PBC in particular (Table 2). Recently, OCA was approved for use in combination with UDCA for the treatment of PBC [20], despite data suggesting that OCA has negative side effects exacerbating biliary injury in animal models of obstructive cholestasis [28]. In fact, in May 2021, Food and Drug Administration (FDA) published a safety communication that can be found on fda.gov, stating that FDA restricts use of OCA in PBC patients with advanced cirrhosis, due to risk of serious liver injury.
Table 2.
Clinical trials for testing FXR ligands or FXR-related targets in PBC/PSC patients, as listed in clinicaltrials.gov database.
A recent review by Jiang et al. describes a full spectrum of small molecules including agonists, partial agonists, and antagonists of FXR, designed to be applied for BA-related liver diseases [58]. The article reveals not only a multitude of various 3D structures of the ligand binding domain of FXR upon binding different types of ligands, but also the structures of steroidal agonists (e.g., OCA, EDP-305, BAR502), nonsteroidal agonists (e.g., GW4064, Nidufexor, Cilofexor, Tropifexor/LJN452), partial agonist (TERN-101, DM175), as well as natural (ivermectin, tuberatolites,) and synthetic (T3, DY268, FLG249) antagonists of FXR [59]. While some of these ligands have shown positive results in clinical studies for treating cholestatic liver diseases, there are still challenges with negative side effects such as high incidence of increased serum LDL, reduced HDL, and pruritus [55,59]. New strategies have been developed to produce more efficient drugs with less adverse symptoms, for example a dual FXR and TGR5 agonist which regulates pathways from both receptors [58,60].
Besides its ability to control the transactivation of genes involved in BA biosynthesis and transport along the enterohepatic tract, and to contribute to liver regeneration and growth, FXR has been proved to counterbalance nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB)—mediated transcription of proinflammatory cytokines, having a role in hepatic repair in liver fibrosis [61,62,63]. Even though FXR is expressed also in HSC and cholangiocytes, it is critical in hepatocytes since BA-induced hepatocyte death activates profibrogenic factors such as transforming growth factor beta 1 (TGF-β1), platelet derived growth factor (PDGF), connective tissue growth factor (CTGF) which then act on quiescent HSC inducing their activation to proliferate and produce excessive extracellular matrix proteins and fibrosis [64,65]. Several published studies revealed that semi-synthesis and non-steroid agonist of FXR, e.g., GW4064 and WAY-362450, were able to mitigate liver inflammation and fibrosis in animal models of cholestasis [22,66,67].
FXR is expressed not only in the liver and entire gastrointestinal tract, but also at lower levels in kidneys, adrenals and even the brain [68,69,70,71]. It is important to underscore that depending on the specific expression of FXR within the body, its activation versus inhibition may favor or impair certain aspects of cholestasis and liver disease-associated syndromes. A good example is the evidence for FXR being expressed in cortical neurons and having role in molecular signaling of BA which induce neurological complications of liver disease [71,72]. It has been demonstrated in animal models of hepatic encephalopathy, that strategies to inhibit brain FXR signaling prevented accumulation of cholesterol in the cortex and neurologic decline [71]. Several dysfunctions in BA metabolism have been associated with neurodegenerative and neurological disorders, and therapies using BA with low hydrophobicity such as conjugated BA tauroursodeoxycholic acid (TUDCA) and UDCA which are considered FXR antagonists, have been found to be neuroprotective [73]. Therefore, future drugs aimed to modulate FXR activity for beneficial effects on the liver in hepatic cholestasis and fibrosis, will need to be designed to avoid unwanted side effects on other tissues and organs such as the brain.
2.2. Targeting FXR-Fibroblast Growth Factor (FGF) 15/19 Enterohepatic Pathway
The physiological role of FXR as master regulator of BA metabolism and homeostasis, was clearly demonstrated in FXR−/− mice, which had abnormally large BA pool size and moreover, exhibited severe hepatotoxicity when challenged with cholic acid-rich diet [5]. It was determined that FXR is highly expressed in certain segments of the intestine, i.e., ileum and colon where it regulates the transcription of BA transporters and also hormones with role in BA homeostasis such as FGF15 [37,38]. As shown in Figure 1, BA-activated FXR in ileum upregulates FGF15/19 which is secreted into the portal circulation and acts on the liver where it inhibits BA synthesis by repressing Cyp7a1 gene in hepatocytes. FGF15/19 acts through a complex of FGF receptor 4 (FGFR4) and βKlotho protein which is a single transmembrane receptor with tyrosine kinase activity [74,75].
FXR gene deletion in mice demonstrated that FXR nuclear receptor plays a role in regulating hepatic inflammation, since FXR−/− mice had high levels of proinflammatory cytokines such as interleukin 1 beta (IL-1β) and were prone to developing liver cancer due to abnormal expression of β-catenin and c-Myc oncogenes [76,77,78]. As mentioned by other reviews on the large array of FXR functions, it is interesting that selective intestinal FXR reactivation in FXR−/− mice, protected against hepatocellular carcinoma [63]. Thus, intestinal FXR restores the control of FGF15 pathway-mediated BA synthesis, and ensures limitation of BA overload and prevention of hepatic inflammation and carcinogenesis [79].
It is presumed that the human ortholog of mouse FGF15 is FGF19, based on 53% amino acid identity; however, there is no definitive consensus on the matter [38]. It has been shown that in humans, serum FGF19 reaches a peak 90–120 min after postprandial release of BA, but before the repression of BA synthesis. Clinical studies indicated that treatment of healthy volunteers with BA sequestrants such as cholestyramine, reduced circulating FGF19, and conversely, administration of FGF19 analogues repressed BA synthesis. Thus, it was confirmed the negative feedback loop of FXR/FGF19 within ileum and the liver, to maintain BA homeostasis. Interestingly, patients suffering from BA-caused diarrhea who overproduce BA, have low circulating FGF19, suggesting a dysfunction of FGF19 and inability to inhibit CYP7A1 and BA synthesis.
The use of FGF19 analogues for the treatment of liver cholestasis appears as an attractive venue to develop new therapeutics, however FGF19 has been shown to promote cell growth and carcinogenesis [80]. A new therapeutic agent is NGM282, an FGF19 nontumorigenic variant that had been described to be hepatoprotective in nonalcoholic steatohepatitis [35], and is in clinical trial for PSC [36] and PBC therapy (NCT02135536) [80].
2.3. Targeting Hydrophobic BA with Role in Hepatocellular Death, Activation of HSC and Fibrogenesis
Cholestatic liver diseases are characterized by chronic hepatic and systemic accumulation of total BA and especially hydrophobic bile salts due to a dramatic dysregulation of BA homeostasis [81]. Most of the cholestasis syndromes are correlated with dysfunctions of genes encoding for transporters of BA along the hepato-biliary tract, such as ATP8B1 coding for phospholipid flippase [82], and ABCB11 for bile salt export pump or BSEP [83,84]. Early studies on the role of excessive BA in liver injury, pointed out that hydrophobic BA such CDCA and GCDC are toxic and induce hepatocellular death by apoptosis [85,86].
