
Role of Nrf2 in Synaptic Plasticity and Memory in Alzheimer’s Disease



{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Nrf2 and its Role in Alzheimer’s Disease
3. Synaptic Plasticity and Reactive Oxygen Species
4. Memory and Reactive Oxygen Species
5. The Role of Nrf2 in Synapse Plasticity
6. The Role of Nrf2 in Memory
7. The Role and Regulation of NF-κB
8. Cross-Talk between the NF-ĸB and Nrf2 Signaling Pathways
9. Anti-Inflammatory Role of the Nrf2/HO-1 Signaling Pathway
10. Nrf2 Activating Compounds
11. Nrf2 and NF-κB Crosstalk with Other Transcription Factors
12. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Adlimoghaddam, A.; Roy, B.; Albensi, B.C. Future Trends and the Economic Burden of Dementia in Manitoba: Comparison with the Rest of Canada and the World. Neuroepidemiology 2018, 51, 71–81. [Google Scholar] [CrossRef]
- Adlimoghaddam, A.; Neuendorff, M.; Roy, B.; Albensi, B.C. A review of clinical treatment considerations of donepezil in severe Alzheimer’s disease. CNS Neurosci. Ther. 2018, 24, 876–888. [Google Scholar] [CrossRef] [Green Version]
- Glabe, C.C. Amyloid accumulation and pathogensis of Alzheimer’s disease: Significance of monomeric, oligomeric and fibrillar Aβ. Alzhaimer’s Dis. 2005, 38, 167–177. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [Green Version]
- Muralidar, S.; Ambi, S.V.; Sekaran, S.; Thirumalai, D.; Palaniappan, B. Role of tau protein in Alzheimer’s disease: The prime pathological player. Int. J. Biol. Macromol. 2020. [Google Scholar] [CrossRef]
- Adlimoghaddam, A.; Snow, W.M.; Stortz, G.; Perez, C.; Djordjevic, J.; Goertzen, A.L.; Ko, J.H.; Albensi, B.C. Regional hypometabolism in the 3× Tg mouse model of Alzheimer’s disease. Neurobiol. Dis. 2019, 127, 264–277. [Google Scholar] [CrossRef]
- Ellis, R.J.; Olichney, J.M.; Thal, L.J.; Mirra, S.S.; Morris, J.C.; Beekly, D.; Heyman, A. Cerebral amyloid angiopathy in the brains of patients with Alzheimer’s disease: The CERAD experience, Part XV. Neurology 1996, 46, 1592–1596. [Google Scholar] [CrossRef] [PubMed]
- Price, J.L.; Ko, A.I.; Wade, M.J.; Tsou, S.K.; McKeel, D.W.; Morris, J.C. Neuron number in the entorhinal cortex and CA1 in preclinical Alzheimer disease. Arch. Neurol. 2001, 58, 1395–1402. [Google Scholar] [CrossRef]
- Qu, Z.; Sun, J.; Zhang, W.; Yu, J.; Zhuang, C. Transcription factor NRF2 as a promising therapeutic target for Alzheimer’s disease. Free Radic. Biol. Med. 2020, 159, 87–102. [Google Scholar] [CrossRef] [PubMed]
- Morroni, F.; Sita, G.; Graziosi, A.; Turrini, E.; Fimognari, C.; Tarozzi, A.; Hrelia, P. Neuroprotective Effect of Caffeic Acid Phenethyl Ester in A Mouse Model of Alzheimer’s Disease Involves Nrf2/HO-1 Pathway. Aging Dis. 2018, 9, 605–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abramov, A.Y.; Potapova, E.V.; Dremin, V.V.; Dunaev, A.V. Interaction of Oxidative Stress and Misfolded Proteins in the Mechanism of Neurodegeneration. Life 2020, 10, 101. [Google Scholar] [CrossRef]
- Massaad, C.A.; Klann, E. Reactive oxygen species in the regulation of synaptic plasticity and memory. Antioxid. Redox Signal. 2011, 14, 2013–2054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, R.X.; Correia, S.C.; Zhu, X.; Lee, H.G.; Petersen, R.B.; Nunomura, A.; Smith, M.A.; Perry, G.; Moreira, P.I. Nuclear and mitochondrial DNA oxidation in Alzheimer’s disease. Free Radic. Res. 2012, 46, 565–576. [Google Scholar] [CrossRef]
- Beal, M.F. Oxidatively modified proteins in aging and disease. Free Radic. Biol. Med. 2002, 32, 797–803. [Google Scholar] [CrossRef]
- Jiang, T.; Sun, Q.; Chen, S. Oxidative stress: A major pathogenesis and potential therapeutic target of antioxidative agents in Parkinson’s disease and Alzheimer’s disease. Prog. Neurobiol. 2016, 147, 1–19. [Google Scholar] [CrossRef] [PubMed]
- White, A.R.; Du, T.; Laughton, K.M.; Volitakis, I.; Sharples, R.A.; Xilinas, M.E.; Hoke, D.E.; Holsinger, R.M.; Evin, G.; Cherny, R.A.; et al. Degradation of the Alzheimer disease amyloid beta-peptide by metal-dependent up-regulation of metalloprotease activity. J. Biol. Chem. 2006, 281, 17670–17680. [Google Scholar] [CrossRef] [Green Version]
- Poprac, P.; Jomova, K.; Simunkova, M.; Kollar, V.; Rhodes, C.J.; Valko, M. Targeting Free Radicals in Oxidative Stress-Related Human Diseases. Trends Pharmacol. Sci. 2017, 38, 592–607. [Google Scholar] [CrossRef]
- Cullinan, S.B.; Gordan, J.D.; Jin, J.; Harper, J.W.; Diehl, J.A. The Keap1-BTB protein is an adaptor that bridges Nrf2 to a Cul3-based E3 ligase: Oxidative stress sensing by a Cul3-Keap1 ligase. Mol. Cell. Biol. 2004, 24, 8477–8486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, A.; Kang, M.I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell. Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.D.; Lo, S.C.; Cross, J.V.; Templeton, D.J.; Hannink, M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol. Cell. Biol. 2004, 24, 10941–10953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furukawa, M.; Xiong, Y. BTB protein Keap1 targets antioxidant transcription factor Nrf2 for ubiquitination by the Cullin 3-Roc1 ligase. Mol. Cell. Biol. 2005, 25, 162–171. [Google Scholar] [CrossRef] [Green Version]
