
Identification of Novel Positive Allosteric Modulators of Neurotrophin Receptors for the Treatment of Cognitive Dysfunction


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Animal Welfare
2.3. TrkA PathHunter® Cell-Based Assay and High Throughput Screening
2.4. Immunoprecipitation and Kinase Activity Assay of Hemagglutinin (HA)-Tagged Full Length TrkA
2.5. Affinity Labeling and Immuno Precipitation/Streptavidin Adsorption
2.6. Surface Plasmon Resonance Studies
2.7. Long Term Potentiation and Electrophysiology
2.8. In Vivo Microdialysis
2.9. Western Blot Analysis
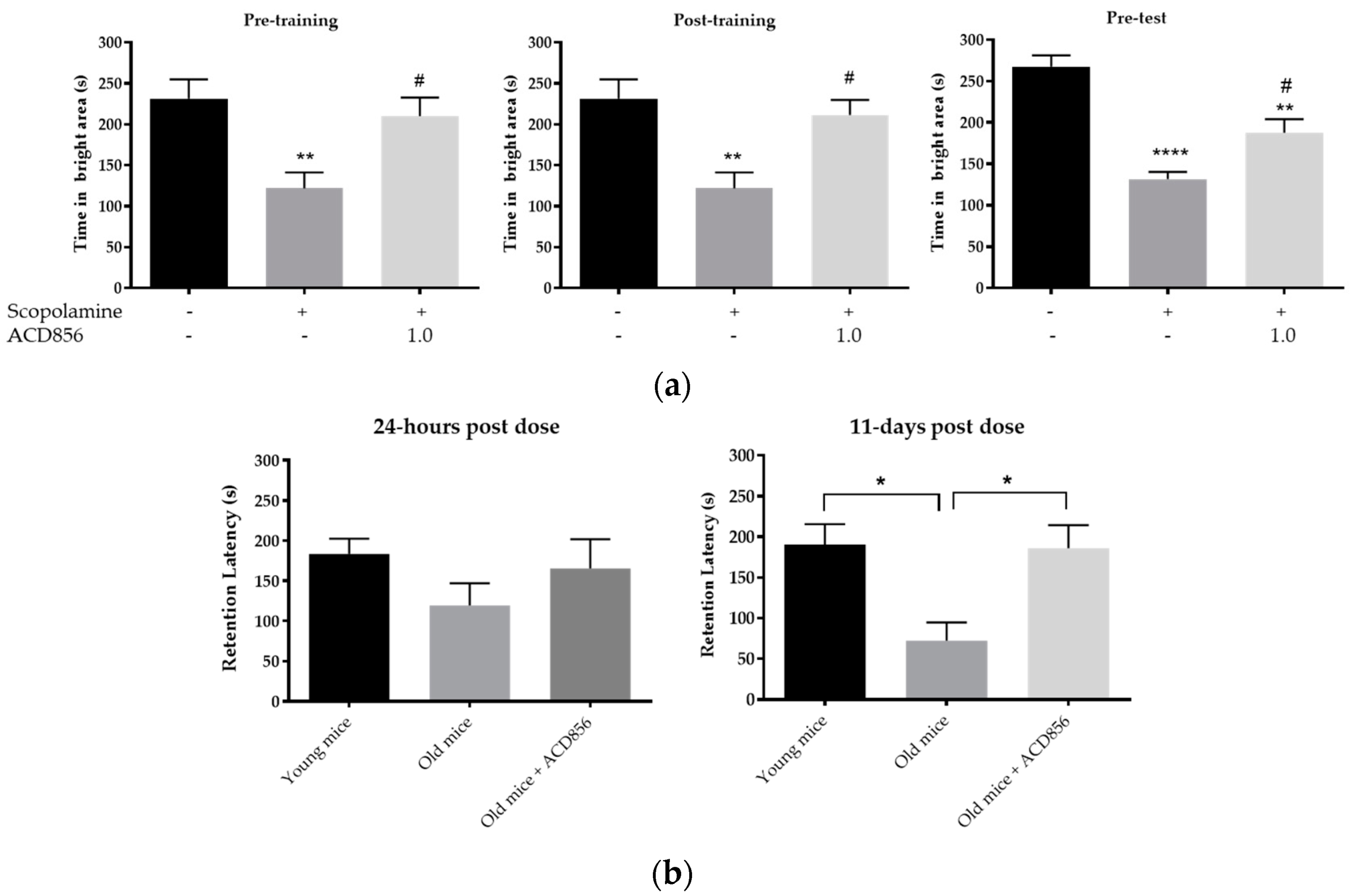
2.10. Morris Water Maze
2.11. Passive Avoidance
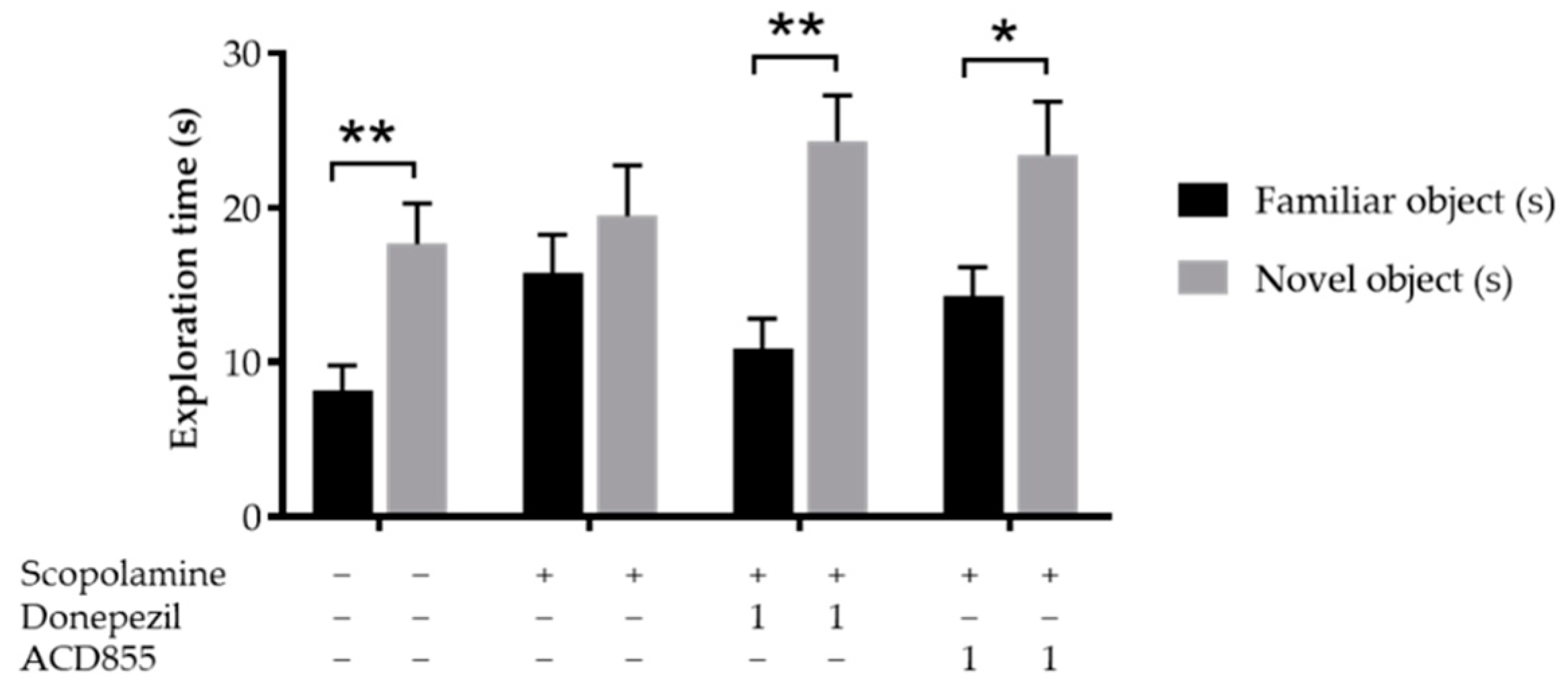
2.12. Novel Object Recognition
2.13. Statistical Analysis
3. Results
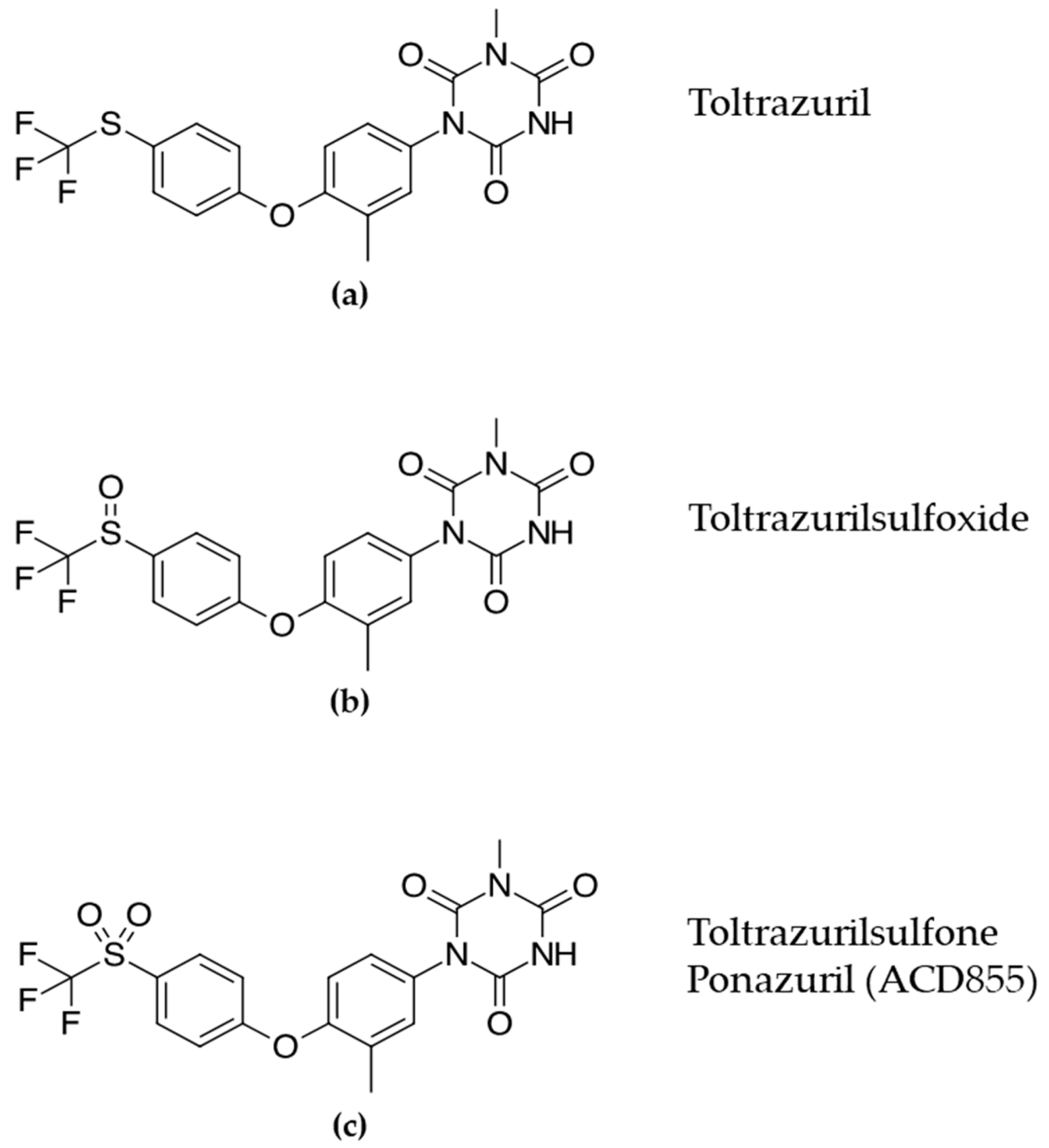
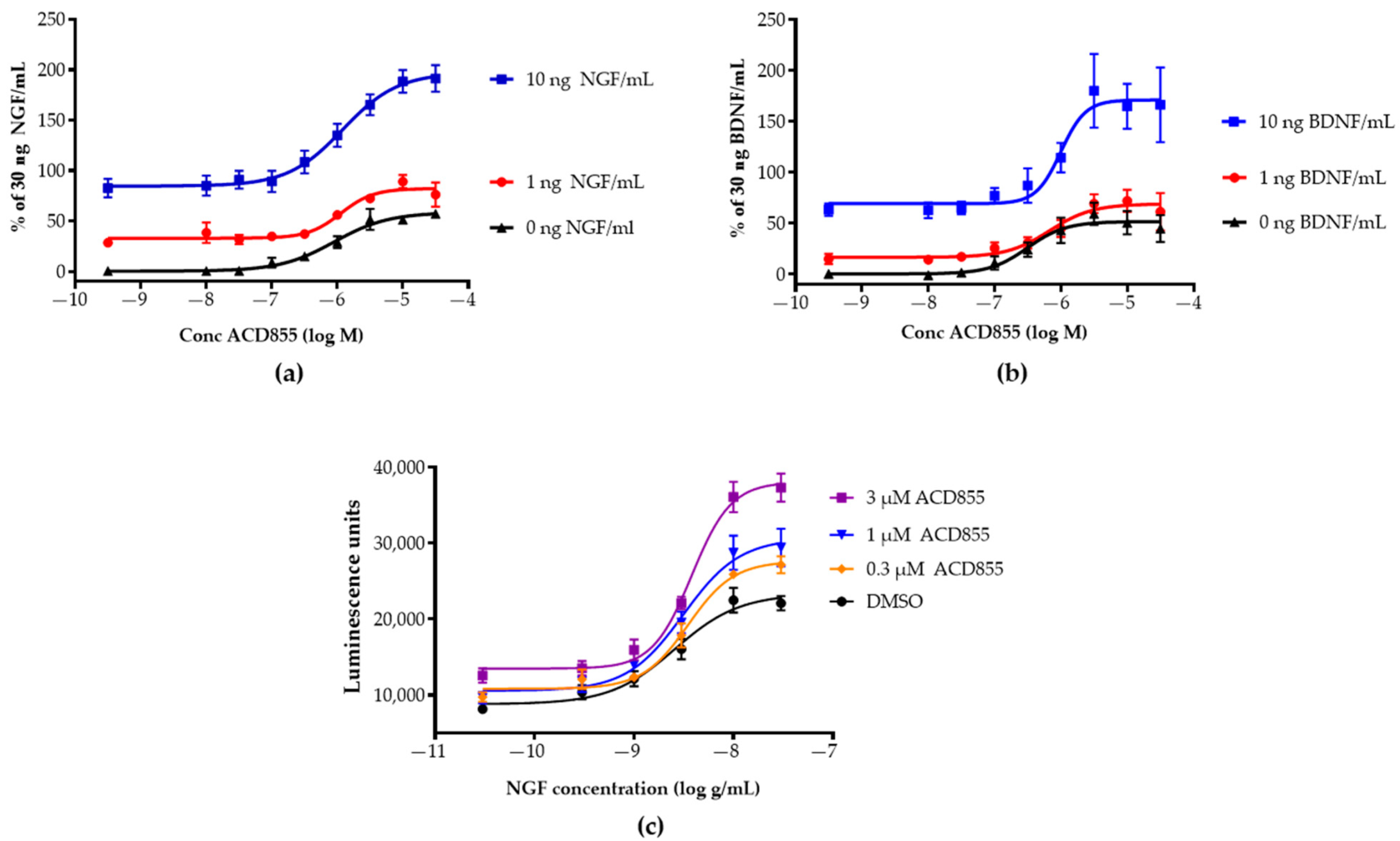
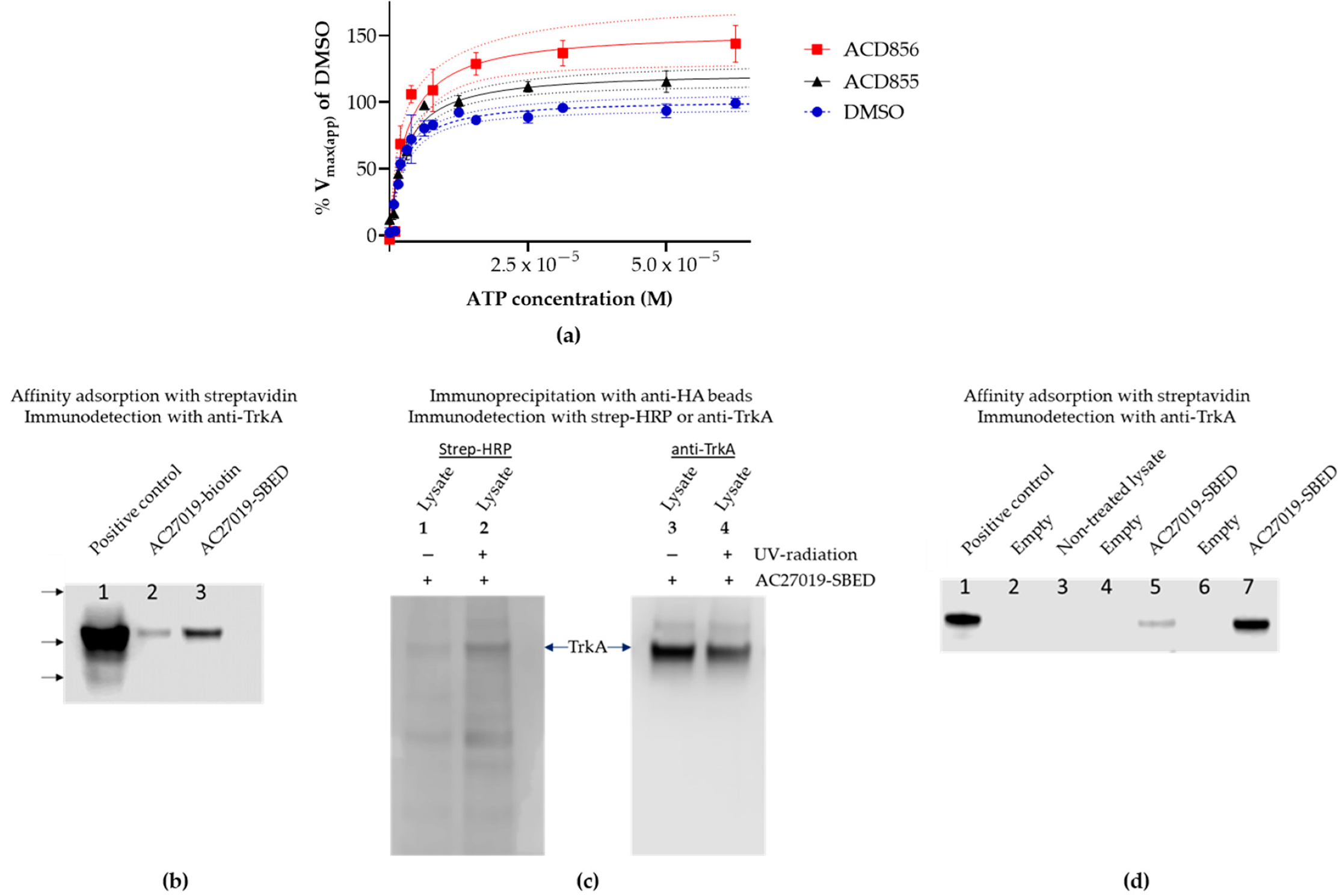
3.1. Screening and in Vitro Characterization
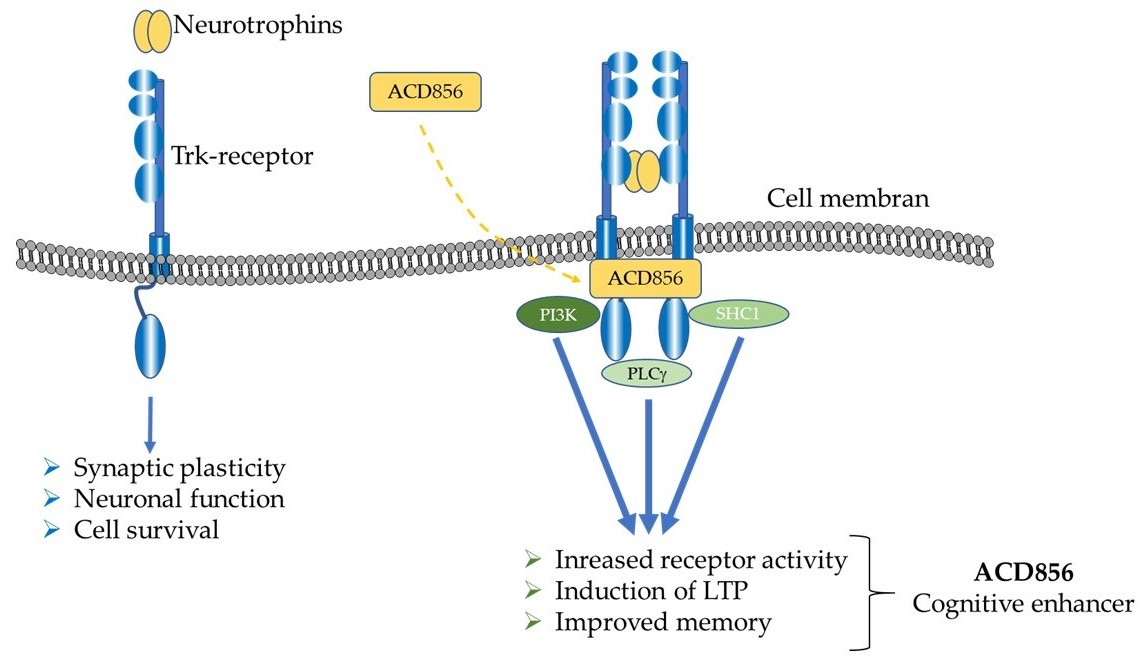
3.2. Mechanism of Action Studies
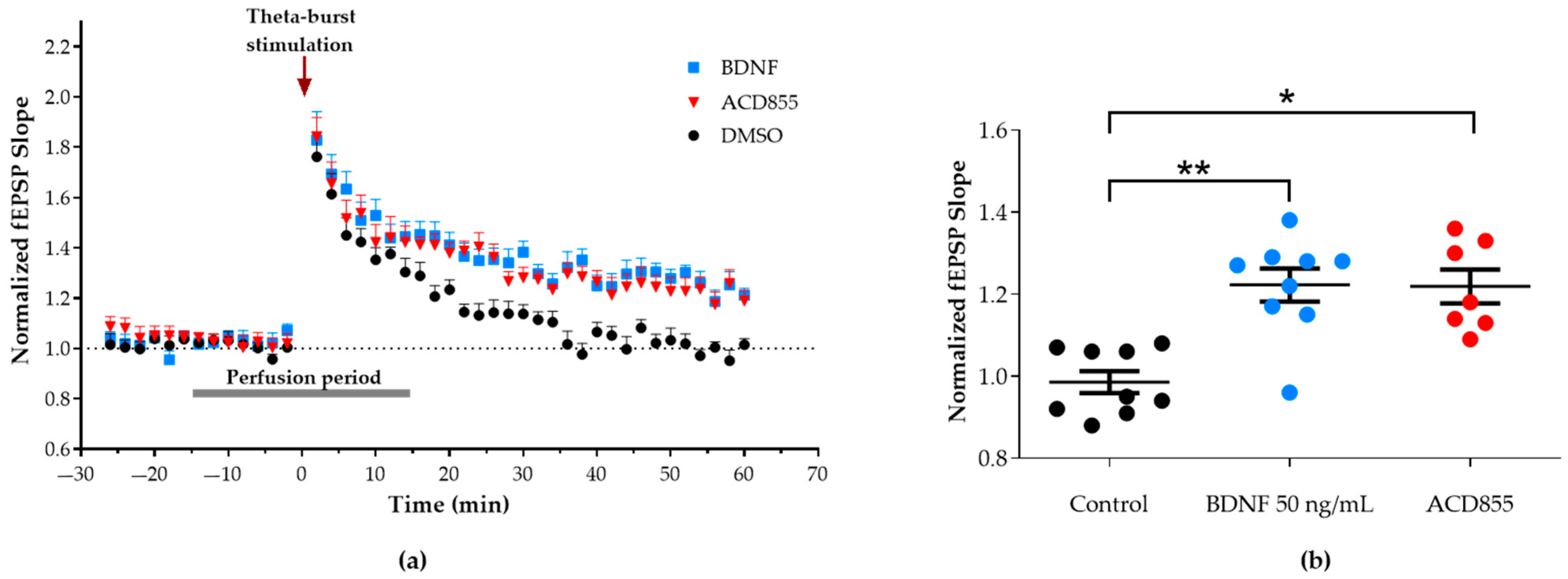
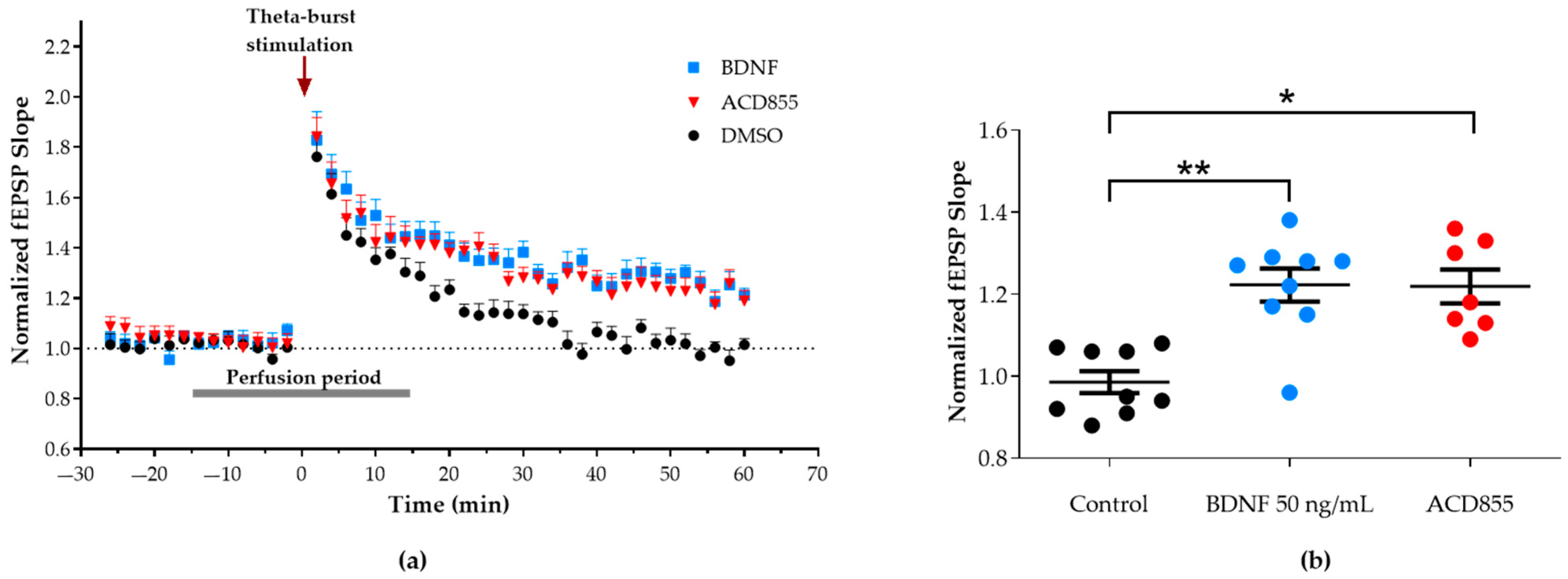
3.3. Facilitation of Long-Term Potentiation in Rat Hippocampal Slices
3.4. Microdialysis
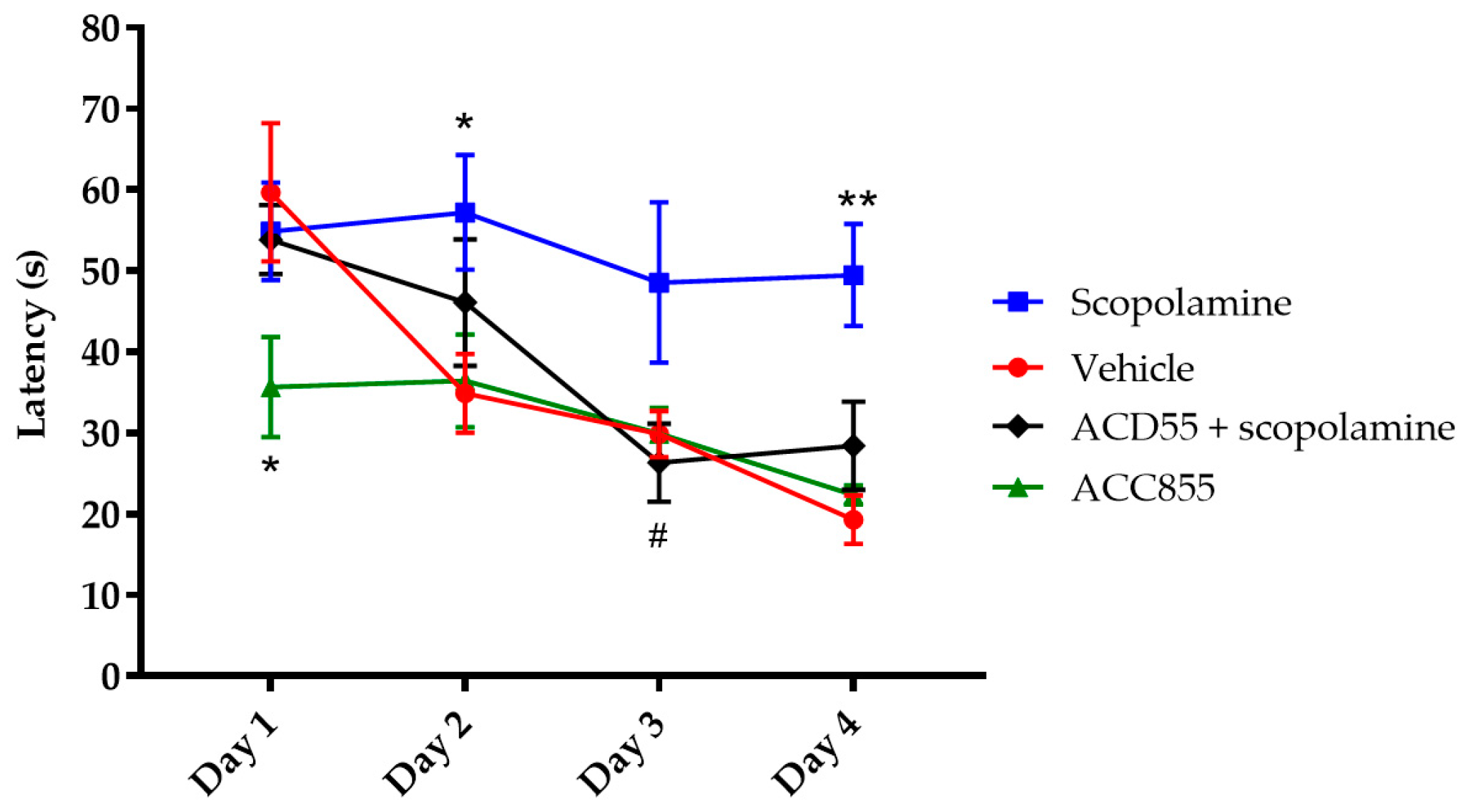
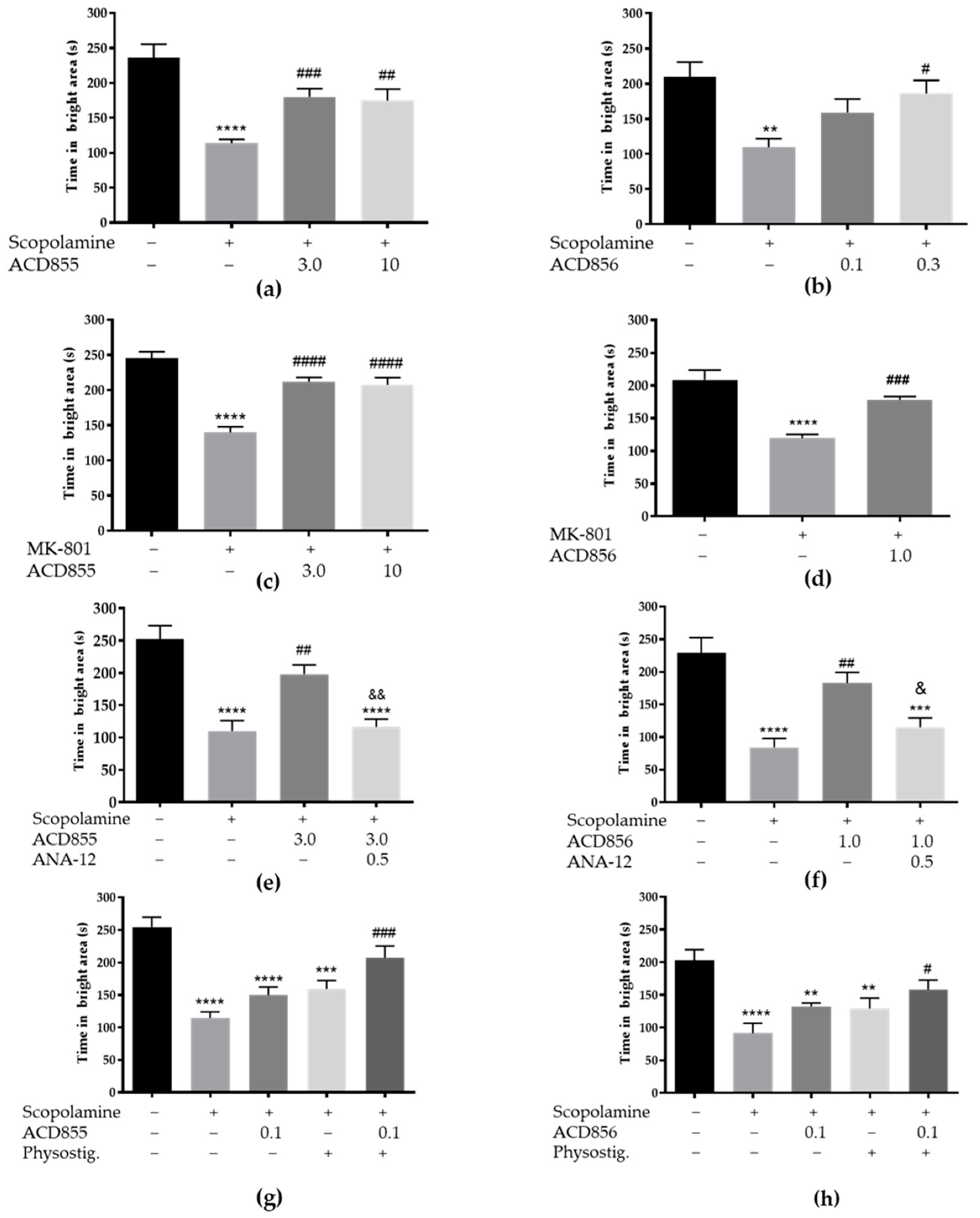
3.5. In Vivo Behavioral Cognitive Models
4. Discussion
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Levi-Montalcini, R. Effects of mouse tumor transplantation on the nervous system. Ann. N. Y. Acad. Sci. 1952, 55, 330–344. [Google Scholar] [CrossRef] [PubMed]
- Barde, Y.A.; Edgar, D.; Thoenen, H. Purification of a new neurotrophic factor from mammalian brain. EMBO J. 1982, 1, 549–553. [Google Scholar] [CrossRef]
- Ernfors, P.; Ibáñez, C.F.; Ebendal, T.; Olson, L.; Persson, H. Molecular cloning and neurotrophic activities of a protein with structural similarities to nerve growth factor: Developmental and topographical expression in the brain. Proc. Natl. Acad. Sci. USA 1990, 87, 5454–5458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maisonpierre, P.; Belluscio, L.; Squinto, S.; Ip, N.; Furth, M.; Lindsay, R.; Yancopoulos, G. Neurotrophin-3: A neurotrophic factor related to NGF and BDNF. Science 1990, 247, 1446–1451. [Google Scholar] [CrossRef] [PubMed]
- Berkemeier, L.R.; Winslow, J.W.; Kaplan, D.R.; Nikolics, K.; Goeddel, D.V.; Rosenthal, A. Neurotrophin-5: A novel neurotrophic factor that activates trk and trkB. Neuron 1991, 7, 857–866. [Google Scholar] [CrossRef]
- Chao, M.V. Neurotrophins and their receptors: A convergence point for many signalling pathways. Nat. Rev. Neurosci. 2003, 4, 299. [Google Scholar] [CrossRef] [PubMed]
- Leal, G.; Afonso, P.M.; Salazar, I.L.; Duarte, C.B. Regulation of hippocampal synaptic plasticity by BDNF. Brain Res. 2015, 1621, 82–101. [Google Scholar] [CrossRef]
- Arancibia, S.; Silhol, M.; Moulière, F.; Meffre, J.; Höllinger, I.; Maurice, T.; Tapia-Arancibia, L. Protective effect of BDNF against beta-amyloid induced neurotoxicity in vitro and in vivo in rats. Neurobiol. Dis. 2008, 31, 316–326. [Google Scholar] [CrossRef]
- Jiao, S.S.; Shen, L.L.; Zhu, C.; Bu, X.L.; Liu, Y.H.; Liu, C.H.; Yao, X.Q.; Zhang, L.L.; Zhou, H.D.; Walker, D.G.; et al. Brain-derived neurotrophic factor protects against tau-related neurodegeneration of Alzheimer’s disease. Transl. Psychiatry 2016, 6, e907. [Google Scholar] [CrossRef]