Liver injury caused by cholestasis involves a multitude of signaling pathways starting with hepatocyte damage or death, activation of Kupffer cells, recruitment of circulating monocytes and macrophages, activation of cholangiocyte and HSC. Therefore, for long time it has been difficult to assess the direct effect of hydrophobic BA on liver fibrosis, in vivo. However, recently, Hohenester et al. have used Atp8b1G308V/G308V mice which have normal hepatic function except when are being fed high amounts of toxic BA and develop cholestasis [87]. This particular mouse model has been created after the G308V/G308V mutation of Atp8b1 gene was identified in humans susceptible to cholestasis [88]. Upon feeding on GCDC-rich diet, the Atp8b1G308V/G308V mice exhibited pro-fibrotic markers including deposition of excess collagen, proliferation and activation of HSC. Interestingly, treatment of HSC isolated from Atp8b1G308V/G308V mice, with CDCA hydrophobic BA, increased BrdU incorporation in proliferating cells and the expression of alpha smooth muscle actin (αSMA), while a more hydrophilic BA, UDCA did not induce profibrogenic effects [87]. It has been suggested that CDCA was able to induce epidermal growth factor receptor (EGFR)/mitogen-activated protein kinases (MEK)-dependent signaling pathway resulting in activation of extracellular signal-regulated kinase (ERK) followed by collagen deposition and cell proliferation [87]. It has been demonstrated that inhibitors of EGFR/MEK/ERK (i.e., AG1478, UO126 and PD98059, respectively) impaired HSC proliferation and collagen secretion induced by CDCA [87]. Thus, this particular signaling pathway in HSC may be target for future drugs, designed to inhibit EGF-induced proliferation of HSC and to lower liver fibrosis in cholestatic patients.
A strong emphasis is placed on studying potential drugs targeting BA biosynthesis, metabolism, and excretion. Rifampicin has been used firstly as an antimicrobial drug, and later on for the treatment of pruritus caused by biliary cholestasis, based on clinical observations. Later on, it was established that rifampicin (also known as Rifadin, Rimactane commercial names), is a drug that upregulates the expression of detoxification enzymes involved in BA metabolism [41,47,89]. Mechanistic studies indicated that rifampicin induces a number of drug-metabolizing enzymes (DME), with a prominent effect on the expression of cytochrome P450 (CYP) 3A4 in the liver [44]. In a study in which rifampicin was administered to BDL-rats and to human hepatocytes pre-treated with GCDCA, it was determined that phase I and II DME (including CYP3A4, CYP7A1, UDP-glucuronosyltransferase 2B4) and BA transporters (such as MRP4, BSEP) were increased while the import of BA into hepatocytes via Na+ taurocholate co-transporting polypeptide, NTCP, was reduced [90]. Clinical studies indicated that rifampicin was effective in treating cholestasis, and analysis of BA excretion suggested that rifampicin stimulated elimination of 6α-,1β, 2β, and 4β-hydroxyl BA in cholestatic patients during the treatment [91]. A study that looked at the effect of rifampicin on anion exchanger 2 (AE2) which mediates Cl−/HCO3− exchange, concluded that the increase in bile flow induced by rifampicin is mainly due to increased HCO3− excretion mediated by increased expression of AE2 [51]. Various mechanisms underlying the choleretic effects of rifampicin are still to be investigated.
Based on publications in reference to metabolites in the mevalonate pathway being related to the hepatic metabolism of BA, a new study demonstrates that conditional deletion of the enzyme that synthetizes geranylgeranyl pyrophosphate (GGPP) from farnesyl pyrophosphate (FPP) in the liver, decreases the level of hepatic BA [48]. This discovery may lead to designing efficient inhibitors of geranylgeranyl diphosphate synthase (GGPPS) to be applied as medication for the alleviation of obstructive cholestatic disease [48].
More recent investigations focused on a possible role of the short form of augmenter of liver regeneration (sfALR) in preventing BA synthesis and BA-induced apoptosis in hepatocytes [40,92]. ALR, also known as hepatopoietin, was discovered as a growth factor with role in hepatocyte proliferation after liver injury, and then shown to be expressed ubiquitously in all organs and exclusively in hepatocytes, in the liver [39]. An initial study on the effect of toxic BA on ALR expression, revealed that even though Alr gene is upregulated at transcriptional level by early growth response-1 protein (Egr-1), BA suppress ALR transcription independently of Egr-1, but via hepatocyte nuclear factor 4-alpha (HNF4α)/SHP pathway [92]. It has been shown that human hepatoma cells with high expression of sfALR in the cytoplasm, exhibited reduced Cyp7a1 mRNA level and lower production of BA, and this was attributed to activation of signal transducer and activator of transcription 3 (STAT3) [39]. Overexpression of sfALR in hepatoma cells caused significant reduction in GCDA-induced apoptosis. Consistent with sfARL being an inhibitor of BA effects on hepatocytes, it was also found that human cholestatic liver samples had low mRNAs of ALR and forkhead box protein A2 (FOXA2) also known as hepatocyte nuclear factor 3α (HNF-3β), a positive transactivator of ALR gene [39]. FOXA2 has important roles to enable access to closed chromatin and displace linker histones, promoting cells type specification [93,94]. These data suggest that ALR, FOXA2 and STAT3 may be drug targets for decreasing BA biosynthesis and subsequent fibrogenesis in cholestatic patients.
3. Targeting FXR in Portal Hypertension Associated with Cholestasis-Induced Cirrhosis
Cirrhosis is the final stage of chronic cholestasis, in which hepatic fibrosis is very advanced so that most of the liver functions are impaired [95]. Thus, advanced hepatic inflammation, biliary and periportal fibrosis, loss of tissue homeostasis followed by abnormal remodeling are conducive to late permanent dysfunctional state of cirrhosis [96]. Portal hypertension (PHT) is commonly seen in patients with cholestatic liver cirrhosis especially in the stage of decompensation when liver injury is associated with complications such as ascites, hepatic encephalopathy, or variceal bleeding [97]. FXR has been found to be a promising target for the treatment of portal hypertension. Thus, in liver fibrosis, the blood vessels are more constricted compared to normal liver due to a significant decrease of FXR activation caused by ROS and proinflammatory cytokines, resulting in suppressed endothelial nitric oxide synthase (eNOS) and nitric oxide (NO) [98]. Vascular FXR in general was demonstrated to be an important regulatory factor, since pharmacological and genetic activation of FXR stimulated eNOS promotor activity [98]. For the treatment of portal hypertension in particular, Mookerjee et al. studied the effect of an FXR agonist, OCA, on dimethylarginine-dimethylaminohydrolase1 (DDAH-1), a marker of portal pressure that is expressed in hepatocytes and downregulated in cirrhosis [99]. It was shown that asymmetric-dimethylarginine (ADMA), an eNOS inhibitor which is metabolized by DDAH-1 was significantly reduced upon treatment with OCA in an animal model of cholestatic cirrhosis, due to rescue of DDAH-1 expression via activation of FXR [99].
Another interesting study was performed on PX20606 (PX), a nonsteroidal agonist of FXR, in regard to its effect on portal hypertension besides liver fibrosis in experimental models of non-cirrhotic (partial portal vein ligation or PPVL) and cirrhotic (carbon tetrachloride, CCl4) models [100]. PX was able to decrease portal pressure markers in both non-cirrhotic and cirrhotic rats, confirming that FXR has critical role in the regulation of eNOS and portal pressure in the liver.