- Wakabayashi, N.; Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Kang, M.I.; Kobayashi, A.; Yamamoto, M.; Kensler, T.W.; Talalay, P. Protection against electrophile and oxidant stress by induction of the phase 2 response: Fate of cysteines of the Keap1 sensor modified by inducers. Proc. Natl. Acad. Sci. USA 2004, 101, 2040–2045. [Google Scholar] [CrossRef] [Green Version]
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Cole, R.N.; Itoh, K.; Wakabayashi, N.; Katoh, Y.; Yamamoto, M.; Talalay, P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl. Acad. Sci. USA 2002, 99, 11908–11913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Wakabayashi, N. Keap1, the sensor for electrophiles and oxidants that regulates the phase 2 response, is a zinc metalloprotein. Biochemistry 2005, 44, 6889–6899. [Google Scholar] [CrossRef]
- Eggler, A.L.; Liu, G.; Pezzuto, J.M.; van Breemen, R.B.; Mesecar, A.D. Modifying specific cysteines of the electrophile-sensing human Keap1 protein is insufficient to disrupt binding to the Nrf2 domain Neh2. Proc. Natl. Acad. Sci. USA 2005, 102, 10070–10075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, K.I.; Kobayashi, A.; Katsuoka, F.; Yamamoto, M. Two-site substrate recognition model for the Keap1-Nrf2 system: A hinge and latch mechanism. Biol. Chem. 2006, 387, 1311–1320. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Kang, M.I.; Watai, Y.; Tong, K.I.; Shibata, T.; Uchida, K.; Yamamoto, M. Oxidative and electrophilic stresses activate Nrf2 through inhibition of ubiquitination activity of Keap1. Mol. Cell. Biol. 2006, 26, 221–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidlin, C.J.; Dodson, M.B.; Madhavan, L.; Zhang, D.D. Redox regulation by NRF2 in aging and disease. Free Radic. Biol. Med. 2019, 134, 702–707. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Lauderback, C.M. Lipid peroxidation and protein oxidation in Alzheimer’s disease brain: Potential causes and consequences involving amyloid beta-peptide-associated free radical oxidative stress. Free Radic. Biol. Med. 2002, 32, 1050–1060. [Google Scholar] [CrossRef]
- Hensley, K.; Hall, N.; Subramaniam, R.; Cole, P.; Harris, M.; Aksenov, M.; Aksenova, M.; Gabbita, S.P.; Wu, J.F.; Carney, J.M.; et al. Brain regional correspondence between Alzheimer’s disease histopathology and biomarkers of protein oxidation. J. Neurochem. 1995, 65, 2146–2156. [Google Scholar] [CrossRef]
- Liu, Z.; Zhou, T.; Ziegler, A.C.; Dimitrion, P.; Zuo, L. Oxidative Stress in Neurodegenerative Diseases: From Molecular Mechanisms to Clinical Applications. Oxid. Med. Cell. Longev. 2017, 2017, 2525967. [Google Scholar] [CrossRef]
- Ferreiro, E.; Oliveira, C.R.; Pereira, C.M. The release of calcium from the endoplasmic reticulum induced by amyloid-beta and prion peptides activates the mitochondrial apoptotic pathway. Neurobiol. Dis. 2008, 30, 331–342. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, T.; Lipton, S.A. Redox modulation by S-nitrosylation contributes to protein misfolding, mitochondrial dynamics, and neuronal synaptic damage in neurodegenerative diseases. Cell Death Differ. 2011, 18, 1478–1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, T.; Lipton, S.A. Preventing Ca2+-mediated nitrosative stress in neurodegenerative diseases: Possible pharmacological strategies. Cell Calcium 2010, 47, 190–197. [Google Scholar] [CrossRef] [Green Version]
- Shelat, P.B.; Chalimoniuk, M.; Wang, J.H.; Strosznajder, J.B.; Lee, J.C.; Sun, A.Y.; Simonyi, A.; Sun, G.Y. Amyloid beta peptide and NMDA induce ROS from NADPH oxidase and AA release from cytosolic phospholipase A2 in cortical neurons. J. Neurochem. 2008, 106, 45–55. [Google Scholar] [CrossRef]
- Ramsey, C.P.; Glass, C.A.; Montgomery, M.B.; Lindl, K.A.; Ritson, G.P.; Chia, L.A.; Hamilton, R.L.; Chu, C.T.; Jordan-Sciutto, K.L. Expression of Nrf2 in neurodegenerative diseases. J. Neuropathol. Exp. Neurol. 2007, 66, 75–85. [Google Scholar] [CrossRef]
- Stack, C.; Jainuddin, S.; Elipenahli, C.; Gerges, M.; Starkova, N.; Starkov, A.A.; Jove, M.; Portero-Otin, M.; Launay, N.; Pujol, A.; et al. Methylene blue upregulates Nrf2/ARE genes and prevents tau-related neurotoxicity. Hum. Mol. Genet. 2014, 23, 3716–3732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Cabrera, M.C.; Salvador-Pascual, A.; Cabo, H.; Ferrando, B.; Vina, J. Redox modulation of mitochondriogenesis in exercise. Does antioxidant supplementation blunt the benefits of exercise training? Free Radic. Biol. Med. 2015, 86, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Santos, A.L.; Sinha, S.; Lindner, A.B. The Good, the Bad, and the Ugly of ROS: New Insights on Aging and Aging-Related Diseases from Eukaryotic and Prokaryotic Model Organisms. Oxid. Med. Cell. Longev. 2018, 2018, 1941285. [Google Scholar] [CrossRef] [PubMed]