- Kemppainen, S.; Rantamäki, T.; Jerónimo-Santos, A.; Lavasseur, G.; Autio, H.; Karpova, N.; Kärkkäinen, E.; Stavén, S.; Miranda, H.; Outeiro, T.F.; et al. Impaired TrkB receptor signaling contributes to memory impairment in APP/PS1 mice. Neurobiol. Aging 2012, 33, 1122.e23–1122.e39. [Google Scholar] [CrossRef]
- Nagahara, A.H.; Merrill, D.A.; Coppola, G.; Tsukada, S.; Schroeder, B.E.; Shaked, G.M.; Wang, L.; Blesch, A.; Kim, A.; Conner, J.M.; et al. Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer’s disease. Nat. Med. 2009, 15, 331–337. [Google Scholar] [CrossRef] [Green Version]
- Boots, E.A.; Schultz, S.A.; Clark, L.R.; Racine, A.M.; Darst, B.F.; Koscik, R.L.; Carlsson, C.M.; Gallagher, C.L.; Hogan, K.J.; Bendlin, B.B.; et al. BDNF Val66Met predicts cognitive decline in the Wisconsin Registry for Alzheimer’s Prevention. Neurology 2017, 88, 2098–2106. [Google Scholar] [CrossRef] [Green Version]
- Lim, Y.Y.; Hassenstab, J.; Cruchaga, C.; Goate, A.; Fagan, A.M.; Benzinger, T.L.; Maruff, P.; Snyder, P.J.; Masters, C.L.; Allegri, R.; et al. BDNF Val66Met moderates memory impairment, hippocampal function and tau in preclinical autosomal dominant Alzheimer’s disease. Brain 2016, 139, 2766–2777. [Google Scholar] [CrossRef] [Green Version]
- Egan, M.F.; Kojima, M.; Callicott, J.H.; Goldberg, T.E.; Kolachana, B.S.; Bertolino, A.; Zaitsev, E.; Gold, B.; Goldman, D.; Dean, M.; et al. The BDNF val66met Polymorphism Affects Activity-Dependent Secretion of BDNF and Human Memory and Hippocampal Function. Cell 2003, 112, 257–269. [Google Scholar] [CrossRef] [Green Version]
- Li, G.D.; Bi, R.; Zhang, D.F.; Xu, M.; Luo, R.; Wang, D.; Fang, Y.; Li, T.; Zhang, C.; Yao, Y.G. Female-specific effect of the BDNF gene on Alzheimer’s disease. Neurobiol. Aging 2017, 53, e11–e192. [Google Scholar] [CrossRef] [Green Version]
- Ward, D.D.; Summers, M.J.; Saunders, N.L.; Janssen, P.; Stuart, K.E.; Vickers, J.C. APOE and BDNF Val66Met polymorphisms combine to influence episodic memory function in older adults. Behav. Brain Res. 2014, 271, 309–315. [Google Scholar] [CrossRef]
- Franzmeier, N.; Ren, J.; Damm, A.; Monté-Rubio, G.; Boada, M.; Ruiz, A.; Ramirez, A.; Jessen, F.; Düzel, E.; Rodríguez Gómez, O.; et al. The BDNF(Val66Met) SNP modulates the association between beta-amyloid and hippocampal disconnection in Alzheimer’s disease. Mol. Psychiatry 2021, 26, 614–628. [Google Scholar] [CrossRef]
- Lim, Y.; Villemagne, V.L.; Laws, S.M.; Ames, D.; Pietrzak, R.H.; Ellis, K.A.; Harrington, K.D.; Bourgeat, P.; Salvado, O.; Darby, D.; et al. BDNF Val66Met, Aβ amyloid, and cognitive decline in preclinical Alzheimer’s disease. Neurobiol. Aging 2013, 34, 2457–2464. [Google Scholar] [CrossRef]
- Turnbull, M.T.; Boskovic, Z.; Coulson, E.J. Acute Down-regulation of BDNF Signaling Does Not Replicate Exacerbated Amyloid-β Levels and Cognitive Impairment Induced by Cholinergic Basal Forebrain Lesion. Front. Mol. Neurosci. 2018, 11, 51. [Google Scholar] [CrossRef] [Green Version]
- Mesulam, M. The cholinergic lesion of Alzheimer’s disease: Pivotal factor or side show? Learn. Mem. 2004, 11, 43–49. [Google Scholar] [CrossRef] [Green Version]