4. FXR Involvement in Autophagy during Cholestasis
Recent studies on hepatic autophagy revealed an important function of this process in maintaining the overall homeostasis of the liver. Autophagy is an evolutionary conserved mechanism of lysosome-dependent degradation of intracellular components, with multiple functions including cell energy homeostasis, organelle turnover, clearance of aggregated materials inside cells and defense against intracellular pathogens [101]. Deficiencies in autophagy result in several pathologies associated with hepatomegaly, liver inflammation and fibrosis and even carcinogenesis [101]. It was first demonstrated that in BDL mice, cholestasis was associated with hepatocyte autophagy activation [102]. In these studies, suppression of autophagy with chloroquine increased hepatocyte apoptosis, while activation of autophagy with rapamycin decreased cholestatic liver injury, and it was concluded that autophagy benefited hepatocyte survival via modulation of reactive oxygen species (ROS) [102]. Later on, it was found that the accumulation of a protein p62/SQSTM1 (sequestosome 1) disabled the ubiquitination and degradation of nuclear factor-erythroid 2-related factor 2 (NRF2), leading to liver injury [103,104,105]. Generally, NRF2 is known as a transcription factor that targets genes with role in adaptive protection against oxidative stress in cells [106]. It was also suggested that NRF2 is involved in the regulation of autophagic processes in response to oxidative stress, functioning in a negative feedback loop in opposition to AMP-activated protein kinase (AMPK), which is critical for autophagy induction via mTOR downregulation (mTOR or mammalian target of rapamycin, has role in cell growth and autophagy) [107]. Recently, it was determined that increased NRF2 due to defective autophagy, causes a larger release of high mobility group box 1 (HMGB1), a protein released during necrosis) from hepatocytes and consequently, enhanced the ductular reaction [105]. Moreover, NRF2 is related to the dysfunction of BA synthesis, secretion and regulation, affecting the expression of FXR, the main nuclear receptor that regulates BA metabolism and transport within the liver [101]. Thus, Khambu et al. [101] demonstrated that mice deficient in autophagy due to a lack of Atg7 and Atg5 genes, exhibited hepatic cholestasis characterized by increased serum and liver BA loads, biliary hyperplasia, and suppressed BA transporters such as BSEP which are transactivated by FXR. Interestingly, deletion of Nrf2 gene in autophagy-deficient mice, rescued FXR suppression and reversed the cholestatic injuries [101]. The authors concluded that there is a regulatory loop between FXR and autophagy, in which BA can suppress autophagy, and deficiency in autophagy downregulates FXR via NRF2 expression [101]. This study suggests that several targets including AMPK, NRF2, autophagy regulators and FXR are to be considered for developing novel therapies for liver cholestatis and fibrosis. Thus, betulinic acid [53] (a natural pentacyclic triterpenoid), 5-aminoimidazole-4-carboxamide-1β-D-ribofuranoside (AICAR) [108], metformin [109,110], and GSK621 [111] are AMPK activators known for beneficial effects related to several diseases including diabetes, obesity, chronic inflammation, and cancer, and deserve to be investigated in the context of cholestasis [112]. In fact, recent studies identified the AMPK/FXR axis as having critical role in cholestatic liver injuries [113]. NRF2 transcription factor has been considered to be almost exclusively positive in promoting cell survival under detrimental conditions due to increased reactive oxygen radicals, via activation of target genes bearing antioxidant response element (ARE) in their promoters [31]. However, there are also negative effects related to NRF2, for example, excessive upregulation of NRF2 pathway can result in cell dysfunction or help cancer cell survival and chemotherapy resistance [29]. A quest for NRF2 antagonists led to the identification of brusatol, a quassinoid isolated from an evergreen shrub Brucea javanica, to decrease the level of NRF2 in a series of cancer cell lines [29,30]. The effect of such NRF2 inhibitors on models of cholestatic liver injuries, are still to be tested.
The schematic representation in Figure 2 summarizes the main signaling pathways involved in the regulatory process of autophagy in relation to hepatocyte injuries due oxidative stress. The signaling pathways initiated by BA through FXR, HNF4α, and other transcription factors and the effects of BA on various homeostatic processes such as autophagy are still to be understood (i.e., how increased BA in the liver due to biliary obstruction, influence cell energy homeostasis, autophagy and lysosome functions). Data from studies on the effect of FXR agonist on liver cholestasis, have been controversial, probably due to a poor understanding of the FXR role in autophagy.
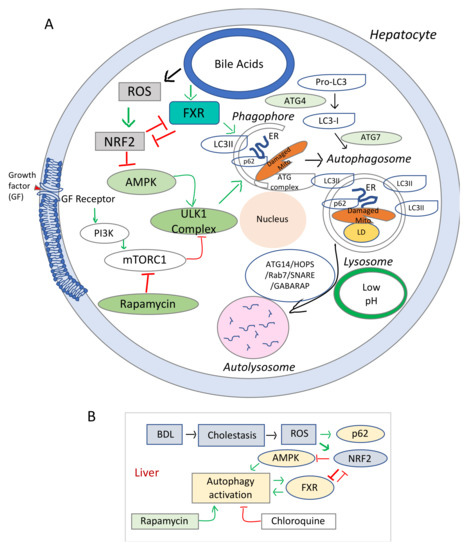
Figure 2.
Diagram of signaling pathways involved in the dysregulation of autophagy and cell growth in cholestasis and liver fibrosis. (A), Schematic representation of the elements involved in the autophagy dysfunction in chronic oxidative stress. mTOR (mechanistic target of rapamycin) is a serine/threonine protein kinase, component of two distinct cellular complexes termed mTOR complex 1 (mTORC1) and mTORC2 where a multitude of signals such as nutrients, growth, energy are sensed and processed [114]. mTORC 1 downregulates autophagy (a catabolic process where macromolecules are sequestered in double membrane bound autophagosomes that fuse with lysosomes to allow their enzymatic breakdown). When there is sufficient energy, mTORC1 is active and through phosphorylation and destabilization of autophagy promoting complexes (ULK1 and other proteins), it ensures that autophagy is inhibited. During conditions of starvation, mTORC1 is inactive, it allows the phagosome to be formed. In addition, due to low energy level, AMPK gets activated and phosphorylates ULK1. Sequestome 1 (SQSTM1) or p62 is an active component of the autophagic process, and acts as a scaffold on lysosomal membranes for mTOCR1. During autophagy, p62 is incorporated into autophagosome and degraded, thus limiting mTORC1 activity. LC3 or microtubule-associated proteins 1A/1B light chain 3B is a biomarker of autophagosome. (B), Diagram of the interaction of cholestasis-induced ROS with autophagy in hepatocytes. While stimulating p62 and phagosome formation, ROS inhibit AMPK-mediated energy production needed for the autophagic flux, via NRF2 upregulation. Stimulation of autophagy by rapamycin and overexpression of FXR in autophagy-deficient cells counteract the effects of NRF2 reducing ROS-mediated hepatic damage.
In summary, recent studies show that increased ROS as result of BA toxicity in cholestasis, have negative effects on autophagy, and more drugs are to be designed to address the signaling pathways of FXR in connection to autophagy.
5. Conclusions
Currently, there are only few therapies for the treatment of cholestasis and liver fibrosis, including UDCA, nor-UDCA, OCA, and rifampicin. The first-line therapy in most cholestasis disorders is treatment with UDCA, the hydrophilic BA which enhances hepatobiliary flow, being effective in approximately 50% of patients of most forms of cholestasis. Recently OCA was approved as a second-line of therapy for patients who do not respond to UDCA treatment. Another FXR agonist, i.e., GW4064 which demonstrated decreased BA synthesis in preclinical studies, was not approved for clinical trials due to the short terminal half-life of the drug. Rifampicin (Rifadin, Rimactane commercial names) is a drug that upregulates the expression of detoxification enzymes involved in BA metabolism.