- Abate, G.; Vezzoli, M.; Sandri, M.; Rungratanawanich, W.; Memo, M.; Uberti, D. Mitochondria and cellular redox state on the route from ageing to Alzheimer’s disease. Mech. Ageing Dev. 2020, 192, 111385. [Google Scholar] [CrossRef]
- Antunes, F.; Brito, P.M. Quantitative biology of hydrogen peroxide signaling. Redox Biol. 2017, 13, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Cheyne, J.E.; Montgomery, J.M. The cellular and molecular basis of in vivo synaptic plasticity in rodents. Am. J. Physiol. Cell Physiol. 2020, 318, C1264–C1283. [Google Scholar] [CrossRef]
- Bindokas, V.P.; Jordan, J.; Lee, C.C.; Miller, R.J. Superoxide production in rat hippocampal neurons: Selective imaging with hydroethidine. J. Neurosci. 1996, 16, 1324–1336. [Google Scholar] [CrossRef] [PubMed]
- Kanterewicz, B.I.; Knapp, L.T.; Klann, E. Stimulation of p42 and p44 mitogen-activated protein kinases by reactive oxygen species and nitric oxide in hippocampus. J. Neurochem. 1998, 70, 1009–1016. [Google Scholar] [CrossRef] [PubMed]
- Knapp, L.T.; Klann, E. Role of reactive oxygen species in hippocampal long-term potentiation: Contributory or inhibitory? J. Neurosci. Res. 2002, 70, 1–7. [Google Scholar] [CrossRef]
- Kishida, K.T.; Hoeffer, C.A.; Hu, D.; Pao, M.; Holland, S.M.; Klann, E. Synaptic plasticity deficits and mild memory impairments in mouse models of chronic granulomatous disease. Mol. Cell. Biol. 2006, 26, 5908–5920. [Google Scholar] [CrossRef] [Green Version]
- Kamsler, A.; Segal, M. Paradoxical actions of hydrogen peroxide on long-term potentiation in transgenic superoxide dismutase-1 mice. J. Neurosci. 2003, 23, 10359–10367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiels, E.; Urban, N.N.; Gonzalez-Burgos, G.R.; Kanterewicz, B.I.; Barrionuevo, G.; Chu, C.T.; Oury, T.D.; Klann, E. Impairment of long-term potentiation and associative memory in mice that overexpress extracellular superoxide dismutase. J. Neurosci. 2000, 20, 7631–7639. [Google Scholar] [CrossRef] [Green Version]
- Betteridge, D.J. What is oxidative stress? Metabolism 2000, 49, 3–8. [Google Scholar] [CrossRef]
- Jones, D.P. Redefining oxidative stress. Antioxid. Redox Signal. 2006, 8, 1865–1879. [Google Scholar] [CrossRef] [PubMed]
- Tsunekawa, H.; Noda, Y.; Mouri, A.; Yoneda, F.; Nabeshima, T. Synergistic effects of selegiline and donepezil on cognitive impairment induced by amyloid beta (25–35). Behav. Brain Res. 2008, 190, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Bienert, G.P.; Moller, A.L.; Kristiansen, K.A.; Schulz, A.; Moller, I.M.; Schjoerring, J.K.; Jahn, T.P. Specific aquaporins facilitate the diffusion of hydrogen peroxide across membranes. J. Biol. Chem. 2007, 282, 1183–1192. [Google Scholar] [CrossRef] [Green Version]
- Szu, J.I.; Binder, D.K. The Role of Astrocytic Aquaporin-4 in Synaptic Plasticity and Learning and Memory. Front. Integr. Neurosci. 2016, 10, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsuki, H.; Nakanishi, C.; Saito, H.; Matsuki, N. Biphasic effect of hydrogen peroxide on field potentials in rat hippocampal slices. Eur. J. Pharmacol. 1997, 337, 213–218. [Google Scholar] [CrossRef]
- Lin, H.H.; Chen, C.H.; Hsieh, W.K.; Chiu, T.H.; Lai, C.C. Hydrogen peroxide increases the activity of rat sympathetic preganglionic neurons in vivo and in vitro. Neuroscience 2003, 121, 641–647. [Google Scholar] [CrossRef]
- Kamsler, A.; Segal, M. Hydrogen peroxide modulation of synaptic plasticity. J. Neurosci. 2003, 23, 269–276. [Google Scholar] [CrossRef]
- Gahtan, E.; Auerbach, J.M.; Groner, Y.; Segal, M. Reversible impairment of long-term potentiation in transgenic Cu/Zn-SOD mice. Eur. J. Neurosci. 1998, 10, 538–544. [Google Scholar] [CrossRef]
- Levin, E.D.; Christopher, N.C.; Crapo, J.D. Memory decline of aging reduced by extracellular superoxide dismutase overexpression. Behav. Genet. 2005, 35, 447–453. [Google Scholar] [CrossRef]
- Hu, D.; Cao, P.; Thiels, E.; Chu, C.T.; Wu, G.Y.; Oury, T.D.; Klann, E. Hippocampal long-term potentiation, memory, and longevity in mice that overexpress mitochondrial superoxide dismutase. Neurobiol. Learn. Mem. 2007, 87, 372–384. [Google Scholar] [CrossRef] [Green Version]
- Robledinos-Anton, N.; Rojo, A.I.; Ferreiro, E.; Nunez, A.; Krause, K.H.; Jaquet, V.; Cuadrado, A. Transcription factor NRF2 controls the fate of neural stem cells in the subgranular zone of the hippocampus. Redox Biol. 2017, 13, 393–401. [Google Scholar] [CrossRef]
