
HDL in Atherosclerotic Cardiovascular Disease: In Search of a Role

 , , ,
, , ,  and
and 
Abstract
:1. Introduction
2. The Role of HDL in Lipid Metabolism
3. Association between HDL-C and Cardiovascular Risk: Epidemiology
4. Increasing Cholesterol in HDL: Pharmacological Trials
5. HDL Cholesterol Content and Cardiovascular Events: Results from Genetic Studies
6. Targeting HDL Function Rather Than HDL Cholesterol Content
7. HDL Function/Dysfunction beyond the Reverse Cholesterol Transport
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Camont, L.; Chapman, M.J.; Kontush, A. Biological activities of HDL subpopulations and their relevance to cardiovascular disease. Trends Mol. Med. 2011, 17, 594–603. [Google Scholar] [CrossRef]
- Kostara, C.E.; Tsimihodimos, V.; Elisaf, M.S.; Bairaktari, E.T. NMR-Based Lipid Profiling of High Density Lipoprotein Particles in Healthy Subjects with Low, Normal, and Elevated HDL-Cholesterol. J. Proteome Res. 2017, 16, 1605–1616. [Google Scholar] [CrossRef] [PubMed]
- Kontush, A.; Lindahl, M.; Lhomme, M.; Calabresi, L.; Chapman, M.J.; Davidson, W.S. Structure of HDL: Particle subclasses and molecular components. Handb. Exp. Pharmacol. 2015, 224, 3–51. [Google Scholar] [CrossRef] [Green Version]
- Helgadottir, A.; Sulem, P.; Thorgeirsson, G.; Gretarsdottir, S.; Thorleifsson, G.; Jensson, B.O.; Arnadottir, G.A.; Olafsson, I.; Eyjolfsson, G.I.; Sigurdardottir, O.; et al. Rare SCARB1 mutations associate with high-density lipoprotein cholesterol but not with coronary artery disease. Eur. Heart J. 2018, 39, 2172–2178. [Google Scholar] [CrossRef] [PubMed]
- Zanoni, P.; Khetarpal, S.A.; Larach, D.B.; Hancock-Cerutti, W.F.; Millar, J.S.; Cuchel, M.; Der Ohannessian, S.; Kontush, A.; Surendran, P.; Saleheen, D.; et al. Rare variant in scavenger receptor BI raises HDL cholesterol and increases risk of coronary heart disease. Science 2016, 351, 1166–1171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pirillo, A.; Norata, G.D.; Catapano, A.L. High-density lipoprotein subfractions—What the clinicians need to know. Cardiology 2013, 124, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Qiu, C.; Zhao, X.; Zhou, Q.; Zhang, Z. High-density lipoprotein cholesterol efflux capacity is inversely associated with cardiovascular risk: A systematic review and meta-analysis. Lipids Health Dis. 2017, 16, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baragetti, A.; Ossoli, A.; Strazzella, A.; Simonelli, S.; Baragetti, I.; Grigore, L.; Pellegatta, F.; Catapano, A.L.; Norata, G.D.; Calabresi, L. Low Plasma Lecithin: Cholesterol Acyltransferase (LCAT) Concentration Predicts Chronic Kidney Disease. J. Clin. Med. 2020, 9, 2289. [Google Scholar] [CrossRef]
- Kronenberg, F. HDL in CKD-The Devil Is in the Detail. J. Am. Soc. Nephrol. JASN 2018, 29, 1356–1371. [Google Scholar] [CrossRef]
- Bonacina, F.; Pirillo, A.; Catapano, A.L.; Norata, G.D. Cholesterol membrane content has a ubiquitous evolutionary function in immune cell activation: The role of HDL. Curr. Opin. Lipidol. 2019, 30, 462–469. [Google Scholar] [CrossRef]
- Drew, B.G.; Rye, K.A.; Duffy, S.J.; Barter, P.; Kingwell, B.A. The emerging role of HDL in glucose metabolism. Nat. Rev. Endocrinol. 2012, 8, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Van Linthout, S.; Spillmann, F.; Schultheiss, H.P.; Tschope, C. High-density lipoprotein at the interface of type 2 diabetes mellitus and cardiovascular disorders. Curr. Pharm. Des. 2010, 16, 1504–1516. [Google Scholar] [CrossRef] [Green Version]
- Castelli, W.P.; Garrison, R.J.; Wilson, P.W.; Abbott, R.D.; Kalousdian, S.; Kannel, W.B. Incidence of coronary heart disease and lipoprotein cholesterol levels. The Framingham Study. JAMA 1986, 256, 2835–2838. [Google Scholar] [CrossRef]
- Di Angelantonio, E.; Sarwar, N.; Perry, P.; Kaptoge, S.; Ray, K.K.; Thompson, A.; Wood, A.M.; Lewington, S.; Sattar, N. Major lipids, apolipoproteins, and risk of vascular disease. JAMA 2009, 302, 1993–2000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, D.J.; Probstfield, J.L.; Garrison, R.J.; Neaton, J.D.; Castelli, W.P.; Knoke, J.D.; Jacobs, D.R., Jr.; Bangdiwala, S.; Tyroler, H.A. High-density lipoprotein cholesterol and cardiovascular disease. Four Prospect. Am. Stud. Circ. 1989, 79, 8–15. [Google Scholar] [CrossRef] [Green Version]
- Castelli, W.P. Cholesterol and lipids in the risk of coronary artery disease—The Framingham Heart Study. Can. J. Cardiol. 1988, 4, 5A–10A. [Google Scholar]
- Rohatgi, A.; Khera, A.; Berry, J.D.; Givens, E.G.; Ayers, C.R.; Wedin, K.E.; Neeland, I.J.; Yuhanna, I.S.; Rader, D.R.; de Lemos, J.A.; et al. HDL cholesterol efflux capacity and incident cardiovascular events. N. Engl. J. Med. 2014, 371, 2383–2393. [Google Scholar] [CrossRef] [Green Version]
- Wilkins, J.T.; Ning, H.; Stone, N.J.; Criqui, M.H.; Zhao, L.; Greenland, P.; Lloyd-Jones, D.M. Coronary heart disease risks associated with high levels of HDL cholesterol. J. Am. Heart Assoc. 2014, 3, e000519. [Google Scholar] [CrossRef] [Green Version]
- Madsen, C.M.; Varbo, A.; Nordestgaard, B.G. Extreme high high-density lipoprotein cholesterol is paradoxically associated with high mortality in men and women: Two prospective cohort studies. Eur. Heart J. 2017, 38, 2478–2486. [Google Scholar] [CrossRef] [Green Version]
- Mazidi, M.; Mikhailidis, D.P.; Banach, M. Associations between risk of overall mortality, cause-specific mortality and level of inflammatory factors with extremely low and high high-density lipoprotein cholesterol levels among American adults. Int. J. Cardiol. 2019, 276, 242–247. [Google Scholar] [CrossRef]