Author Contributions
Conceptualization, A.D.P. and S.D.; resources, S.D.; writing—original draft preparation, A.D.P.; writing—review and editing, S.D. and A.D.P.; supervision, S.D.; project administration, S.D.; funding acquisition, S.D. All authors have read and agreed to the published version of the manuscript.
Funding
This study was funded by NIH R01 awards (DK082435 and DK112803) to Sharon DeMorrow.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgments
This work was completed with support from the Veterans Health Administration and with resources and the use of facilities at the Central Texas Veterans Health Care System, Temple, Texas. The contents do not represent the views of the U.S. Department of Veterans Affairs or the United States Government.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Asrani, S.K.; Devarbhavi, H.; Eaton, J.; Kamath, P.S. Burden of liver diseases in the world. J. Hepatol. 2019, 70, 151–171. [Google Scholar] [CrossRef] [PubMed]
- Samant, H.; Manatsathit, W.; Dies, D.; Shokouh-Amiri, H.; Zibari, G.; Boktor, M.; Alexander, J.S. Cholestatic liver diseases: An era of emerging therapies. World J. Clin. Cases 2019, 7, 1571–1581. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, A.F.; Hagey, L.R. Bile acids: Chemistry, pathochemistry, biology, pathobiology, and therapeutics. Cell Mol. Life Sci. 2008, 65, 2461–2483. [Google Scholar] [CrossRef] [PubMed]
- Keitel, V.; Kubitz, R.; Haussinger, D. Endocrine and paracrine role of bile acids. World J. Gastroenterol. 2008, 14, 5620–5629. [Google Scholar] [CrossRef]
- Sinal, C.J.; Tohkin, M.; Miyata, M.; Ward, J.M.; Lambert, G.; Gonzalez, F.J. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell 2000, 102, 731–744. [Google Scholar] [CrossRef]
- Jiao, Y.; Lu, Y.; Li, X.Y. Farnesoid X receptor: A master regulator of hepatic triglyceride and glucose homeostasis. Acta Pharmacol. Sin. 2015, 36, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lee, F.Y.; Barrera, G.; Lee, H.; Vales, C.; Gonzalez, F.J.; Willson, T.M.; Edwards, P.A. Activation of the nuclear receptor FXR improves hyperglycemia and hyperlipidemia in diabetic mice. Proc. Natl. Acad. Sci. USA 2006, 103, 1006–1011. [Google Scholar] [CrossRef]
- Makishima, M.; Okamoto, A.Y.; Repa, J.J.; Tu, H.; Learned, R.M.; Luk, A.; Hull, M.V.; Lustig, K.D.; Mangelsdorf, D.J.; Shan, B. Identification of a nuclear receptor for bile acids. Science 1999, 284, 1362–1365. [Google Scholar] [CrossRef]
- Parks, D.J.; Blanchard, S.G.; Bledsoe, R.K.; Chandra, G.; Consler, T.G.; Kliewer, S.A.; Stimmel, J.B.; Willson, T.M.; Zavacki, A.M.; Moore, D.D.; et al. Bile acids: Natural ligands for an orphan nuclear receptor. Science 1999, 284, 1365–1368. [Google Scholar] [CrossRef]
- Fuchs, C.D.; Schwabl, P.; Reiberger, T.; Trauner, M. Liver Capsule: FXR agonists against liver disease. Hepatology 2016, 64, 1773. [Google Scholar] [CrossRef]
- Kanaya, E.; Shiraki, T.; Jingami, H. The nuclear bile acid receptor FXR is activated by PGC-1alpha in a ligand-dependent manner. Biochem. J. 2004, 382, 913–921. [Google Scholar] [CrossRef]
- Pathak, P.; Liu, H.; Boehme, S.; Xie, C.; Krausz, K.W.; Gonzalez, F.; Chiang, J.Y.L. Farnesoid X receptor induces Takeda G-protein receptor 5 cross-talk to regulate bile acid synthesis and hepatic metabolism. J. Biol. Chem. 2017, 292, 11055–11069. [Google Scholar] [CrossRef]
- Marzolini, C.; Tirona, R.G.; Gervasini, G.; Poonkuzhali, B.; Assem, M.; Lee, W.; Leake, B.F.; Schuetz, J.D.; Schuetz, E.G.; Kim, R.B. A common polymorphism in the bile acid receptor farnesoid X receptor is associated with decreased hepatic target gene expression. Mol. Endocrinol. 2007, 21, 1769–1780. [Google Scholar] [CrossRef] [PubMed]
- Van Mil, S.W.; Milona, A.; Dixon, P.H.; Mullenbach, R.; Geenes, V.L.; Chambers, J.; Shevchuk, V.; Moore, G.E.; Lammert, F.; Glantz, A.G.; et al. Functional variants of the central bile acid sensor FXR identified in intrahepatic cholestasis of pregnancy. Gastroenterology 2007, 133, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Bull, L.N.; van Eijk, M.J.; Pawlikowska, L.; DeYoung, J.A.; Juijn, J.A.; Liao, M.; Klomp, L.W.; Lomri, N.; Berger, R.; Scharschmidt, B.F.; et al. A gene encoding a P-type ATPase mutated in two forms of hereditary cholestasis. Nat. Genet. 1998, 18, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Frankenberg, T.; Miloh, T.; Chen, F.Y.; Ananthanarayanan, M.; Sun, A.Q.; Balasubramaniyan, N.; Arias, I.; Setchell, K.D.; Suchy, F.J.; Shneider, B.L. The membrane protein ATPase class I type 8B member 1 signals through protein kinase C zeta to activate the farnesoid X receptor. Hepatology 2008, 48, 1896–1905. [Google Scholar] [CrossRef]
- Setchell, K.D.; Schwarz, M.; O’Connell, N.C.; Lund, E.G.; Davis, D.L.; Lathe, R.; Thompson, H.R.; Weslie Tyson, R.; Sokol, R.J.; Russell, D.W. Identification of a new inborn error in bile acid synthesis: Mutation of the oxysterol 7alpha-hydroxylase gene causes severe neonatal liver disease. J. Clin. Investig. 1998, 102, 1690–1703. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.Y. Regulation of bile acid synthesis: Pathways, nuclear receptors, and mechanisms. J. Hepatol. 2004, 40, 539–551. [Google Scholar] [CrossRef]
- Cornelison, J.L.; Cato, M.L.; Johnson, A.M.; D’Agostino, E.H.; Melchers, D.; Patel, A.B.; Mays, S.G.; Houtman, R.; Ortlund, E.A.; Jui, N.T. Development of a new class of liver receptor homolog-1 (LRH-1) agonists by photoredox conjugate addition. Bioorg. Med. Chem. Lett. 2020, 30, 127293. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.S., Jr. Use of Obeticholic Acid in Patients with Primary Biliary Cholangitis. Gastroenterol. Hepatol. 2018, 14, 654–657. [Google Scholar]