- Rojo, A.I.; Pajares, M.; Rada, P.; Nunez, A.; Nevado-Holgado, A.J.; Killik, R.; Van Leuven, F.; Ribe, E.; Lovestone, S.; Yamamoto, M.; et al. NRF2 deficiency replicates transcriptomic changes in Alzheimer’s patients and worsens APP and TAU pathology. Redox Biol. 2017, 13, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Tarantini, S.; Valcarcel-Ares, M.N.; Yabluchanskiy, A.; Tucsek, Z.; Hertelendy, P.; Kiss, T.; Gautam, T.; Zhang, X.A.; Sonntag, W.E.; de Cabo, R.; et al. Nrf2 Deficiency Exacerbates Obesity-Induced Oxidative Stress, Neurovascular Dysfunction, Blood-Brain Barrier Disruption, Neuroinflammation, Amyloidogenic Gene Expression, and Cognitive Decline in Mice, Mimicking the Aging Phenotype. J. Gerontol. A 2018, 73, 853–863. [Google Scholar] [CrossRef]
- Zweig, J.A.; Caruso, M.; Brandes, M.S.; Gray, N.E. Loss of NRF2 leads to impaired mitochondrial function, decreased synaptic density and exacerbated age-related cognitive deficits. Exp. Gerontol. 2020, 131, 110767. [Google Scholar] [CrossRef] [PubMed]
- Jo, J.H.; Park, E.J.; Lee, J.K.; Jung, M.W.; Lee, C.J. Lipopolysaccharide inhibits induction of long-term potentiation and depression in the rat hippocampal CA1 area. Eur. J. Pharmacol. 2001, 422, 69–76. [Google Scholar] [CrossRef]
- Paraiso, H.C.; Kuo, P.C.; Curfman, E.T.; Moon, H.J.; Sweazey, R.D.; Yen, J.H.; Chang, F.L.; Yu, I.C. Dimethyl fumarate attenuates reactive microglia and long-term memory deficits following systemic immune challenge. J. Neuroinflamm. 2018, 15, 100. [Google Scholar] [CrossRef]
- Khajevand-Khazaei, M.R.; Ziaee, P.; Motevalizadeh, S.A.; Rohani, M.; Afshin-Majd, S.; Baluchnejadmojarad, T.; Roghani, M. Naringenin ameliorates learning and memory impairment following systemic lipopolysaccharide challenge in the rat. Eur. J. Pharmacol. 2018, 826, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Wang, Z.Y.; Xie, J.W.; Wang, T.; Wang, X.; Xu, Y.; Cai, J.H. Dl-3-n-butylphthalide-induced upregulation of antioxidant defense is involved in the enhancement of cross talk between CREB and Nrf2 in an Alzheimer’s disease mouse model. Neurobiol. Aging 2016, 38, 32–46. [Google Scholar] [CrossRef]
- Xu, P.; Wang, K.; Lu, C.; Dong, L.; Gao, L.; Yan, M.; Aibai, S.; Yang, Y.; Liu, X. The Protective Effect of Lavender Essential Oil and Its Main Component Linalool against the Cognitive Deficits Induced by D-Galactose and Aluminum Trichloride in Mice. Evid. Based Complement. Altern. Med. 2017, 2017, 7426538. [Google Scholar] [CrossRef]
- Lu, Y.; Christian, K.; Lu, B. BDNF: A key regulator for protein synthesis-dependent LTP and long-term memory? Neurobiol. Learn. Mem. 2008, 89, 312–323. [Google Scholar] [CrossRef] [Green Version]
- Aicardi, G.; Argilli, E.; Cappello, S.; Santi, S.; Riccio, M.; Thoenen, H.; Canossa, M. Induction of long-term potentiation and depression is reflected by corresponding changes in secretion of endogenous brain-derived neurotrophic factor. Proc. Natl. Acad. Sci. USA 2004, 101, 15788–15792. [Google Scholar] [CrossRef] [Green Version]
- Balkowiec, A.; Katz, D.M. Cellular mechanisms regulating activity-dependent release of native brain-derived neurotrophic factor from hippocampal neurons. J. Neurosci. 2002, 22, 10399–10407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gartner, A.; Staiger, V. Neurotrophin secretion from hippocampal neurons evoked by long-term-potentiation-inducing electrical stimulation patterns. Proc. Natl. Acad. Sci. USA 2002, 99, 6386–6391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartmann, M.; Heumann, R.; Lessmann, V. Synaptic secretion of BDNF after high-frequency stimulation of glutamatergic synapses. EMBO J. 2001, 20, 5887–5897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lever, I.J.; Bradbury, E.J.; Cunningham, J.R.; Adelson, D.W.; Jones, M.G.; McMahon, S.B.; Marvizon, J.C.; Malcangio, M. Brain-derived neurotrophic factor is released in the dorsal horn by distinctive patterns of afferent fiber stimulation. J. Neurosci. 2001, 21, 4469–4477. [Google Scholar] [CrossRef]
- Bouvier, E.; Brouillard, F.; Molet, J.; Claverie, D.; Cabungcal, J.H.; Cresto, N.; Doligez, N.; Rivat, C.; Do, K.Q.; Bernard, C.; et al. Nrf2-dependent persistent oxidative stress results in stress-induced vulnerability to depression. Mol. Psychiatry 2017, 22, 1795. [Google Scholar] [CrossRef] [Green Version]
- Sunkaria, A.; Bhardwaj, S.; Yadav, A.; Halder, A.; Sandhir, R. Sulforaphane attenuates postnatal proteasome inhibition and improves spatial learning in adult mice. J. Nutr. Biochem. 2018, 51, 69–79. [Google Scholar] [CrossRef]