- Mackey, R.H.; Greenland, P.; Goff, D.C., Jr.; Lloyd-Jones, D.; Sibley, C.T.; Mora, S. High-density lipoprotein cholesterol and particle concentrations, carotid atherosclerosis, and coronary events: MESA (multi-ethnic study of atherosclerosis). J. Am. Coll. Cardiol. 2012, 60, 508–516. [Google Scholar] [CrossRef] [Green Version]
- Ko, D.T.; Alter, D.A.; Guo, H.; Koh, M.; Lau, G.; Austin, P.C.; Booth, G.L.; Hogg, W.; Jackevicius, C.A.; Lee, D.S.; et al. High-Density Lipoprotein Cholesterol and Cause-Specific Mortality in Individuals Without Previous Cardiovascular Conditions: The CANHEART Study. J. Am. Coll. Cardiol. 2016, 68, 2073–2083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Steeg, W.A.; Holme, I.; Boekholdt, S.M.; Larsen, M.L.; Lindahl, C.; Stroes, E.S.; Tikkanen, M.J.; Wareham, N.J.; Faergeman, O.; Olsson, A.G.; et al. High-density lipoprotein cholesterol, high-density lipoprotein particle size, and apolipoprotein A-I: Significance for cardiovascular risk: The IDEAL and EPIC-Norfolk studies. J. Am. Coll. Cardiol. 2008, 51, 634–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, R.V.; Wittrup, H.H.; Tybjaerg-Hansen, A.; Steffensen, R.; Schnohr, P.; Nordestgaard, B.G. Hepatic lipase mutations, elevated high-density lipoprotein cholesterol, and increased risk of ischemic heart disease: The Copenhagen City Heart Study. J. Am. Coll. Cardiol. 2003, 41, 1972–1982. [Google Scholar] [CrossRef] [Green Version]
- Singh, K.; Rohatgi, A. Examining the paradox of high high-density lipoprotein and elevated cardiovascular risk. J. Thorac. Dis. 2018, 10, 109–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Investigators, A.-H.; Boden, W.E.; Probstfield, J.L.; Anderson, T.; Chaitman, B.R.; Desvignes-Nickens, P.; Koprowicz, K.; McBride, R.; Teo, K.; Weintraub, W. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N. Engl. J. Med. 2011, 365, 2255–2267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Group, H.T.C.; Landray, M.J.; Haynes, R.; Hopewell, J.C.; Parish, S.; Aung, T.; Tomson, J.; Wallendszus, K.; Craig, M.; Jiang, L.; et al. Effects of extended-release niacin with laropiprant in high-risk patients. N. Engl. J. Med. 2014, 371, 203–212. [Google Scholar] [CrossRef] [Green Version]
- Ritsch, A.; Duerr, A.; Kahler, P.; Hunjadi, M.; Stojakovic, T.; Silbernagel, G.; Scharnagl, H.; Kleber, M.E.; Marz, W. Cholesterol Efflux Capacity and Cardiovascular Disease: The Ludwigshafen Risk and Cardiovascular Health (LURIC) Study. Biomedicines 2020, 8, 524. [Google Scholar] [CrossRef] [PubMed]
- Hunjadi, M.; Lamina, C.; Kahler, P.; Bernscherer, T.; Viikari, J.; Lehtimaki, T.; Kahonen, M.; Hurme, M.; Juonala, M.; Taittonen, L.; et al. HDL cholesterol efflux capacity is inversely associated with subclinical cardiovascular risk markers in young adults: The cardiovascular risk in Young Finns study. Sci. Rep. 2020, 10, 19223. [Google Scholar] [CrossRef]
- Ida, S.; Kaneko, R.; Murata, K. Efficacy and safety of pemafibrate administration in patients with dyslipidemia: A systematic review and meta-analysis. Cardiovasc. Diabetol. 2019, 18, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Briel, M.; Ferreira-Gonzalez, I.; You, J.J.; Karanicolas, P.J.; Akl, E.A.; Wu, P.; Blechacz, B.; Bassler, D.; Wei, X.; Sharman, A.; et al. Association between change in high density lipoprotein cholesterol and cardiovascular disease morbidity and mortality: Systematic review and meta-regression analysis. BMJ 2009, 338, b92. [Google Scholar] [CrossRef] [Green Version]
- McTaggart, F.; Jones, P. Effects of statins on high-density lipoproteins: A potential contribution to cardiovascular benefit. Cardiovasc. Drugs Ther. 2008, 22, 321–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapman, M.J. Are the effects of statins on HDL-cholesterol clinically relevant? Eur. Heart J. Suppl. 2004, 6, C58–C63. [Google Scholar] [CrossRef]
- Dormandy, J.A.; Charbonnel, B.; Eckland, D.J.; Erdmann, E.; Massi-Benedetti, M.; Moules, I.K.; Skene, A.M.; Tan, M.H.; Lefebvre, P.J.; Murray, G.D.; et al. Secondary prevention of macrovascular events in patients with type 2 diabetes in the PROactive Study (PROspective pioglitAzone Clinical Trial In macroVascular Events): A randomised controlled trial. Lancet 2005, 366, 1279–1289. [Google Scholar] [CrossRef]
- Hulley, S.; Grady, D.; Bush, T.; Furberg, C.; Herrington, D.; Riggs, B.; Vittinghoff, E. Randomized trial of estrogen plus progestin for secondary prevention of coronary heart disease in postmenopausal women. Heart and Estrogen/progestin Replacement Study (HERS) Research Group. JAMA 1998, 280, 605–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brousseau, M.E.; Schaefer, E.J.; Wolfe, M.L.; Bloedon, L.T.; Digenio, A.G.; Clark, R.W.; Mancuso, J.P.; Rader, D.J. Effects of an inhibitor of cholesteryl ester transfer protein on HDL cholesterol. N. Engl. J. Med. 2004, 350, 1505–1515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forrest, M.J.; Bloomfield, D.; Briscoe, R.J.; Brown, P.N.; Cumiskey, A.M.; Ehrhart, J.; Hershey, J.C.; Keller, W.J.; Ma, X.; McPherson, H.E.; et al. Torcetrapib-induced blood pressure elevation is independent of CETP inhibition and is accompanied by increased circulating levels of aldosterone. Br. J. Pharmacol. 2008, 154, 1465–1473. [Google Scholar] [CrossRef] [PubMed]
- Bots, M.L.; Visseren, F.L.; Evans, G.W.; Riley, W.A.; Revkin, J.H.; Tegeler, C.H.; Shear, C.L.; Duggan, W.T.; Vicari, R.M.; Grobbee, D.E.; et al. Torcetrapib and carotid intima-media thickness in mixed dyslipidaemia (RADIANCE 2 study): A randomised, double-blind trial. Lancet 2007, 370, 153–160. [Google Scholar] [CrossRef]