- Fiorucci, S.; Clerici, C.; Antonelli, E.; Orlandi, S.; Goodwin, B.; Sadeghpour, B.M.; Sabatino, G.; Russo, G.; Castellani, D.; Willson, T.M.; et al. Protective effects of 6-ethyl chenodeoxycholic acid, a farnesoid X receptor ligand, in estrogen-induced cholestasis. J. Pharmacol. Exp. Ther. 2005, 313, 604–612. [Google Scholar] [CrossRef] [PubMed]
- Flatt, B.; Martin, R.; Wang, T.L.; Mahaney, P.; Murphy, B.; Gu, X.H.; Foster, P.; Li, J.; Pircher, P.; Petrowski, M.; et al. Discovery of XL335 (WAY-362450), a highly potent, selective, and orally active agonist of the farnesoid X receptor (FXR). J. Med. Chem. 2009, 52, 904–907. [Google Scholar] [CrossRef] [PubMed]
- Gatselis, N.K.; Goet, J.C.; Zachou, K.; Lammers, W.J.; Janssen, H.L.A.; Hirschfield, G.; Corpechot, C.; Lindor, K.D.; Invernizzi, P.; Mayo, M.J.; et al. Factors Associated With Progression and Outcomes of Early Stage Primary Biliary Cholangitis. Clin. Gastroenterol. Hepatol. 2020, 18, 684–692e686. [Google Scholar] [CrossRef] [PubMed]
- Gochanour, E.M.; Kowdley, K.V. Investigational drugs in early phase development for primary biliary cholangitis. Expert Opin. Investig. Drugs 2021, 30, 131–141. [Google Scholar] [CrossRef]
- Kowdley, K.V.; Vuppalanchi, R.; Levy, C.; Floreani, A.; Andreone, P.; LaRusso, N.F.; Shrestha, R.; Trotter, J.; Goldberg, D.; Rushbrook, S.; et al. A randomized, placebo-controlled, phase II study of obeticholic acid for primary sclerosing cholangitis. J. Hepatol. 2020, 73, 94–101. [Google Scholar] [CrossRef]
- Ma, Y.; Huang, Y.; Yan, L.; Gao, M.; Liu, D. Synthetic FXR agonist GW4064 prevents diet-induced hepatic steatosis and insulin resistance. Pharm. Res. 2013, 30, 1447–1457. [Google Scholar] [CrossRef]
- Shah, R.A.; Kowdley, K.V. Current and potential treatments for primary biliary cholangitis. Lancet Gastroenterol. Hepatol. 2020, 5, 306–315. [Google Scholar] [CrossRef]
- Van Golen, R.F.; Olthof, P.B.; Lionarons, D.A.; Reiniers, M.J.; Alles, L.K.; Uz, Z.; de Haan, L.; Ergin, B.; de Waart, D.R.; Maas, A.; et al. FXR agonist obeticholic acid induces liver growth but exacerbates biliary injury in rats with obstructive cholestasis. Sci. Rep. 2018, 8, 16529. [Google Scholar] [CrossRef]
- Ren, D.; Villeneuve, N.F.; Jiang, T.; Wu, T.; Lau, A.; Toppin, H.A.; Zhang, D.D. Brusatol enhances the efficacy of chemotherapy by inhibiting the Nrf2-mediated defense mechanism. Proc. Natl. Acad. Sci. USA 2011, 108, 1433–1438. [Google Scholar] [CrossRef] [PubMed]
- Ye, R.; Dai, N.; He, Q.; Guo, P.; Xiang, Y.; Zhang, Q.; Hong, Z.; Zhang, Q. Comprehensive anti-tumor effect of Brusatol through inhibition of cell viability and promotion of apoptosis caused by autophagy via the PI3K/Akt/mTOR pathway in hepatocellular carcinoma. Biomed. Pharmacother. 2018, 105, 962–973. [Google Scholar] [CrossRef]
- Zhang, D.D. Mechanistic studies of the Nrf2-Keap1 signaling pathway. Drug Metab. Rev. 2006, 38, 769–789. [Google Scholar] [CrossRef]
- Halasi, M.; Wang, M.; Chavan, T.S.; Gaponenko, V.; Hay, N.; Gartel, A.L. ROS inhibitor N-acetyl-L-cysteine antagonizes the activity of proteasome inhibitors. Biochem. J. 2013, 454, 201–208. [Google Scholar] [CrossRef]
- Lu, H.; Hu, H.; Yang, Y.; Li, S. The inhibition of reactive oxygen species (ROS) by antioxidants inhibits the release of an autophagy marker in ectopic endometrial cells. Taiwan J. Obstet. Gynecol. 2020, 59, 256–261. [Google Scholar] [CrossRef] [PubMed]
- Orr, A.L.; Ashok, D.; Sarantos, M.R.; Shi, T.; Hughes, R.E.; Brand, M.D. Inhibitors of ROS production by the ubiquinone-binding site of mitochondrial complex I identified by chemical screening. Free Radic. Biol. Med. 2013, 65, 1047–1059. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.A.; Rinella, M.E.; Abdelmalek, M.F.; Trotter, J.F.; Paredes, A.H.; Arnold, H.L.; Kugelmas, M.; Bashir, M.R.; Jaros, M.J.; Ling, L.; et al. NGM282 for treatment of non-alcoholic steatohepatitis: A multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 2018, 391, 1174–1185. [Google Scholar] [CrossRef]
- Hirschfield, G.M.; Chazouilleres, O.; Drenth, J.P.; Thorburn, D.; Harrison, S.A.; Landis, C.S.; Mayo, M.J.; Muir, A.J.; Trotter, J.F.; Leeming, D.J.; et al. Effect of NGM282, an FGF19 analogue, in primary sclerosing cholangitis: A multicenter, randomized, double-blind, placebo-controlled phase II trial. J. Hepatol. 2019, 70, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, T.; Choi, M.; Moschetta, A.; Peng, L.; Cummins, C.L.; McDonald, J.G.; Luo, G.; Jones, S.A.; Goodwin, B.; Richardson, J.A.; et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005, 2, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Kliewer, S.A.; Mangelsdorf, D.J. Bile Acids as Hormones: The FXR-FGF15/19 Pathway. Dig. Dis. 2015, 33, 327–331. [Google Scholar] [CrossRef]
- Gandhi, C.R. Augmenter of liver regeneration. Fibrogenes. Tissue Repair 2012, 5, 10. [Google Scholar] [CrossRef]
- Ibrahim, S.; Dayoub, R.; Krautbauer, S.; Liebisch, G.; Wege, A.K.; Melter, M.; Weiss, T.S. Bile acid-induced apoptosis and bile acid synthesis are reduced by over-expression of Augmenter of Liver Regeneration (ALR) in a STAT3-dependent mechanism. Exp. Cell Res. 2019, 374, 189–197. [Google Scholar] [CrossRef]
- Dyson, J.; Jones, D. Diagnosis and management of patients with primary biliary cirrhosis. Clin. Liver Dis. 2014, 3, 52–55. [Google Scholar] [CrossRef]
- Dyson, J.K.; Elsharkawy, A.M.; Lamb, C.A.; Al-Rifai, A.; Newton, J.L.; Jones, D.E.; Hudson, M. Fatigue in primary sclerosing cholangitis is associated with sympathetic over-activity and increased cardiac output. Liver Int. 2015, 35, 1633–1641. [Google Scholar] [CrossRef]
- Griffiths, L.; Dyson, J.K.; Jones, D.E. The new epidemiology of primary biliary cirrhosis. Semin. Liver Dis. 2014, 34, 318–328. [Google Scholar] [CrossRef]