- Triaca, V.; Calissano, P. Impairment of the nerve growth factor pathway driving amyloid accumulation in cholinergic neurons: The incipit of the Alzheimer’s disease story? Neural Regen. Res. 2016, 11, 1553–1556. [Google Scholar] [CrossRef]
- Triaca, V.; Sposato, V.; Bolasco, G.; Ciotti, M.T.; Pelicci, P.; Bruni, A.C.; Cupidi, C.; Maletta, R.; Feligioni, M.; Nisticò, R.; et al. NGF controls APP cleavage by downregulating APP phosphorylation at Thr668: Relevance for Alzheimer’s disease. Aging Cell 2016, 15, 661–672. [Google Scholar] [CrossRef] [Green Version]
- Canu, N.; Pagano, I.; La Rosa, L.R.; Pellegrino, M.; Ciotti, M.T.; Mercanti, D.; Moretti, F.; Sposato, V.; Triaca, V.; Petrella, C.; et al. Association of TrkA and APP Is Promoted by NGF and Reduced by Cell Death-Promoting Agents. Front. Mol. Neurosci. 2017, 10, 15. [Google Scholar] [CrossRef] [PubMed]
- Eriksdotter-Jönhagen, M.; Linderoth, B.; Lind, G.; Aladellie, L.; Almkvist, O.; Andreasen, N.; Blennow, K.; Bogdanovic, N.; Jelic, V.; Kadir, A.; et al. Encapsulated Cell Biodelivery of Nerve Growth Factor to the Basal Forebrain in Patients with Alzheimer’s Disease. Dement. Geriatr. Cogn. Disord. 2012, 33, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Karami, A.; Eyjolfsdottir, H.; Vijayaraghavan, S.; Lind, G.; Almqvist, P.; Kadir, A.; Linderoth, B.; Andreasen, N.; Blennow, K.; Wall, A.; et al. Changes in CSF cholinergic biomarkers in response to cell therapy with NGF in patients with Alzheimer’s disease. Alzheimers Dement. 2015, 11, 1316–1328. [Google Scholar] [CrossRef] [PubMed]
- Eyjolfsdottir, H.; Eriksdotter, M.; Linderoth, B.; Lind, G.; Juliusson, B.; Kusk, P.; Almkvist, O.; Andreasen, N.; Blennow, K.; Ferreira, D.; et al. Targeted delivery of nerve growth factor to the cholinergic basal forebrain of Alzheimer’s disease patients: Application of a second-generation encapsulated cell biodelivery device. Alzheimers Res. Ther. 2016, 8, 30. [Google Scholar] [CrossRef] [Green Version]
- Mitra, S.; Behbahani, H.; Eriksdotter, M. Innovative Therapy for Alzheimer’s Disease-With Focus on Biodelivery of NGF. Front. Neurosci. 2019, 13, 38. [Google Scholar] [CrossRef] [Green Version]
- LeSauteur, L.; Wei, L.; Gibbs, B.F.; Saragovi, H.U. Small Peptide Mimics of Nerve Growth Factor Bind TrkA Receptors and Affect Biological Responses. J. Biol. Chem. 1995, 270, 6564–6569. [Google Scholar] [CrossRef] [Green Version]
- Jain, P.; Li, R.; Lama, T.; Saragovi, H.U.; Cumberlidge, G.; Meerovitch, K. An NGF mimetic, MIM-D3, stimulates conjunctival cell glycoconjugate secretion and demonstrates therapeutic efficacy in a rat model of dry eye. Exp. Eye Res. 2011, 93, 503–512. [Google Scholar] [CrossRef]
- Colangelo, A.; Bianco, M.; Vitagliano, L.; Cavaliere, C.; Cirillo, G.; Gioia, L.; Diana, D.; Colombo, D.; Redaelli, C.; Zaccaro, L.; et al. A New Nerve Growth Factor-Mimetic Peptide Active on Neuropathic Pain in Rats. J. Neurosci. 