- Kastelein, J.J.; van Leuven, S.I.; Burgess, L.; Evans, G.W.; Kuivenhoven, J.A.; Barter, P.J.; Revkin, J.H.; Grobbee, D.E.; Riley, W.A.; Shear, C.L.; et al. Effect of torcetrapib on carotid atherosclerosis in familial hypercholesterolemia. N. Engl. J. Med. 2007, 356, 1620–1630. [Google Scholar] [CrossRef] [PubMed]
- Nissen, S.E.; Tardif, J.C.; Nicholls, S.J.; Revkin, J.H.; Shear, C.L.; Duggan, W.T.; Ruzyllo, W.; Bachinsky, W.B.; Lasala, G.P.; Tuzcu, E.M.; et al. Effect of torcetrapib on the progression of coronary atherosclerosis. N. Engl. J. Med. 2007, 356, 1304–1316. [Google Scholar] [CrossRef]
- Stein, E.A.; Roth, E.M.; Rhyne, J.M.; Burgess, T.; Kallend, D.; Robinson, J.G. Safety and tolerability of dalcetrapib (RO4607381/JTT-705): Results from a 48-week trial. Eur. Heart J. 2010, 31, 480–488. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, G.G.; Olsson, A.G.; Abt, M.; Ballantyne, C.M.; Barter, P.J.; Brumm, J.; Chaitman, B.R.; Holme, I.M.; Kallend, D.; Leiter, L.A.; et al. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N. Engl. J. Med. 2012, 367, 2089–2099. [Google Scholar] [CrossRef] [Green Version]
- Tardif, J.C.; Rheaume, E.; Lemieux Perreault, L.P.; Gregoire, J.C.; Feroz Zada, Y.; Asselin, G.; Provost, S.; Barhdadi, A.; Rhainds, D.; L’Allier, P.L.; et al. Pharmacogenomic determinants of the cardiovascular effects of dalcetrapib. Circ. Cardiovasc. Genet. 2015, 8, 372–382. [Google Scholar] [CrossRef] [Green Version]
- Tardif, J.C.; Dube, M.P.; Pfeffer, M.A.; Waters, D.D.; Koenig, W.; Maggioni, A.P.; McMurray, J.J.V.; Mooser, V.; White, H.D.; Heinonen, T.; et al. Study design of Dal-GenE, a pharmacogenetic trial targeting reduction of cardiovascular events with dalcetrapib. Am. Heart J. 2020, 222, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, S.J.; Brewer, H.B.; Kastelein, J.J.; Krueger, K.A.; Wang, M.D.; Shao, M.; Hu, B.; McErlean, E.; Nissen, S.E. Effects of the CETP inhibitor evacetrapib administered as monotherapy or in combination with statins on HDL and LDL cholesterol: A randomized controlled trial. JAMA 2011, 306, 2099–2109. [Google Scholar] [CrossRef] [Green Version]
- Bloomfield, D.; Carlson, G.L.; Sapre, A.; Tribble, D.; McKenney, J.M.; Littlejohn, T.W., 3rd; Sisk, C.M.; Mitchel, Y.; Pasternak, R.C. Efficacy and safety of the cholesteryl ester transfer protein inhibitor anacetrapib as monotherapy and coadministered with atorvastatin in dyslipidemic patients. Am. Heart J. 2009, 157, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Lincoff, A.M.; Nicholls, S.J.; Riesmeyer, J.S.; Barter, P.J.; Brewer, H.B.; Fox, K.A.A.; Gibson, C.M.; Granger, C.; Menon, V.; Montalescot, G.; et al. Evacetrapib and Cardiovascular Outcomes in High-Risk Vascular Disease. N. Engl. J. Med. 2017, 376, 1933–1942. [Google Scholar] [CrossRef] [PubMed]
- Group, R.C.; Bowman, L.; Chen, F.; Sammons, E.; Hopewell, J.C.; Wallendszus, K.; Stevens, W.; Valdes-Marquez, E.; Wiviott, S.; Cannon, C.P.; et al. Randomized Evaluation of the Effects of Anacetrapib through Lipid-modification (REVEAL)-A large-scale, randomized, placebo-controlled trial of the clinical effects of anacetrapib among people with established vascular disease: Trial design, recruitment, and baseline characteristics. Am. Heart J. 2017, 187, 182–190. [Google Scholar] [CrossRef]
- Guo, S.X.; Yao, M.H.; Ding, Y.S.; Zhang, J.Y.; Yan, Y.Z.; Liu, J.M.; Zhang, M.; Rui, D.S.; Niu, Q.; He, J.; et al. Associations of Cholesteryl Ester Transfer Protein TaqIB Polymorphism with the Composite Ischemic Cardiovascular Disease Risk and HDL-C Concentrations: A Meta-Analysis. Int. J. Environ. Res. Public Health 2016, 13, 882. [Google Scholar] [CrossRef] [PubMed]
- Ference, B.A.; Kastelein, J.J.P.; Ginsberg, H.N.; Chapman, M.J.; Nicholls, S.J.; Ray, K.K.; Packard, C.J.; Laufs, U.; Brook, R.D.; Oliver-Williams, C.; et al. Association of Genetic Variants Related to CETP Inhibitors and Statins with Lipoprotein Levels and Cardiovascular Risk. JAMA 2017, 318, 947–956. [Google Scholar] [CrossRef] [Green Version]
- Taheri, H.; Filion, K.B.; Windle, S.B.; Reynier, P.; Eisenberg, M.J. Cholesteryl Ester Transfer Protein Inhibitors and Cardiovascular Outcomes: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Cardiology 2020, 145, 236–250. [Google Scholar] [CrossRef] [PubMed]
- Rashid, S. Lower LDL is better—Can this be achieved with CETP inhibition therapy? Expert Rev. Cardiovasc. Ther. 2020, 18, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Fan, J.; Liu, J.; Wang, W.; Wang, M.; Sun, J.; Liu, J.; Xie, W.; Zhao, F.; Li, Y.; et al. Cholesterol-overloaded HDL particles are independently associated with progression of carotid atherosclerosis in a cardiovascular disease-free population: A community-based cohort study. J. Am. Coll. Cardiol. 2015, 65, 355–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicholls, S.J.; Ray, K.K.; Ballantyne, C.M.; Beacham, L.A.; Miller, D.L.; Ruotolo, G.; Nissen, S.E.; Riesmeyer, J.S.; Investigators, A. Comparative effects of cholesteryl ester transfer protein inhibition, statin or ezetimibe on lipid factors: The ACCENTUATE trial. Atherosclerosis 2017, 261, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Antiochos, P.; Marques-Vidal, P.; Virzi, J.; Pagano, S.; Satta, N.; Hartley, O.; Montecucco, F.; Mach, F.; Kutalik, Z.; Waeber, G.; et al. Impact of CD14 Polymorphisms on Anti-Apolipoprotein A-1 IgG-Related Coronary Artery Disease Prediction in the General Population. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 2342–2349. [Google Scholar] [CrossRef] [Green Version]