- Kremer, A.E.; Bolier, R.; van Dijk, R.; Oude Elferink, R.P.; Beuers, U. Advances in pathogenesis and management of pruritus in cholestasis. Dig. Dis. 2014, 32, 637–645. [Google Scholar] [CrossRef]
- Kremer, A.E.; Feramisco, J.; Reeh, P.W.; Beuers, U.; Oude Elferink, R.P. Receptors, cells and circuits involved in pruritus of systemic disorders. Biochim. Biophys. Acta 2014, 1842, 869–892. [Google Scholar] [CrossRef] [PubMed]
- Lilja, J.J.; Niemi, M.; Neuvonen, P.J. Rifampicin reduces plasma concentrations of celiprolol. Eur. J. Clin. Pharmacol. 2004, 59, 819–824. [Google Scholar] [CrossRef] [PubMed]
- Niemi, M.; Backman, J.T.; Fromm, M.F.; Neuvonen, P.J.; Kivisto, K.T. Pharmacokinetic interactions with rifampicin: Clinical relevance. Clin. Pharmacokinet. 2003, 42, 819–850. [Google Scholar] [CrossRef]
- Jia, W.J.; Tang, Q.L.; Jiang, S.; Sun, S.Q.; Xue, B.; Qiu, Y.D.; Li, C.J.; Mao, L. Conditional loss of geranylgeranyl diphosphate synthase alleviates acute obstructive cholestatic liver injury by regulating hepatic bile acid metabolism. FEBS J. 2020, 287, 3328–3345. [Google Scholar] [CrossRef]
- Seki, Y.; Mizuochi, T.; Kimura, A.; Takahashi, T.; Ohtake, A.; Hayashi, S.; Morimura, T.; Ohno, Y.; Hoshina, T.; Ihara, K.; et al. Two neonatal cholestasis patients with mutations in the SRD5B1 (AKR1D1) gene: Diagnosis and bile acid profiles during chenodeoxycholic acid treatment. J. Inherit. Metab. Dis. 2013, 36, 565–573. [Google Scholar] [CrossRef]
- Tan, H.; Xu, C.; Zeng, H.; Wang, Y.; Li, Y.; Fan, X.; Chen, P.; Jiang, Y.; Chen, X.; Huang, M.; et al. SUMOylation of pregnane X receptor suppresses rifampicin-induced CYP3A4 and P-gp expression and activity in LS174T cells. J. Pharmacol. Sci. 2016, 130, 66–71. [Google Scholar] [CrossRef][Green Version]
- Wang, W.; Ren, X.; Cai, Y.; Chen, L.; Zhang, W.; Xu, J. Rifampicin Induces Bicarbonate-Rich Choleresis in Rats: Involvement of Anion Exchanger 2. Dig. Dis. Sci. 2016, 61, 126–136. [Google Scholar] [CrossRef]
- Kim, J.; Yang, G.; Kim, Y.; Kim, J.; Ha, J. AMPK activators: Mechanisms of action and physiological activities. Exp. Mol. Med. 2016, 48, e224. [Google Scholar] [CrossRef] [PubMed]
- Song, T.J.; Park, C.H.; In, K.R.; Kim, J.B.; Kim, J.H.; Kim, M.; Chang, H.J. Antidiabetic effects of betulinic acid mediated by the activation of the AMP-activated protein kinase pathway. PLoS ONE 2021, 16, e0249109. [Google Scholar] [CrossRef]
- Koppula, S.; Kumar, H.; Kim, I.S.; Choi, D.K. Reactive oxygen species and inhibitors of inflammatory enzymes, NADPH oxidase, and iNOS in experimental models of Parkinson’s disease. Mediat. Inflamm. 2012, 2012, 823902. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.Y.L.; Ferrell, J.M. Bile acid receptors FXR and TGR5 signaling in fatty liver diseases and therapy. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 318, G554–G573. [Google Scholar] [CrossRef]
- Watanabe, M.; Houten, S.M.; Wang, L.; Moschetta, A.; Mangelsdorf, D.J.; Heyman, R.A.; Moore, D.D.; Auwerx, J. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J. Clin. Investig. 2004, 113, 1408–1418. [Google Scholar] [CrossRef]
- Hegade, V.S.; Speight, R.A.; Etherington, R.E.; Jones, D.E. Novel bile acid therapeutics for the treatment of chronic liver diseases. Therap. Adv. Gastroenterol. 2016, 9, 376–391. [Google Scholar] [CrossRef]
- Jiang, L.; Zhang, H.; Xiao, D.; Wei, H.; Chen, Y. Farnesoid X receptor (FXR): Structures and ligands. Comput. Struct. Biotechnol. J. 2021, 19, 2148–2159. [Google Scholar] [CrossRef]
- Hirschfield, G.M.; Mason, A.; Luketic, V.; Lindor, K.; Gordon, S.C.; Mayo, M.; Kowdley, K.V.; Vincent, C.; Bodhenheimer, H.C., Jr.; Pares, A.; et al. Efficacy of obeticholic acid in patients with primary biliary cirrhosis and inadequate response to ursodeoxycholic acid. Gastroenterology 2015, 148, 751–761e758. [Google Scholar] [CrossRef]
- Wang, X.X.; Wang, D.; Luo, Y.; Myakala, K.; Dobrinskikh, E.; Rosenberg, A.Z.; Levi, J.; Kopp, J.B.; Field, A.; Hill, A.; et al. FXR/TGR5 Dual Agonist Prevents Progression of Nephropathy in Diabetes and Obesity. J. Am. Soc. Nephrol. 2018, 29, 118–137. [Google Scholar] [CrossRef]
- Gadaleta, R.M.; Oldenburg, B.; Willemsen, E.C.; Spit, M.; Murzilli, S.; Salvatore, L.; Klomp, L.W.; Siersema, P.D.; van Erpecum, K.J.; van Mil, S.W. Activation of bile salt nuclear receptor FXR is repressed by pro-inflammatory cytokines activating NF-kappaB signaling in the intestine. Biochim. Biophys. Acta 2011, 1812, 851–858. [Google Scholar] [CrossRef]
- Wang, Y.D.; Chen, W.D.; Wang, M.; Yu, D.; Forman, B.M.; Huang, W. Farnesoid X receptor antagonizes nuclear factor kappaB in hepatic inflammatory response. Hepatology 2008, 48, 1632–1643. [Google Scholar] [CrossRef]
- Cariello, M.; Piccinin, E.; Garcia-Irigoyen, O.; Sabba, C.; Moschetta, A. Nuclear receptor FXR, bile acids and liver damage: Introducing the progressive familial intrahepatic cholestasis with FXR mutations. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1308–1318. [Google Scholar] [CrossRef] [PubMed]
- Mederacke, I.; Hsu, C.C.; Troeger, J.S.; Huebener, P.; Mu, X.; Dapito, D.H.; Pradere, J.P.; Schwabe, R.F. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat. Commun. 2013, 4, 2823. [Google Scholar] [CrossRef] [PubMed]
- Fickert, P.; Fuchsbichler, A.; Moustafa, T.; Wagner, M.; Zollner, G.; Halilbasic, E.; Stoger, U.; Arrese, M.; Pizarro, M.; Solis, N.; et al. Farnesoid X receptor critically determines the fibrotic response in mice but is expressed to a low extent in human hepatic stellate cells and periductal myofibroblasts. Am. J. Pathol. 2009, 175, 2392–2405. [Google Scholar] [CrossRef]
- Fiorucci, S.; Rizzo, G.; Antonelli, E.; Renga, B.; Mencarelli, A.; Riccardi, L.; Orlandi, S.; Pruzanski, M.; Morelli, A.; Pellicciari, R. A farnesoid x receptor-small heterodimer partner regulatory cascade modulates tissue metalloproteinase inhibitor-1 and matrix metalloprotease expression in hepatic stellate cells and promotes resolution of liver fibrosis. J. Pharmacol. Exp. Ther. 2005, 314, 584–595. [Google Scholar] [CrossRef]