- Kubo, E.; Chhunchha, B.; Singh, P.; Sasaki, H.; Singh, D.P. Sulforaphane reactivates cellular antioxidant defense by inducing Nrf2/ARE/Prdx6 activity during aging and oxidative stress. Sci. Rep. 2017, 7, 14130. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Lee, S.; Choi, B.R.; Yang, H.; Hwang, Y.; Park, J.H.; LaFerla, F.M.; Han, J.S.; Lee, K.W.; Kim, J. Sulforaphane epigenetically enhances neuronal BDNF expression and TrkB signaling pathways. Mol. Nutr. Food Res. 2017, 61. [Google Scholar] [CrossRef]
- Spagnuolo, M.S.; Bergamo, P.; Crescenzo, R.; Iannotta, L.; Treppiccione, L.; Iossa, S.; Cigliano, L. Brain Nrf2 pathway, autophagy, and synaptic function proteins are modulated by a short-term fructose feeding in young and adult rats. Nutr. Neurosci. 2020, 23, 309–320. [Google Scholar] [CrossRef]
- Habas, A.; Hahn, J.; Wang, X.; Margeta, M. Neuronal activity regulates astrocytic Nrf2 signaling. Proc. Natl. Acad. Sci. USA 2013, 110, 18291–18296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abou El-Ezz, D.; Maher, A.; Sallam, N.; El-Brairy, A.; Kenawy, S. Trans-cinnamaldehyde Modulates Hippocampal Nrf2 Factor and Inhibits Amyloid Beta Aggregation in LPS-Induced Neuroinflammation Mouse Model. Neurochem. Res. 2018, 43, 2333–2342. [Google Scholar] [CrossRef] [PubMed]
- Derave, W.; Everaert, I.; Beeckman, S.; Baguet, A. Muscle carnosine metabolism and beta-alanine supplementation in relation to exercise and training. Sports Med. 2010, 40, 247–263. [Google Scholar] [CrossRef] [Green Version]
- Ahshin-Majd, S.; Zamani, S.; Kiamari, T.; Kiasalari, Z.; Baluchnejadmojarad, T.; Roghani, M. Carnosine ameliorates cognitive deficits in streptozotocin-induced diabetic rats: Possible involved mechanisms. Peptides 2016, 86, 102–111. [Google Scholar] [CrossRef]
- Ali, T.; Kim, T.; Rehman, S.U.; Khan, M.S.; Amin, F.U.; Khan, M.; Ikram, M.; Kim, M.O. Natural Dietary Supplementation of Anthocyanins via PI3K/Akt/Nrf2/HO-1 Pathways Mitigate Oxidative Stress, Neurodegeneration, and Memory Impairment in a Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2018, 55, 6076–6093. [Google Scholar] [CrossRef] [PubMed]
- Kiasalari, Z.; Heydarifard, R.; Khalili, M.; Afshin-Majd, S.; Baluchnejadmojarad, T.; Zahedi, E.; Sanaierad, A.; Roghani, M. Ellagic acid ameliorates learning and memory deficits in a rat model of Alzheimer’s disease: An exploration of underlying mechanisms. Psychopharmacology 2017, 234, 1841–1852. [Google Scholar] [CrossRef]
- Song, J.; Hur, B.E.; Bokara, K.K.; Yang, W.; Cho, H.J.; Park, K.A.; Lee, W.T.; Lee, K.M.; Lee, J.E. Agmatine improves cognitive dysfunction and prevents cell death in a streptozotocin-induced Alzheimer rat model. Yonsei Med. J. 2014, 55, 689–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saeed, K.; Shah, S.A.; Ullah, R.; Alam, S.I.; Park, J.S.; Saleem, S.; Jo, M.H.; Kim, M.W.; Hahm, J.R.; Kim, M.O. Quinovic Acid Impedes Cholesterol Dyshomeostasis, Oxidative Stress, and Neurodegeneration in an Amyloid-beta-Induced Mouse Model. Oxid. Med. Cell. Longev. 2020, 2020, 9523758. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Huang, J.; Chen, Y.; Shang, H.; Zhang, W.; Yu, J.; He, L.; Xing, C.; Zhuang, C. Direct inhibition of Keap1-Nrf2 Protein-Protein interaction as a potential therapeutic strategy for Alzheimer’s disease. Bioorg. Chem. 2020, 103, 104172. [Google Scholar] [CrossRef]
- Ren, P.; Chen, J.; Li, B.; Zhang, M.; Yang, B.; Guo, X.; Chen, Z.; Cheng, H.; Wang, P.; Wang, S.; et al. Nrf2 Ablation Promotes Alzheimer’s Disease-Like Pathology in APP/PS1 Transgenic Mice: The Role of Neuroinflammation and Oxidative Stress. Oxid. Med. Cell. Longev. 2020, 2020, 3050971. [Google Scholar] [CrossRef]
- Branca, C.; Ferreira, E.; Nguyen, T.V.; Doyle, K.; Caccamo, A.; Oddo, S. Genetic reduction of Nrf2 exacerbates cognitive deficits in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2017, 26, 4823–4835. [Google Scholar] [CrossRef]
- Dong, Y.; Stewart, T.; Bai, L.; Li, X.; Xu, T.; Iliff, J.; Shi, M.; Zheng, D.; Yuan, L.; Wei, T.; et al. Coniferaldehyde attenuates Alzheimer’s pathology via activation of Nrf2 and its targets. Theranostics 2020, 10, 179–200. [Google Scholar] [CrossRef]
- Ding, Y.; Bao, X.; Lao, L.; Ling, Y.; Wang, Q.; Xu, S. p-Hydroxybenzyl Alcohol Prevents Memory Deficits by Increasing Neurotrophic Factors and Decreasing Inflammatory Factors in a Mice Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2019, 67, 1007–1019. [Google Scholar] [CrossRef]
- Jiao, W.; Wang, Y.; Kong, L.; Ou-Yang, T.; Meng, Q.; Fu, Q.; Hu, Z. CART peptide activates the Nrf2/HO-1 antioxidant pathway and protects hippocampal neurons in a rat model of Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2018, 501, 1016–1022. [Google Scholar] [CrossRef]
- Chen, L.; Shi, R.; She, X.; Gu, C.; Chong, L.; Zhang, L.; Li, R. Mineralocorticoid receptor antagonist-mediated cognitive improvement in a mouse model of Alzheimer’s type: Possible involvement of BDNF-H2 S-Nrf2 signaling. Fundam. Clin. Pharmacol. 2020, 34, 697–707. [Google Scholar] [CrossRef]