- Antiochos, P.; Marques-Vidal, P.; Virzi, J.; Pagano, S.; Satta, N.; Bastardot, F.; Hartley, O.; Montecucco, F.; Mach, F.; Waeber, G.; et al. Association between anti-apolipoprotein A-1 antibodies and cardiovascular disease in the general population. Results from the CoLaus study. Thromb. Haemost. 2016, 116, 764–771. [Google Scholar] [CrossRef] [Green Version]
- Antiochos, P.; Marques-Vidal, P.; Virzi, J.; Pagano, S.; Satta, N.; Hartley, O.; Montecucco, F.; Mach, F.; Kutalik, Z.; Waeber, G.; et al. Anti-Apolipoprotein A-1 IgG Predict All-Cause Mortality and Are Associated with Fc Receptor-Like 3 Polymorphisms. Front. Immunol. 2017, 8, 437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carbone, F.; Satta, N.; Montecucco, F.; Virzi, J.; Burger, F.; Roth, A.; Roversi, G.; Tamborino, C.; Casetta, I.; Seraceni, S.; et al. Anti-ApoA-1 IgG serum levels predict worse poststroke outcomes. Eur. J. Clin. Investig. 2016, 46, 805–817. [Google Scholar] [CrossRef]
- Vuilleumier, N.; Montecucco, F.; Spinella, G.; Pagano, S.; Bertolotto, M.; Pane, B.; Pende, A.; Galan, K.; Roux-Lombard, P.; Combescure, C.; et al. Serum levels of anti-apolipoprotein A-1 auto-antibodies and myeloperoxidase as predictors of major adverse cardiovascular events after carotid endarterectomy. Thromb. Haemost. 2013, 109, 706–715. [Google Scholar] [CrossRef] [Green Version]
- Anderson, J.L.C.; Pagano, S.; Virzi, J.; Dullaart, R.P.F.; Annema, W.; Kuipers, F.; Bakker, S.J.L.; Vuilleumier, N.; Tietge, U.J.F. Autoantibodies to Apolipoprotein A-1 as Independent Predictors of Cardiovascular Mortality in Renal Transplant Recipients. J. Clin. Med. 2019, 8, 948. [Google Scholar] [CrossRef] [Green Version]
- Vuilleumier, N.; Pagano, S.; Combescure, C.; Gencer, B.; Virzi, J.; Raber, L.; Carballo, D.; Carballo, S.; Nanchen, D.; Rodondi, N.; et al. Non-Linear Relationship between Anti-Apolipoprotein A-1 IgGs and Cardiovascular Outcomes in Patients with Acute Coronary Syndromes. J. Clin. Med. 2019, 8, 1002. [Google Scholar] [CrossRef] [Green Version]
- Batuca, J.R.; Amaral, M.C.; Favas, C.; Paula, F.S.; Ames, P.R.J.; Papoila, A.L.; Delgado Alves, J. Extended-release niacin increases anti-apolipoprotein A-I antibodies that block the antioxidant effect of high-density lipoprotein-cholesterol: The EXPLORE clinical trial. Br. J. Clin. Pharm. 2017, 83, 1002–1010. [Google Scholar] [CrossRef] [Green Version]
- Pagano, S.; Satta, N.; Werling, D.; Offord, V.; de Moerloose, P.; Charbonney, E.; Hochstrasser, D.; Roux-Lombard, P.; Vuilleumier, N. Anti-apolipoprotein A-1 IgG in patients with myocardial infarction promotes inflammation through TLR2/CD14 complex. J. Intern. Med. 2012, 272, 344–357. [Google Scholar] [CrossRef]
- Montecucco, F.; Vuilleumier, N.; Pagano, S.; Lenglet, S.; Bertolotto, M.; Braunersreuther, V.; Pelli, G.; Kovari, E.; Pane, B.; Spinella, G.; et al. Anti-Apolipoprotein A-1 auto-antibodies are active mediators of atherosclerotic plaque vulnerability. Eur. Heart J. 2011, 32, 412–421. [Google Scholar] [CrossRef] [Green Version]
- Vuilleumier, N.; Bas, S.; Pagano, S.; Montecucco, F.; Guerne, P.A.; Finckh, A.; Lovis, C.; Mach, F.; Hochstrasser, D.; Roux-Lombard, P.; et al. Anti-apolipoprotein A-1 IgG predicts major cardiovascular events in patients with rheumatoid arthritis. Arthritis Rheum. 2010, 62, 2640–2650. [Google Scholar] [CrossRef] [PubMed]
- Barter, P.J.; Caulfield, M.; Eriksson, M.; Grundy, S.M.; Kastelein, J.J.; Komajda, M.; Lopez-Sendon, J.; Mosca, L.; Tardif, J.C.; Waters, D.D.; et al. Effects of torcetrapib in patients at high risk for coronary events. N. Engl. J. Med. 2007, 357, 2109–2122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Group, H.T.R.C.; Bowman, L.; Hopewell, J.C.; Chen, F.; Wallendszus, K.; Stevens, W.; Collins, R.; Wiviott, S.D.; Cannon, C.P.; Braunwald, E.; et al. Effects of Anacetrapib in Patients with Atherosclerotic Vascular Disease. N. Engl. J. Med. 2017, 377, 1217–1227. [Google Scholar] [CrossRef]
- Qasim, A.; Rader, D.J. Human genetics of variation in high-density lipoprotein cholesterol. Curr. Atheroscler. Rep. 2006, 8, 198–205. [Google Scholar] [CrossRef]
- Frikke-Schmidt, R.; Nordestgaard, B.G.; Stene, M.C.; Sethi, A.A.; Remaley, A.T.; Schnohr, P.; Grande, P.; Tybjaerg-Hansen, A. Association of loss-of-function mutations in the ABCA1 gene with high-density lipoprotein cholesterol levels and risk of ischemic heart disease. JAMA 2008, 299, 2524–2532. [Google Scholar] [CrossRef]
- Kuivenhoven, J.A.; Pritchard, H.; Hill, J.; Frohlich, J.; Assmann, G.; Kastelein, J. The molecular pathology of lecithin:cholesterol acyltransferase (LCAT) deficiency syndromes. J. Lipid Res. 1997, 38, 191–205. [Google Scholar] [CrossRef]
- Voight, B.F.; Peloso, G.M.; Orho-Melander, M.; Frikke-Schmidt, R.; Barbalic, M.; Jensen, M.K.; Hindy, G.; Holm, H.; Ding, E.L.; Johnson, T.; et al. Plasma HDL cholesterol and risk of myocardial infarction: A mendelian randomisation study. Lancet 2012, 380, 572–580. [Google Scholar] [CrossRef] [Green Version]
- Haase, C.L.; Tybjaerg-Hansen, A.; Qayyum, A.A.; Schou, J.; Nordestgaard, B.G.; Frikke-Schmidt, R. LCAT, HDL cholesterol and ischemic cardiovascular disease: A Mendelian randomization study of HDL cholesterol in 54,500 individuals. J. Clin. Endocrinol. Metab. 2012, 97, E248–E256. [Google Scholar] [CrossRef] [Green Version]