- Liu, H.M.; Lee, T.Y.; Liao, J.F. GW4064 attenuates lipopolysaccharideinduced hepatic inflammation and apoptosis through inhibition of the Tolllike receptor 4mediated p38 mitogenactivated protein kinase signaling pathway in mice. Int. J. Mol. Med. 2018, 41, 1455–1462. [Google Scholar] [CrossRef]
- Forman, B.M.; Goode, E.; Chen, J.; Oro, A.E.; Bradley, D.J.; Perlmann, T.; Noonan, D.J.; Burka, L.T.; McMorris, T.; Lamph, W.W.; et al. Identification of a nuclear receptor that is activated by farnesol metabolites. Cell 1995, 81, 687–693. [Google Scholar] [CrossRef]
- Lu, T.T.; Repa, J.J.; Mangelsdorf, D.J. Orphan nuclear receptors as eLiXiRs and FiXeRs of sterol metabolism. J. Biol. Chem. 2001, 276, 37735–37738. [Google Scholar] [CrossRef]
- Wang, H.; Chen, J.; Hollister, K.; Sowers, L.C.; Forman, B.M. Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol. Cell 1999, 3, 543–553. [Google Scholar] [CrossRef]
- McMillin, M.; Grant, S.; Frampton, G.; Petrescu, A.D.; Kain, J.; Williams, E.; Haines, R.; Canady, L.; DeMorrow, S. FXR-Mediated Cortical Cholesterol Accumulation Contributes to the Pathogenesis of Type a Hepatic Encephalopathy. Cell Mol. Gastroenterol. Hepatol. 2018, 6, 47–63. [Google Scholar] [CrossRef]
- DeMorrow, S. Bile Acids in Hepatic Encephalopathy. J. Clin. Exp. Hepatol. 2019, 9, 117–124. [Google Scholar] [CrossRef]
- Grant, S.M.; DeMorrow, S. Bile Acid Signaling in Neurodegenerative and Neurological Disorders. Int. J. Mol. Sci. 2020, 21, 5982. [Google Scholar] [CrossRef]
- Yu, C.; Wang, F.; Kan, M.; Jin, C.; Jones, R.B.; Weinstein, M.; Deng, C.X.; McKeehan, W.L. Elevated cholesterol metabolism and bile acid synthesis in mice lacking membrane tyrosine kinase receptor FGFR4. J. Biol. Chem. 2000, 275, 15482–15489. [Google Scholar] [CrossRef]
- Ito, S.; Fujimori, T.; Furuya, A.; Satoh, J.; Nabeshima, Y.; Nabeshima, Y. Impaired negative feedback suppression of bile acid synthesis in mice lacking betaKlotho. J. Clin. Investig. 2005, 115, 2202–2208. [Google Scholar] [CrossRef]
- Yang, F.; Huang, X.; Yi, T.; Yen, Y.; Moore, D.D.; Huang, W. Spontaneous development of liver tumors in the absence of the bile acid receptor farnesoid X receptor. Cancer Res. 2007, 67, 863–867. [Google Scholar] [CrossRef]
- Wolfe, A.; Thomas, A.; Edwards, G.; Jaseja, R.; Guo, G.L.; Apte, U. Increased activation of the Wnt/beta-catenin pathway in spontaneous hepatocellular carcinoma observed in farnesoid X receptor knockout mice. J. Pharmacol. Exp. Ther. 2011, 338, 12–21. [Google Scholar] [CrossRef]
- Kim, I.; Morimura, K.; Shah, Y.; Yang, Q.; Ward, J.M.; Gonzalez, F.J. Spontaneous hepatocarcinogenesis in farnesoid X receptor-null mice. Carcinogenesis 2007, 28, 940–946. [Google Scholar] [CrossRef]
- Degirolamo, C.; Modica, S.; Vacca, M.; Di Tullio, G.; Morgano, A.; D’Orazio, A.; Kannisto, K.; Parini, P.; Moschetta, A. Prevention of spontaneous hepatocarcinogenesis in farnesoid X receptor-null mice by intestinal-specific farnesoid X receptor reactivation. Hepatology 2015, 61, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Peeraphatdit, T.B.; Simonetto, D.A.; Shah, V.H. Exploring new treatment paradigms for alcoholic hepatitis by extrapolating from NASH and cholestasis. J. Hepatol. 2018, 69, 275–277. [Google Scholar] [CrossRef]
- Geerts, A. History, heterogeneity, developmental biology, and functions of quiescent hepatic stellate cells. Semin. Liver Dis. 2001, 21, 311–335. [Google Scholar] [CrossRef] [PubMed]
- Dawson, P.A. Liver disease without flipping: New functions of ATP8B1, the protein affected in familial intrahepatic cholestasis type 1. Hepatology 2010, 51, 1885–1887. [Google Scholar] [CrossRef] [PubMed]
- Stieger, B. Role of the bile salt export pump, BSEP, in acquired forms of cholestasis. Drug Metab. Rev. 2010, 42, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Soroka, C.J.; Boyer, J.L. Biosynthesis and trafficking of the bile salt export pump, BSEP: Therapeutic implications of BSEP mutations. Mol. Asp. Med. 2014, 37, 3–14. [Google Scholar] [CrossRef]
- Kwo, P.; Patel, T.; Bronk, S.F.; Gores, G.J. Nuclear serine protease activity contributes to bile acid-induced apoptosis in hepatocytes. Am. J. Physiol. 1995, 268, G613–G621. [Google Scholar] [CrossRef]
- Faubion, W.A.; Guicciardi, M.E.; Miyoshi, H.; Bronk, S.F.; Roberts, P.J.; Svingen, P.A.; Kaufmann, S.H.; Gores, G.J. Toxic bile salts induce rodent hepatocyte apoptosis via direct activation of Fas. J. Clin. Investig. 1999, 103, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Hohenester, S.; Kanitz, V.; Kremer, A.E.; Paulusma, C.C.; Wimmer, R.; Kuehn, H.; Denk, G.; Horst, D.; Elferink, R.O.; Beuers, U. Glycochenodeoxycholate Promotes Liver Fibrosis in Mice with Hepatocellular Cholestasis. Cells 2020, 9, 281. [Google Scholar] [CrossRef]
- Pawlikowska, L.; Groen, A.; Eppens, E.F.; Kunne, C.; Ottenhoff, R.; Looije, N.; Knisely, A.S.; Killeen, N.P.; Bull, L.N.; Elferink, R.P.; et al. A mouse genetic model for familial cholestasis caused by ATP8B1 mutations reveals perturbed bile salt homeostasis but no impairment in bile secretion. Hum. Mol. Genet. 2004, 13, 881–892. [Google Scholar] [CrossRef]
- Tandon, P.; Rowe, B.H.; Vandermeer, B.; Bain, V.G. The efficacy and safety of bile Acid binding agents, opioid antagonists, or rifampin in the treatment of cholestasis-associated pruritus. Am. J. Gastroenterol. 2007, 102, 1528–1536. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, R.; Cruz, A.; Ferrin, G.; Lopez-Cillero, P.; Briceno, J.; Gomez, M.A.; Rufian, S.; Padillo, J.; De la Mata, M.; Marin, J.J.; et al. Cytoprotective properties of rifampicin are related to the regulation of detoxification system and bile acid transporter expression during hepatocellular injury induced by hydrophobic bile acids. J. Hepatobiliary Pancreat. Sci. 2011, 18, 740–750. [Google Scholar] [CrossRef]