- Power, J.H.; Asad, S.; Chataway, T.K.; Chegini, F.; Manavis, J.; Temlett, J.A.; Jensen, P.H.; Blumbergs, P.C.; Gai, W.P. Peroxiredoxin 6 in human brain: Molecular forms, cellular distribution and association with Alzheimer’s disease pathology. Acta Neuropathol. 2008, 115, 611–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, H.M.; Jin, P.; Han, J.Y.; Lee, M.S.; Han, S.B.; Oh, K.W.; Hong, S.H.; Jung, E.Y.; Hong, J.T. Acceleration of the development of Alzheimer’s disease in amyloid beta-infused peroxiredoxin 6 overexpression transgenic mice. Mol. Neurobiol. 2013, 48, 941–951. [Google Scholar] [CrossRef] [PubMed]
- Leiros, M.; Alonso, E.; Rateb, M.E.; Houssen, W.E.; Ebel, R.; Jaspars, M.; Alfonso, A.; Botana, L.M. Gracilins: Spongionella-derived promising compounds for Alzheimer disease. Neuropharmacology 2015, 93, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tian, Q.; Li, Z.; Dang, M.; Lin, Y.; Hou, X. Activation of Nrf2 signaling by sitagliptin and quercetin combination against beta-amyloid induced Alzheimer’s disease in rats. Drug Dev. Res. 2019, 80, 837–845. [Google Scholar] [CrossRef]
- Nakhate, K.T.; Bharne, A.P.; Verma, V.S.; Aru, D.N.; Kokare, D.M. Plumbagin ameliorates memory dysfunction in streptozotocin induced Alzheimer’s disease via activation of Nrf2/ARE pathway and inhibition of beta-secretase. Biomed. Pharmacother. 2018, 101, 379–390. [Google Scholar] [CrossRef]
- Han, Y.; Nan, S.; Fan, J.; Chen, Q.; Zhang, Y. Inonotus obliquus polysaccharides protect against Alzheimer’s disease by regulating Nrf2 signaling and exerting antioxidative and antiapoptotic effects. Int. J. Biol. Macromol. 2019, 131, 769–778. [Google Scholar] [CrossRef]
- Ma, B.; Lucas, B.; Capacci, A.; Lin, E.Y.; Jones, J.H.; Dechantsreiter, M.; Enyedy, I.; Marcotte, D.; Xiao, G.; Li, B.; et al. Design, synthesis and identification of novel, orally bioavailable non-covalent Nrf2 activators. Bioorg. Med. Chem. Lett. 2020, 30, 126852. [Google Scholar] [CrossRef]
- Weissig, V. Drug Development for the Therapy of Mitochondrial Diseases. Trends Mol. Med. 2020, 26, 40–57. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhou, Q.; Zhou, C.L. RTA-408 protects against propofol-induced cognitive impairment in neonatal mice via the activation of Nrf2 and the inhibition of NF-kappaB p65 nuclear translocation. Brain Behav. 2021, 11, e01918. [Google Scholar] [CrossRef]
- Tsai, T.H.; Lin, S.H.; Wu, C.H.; Tsai, Y.C.; Yang, S.F.; Lin, C.L. Mechanisms and therapeutic implications of RTA 408, an activator of Nrf2, in subarachnoid hemorrhage-induced delayed cerebral vasospasm and secondary brain injury. PLoS ONE 2020, 15, e0240122. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.J.; Pareek, T.K.; Liu, Q.; Letterio, J.J. A unique tolerizing dendritic cell phenotype induced by the synthetic triterpenoid CDDO-DFPA (RTA-408) is protective against EAE. Sci. Rep. 2017, 7, 9886. [Google Scholar] [CrossRef] [Green Version]
- Han, P.; Qin, Z.; Tang, J.; Xu, Z.; Li, R.; Jiang, X.; Yang, C.; Xing, Q.; Qi, X.; Tang, M.; et al. RTA-408 Protects Kidney from Ischemia-Reperfusion Injury in Mice via Activating Nrf2 and Downstream GSH Biosynthesis Gene. Oxid. Med. Cell. Longev. 2017, 2017, 7612182. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Suo, C.J.; Ruan, Y.S.; Tan, R.Y.; Zhang, W.; Niu, T.L. Effect and related mechanisms of RTA-408 on rat vascular smooth muscle cell calcification induced by advanced glycation end products. Zhonghua Xin Xue Guan Bing Za Zhi 2018, 46, 475–479. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Xie, Z.; Hu, B.; Zhang, B.; Ma, Y.; Pan, X.; Huang, H.; Wang, J.; Zhao, X.; Jie, Z.; et al. The Nrf2 activator RTA-408 attenuates osteoclastogenesis by inhibiting STING dependent NF-kappab signaling. Redox Biol. 2020, 28, 101309. [Google Scholar] [CrossRef]
- Esmaeilzade, B.; Artimani, T.; Amiri, I.; Najafi, R.; Shahidi, S.; Sabec, M.; Farzadinia, P.; Zare, M.; Zahiri, M.; Soleimani Asl, S. Dimethyloxalylglycine preconditioning enhances protective effects of bone marrow-derived mesenchymal stem cells in Aβ-induced Alzheimer disease. Physiol. Behav. 2019, 199, 265–272. [Google Scholar] [CrossRef]
- Tian, Y.; Wang, W.; Xu, L.; Li, H.; Wei, Y.; Wu, Q.; Jia, J. Activation of Nrf2/ARE pathway alleviates the cognitive deficits in PS1V97L-Tg mouse model of Alzheimer’s disease through modulation of oxidative stress. J. Neurosci. Res. 2019, 97, 492–505. [Google Scholar] [CrossRef]