- Johannsen, T.H.; Kamstrup, P.R.; Andersen, R.V.; Jensen, G.B.; Sillesen, H.; Tybjaerg-Hansen, A.; Nordestgaard, B.G. Hepatic lipase, genetically elevated high-density lipoprotein, and risk of ischemic cardiovascular disease. J. Clin. Endocrinol. Metab. 2009, 94, 1264–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haase, C.L.; Tybjaerg-Hansen, A.; Grande, P.; Frikke-Schmidt, R. Genetically elevated apolipoprotein A-I, high-density lipoprotein cholesterol levels, and risk of ischemic heart disease. J. Clin. Endocrinol. Metab. 2010, 95, E500–E510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmes, M.V.; Asselbergs, F.W.; Palmer, T.M.; Drenos, F.; Lanktree, M.B.; Nelson, C.P.; Dale, C.E.; Padmanabhan, S.; Finan, C.; Swerdlow, D.I.; et al. Mendelian randomization of blood lipids for coronary heart disease. Eur. Heart J. 2015, 36, 539–550. [Google Scholar] [CrossRef] [Green Version]
- Johannsen, T.H.; Frikke-Schmidt, R.; Schou, J.; Nordestgaard, B.G.; Tybjaerg-Hansen, A. Genetic inhibition of CETP, ischemic vascular disease and mortality, and possible adverse effects. J. Am. Coll. Cardiol. 2012, 60, 2041–2048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blauw, L.L.; Noordam, R.; Soidinsalo, S.; Blauw, C.A.; Li-Gao, R.; de Mutsert, R.; Berbee, J.F.P.; Wang, Y.; van Heemst, D.; Rosendaal, F.R.; et al. Mendelian randomization reveals unexpected effects of CETP on the lipoprotein profile. Eur. J. Hum. Genet. EJHG 2019, 27, 422–431. [Google Scholar] [CrossRef] [Green Version]
- Mora, S.; Glynn, R.J.; Ridker, P.M. High-density lipoprotein cholesterol, size, particle number, and residual vascular risk after potent statin therapy. Circulation 2013, 128, 1189–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, L.; Fu, M. The relationship between high density lipoprotein subclass profile and plasma lipids concentrations. Lipids Health Dis. 2010, 9, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Guey, L.T.; Pullinger, C.R.; Ishida, B.Y.; O’Connor, P.M.; Zellner, C.; Francone, O.L.; Laramie, J.M.; Naya-Vigne, J.M.; Siradze, K.A.; Deedwania, P.; et al. Relation of increased prebeta-1 high-density lipoprotein levels to risk of coronary heart disease. Am. J. Cardiol. 2011, 108, 360–366. [Google Scholar] [CrossRef] [PubMed]
- Sethi, A.A.; Sampson, M.; Warnick, R.; Muniz, N.; Vaisman, B.; Nordestgaard, B.G.; Tybjaerg-Hansen, A.; Remaley, A.T. High pre-beta1 HDL concentrations and low lecithin: Cholesterol acyltransferase activities are strong positive risk markers for ischemic heart disease and independent of HDL-cholesterol. Clin. Chem. 2010, 56, 1128–1137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, M.; Wada, H.; Maeda, S.; Saito, K.; Minatoguchi, S.; Saito, K.; Seishima, M. Increased plasma lipid-poor apolipoprotein A-I in patients with coronary artery disease. Clin. Chem. 2005, 51, 132–137. [Google Scholar] [CrossRef] [Green Version]
- Baragetti, A.; Norata, G.D.; Sarcina, C.; Rastelli, F.; Grigore, L.; Garlaschelli, K.; Uboldi, P.; Baragetti, I.; Pozzi, C.; Catapano, A.L. High density lipoprotein cholesterol levels are an independent predictor of the progression of chronic kidney disease. J. Intern. Med. 2013, 274, 252–262. [Google Scholar] [CrossRef]
- Calabresi, L.; Simonelli, S.; Conca, P.; Busnach, G.; Cabibbe, M.; Gesualdo, L.; Gigante, M.; Penco, S.; Veglia, F.; Franceschini, G. Acquired lecithin:cholesterol acyltransferase deficiency as a major factor in lowering plasma HDL levels in chronic kidney disease. J. Intern. Med. 2015, 277, 552–561. [Google Scholar] [CrossRef]
- Zeljkovic, A.; Vekic, J.; Spasojevic-Kalimanovska, V.; Jelic-Ivanovic, Z.; Bogavac-Stanojevic, N.; Gulan, B.; Spasic, S. LDL and HDL subclasses in acute ischemic stroke: Prediction of risk and short-term mortality. Atherosclerosis 2010, 210, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Lamarche, B.; Moorjani, S.; Cantin, B.; Dagenais, G.R.; Lupien, P.J.; Despres, J.P. Associations of HDL2 and HDL3 subfractions with ischemic heart disease in men. Prospective results from the Quebec Cardiovascular Study. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 1098–1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mueller, O.; Chang, E.; Deng, D.; Franz, T.; Jing, D.; Kincaid, R.; Konigshofer, Y.; Kratzmeier, M.; McNulty, M.; Qian, H.; et al. PROCAM Study: Risk prediction for myocardial infarction using microfluidic high-density lipoprotein (HDL) subfractionation is independent of HDL cholesterol. Clin. Chem. Lab. Med. 2008, 46, 490–498. [Google Scholar] [CrossRef]
- Drexel, H.; Amann, F.W.; Rentsch, K.; Neuenschwander, C.; Luethy, A.; Khan, S.I.; Follath, F. Relation of the level of high-density lipoprotein subfractions to the presence and extent of coronary artery disease. Am. J. Cardiol. 1992, 70, 436–440. [Google Scholar] [CrossRef]
- Rosenbaum, D.; Hansel, B.; Bonnefont-Rousselot, D.; Bittar, R.; Girerd, X.; Giral, P.; Bruckert, E. Waist circumference is a strong and independent determinant of the distribution of HDL subfractions in overweight patients with cardiovascular risk factors. Diabetes Vasc. Dis. Res. 2012, 9, 153–159. [Google Scholar] [CrossRef]
- Tian, L.; Jia, L.; Mingde, F.; Tian, Y.; Xu, Y.; Tian, H.; Yang, Y. Alterations of high density lipoprotein subclasses in obese subjects. Lipids 2006, 41, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Xian, X.; Ma, Y.; Yang, D.D.; Huang, W.; Wang, Y.; Mueller, O.; Chang, E.; Konigshofer, Y.; Van-Cleve, M.; Yang, J.; et al. Reduced high-density lipoprotein 2b in non-obese type 2 diabetic patients analysed by a microfluidic chip method in a case-control study. Biomark. Biochem. Indic. Expo. Response Susceptibility Chem. 2009, 14, 619–623. [Google Scholar] [CrossRef]