2008, 28, 2698–2709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gudasheva, T.A.; Povarnina, P.; Antipova, T.A.; Firsova, Y.N.; Konstantinopolsky, M.A.; Seredenin, S.B. Dimeric dipeptide mimetics of the nerve growth factor Loop 4 and Loop 1 activate TRKA with different patterns of intracellular signal transduction. J. Biomed. Sci. 2015, 22, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gudasheva, T.A.; Povarnina, P.; Logvinov, I.O.; Antipova, T.A.; Seredenin, S.B. Mimetics of brain-derived neurotrophic factor loops 1 and 4 are active in a model of ischemic stroke in rats. Drug Des. Dev. Ther. 2016, 10, 3545–3553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maliartchouk, S.; Feng, Y.; Ivanisevic, L.; Debeir, T.; Cuello, A.C.; Burgess, K.; Saragovi, H.U. A designed peptidomimetic agonistic ligand of TrkA nerve growth factor receptors. Mol. Pharmacol. 2000, 57, 385–391. [Google Scholar]
- Pattarawarapan, M.; Zaccaro, M.C.; Saragovi, U.H.; Burgess, K. New templates for syntheses of ring-fused, C (10) beta-turn peptidomimetics leading to the first reported small-molecule mimic of neurotrophin-3. J. Med. Chem. 2002, 45, 4387–4390. [Google Scholar] [CrossRef]
- Xie, Y.; Tisi, M.A.; Yeo, T.T.; Longo, F.M. Nerve Growth Factor (NGF) Loop 4 Dimeric Mimetics Activate ERK and AKT and Promote NGF-like Neurotrophic Effects. J. Biol. Chem. 2000, 275, 29868–29874. [Google Scholar] [CrossRef] [Green Version]
- Jang, S.W.; Liu, X.; Yepes, M.; Shepherd, K.R.; Miller, G.W.; Liu, Y.; Wilson, W.D.; Xiao, G.; Blanchi, B.; Sun, Y.E.; et al. A selective TrkB agonist with potent neurotrophic activities by 7,8-dihydroxyflavone. Proc. Natl. Acad. Sci. USA 2010, 107, 2687–2692. [Google Scholar] [CrossRef] [Green Version]
- Jang, S.-W.; Okada, M.; Sayeed, I.; Xiao, G.; Stein, D.; Jin, P.; Ye, K. Gambogic amide, a selective agonist for TrkA receptor that possesses robust neurotrophic activity, prevents neuronal cell death. Proc. Natl. Acad. Sci. USA 2007, 104, 16329–16334. [Google Scholar] [CrossRef] [Green Version]
- Jang, S.W.; Liu, X.; Chan, C.B.; Weinshenker, D.; Hall, R.A.; Xiao, G.; Ye, K. Amitriptyline is a TrkA and TrkB receptor agonist that promotes TrkA/TrkB heterodimerization and has potent neurotrophic activity. Chem. Biol. 2009, 16, 644–656. [Google Scholar] [CrossRef] [Green Version]
- Massa, S.M.; Yang, T.; Xie, Y.; Shi, J.; Bilgen, M.; Joyce, J.N.; Nehama, D.; Rajadas, J.; Longo, F.M. Small molecule BDNF mimetics activate TrkB signaling and prevent neuronal degeneration in rodents. J. Clin. Investig. 2010, 120, 1774–1785. [Google Scholar] [CrossRef] [Green Version]
- Holmes, M.; Maysinger, D.; Foerster, A.; Pertens, E.; Barlas, C.; Diamond, J. Neotrofin, a novel purine that induces NGF-dependent nociceptive nerve sprouting but not hyperalgesia in adult rat skin. Mol. Cell. Neurosci. 2003, 24, 568–580. [Google Scholar] [CrossRef]
- Grundman, M.; Capparelli, E.; Kim, H.T.; Morris, J.C.; Farlow, M.; Rubin, E.H.; Heidebrink, J.; Hake, A.; Ho, G.; Schultz, A.N.; et al. A multicenter, randomized, placebo controlled, multiple-dose, safety and pharmacokinetic study of AIT-082 (Neotrofin) in mild Alzheimer’s disease patients. Life Sci. 2003, 73, 539–553. [Google Scholar] [CrossRef]
- Wilkie, N.; Wingrove, P.B.; Bilsland, J.G.; Young, L.; Harper, S.J.; Hefti, F.; Ellis, S.; Pollack, S.J. The non-peptidyl fungal metabolite L-783,281 activates TRK neurotrophin receptors. J. Neurochem. 2001, 78, 1135–1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, B.; Pirrung, M.C.; Deng, L.; Li, Z.; Liu, Y.; Webster, N.J.G. Neuroprotection by Small Molecule Activators of the Nerve Growth Factor Receptor. J. Pharmacol. Exp. Ther. 2007, 322, 59–69. [Google Scholar] [CrossRef] [Green Version]