- Mizuochi, T.; Kimura, A.; Tanaka, A.; Muto, A.; Nittono, H.; Seki, Y.; Takahashi, T.; Kurosawa, T.; Kage, M.; Takikawa, H.; et al. Characterization of urinary bile acids in a pediatric BRIC-1 patient: Effect of rifampicin treatment. Clin. Chim. Acta 2012, 413, 1301–1304. [Google Scholar] [CrossRef]
- Ibrahim, S.; Dayoub, R.; Melter, M.; Weiss, T.S. Bile acids down-regulate the expression of Augmenter of Liver Regeneration (ALR) via SHP/HNF4alpha1 and independent of Egr-1. Exp. Mol. Pathol. 2018, 105, 236–242. [Google Scholar] [CrossRef]
- Iwafuchi-Doi, M.; Zaret, K.S. Cell fate control by pioneer transcription factors. Development 2016, 143, 1833–1837. [Google Scholar] [CrossRef]
- Zaret, K.S.; Mango, S.E. Pioneer transcription factors, chromatin dynamics, and cell fate control. Curr. Opin. Genet. Dev. 2016, 37, 76–81. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, G.; Pasta, L.; Morabito, A.; D’Amico, M.; Caltagirone, M.; Malizia, G.; Tine, F.; Giannuoli, G.; Traina, M.; Vizzini, G.; et al. Competing risks and prognostic stages of cirrhosis: A 25-year inception cohort study of 494 patients. Aliment. Pharmacol. Ther. 2014, 39, 1180–1193. [Google Scholar] [CrossRef] [PubMed]
- Yokoda, R.T.; Rodriguez, E.A. Review: Pathogenesis of cholestatic liver diseases. World J. Hepatol. 2020, 12, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Schwabl, P.; Laleman, W. Novel treatment options for portal hypertension. Gastroenterol. Rep. 2017, 5, 90–103. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wilson, A.; Kuruba, R.; Zhang, Q.; Gao, X.; He, F.; Zhang, L.M.; Pitt, B.R.; Xie, W.; Li, S. FXR-mediated regulation of eNOS expression in vascular endothelial cells. Cardiovasc. Res. 2008, 77, 169–177. [Google Scholar] [CrossRef]
- Mookerjee, R.P.; Mehta, G.; Balasubramaniyan, V.; Mohamed Fel, Z.; Davies, N.; Sharma, V.; Iwakiri, Y.; Jalan, R. Hepatic dimethylarginine-dimethylaminohydrolase1 is reduced in cirrhosis and is a target for therapy in portal hypertension. J. Hepatol. 2015, 62, 325–331. [Google Scholar] [CrossRef]
- Schwabl, P.; Hambruch, E.; Seeland, B.A.; Hayden, H.; Wagner, M.; Garnys, L.; Strobel, B.; Schubert, T.L.; Riedl, F.; Mitteregger, D.; et al. The FXR agonist PX20606 ameliorates portal hypertension by targeting vascular remodelling and sinusoidal dysfunction. J. Hepatol. 2017, 66, 724–733. [Google Scholar] [CrossRef]
- Khambu, B.; Li, T.; Yan, S.; Yu, C.; Chen, X.; Goheen, M.; Li, Y.; Lin, J.; Cummings, O.W.; Lee, Y.A.; et al. Hepatic Autophagy Deficiency Compromises Farnesoid X Receptor Functionality and Causes Cholestatic Injury. Hepatology 2019, 69, 2196–2213. [Google Scholar] [CrossRef]
- Gao, L.; Lv, G.; Guo, X.; Jing, Y.; Han, Z.; Zhang, S.; Sun, K.; Li, R.; Yang, Y.; Wei, L. Activation of autophagy protects against cholestasis-induced hepatic injury. Cell Biosci. 2014, 4, 47. [Google Scholar] [CrossRef]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef]
- Ni, H.M.; Woolbright, B.L.; Williams, J.; Copple, B.; Cui, W.; Luyendyk, J.P.; Jaeschke, H.; Ding, W.X. Nrf2 promotes the development of fibrosis and tumorigenesis in mice with defective hepatic autophagy. J. Hepatol. 2014, 61, 617–625. [Google Scholar] [CrossRef]
- Khambu, B.; Huda, N.; Chen, X.; Antoine, D.J.; Li, Y.; Dai, G.; Kohler, U.A.; Zong, W.X.; Waguri, S.; Werner, S.; et al. HMGB1 promotes ductular reaction and tumorigenesis in autophagy-deficient livers. J. Clin. Investig. 2018, 128, 2419–2435. [Google Scholar] [CrossRef]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef]
- Kapuy, O.; Papp, D.; Vellai, T.; Banhegyi, G.; Korcsmaros, T. Systems-Level Feedbacks of NRF2 Controlling Autophagy upon Oxidative Stress Response. Antioxidants 2018, 7, 39. [Google Scholar] [CrossRef]
- Rattan, R.; Giri, S.; Singh, A.K.; Singh, I. 5-Aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside inhibits cancer cell proliferation in vitro and in vivo via AMP-activated protein kinase. J. Biol. Chem. 2005, 280, 39582–39593. [Google Scholar] [CrossRef]
- Viollet, B.; Foretz, M.; Guigas, B.; Horman, S.; Dentin, R.; Bertrand, L.; Hue, L.; Andreelli, F. Activation of AMP-activated protein kinase in the liver: A new strategy for the management of metabolic hepatic disorders. J. Physiol. 2006, 574, 41–53. [Google Scholar] [CrossRef]
- Guigas, B.; Bertrand, L.; Taleux, N.; Foretz, M.; Wiernsperger, N.; Vertommen, D.; Andreelli, F.; Viollet, B.; Hue, L. 5-Aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside and metformin inhibit hepatic glucose phosphorylation by an AMP-activated protein kinase-independent effect on glucokinase translocation. Diabetes 2006, 55, 865–874. [Google Scholar] [CrossRef]
- Wu, Y.H.; Li, Q.; Li, P.; Liu, B. GSK621 activates AMPK signaling to inhibit LPS-induced TNFalpha production. Biochem. Biophys. Res. Commun. 2016, 480, 289–295. [Google Scholar] [CrossRef]
- Li, X.; Liu, R.; Zhang, L.; Jiang, Z. The emerging role of AMP-activated protein kinase in cholestatic liver diseases. Pharmacol. Res. 2017, 125, 105–113. [Google Scholar] [CrossRef]
- Li, T.; Zheng, R.; Xu, L.; Zhou, M.; Wang, X.; Guo, Q.; Ji, H.; Li, L. Picroside II alleviates liver injury induced by alpha-naphthylisothiocyanate through AMPK-FXR pathway. Toxicol. Appl. Pharmacol. 2020, 408, 115248. [Google Scholar] [CrossRef] [PubMed]
- Dunlop, E.A.; Tee, A.R. mTOR and autophagy: A dynamic relationship governed by nutrients and energy. Semin. Cell Dev. Biol. 2014, 36, 121–129. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).