- Chang, W.H.; Chen, M.C.; Cheng, I.H. Antroquinonol Lowers Brain Amyloid-beta Levels and Improves Spatial Learning and Memory in a Transgenic Mouse Model of Alzheimer’s Disease. Sci. Rep. 2015, 5, 15067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanninen, K.; Heikkinen, R.; Malm, T.; Rolova, T.; Kuhmonen, S.; Leinonen, H.; Yla-Herttuala, S.; Tanila, H.; Levonen, A.L.; Koistinaho, M.; et al. Intrahippocampal injection of a lentiviral vector expressing Nrf2 improves spatial learning in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2009, 106, 16505–16510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.Y.; Wang, Z.Y.; Xie, J.W.; Cai, J.H.; Wang, T.; Xu, Y.; Wang, X.; An, L. CD36 upregulation mediated by intranasal LV-NRF2 treatment mitigates hypoxia-induced progression of Alzheimer’s-like pathogenesis. Antioxid. Redox Signal. 2014, 21, 2208–2230. [Google Scholar] [CrossRef] [Green Version]
- Chu, Q.; Zhu, Y.; Cao, T.; Zhang, Y.; Chang, Z.; Liu, Y.; Lu, J.; Zhang, Y. Studies on the Neuroprotection of Osthole on Glutamate-Induced Apoptotic Cells and an Alzheimer’s Disease Mouse Model via Modulation Oxidative Stress. Appl. Biochem. Biotechnol. 2020, 190, 634–644. [Google Scholar] [CrossRef]
- Lipton, S.A.; Rezaie, T.; Nutter, A.; Lopez, K.M.; Parker, J.; Kosaka, K.; Satoh, T.; McKercher, S.R.; Masliah, E.; Nakanishi, N. Therapeutic advantage of pro-electrophilic drugs to activate the Nrf2/ARE pathway in Alzheimer’s disease models. Cell Death Dis. 2016, 7, e2499. [Google Scholar] [CrossRef]
- Wruck, C.J.; Gotz, M.E.; Herdegen, T.; Varoga, D.; Brandenburg, L.O.; Pufe, T. Kavalactones protect neural cells against amyloid beta peptide-induced neurotoxicity via extracellular signal-regulated kinase 1/2-dependent nuclear factor erythroid 2-related factor 2 activation. Mol. Pharmacol. 2008, 73, 1785–1795. [Google Scholar] [CrossRef] [Green Version]
- Fragoulis, A.; Siegl, S.; Fendt, M.; Jansen, S.; Soppa, U.; Brandenburg, L.O.; Pufe, T.; Weis, J.; Wruck, C.J. Oral administration of methysticin improves cognitive deficits in a mouse model of Alzheimer’s disease. Redox Biol. 2017, 12, 843–853. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhao, X.; Lazarovici, P.; Zheng, W. Artemether Activation of AMPK/GSK3beta(ser9)/Nrf2 Signaling Confers Neuroprotection towards beta-Amyloid-Induced Neurotoxicity in 3× Tg Alzheimer’s Mouse Model. Oxid. Med. Cell. Longev. 2019, 2019, 1862437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.; Yang, L.; Tucker, D.; Dong, Y.; Zhu, L.; Duan, R.; Liu, T.C.; Zhang, Q. Beneficial Effects of Exercise Pretreatment in a Sporadic Alzheimer’s Rat Model. Med. Sci. Sports Exerc. 2018, 50, 945–956. [Google Scholar] [CrossRef]
- Lawrence, T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef] [Green Version]
- Eshraghi, M.; Adlimoghaddam, A.; Mahmoodzadeh, A.; Sharifzad, F.; Yasavoli-Sharahi, H.; Lorzadeh, S.; Albensi, B.C.; Ghavami, S. Alzheimer’s Disease Pathogenesis: Role of Autophagy and Mitophagy Focusing in Microglia. Int. J. Mol. Sci. 2021, 22, 3330. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. Signaling to NF-kappaB. Genes Dev. 2004, 18, 2195–2224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albensi, B.C. What Is Nuclear Factor Kappa B (NF-kappaB) Doing in and to the Mitochondrion? Front. Cell Dev. Biol. 2019, 7, 154. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Verma, I.M. NF-kappaB regulation in the immune system. Nat. Rev. Immunol. 2002, 2, 725–734. [Google Scholar] [CrossRef]
- Newton, K.; Dixit, V.M. Signaling in innate immunity and inflammation. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef]
- Mogensen, T.H. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 2009, 22, 240–273. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-kappaB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.C.; Yeh, W.C.; Ohashi, P.S. LPS/TLR4 signal transduction pathway. Cytokine 2008, 42, 145–151. [Google Scholar] [CrossRef]
- Gohda, J.; Matsumura, T.; Inoue, J. Cutting edge: TNFR-associated factor (TRAF) 6 is essential for MyD88-dependent pathway but not toll/IL-1 receptor domain-containing adaptor-inducing IFN-beta (TRIF)-dependent pathway in TLR signaling. J. Immunol. 2004, 173, 2913–2917. [Google Scholar] [CrossRef] [Green Version]
- Sato, S.; Sanjo, H.; Takeda, K.; Ninomiya-Tsuji, J.; Yamamoto, M.; Kawai, T.; Matsumoto, K.; Takeuchi, O.; Akira, S. Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat. Immunol. 2005, 6, 1087–1095. [Google Scholar] [CrossRef]
- Oh, H.; Ghosh, S. NF-kappaB: Roles and regulation in different CD4(+) T-cell subsets. Immunol. Rev. 2013, 252, 41–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.Y.; D’Acquisto, F.; Hayden, M.S.; Shim, J.H.; Ghosh, S. PDK1 nucleates T cell receptor-induced signaling complex for NF-kappaB activation. Science 2005, 308, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Murray, S.E.; Polesso, F.; Rowe, A.M.; Basak, S.; Koguchi, Y.; Toren, K.G.; Hoffmann, A.; Parker, D.C. NF-kappaB-inducing kinase plays an essential T cell-intrinsic role in graft-versus-host disease and lethal autoimmunity in mice. J. Clin. Investig. 2011, 121, 4775–4786. [Google Scholar] [CrossRef]