- Scarpi, D.; Cirelli, D.; Matrone, C.; Castronovo, G.; Rosini, P.; Occhiato, E.G.; Romano, F.; Bartali, L.; Clemente, A.M.; Bottegoni, G.; et al. Low molecular weight, non-peptidic agonists of TrkA receptor with NGF-mimetic activity. Cell Death Dis. 2012, 3, e339. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Tsuchida, Y.; Mizoue, K.; Hanada, K. NG-011 and NG-012, novel potentiators of nerve growth factor. II. The structure determination of NG-011 and NG-012. J. Antibiot. 1992, 45, 1566–1572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, M.; Sakai, N.; Ito, K.; Mizobe, F.; Hanada, K.; Mizoue, K. A novel fungal metabolite NG-061 enhances and mimics neurotrophic effect of nerve growth factor (NGF) on neurite outgrowth in PC12 cells. J. Antibiot. 1999, 52, 224–230. [Google Scholar] [CrossRef] [Green Version]
- Pediaditakis, I.; Efstathopoulos, P.; Prousis, K.C.; Zervou, M.; Arévalo, J.; Alexaki, V.I.; Nikoletopoulou, V.; Karagianni, E.; Potamitis, C.; Tavernarakis, N.; et al. Selective and differential interactions of BNN27, a novel C17-spiroepoxy steroid derivative, with TrkA receptors, regulating neuronal survival and differentiation. Neuropharmacology 2016, 111, 266–282. [Google Scholar] [CrossRef] [Green Version]
- Jeon, S.; Lee, H.; Lee, H.; Park, S.; Gwon, Y.; Kim, H.; Zhang, J.; Shin, C.; Kim, D.; Ryu, J. Oleanolic acid ameliorates cognitive dysfunction caused by cholinergic blockade via TrkB-dependent BDNF signaling. Neuropharmacology 2017, 113, 100–109. [Google Scholar] [CrossRef]
- Nordvall, G.; Forsell, P. Stimulating neurotrophin receptors in the treatment of neurodegenerative disorders. Annu. Rep. Med. Chem. 2014, 49, 59–73. [Google Scholar]
- Casarotto, P.C.; Girych, M.; Fred, S.M.; Kovaleva, V.; Moliner, R.; Enkavi, G.; Biojone, C.; Cannarozzo, C.; Sahu, M.P.; Kaurinkoski, K.; et al. Antidepressant drugs act by directly binding to TRKB neurotrophin receptors. Cell 2021, 184, 1299–1313. [Google Scholar] [CrossRef] [PubMed]
- Forsell, P.; Almqvist, H.; Hillertz, P.; Åkerud, T.; Otrocka, M.; Eisele, L.; Sun, K.; Andersson, H.; Trivedi, S.; Wollberg, A.; et al. The Use of TrkA-PathHunter Assay in High-Throughput Screening to Identify Compounds That Affect Nerve Growth Factor Signaling. J. Biomol. Screen. 2013, 18, 659–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kehr, J.; Dechent, P.; Kato, T.; Ogren, S.O. Simultaneous determination of acetylcholine, choline and physostigmine in microdialysis samples from rat hippocampus by microbore liquid chromatography/electrochemistry on peroxidase redox polymer coated electrodes. J. Neurosci. Methods 1998, 83, 143–150. [Google Scholar] [CrossRef]
- Yoshitake, T.; Yoshitake, S.; Fujino, K.; Nohta, H.; Yamaguchi, M.; Kehr, J. High-sensitive liquid chromatographic method for determination of neuronal release of serotonin, noradrenaline and dopamine monitored by microdialysis in the rat prefrontal cortex. J. Neurosci. Methods 2004, 140, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Kehr, J.; Hu, X.J.; Yoshitake, T.; Scheller, D. Determination of the dopamine agonist rotigotine in microdialysates from the rat brain by microbore column liquid chromatography with electrochemical detection. J. Chromatogr. 2007, 845, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Dahlström, M.; Nordvall, G.; Sundström, E.; Åkesson, E.; Tegerstedt, G.; Eriksdotter, M.; Forsell, P. Identification of amino acid residues of nerve growth factor important for neurite outgrowth in human dorsal root ganglion neurons. Eur. J. Neurosci. 2019, 50, 3487–3501. [Google Scholar] [CrossRef] [Green Version]