- Lagos, K.G.; Filippatos, T.D.; Tsimihodimos, V.; Gazi, I.F.; Rizos, C.; Tselepis, A.D.; Mikhailidis, D.P.; Elisaf, M.S. Alterations in the high density lipoprotein phenotype and HDL-associated enzymes in subjects with metabolic syndrome. Lipids 2009, 44, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Long, S.; Li, C.; Liu, Y.; Chen, Y.; Zeng, Z.; Fu, M. High-density lipoprotein subclass and particle size in coronary heart disease patients with or without diabetes. Lipids Health Dis. 2012, 11, 1–9. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, T.; Nguyen, T.T.; Hallaway, B.J.; Hodge, D.; Bailey, K.; Holmes, D.; Kottke, B.A. The role of lipoprotein A-I and lipoprotein A-I/A-II in predicting coronary artery disease. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 228–231. [Google Scholar] [CrossRef] [PubMed]
- Asztalos, B.F.; Demissie, S.; Cupples, L.A.; Collins, D.; Cox, C.E.; Horvath, K.V.; Bloomfield, H.E.; Robins, S.J.; Schaefer, E.J. LpA-I, LpA-I:A-II HDL and CHD-risk: The Framingham Offspring Study and the Veterans Affairs HDL Intervention Trial. Atherosclerosis 2006, 188, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Marsche, G.; Heine, G.H.; Stadler, J.T.; Holzer, M. Current Understanding of the Relationship of HDL Composition, Structure and Function to Their Cardioprotective Properties in Chronic Kidney Disease. Biomolecules 2020, 10, 1348. [Google Scholar] [CrossRef] [PubMed]
- Syvanne, M.; Kahri, J.; Virtanen, K.S.; Taskinen, M.R. HDLs containing apolipoproteins A-I and A-II (LpA-I:A-II) as markers of coronary artery disease in men with non-insulin-dependent diabetes mellitus. Circulation 1995, 92, 364–370. [Google Scholar] [CrossRef]
- Rasmussen, K.L.; Tybjaerg-Hansen, A.; Nordestgaard, B.G.; Frikke-Schmidt, R. Plasma levels of apolipoprotein E and risk of ischemic heart disease in the general population. Atherosclerosis 2016, 246, 63–70. [Google Scholar] [CrossRef]
- Bennet, A.M.; Di Angelantonio, E.; Ye, Z.; Wensley, F.; Dahlin, A.; Ahlbom, A.; Keavney, B.; Collins, R.; Wiman, B.; de Faire, U.; et al. Association of apolipoprotein E genotypes with lipid levels and coronary risk. JAMA 2007, 298, 1300–1311. [Google Scholar] [CrossRef]
- Zhang, M.D.; Gu, W.; Qiao, S.B.; Zhu, E.J.; Zhao, Q.M.; Lv, S.Z. Apolipoprotein E gene polymorphism and risk for coronary heart disease in the Chinese population: A meta-analysis of 61 studies including 6634 cases and 6393 controls. PLoS ONE 2014, 9, e95463. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Li, H.; Liu, J.; Zhu, D.; Wang, Z.; Chen, A.; Zhao, Q. Meta-analysis of apolipoprotein E gene polymorphism and susceptibility of myocardial infarction. PLoS ONE 2014, 9, e104608. [Google Scholar] [CrossRef] [Green Version]
- Holmes, M.V.; Frikke-Schmidt, R.; Melis, D.; Luben, R.; Asselbergs, F.W.; Boer, J.M.; Cooper, J.; Palmen, J.; Horvat, P.; Engmann, J.; et al. A systematic review and meta-analysis of 130,000 individuals shows smoking does not modify the association of APOE genotype on risk of coronary heart disease. Atherosclerosis 2014, 237, 5–12. [Google Scholar] [CrossRef] [Green Version]
- Kuusisto, S.; Holmes, M.V.; Ohukainen, P.; Kangas, A.J.; Karsikas, M.; Tiainen, M.; Perola, M.; Salomaa, V.; Kettunen, J.; Ala-Korpela, M. Direct Estimation of HDL-Mediated Cholesterol Efflux Capacity from Serum. Clin. Chem. 2019, 65, 1042–1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Favari, E.; Chroni, A.; Tietge, U.J.; Zanotti, I.; Escola-Gil, J.C.; Bernini, F. Cholesterol efflux and reverse cholesterol transport. Handb. Exp. Pharmacol. 2015, 224, 181–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebtehaj, S.; Gruppen, E.G.; Bakker, S.J.L.; Dullaart, R.P.F.; Tietge, U.J.F. HDL (High-Density Lipoprotein) Cholesterol Efflux Capacity Is Associated With Incident Cardiovascular Disease in the General Population. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1874–1883. [Google Scholar] [CrossRef]
- Li, X.M.; Tang, W.H.; Mosior, M.K.; Huang, Y.; Wu, Y.; Matter, W.; Gao, V.; Schmitt, D.; Didonato, J.A.; Fisher, E.A.; et al. Paradoxical association of enhanced cholesterol efflux with increased incident cardiovascular risks. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1696–1705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khera, A.V.; Cuchel, M.; de la Llera-Moya, M.; Rodrigues, A.; Burke, M.F.; Jafri, K.; French, B.C.; Phillips, J.A.; Mucksavage, M.L.; Wilensky, R.L.; et al. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N. Engl. J. Med. 2011, 364, 127–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogura, M.; Hori, M.; Harada-Shiba, M. Association between Cholesterol Efflux Capacity and Atherosclerotic Cardiovascular Disease in Patients with Familial Hypercholesterolemia. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 181–188. [Google Scholar] [CrossRef] [Green Version]
- Khera, A.V.; Demler, O.V.; Adelman, S.J.; Collins, H.L.; Glynn, R.J.; Ridker, P.M.; Rader, D.J.; Mora, S. Cholesterol Efflux Capacity, High-Density Lipoprotein Particle Number, and Incident Cardiovascular Events: An Analysis from the JUPITER Trial (Justification for the Use of Statins in Prevention: An Intervention Trial Evaluating Rosuvastatin). Circulation 2017, 135, 2494–2504. [Google Scholar] [CrossRef]
- Zimetti, F.; Freitas, W.M.; Campos, A.M.; Daher, M.; Adorni, M.P.; Bernini, F.; Sposito, A.C.; Zanotti, I. Cholesterol efflux capacity does not associate with coronary calcium, plaque vulnerability, and telomere length in healthy octogenarians. J. Lipid Res. 2018, 59, 714–721. [Google Scholar] [CrossRef] [Green Version]
- Nicholls, S.J.; Ruotolo, G.; Brewer, H.B.; Kane, J.P.; Wang, M.D.; Krueger, K.A.; Adelman, S.J.; Nissen, S.E.; Rader, D.J. Cholesterol Efflux Capacity and Pre-Beta-1 HDL Concentrations Are Increased in Dyslipidemic Patients Treated with Evacetrapib. J. Am. Coll. Cardiol. 2015, 66, 2201–2210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karjalainen, M.K.; Holmes, M.V.; Wang, Q.; Anufrieva, O.; Kahonen, M.; Lehtimaki, T.; Havulinna, A.S.; Kristiansson, K.; Salomaa, V.; Perola, M.; et al. Apolipoprotein A-I concentrations and risk of coronary artery disease: A Mendelian randomization study. Atherosclerosis 2020, 299, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Nissen, S.E.; Tsunoda, T.; Tuzcu, E.M.; Schoenhagen, P.; Cooper, C.J.; Yasin, M.; Eaton, G.M.; Lauer, M.A.; Sheldon, W.S.; Grines, C.L.; et al. Effect of recombinant ApoA-I Milano on coronary atherosclerosis in patients with acute coronary syndromes: A randomized controlled trial. JAMA 2003, 290, 2292–2300. [Google Scholar] [CrossRef] [PubMed]
- Tardif, J.C.; Gregoire, J.; L’Allier, P.L.; Ibrahim, R.; Lesperance, J.; Heinonen, T.M.; Kouz, S.; Berry, C.; Basser, R.; Lavoie, M.A.; et al. Effects of reconstituted high-density lipoprotein infusions on coronary atherosclerosis: A randomized controlled trial. JAMA 2007, 297, 1675–1682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicholls, S.J.; Andrews, J.; Kastelein, J.J.P.; Merkely, B.; Nissen, S.E.; Ray, K.K.; Schwartz, G.G.; Worthley, S.G.; Keyserling, C.; Dasseux, J.L.; et al. Effect of Serial Infusions of CER-001, a Pre-beta High-Density Lipoprotein Mimetic, on Coronary Atherosclerosis in Patients Following Acute Coronary Syndromes in the CER-001 Atherosclerosis Regression Acute Coronary Syndrome Trial: A Randomized Clinical Trial. JAMA Cardiol. 2018, 3, 815–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicholls, S.J.; Puri, R.; Ballantyne, C.M.; Jukema, J.W.; Kastelein, J.J.P.; Koenig, W.; Wright, R.S.; Kallend, D.; Wijngaard, P.; Borgman, M.; et al. Effect of Infusion of High-Density Lipoprotein Mimetic Containing Recombinant Apolipoprotein A-I Milano on Coronary Disease in Patients with an Acute Coronary Syndrome in the MILANO-PILOT Trial: A Randomized Clinical Trial. JAMA Cardiol. 2018, 3, 806–814. [Google Scholar] [CrossRef] [Green Version]
- Jia, C.; Anderson, J.L.C.; Gruppen, E.G.; Lei, Y.; Bakker, S.J.L.; Dullaart, R.P.F.; Tietge, U.J.F. High-Density Lipoprotein Anti-Inflammatory Capacity and Incident Cardiovascular Events. Circulation 2021, 143, 1935–1945. [Google Scholar] [CrossRef]
- Michael Gibson, C.; Korjian, S.; Tricoci, P.; Daaboul, Y.; Yee, M.; Jain, P.; Alexander, J.H.; Steg, P.G.; Lincoff, A.M.; Kastelein, J.J.; et al. Safety and Tolerability of CSL112, a Reconstituted, Infusible, Plasma-Derived Apolipoprotein A-I, After Acute Myocardial Infarction: The AEGIS-I Trial (ApoA-I Event Reducing in Ischemic Syndromes I). Circulation 2016, 134, 1918–1930. [Google Scholar] [CrossRef]
- Tardif, J.C.; Ballantyne, C.M.; Barter, P.; Dasseux, J.L.; Fayad, Z.A.; Guertin, M.C.; Kastelein, J.J.; Keyserling, C.; Klepp, H.; Koenig, W.; et al. Effects of the high-density lipoprotein mimetic agent CER-001 on coronary atherosclerosis in patients with acute coronary syndromes: A randomized trial. Eur. Heart J. 2014, 35, 3277–3286. [Google Scholar] [CrossRef]
- Nicholls, S.J.; Gordon, A.; Johannson, J.; Ballantyne, C.M.; Barter, P.J.; Brewer, H.B.; Kastelein, J.J.; Wong, N.C.; Borgman, M.R.; Nissen, S.E. ApoA-I induction as a potential cardioprotective strategy: Rationale for the SUSTAIN and ASSURE studies. Cardiovasc. Drugs Ther. 2012, 26, 181–187. [Google Scholar] [CrossRef]
- Nicholls, S.J.; Puri, R.; Wolski, K.; Ballantyne, C.M.; Barter, P.J.; Brewer, H.B.; Kastelein, J.J.; Hu, B.; Uno, K.; Kataoka, Y.; et al. Effect of the BET Protein Inhibitor, RVX-208, on Progression of Coronary Atherosclerosis: Results of the Phase 2b, Randomized, Double-Blind, Multicenter, ASSURE Trial. Am. J. Cardiovasc. Drugs 2016, 16, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Ray, K.K.; Nicholls, S.J.; Buhr, K.A.; Ginsberg, H.N.; Johansson, J.O.; Kalantar-Zadeh, K.; Kulikowski, E.; Toth, P.P.; Wong, N.; Sweeney, M.; et al. Effect of Apabetalone Added to Standard Therapy on Major Adverse Cardiovascular Events in Patients With Recent Acute Coronary Syndrome and Type 2 Diabetes: A Randomized Clinical Trial. JAMA 2020, 323, 1565–1573. [Google Scholar] [CrossRef] [PubMed]
- Baragetti, A.; Bonacina, F.; Catapano, A.L.; Norata, G.D. Effect of Lipids and Lipoproteins on Hematopoietic Cell Metabolism and Commitment in Atherosclerosis. Immunometabolism 2021, 3, e210014. [Google Scholar] [CrossRef] [PubMed]
- Pirillo, A.; Catapano, A.L.; Norata, G.D. HDL in infectious diseases and sepsis. Handb. Exp. Pharmacol. 2015, 224, 483–508. [Google Scholar] [CrossRef] [Green Version]
- Chien, J.Y.; Jerng, J.S.; Yu, C.J.; Yang, P.C. Low serum level of high-density lipoprotein cholesterol is a poor prognostic factor for severe sepsis. Crit. Care Med. 2005, 33, 1688–1693. [Google Scholar] [CrossRef] [PubMed]
- Vanhollebeke, B.; Pays, E. The function of apolipoproteins L. Cell. Mol. Life Sci. CMLS 2006, 63, 1937–1944. [Google Scholar] [CrossRef] [PubMed]
- Parsa, A.; Kao, W.H.; Xie, D.; Astor, B.C.; Li, M.; Hsu, C.Y.; Feldman, H.I.; Parekh, R.S.; Kusek, J.W.; Greene, T.H.; et al. APOL1 risk variants, race, and progression of chronic kidney disease. N. Engl. J. Med. 2013, 369, 2183–2196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holzer, M.; Birner-Gruenberger, R.; Stojakovic, T.; El-Gamal, D.; Binder, V.; Wadsack, C.; Heinemann, A.; Marsche, G. Uremia alters HDL composition and function. J. Am. Soc. Nephrol. JASN 2011, 22, 1631–1641. [Google Scholar] [CrossRef] [Green Version]