- Daumas, S.; Sandin, J.; Chen, K.S.; Kobayashi, D.; Tulloch, J.; Martin, S.J.; Games, D.; Morris, R.G. Faster forgetting contributes to impaired spatial memory in the PDAPP mouse: Deficit in memory retrieval associated with increased sensitivity to interference? Learn. Mem. 2008, 15, 625–632. [Google Scholar] [CrossRef] [Green Version]
- Madjid, N.; Tottie, E.E.; Luttgen, M.; Meister, B.; Sandin, J.; Kuzmin, A.; Stiedl, O.; Ogren, S.O. 5-Hydroxytryptamine 1A receptor blockade facilitates aversive learning in mice: Interactions with cholinergic and glutamatergic mechanisms. J. Pharmacol. Exp. Ther. 2006, 316, 581–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lueptow, L.M. Novel Object Recognition Test for the Investigation of Learning and Memory in Mice. J. Vis. Exp. 2017, 126, e55718. [Google Scholar] [CrossRef]
- Haberkorn, A.; Friis, C.W.; Schulz, H.P.; Meister, G.; Feller, W. Control of an outbreak of mouse coccidiosis in a closed colony. Lab. Anim. 1983, 17, 59–64. [Google Scholar] [CrossRef] [Green Version]
- Gjerde, B.; Helle, O. Efficacy of toltrazuril in the prevention of coccidiosis in naturally infected lambs on pasture. Acta Vet. Scand. 1986, 27, 124–137. [Google Scholar] [CrossRef]
- Mehlhorn, H.; Schmahl, G.; Haberkorn, A. Toltrazuril effective against a broad spectrum of protozoan parasites. Parasitol. Res. 1988, 75, 64–66. [Google Scholar] [CrossRef] [PubMed]
- Furr, M.; Kennedy, T.; MacKay, R.; Reed, S.; Andrews, F.; Bernard, B.; Bain, F.; Byars, D. Efficacy of ponazuril 15% oral paste as a treatment for equine protozoal myeloencephalitis. Vet. Ther. 2001, 2, 215–222. [Google Scholar] [PubMed]
- Kritzner, S.; Sager, H.; Blum, J.; Krebber, R.; Greif, G.; Gottstein, B. An explorative study to assess the efficacy of toltrazuril-sulfone (ponazuril) in calves experimentally infected with Neospora caninum. Ann. Clin. Microbiol. Antimicrob. 2002, 1, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Nordvall, G.; Forsell, P.; Sandin, J. Triazinetrione Derivatives and Their Use as Modulators of Neurotrophin Receptor and Receptor Tyrosine Kinases. U.S. Patent 16/471,923, 16 April 2020. [Google Scholar]
- Dirikolu, L.; Yohn, R.; Garrett, E.F.; Chakkath, T.; Ferguson, D.C. Detection, quantifications and pharmacokinetics of toltrazuril sulfone (Ponazuril) in cattle. J. Vet. Pharmacol. Ther. 2009, 32, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, T.; Campbell, J.; Selzer, V. Safety of ponazuril 15% oral paste in horses. Vet. Ther. 2001, 2, 223–231. [Google Scholar] [PubMed]
- Nordvall, G.; Forsell, P. Triazine Derivatives for Treating Diseases Relating to Neurotrophins and Their Preparation. WO2021038241A1, 4 March 2021. [Google Scholar]
- Nordvall, G.; Forsell, P. Triazine Derivatives for Treating Diseases Relating to Neurotrophins. WO2019162702A1, 29 August 2019. [Google Scholar]