- Ammirati, E.; Bozzolo, E.P.; Contri, R.; Baragetti, A.; Palini, A.G.; Cianflone, D.; Banfi, M.; Uboldi, P.; Bottoni, G.; Scotti, I.; et al. Cardiometabolic and immune factors associated with increased common carotid artery intima-media thickness and cardiovascular dis-ease in patients with systemic lupus erythematosus. Nutr. Metab. Cardio-Vasc. Dis. NMCD 2014, 24, 751–759. [Google Scholar] [CrossRef]
- Baragetti, A.; Ramirez, G.A.; Magnoni, M.; Garlaschelli, K.; Grigore, L.; Berteotti, M.; Scotti, I.; Bozzolo, E.; Berti, A.; Camici, P.G.; et al. Disease trends over time and CD4(+)CCR5(+) T-cells expansion predict carotid atherosclerosis development in patients with systemic lupus erythematosus. Nutr. Metab. Cardiovasc. Dis. NMCD 2018, 28, 53–63. [Google Scholar] [CrossRef] [Green Version]
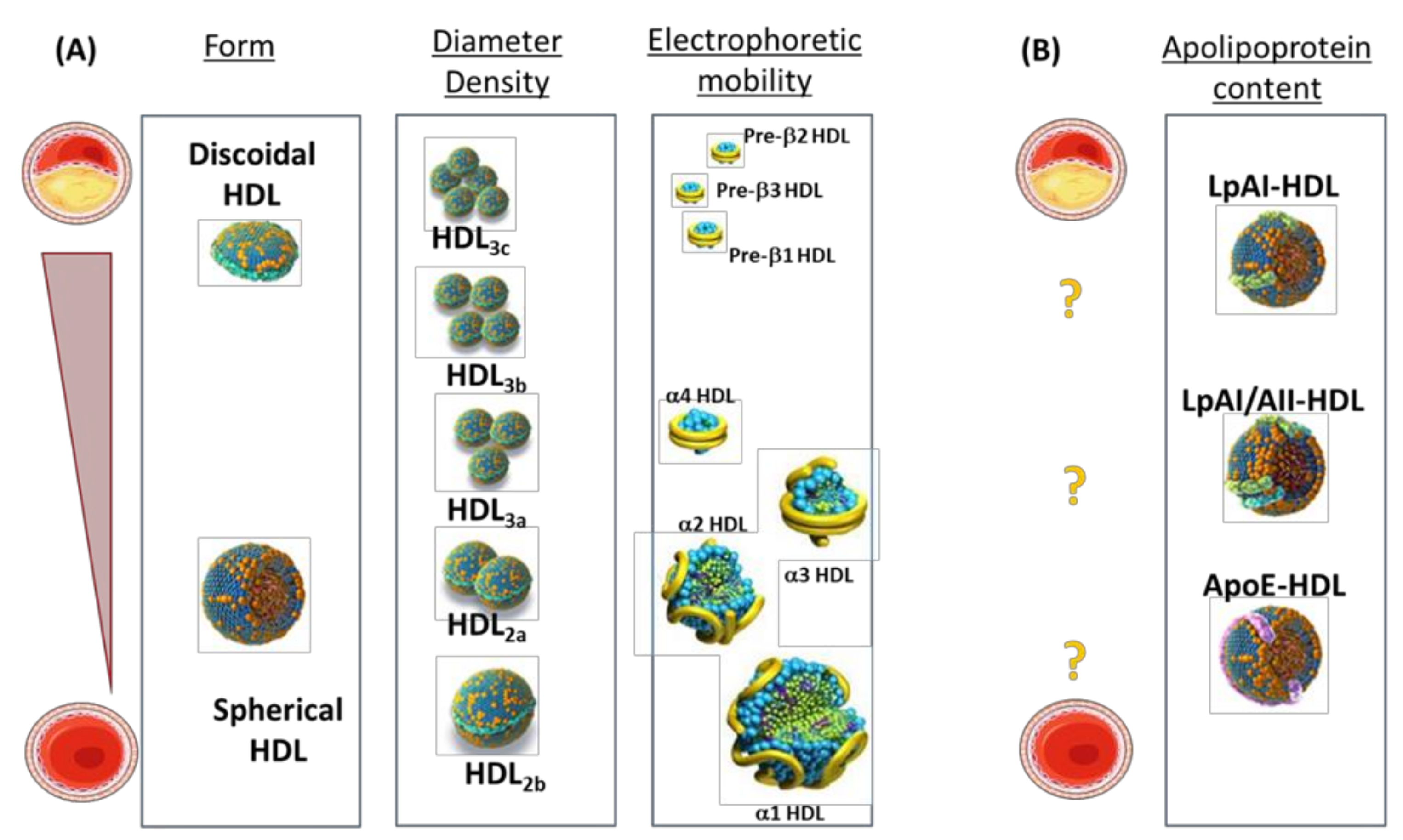

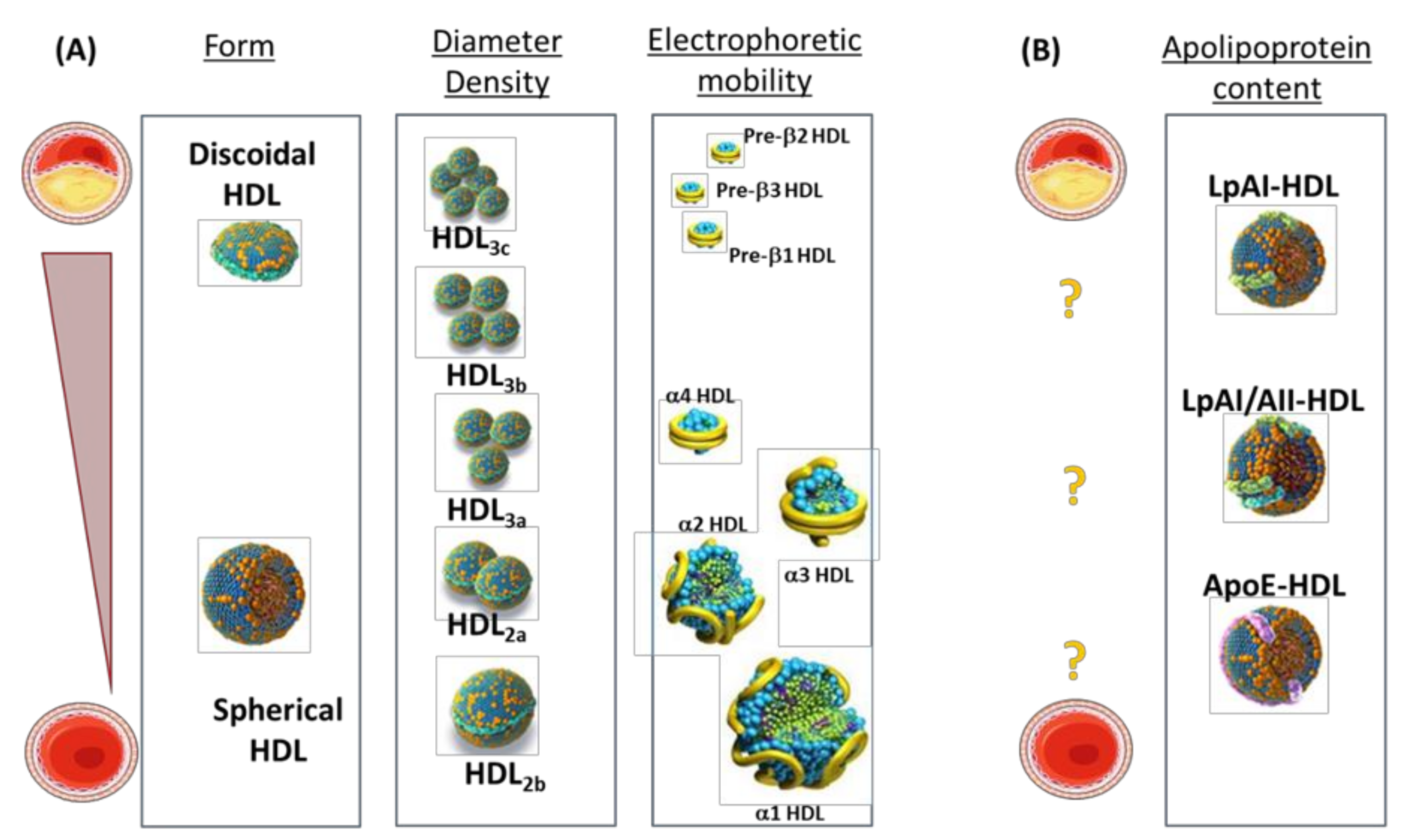{kind=link}
| Ref. | CETP Inhibitor | Trial | Patients | Duration | Effect on Lipids in Treatment Group | CV Results |
|---|---|---|---|---|---|---|
| Barter PJ. N. Engl. J. Med. 2007 [66] | Torcetrapib | ILLUMINATE | 15,067 patients at high cardiovascular risk | 1–2 years | HDL-C: +72% LDL-C: −25% | Increased risk of cardiovascular events (HR 1.25; 95%CI 1.09–1.44; p = 0.001) and death from any cause (HR 1.58; 95%CI 1.14–2.19; p = 0.006) |
| Schwartz GG. N. Engl. J. Med. 2012 [42] | Dalcetrapib | Dal-OUTCOMES | 15,871 patients recently hospitalized for acute coronary syndrome | 31 months (stopped early for futility) | HDL-C: 31 to 40% minimal effect on LDL-C | No effect on the CV composite endpoint (HR 1.04; 95%CI 0.93–1.16; p = 0.52) |
| Lincoff AM. N. Engl. J. Med. 2017 [47] | Evacetrapib | ACCELERATE | 12,092 patients at high cardiovascular risk | 26 months (stopped early for futility) | HDL-C: +133% LDL-C: −31% | No effect on the CV composite endpoint (HR 1.01; 95%CI 0.91–1.11; p = 0.91) |
| Bowman L. N. Engl. J. Med. 2017 [67] | Anacetrapib | REVEAL | 30,449 patients at high cardiovascular risk | 4.1 years | HDL-C: +104% LDL-C: −26% | Decrease in the CV composite endpoint (HR 0.91; 95%CI 0.85–0.97; p = 0.004) |
| Publication | Gene | Effect on HDL-C Levels | CV Outcome | Expected Effect on CV Outcome | Observed Effect on CV Outcome |
|---|---|---|---|---|---|
| Frikke-Schmidt R. 2008 [69] | ABCA1 | −17 mg/dL | ischemic heart disease | HR 1.70 (95%CI 1.57–1.85) | OR 0.93 (95%CI 0.53–1.62) |
| Johannsen TH. 2009 [73] | LIPC | +16% (8 mg/dL) | ischemic heart disease | HR 0.87 (95%CI 0.84–0.90) | OR 1.19 (95%CI 0.76–1.88) |
| Haase CL. 2010 [74] | APOA1 | +7.7% | ischemic heart disease myocardial infarction | HR 0.93 (95%CI 0.91–0.94) HR 0.89 (95%CI 0.86–0.92) | OR 1.10 (95%CI 0.89–1.35) OR 1.14 (95%CI 0.89–1.46) |
| Voight BF. 2012 [71] | LIPG | +5.5 mg/dL | myocardial infarction | OR 0.87 (95%CI 0.84–0.91) | OR 0.99 (95%CI 0.88–1.11) |
| Haase CL. 2012 [72] | LCAT | −13% (8 mg/dL) | myocardial infarction | HR 1.18 (95%CI 1.12–1.24) | OR 0.53 (95%CI 0.92–1.25) |
| Voight BF. 2012 [71] | 14 SNP score | - | myocardial infarction | OR 0.62 (95%CI 0.58–0.66) * | OR 0.93 (95%CI 0.68–1.26) * |
| Holmes MV. 2015 [75] | 19 SNP score | 8.9 mg/dL ^ | myocardial infarction | - | OR 0.91 (95%CI 0.42–1.98) ** |
| 48 SNP score | 3.1 mg/dL ^ | myocardial infarction | - | OR 0.81 (95%CI 0.44–1.46) ** |
| Ref. | HDL Mimetic | Apo A1 | Trial | Patients | Effect on Cholesterol Efflux | CV Outcome | Results |
|---|---|---|---|---|---|---|---|
| Michael Gibson C. 2016 [118] | CSL-112 | wild-type apoA-I | AEGIS-I trial | 1258 patients with a recent acute myocardial infarction | increased apoA-I and ex vivo cholesterol efflux | time to first occurrence of a MACE ** | HR 1.02 (95%CI, 0.57–1.80; p = 0.52) |
| Tardif J-C. 2014 [119] | CER-001 | apoA-I and sphingomyelin | CHI-SQUARE study | 507 patients with a clinical indication for coronary angiography | increased cholesterol mobilization | PAV * | 0.02% with placebo and 0.19%; with CER-001 (difference, p = 0.53) |
| Nicholls SJ. 2018 [116] | MDCO-216 | apoA-I Milano | MILANO-PILOT Trial | 122 post-ACS patients on optimal conventional medical treatment | increased ATP-binding cassette transporter A1-mediated cholesterol efflux | PAV * | −0.94% with placebo and −0.21% with MDCO-216 (difference, 0.73%; 95%CI, −0.07 to 1.52; p = 0.07) |
| Nicholls SJ. JAMA Cardiol. 2018 [115] | CER-001 | apoA-I and sphingomyelin | CARAT study | 293 patients with status post-ACS | increased cholesterol mobilization | PAV* | −0.41% with placebo and −0.09%; with CER-001 (difference 0.32%; p = 0.15) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Casula, M.; Colpani, O.; Xie, S.; Catapano, A.L.; Baragetti, A. HDL in Atherosclerotic Cardiovascular Disease: In Search of a Role. Cells 2021, 10, 1869. https://doi.org/10.3390/cells10081869
Casula M, Colpani O, Xie S, Catapano AL, Baragetti A. HDL in Atherosclerotic Cardiovascular Disease: In Search of a Role. Cells. 2021; 10(8):1869. https://doi.org/10.3390/cells10081869
Chicago/Turabian StyleCasula, Manuela, Ornella Colpani, Sining Xie, Alberico L. Catapano, and Andrea Baragetti. 2021. "HDL in Atherosclerotic Cardiovascular Disease: In Search of a Role" Cells 10, no. 8: 1869. https://doi.org/10.3390/cells10081869
APA StyleCasula, M., Colpani, O., Xie, S., Catapano, A. L., & Baragetti, A. (2021). HDL in Atherosclerotic Cardiovascular Disease: In Search of a Role. Cells, 10(8), 1869. https://doi.org/10.3390/cells10081869