- Nordvall, G.; Forsell, P. 4-Substituted Phenyl-1,3,5-triazine Derivatives as Modulators of Trk Receptors. WO2020002949A1, 2 January 2020. [Google Scholar]
- Nordvall, G.; Forsell, P. 4-Substituted Phenyl-1,3,5-Triazine Derivatives as Modulators of Trk Receptors. WO2020002950A1, 2 January 2020. [Google Scholar]
- Figurov, A.; Pozzo-Miller, L.D.; Olafsson, P.; Wang, T.; Lu, B. Regulation of synaptic responses to high-frequency stimulation and LTP by neurotrophins in the hippocampus. Nature 1996, 381, 706–709. [Google Scholar] [CrossRef]
- Ikegaya, Y.; Ishizaka, Y.; Matsuki, N. BDNF attenuates hippocampal LTD via activation of phospholipase C: Implications for a vertical shift in the frequency-response curve of synaptic plasticity. Eur. J. Neurosci. 2002, 16, 145–148. [Google Scholar] [CrossRef]
- Ji, Y.; Lu, Y.; Yang, F.; Shen, W.; Tang, T.T.; Feng, L.; Duan, S.; Lu, B. Acute and gradual increases in BDNF concentration elicit distinct signaling and functions in neurons. Nat. Neurosci. 2010, 13, 302–309. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Garcia-Ptacek, S.; Jönsson, L.; Anders, W.; Nordström, P.; Eriksdotter, M. Long Term Effects of Cholinesterase Inhibitors on Cognitive Decline and Mortality. Neurology 2021, 96, e2220–e2230. [Google Scholar] [CrossRef]
- Nordstrom, P.; Religa, D.; Wimo, A.; Winblad, B.; Eriksdotter, M. The use of cholinesterase inhibitors and the risk of myocardial infarction and death: A nationwide cohort study in subjects with Alzheimer’s disease. Eur. Heart J. 2013, 34, 2585–2591. [Google Scholar] [CrossRef]
- Tan, E.C.K.; Johnell, K.; Garcia-Ptacek, S.; Haaksma, M.L.; Fastbom, J.; Bell, J.S.; Eriksdotter, M. Acetylcholinesterase inhibitors and risk of stroke and death in people with dementia. Alzheimers Dement. 2018, 14, 944–951. [Google Scholar] [CrossRef] [PubMed]
- Isik, A.T.; Soysal, P.; Stubbs, B.; Solmi, M.; Basso, C.; Maggi, S.; Schofield, P.; Veronese, N.; Mueller, C. Cardiovascular Outcomes of Cholinesterase Inhibitors in Individuals with Dementia: A Meta-Analysis and Systematic Review. J. Am. Geriatr. Soc. 2018, 66, 1805–1811. [Google Scholar] [CrossRef]
- Blanco-Silvente, L.; Castells, X.; Garre-Olmo, J.; Vilalta-Franch, J.; Saez, M.; Barcelo, M.A.; Capella, D. Study of the strength of the evidence and the redundancy of the research on pharmacological treatment for Alzheimer’s disease: A cumulative meta-analysis and trial sequential analysis. Eur. J. Clin. Pharmacol. 2019, 75, 1659–1667. [Google Scholar] [CrossRef] [PubMed]
- Gold, M. Phase II clinical trials of anti-amyloid beta antibodies: When is enough, enough? Alzheimers Dement. 2017, 3, 402–409. [Google Scholar] [CrossRef]
- Counts, S.E.; Nadeem, M.; Wuu, J.; Ginsberg, S.D.; Saragovi, H.U.; Mufson, E.J. Reduction of cortical TrkA but not p75(NTR) protein in early-stage Alzheimer’s disease. Ann. Neurol. 2004, 56, 520–531. [Google Scholar] [CrossRef]
- Tiveron, C.; Fasulo, L.; Capsoni, S.; Malerba, F.; Marinelli, S.; Paoletti, F.; Piccinin, S.; Scardigli, R.; Amato, G.; Brandi, R.; et al. ProNGF\NGF imbalance triggers learning and memory deficits, neurodegeneration and spontaneous epileptic-like discharges in transgenic mice. Cell Death Differ. 2013, 20, 1017–1030. [Google Scholar] [CrossRef] [PubMed]
- Capsoni, S.; Brandi, R.; Arisi, I.; D’Onofrio, M.; Cattaneo, A. A dual mechanism linking NGF/proNGF imbalance and early inflammation to Alzheimer’s disease neurodegeneration in the AD11 anti-NGF mouse model. CNS Neurol. Disord. Drug Targets 2011, 10, 635–647. [Google Scholar] [CrossRef] [PubMed]
- Zou, M.; Guo, G.; Zhao, Y.; Zhang, Q. Detection, quantifications, and pharmacokinetics of ponazuril in healthy swine. J. Vet. Pharmacol. Ther. 2014, 37, 598–602. [Google Scholar] [CrossRef]
- Francis, P.T.; Parsons, C.G.; Jones, R.W. Rationale for combining glutamatergic and cholinergic approaches in the symptomatic treatment of Alzheimer’s disease. Expert Rev. Neurother. 2012, 12, 1351–1365. [Google Scholar] [CrossRef] [Green Version]
- Minichiello, L.; Korte, M.; Wolfer, D.; Kühn, R.; Unsicker, K.; Cestari, V.; Rossi-Arnaud, C.; Lipp, H.-P.; Bonhoeffer, T.; Klein, R. Essential Role for TrkB Receptors in Hippocampus-Mediated Learning. Neuron 1999, 24, 401–414. [Google Scholar] [CrossRef] [Green Version]
- Alonso, M.; Vianna, M.R.M.; Depino, A.M.; Mello e Souza, T.; Pereira, P.; Szapiro, G.; Viola, H.; Pitossi, F.; Izquierdo, I.; Medina, J.H. BDNF–triggered events in the rat hippocampus are required for both short- and long-term memory formation. Hippocampus 2002, 12, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Gooney, M.; Shaw, K.; Kelly, A.; O’Mara, S.; Lynch, M. Long-Term Potentiation and Spatial Learning Are Associated with Increased Phosphorylation of TrkB and Extracellular Signal-Regulated Kinase (ERK) in the Dentate Gyrus: Evidence for a Role for Brain-Derived Neurotrophic Factor. Behav. Neurosci. 2002, 116, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Linnarsson, S.; Björklund, A.; Ernfors, P. Learning Deficit in BDNF Mutant Mice. Eur. J. Neurosci. 1997, 9, 2581–2587. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TrkA | TrkB | TrkC | IGF1R | FGFR1 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| EC50 | Effect | EC50 | Effect | EC50 | Effect | EC50 | Effect | EC50 | Effect | |
| ACD855 | 1.9 | 144% | 3.2 | 319% | 0.8 | 56% | 0.9 | 25% | 0.8 | 44% |
| ACD856 | 0.38 | 185% | 0.29 | 176% | 0.33 | 328% | 0.25 | 104% | 0.19 | 117% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dahlström, M.; Madjid, N.; Nordvall, G.; Halldin, M.M.; Vazquez-Juarez, E.; Lindskog, M.; Sandin, J.; Winblad, B.; Eriksdotter, M.; Forsell, P. Identification of Novel Positive Allosteric Modulators of Neurotrophin Receptors for the Treatment of Cognitive Dysfunction. Cells 2021, 10, 1871. https://doi.org/10.3390/cells10081871
Dahlström M, Madjid N, Nordvall G, Halldin MM, Vazquez-Juarez E, Lindskog M, Sandin J, Winblad B, Eriksdotter M, Forsell P. Identification of Novel Positive Allosteric Modulators of Neurotrophin Receptors for the Treatment of Cognitive Dysfunction. Cells. 2021; 10(8):1871. https://doi.org/10.3390/cells10081871
Chicago/Turabian StyleDahlström, Märta, Nather Madjid, Gunnar Nordvall, Magnus M. Halldin, Erika Vazquez-Juarez, Maria Lindskog, Johan Sandin, Bengt Winblad, Maria Eriksdotter, and Pontus Forsell. 2021. "Identification of Novel Positive Allosteric Modulators of Neurotrophin Receptors for the Treatment of Cognitive Dysfunction" Cells 10, no. 8: 1871. https://doi.org/10.3390/cells10081871