- Li, Y.; Wang, H.; Zhou, X.; Xie, X.; Chen, X.; Jie, Z.; Zou, Q.; Hu, H.; Zhu, L.; Cheng, X.; et al. Cell intrinsic role of NF-kappaB-inducing kinase in regulating T cell-mediated immune and autoimmune responses. Sci. Rep. 2016, 6, 22115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adlimoghaddam, A.; Albensi, B.C. The nuclear factor kappa B (NF-kappaB) signaling pathway is involved in ammonia-induced mitochondrial dysfunction. Mitochondrion 2020, 57, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Adlimoghaddam, A.; Odero, G.G.; Glazner, G.; Turner, R.S.; Albensi, B.C. Nilotinib Improves Bioenergetic Profiling in Brain Astroglia in the 3× Tg Mouse Model of Alzheimer’s Disease. Aging Dis. 2021, 12, 441–465. [Google Scholar] [CrossRef] [PubMed]
- Adlimoghaddam, A.; Sabbir, M.G.; Albensi, B.C. Ammonia as a Potential Neurotoxic Factor in Alzheimer’s Disease. Front. Mol. Neurosci. 2016, 9, 57. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, L.A.; Kaltschmidt, C. NF-kappa B: A crucial transcription factor for glial and neuronal cell function. Trends Neurosci. 1997, 20, 252–258. [Google Scholar] [CrossRef]
- Chen, X.L.; Dodd, G.; Thomas, S.; Zhang, X.; Wasserman, M.A.; Rovin, B.H.; Kunsch, C. Activation of Nrf2/ARE pathway protects endothelial cells from oxidant injury and inhibits inflammatory gene expression. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H1862–H1870. [Google Scholar] [CrossRef] [Green Version]
- Vargas, M.R.; Johnson, J.A. The Nrf2-ARE cytoprotective pathway in astrocytes. Expert Rev. Mol. Med. 2009, 11, e17. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, S.M.; Luo, L.; Namani, A.; Wang, X.J.; Tang, X. Nrf2 signaling pathway: Pivotal roles in inflammation. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 585–597. [Google Scholar] [CrossRef] [PubMed]
- Ahn, K.S.; Aggarwal, B.B. Transcription factor NF-kappaB: A sensor for smoke and stress signals. Ann. N. Y. Acad. Sci. 2005, 1056, 218–233. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, N.; Slocum, S.L.; Skoko, J.J.; Shin, S.; Kensler, T.W. When NRF2 talks, who’s listening? Antioxid. Redox Signal. 2010, 13, 1649–1663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wardyn, J.D.; Ponsford, A.H.; Sanderson, C.M. Dissecting molecular cross-talk between Nrf2 and NF-kappaB response pathways. Biochem. Soc. Trans. 2015, 43, 621–626. [Google Scholar] [CrossRef] [Green Version]
- Tu, W.; Wang, H.; Li, S.; Liu, Q.; Sha, H. The Anti-Inflammatory and Anti-Oxidant Mechanisms of the Keap1/Nrf2/ARE Signaling Pathway in Chronic Diseases. Aging Dis. 2019, 10, 637–651. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.F.; Kuo, H.P.; Liu, M.; Chou, C.K.; Xia, W.; Du, Y.; Shen, J.; Chen, C.T.; Huo, L.; Hsu, M.C.; et al. KEAP1 E3 ligase-mediated downregulation of NF-kappaB signaling by targeting IKKbeta. Mol. Cell 2009, 36, 131–140. [Google Scholar] [CrossRef] [Green Version]
- Bellezza, I.; Tucci, A.; Galli, F.; Grottelli, S.; Mierla, A.L.; Pilolli, F.; Minelli, A. Inhibition of NF-kappaB nuclear translocation via HO-1 activation underlies alpha-tocopheryl succinate toxicity. J. Nutr. Biochem. 2012, 23, 1583–1591. [Google Scholar] [CrossRef]
- Karin, M.; Yamamoto, Y.; Wang, Q.M. The IKK NF-kappa B system: A treasure trove for drug development. Nat. Rev. Drug Discov. 2004, 3, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Ganesh Yerra, V.; Negi, G.; Sharma, S.S.; Kumar, A. Potential therapeutic effects of the simultaneous targeting of the Nrf2 and NF-kappaB pathways in diabetic neuropathy. Redox Biol. 2013, 1, 394–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.H.; Qu, J.; Shen, X. NF-kappaB/p65 antagonizes Nrf2-ARE pathway by depriving CBP from Nrf2 and facilitating recruitment of HDAC3 to MafK. Biochim. Biophys. Acta BBA Mol. Cell Res. 2008, 1783, 713–727. [Google Scholar] [CrossRef] [Green Version]
- Thimmulappa, R.K.; Mai, K.H.; Srisuma, S.; Kensler, T.W.; Yamamoto, M.; Biswal, S. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res. 2002, 62, 5196–5203. [Google Scholar]
- Saddawi-Konefka, R.; Seelige, R.; Gross, E.T.; Levy, E.; Searles, S.C.; Washington, A., Jr.; Santosa, E.K.; Liu, B.; O’Sullivan, T.E.; Harismendy, O.; et al. Nrf2 Induces IL-17D to Mediate Tumor and Virus Surveillance. Cell Rep. 2016, 16, 2348–2358. [Google Scholar] [CrossRef] [Green Version]
- Ishii, T.; Itoh, K.; Ruiz, E.; Leake, D.S.; Unoki, H.; Yamamoto, M.; Mann, G.E. Role of Nrf2 in the regulation of CD36 and stress protein expression in murine macrophages: Activation by oxidatively modified LDL and 4-hydroxynonenal. Circ. Res. 2004, 94, 609–616. [Google Scholar] [CrossRef] [Green Version]
- Rojo, A.I.; McBean, G.; Cindric, M.; Egea, J.; Lopez, M.G.; Rada, P.; Zarkovic, N.; Cuadrado, A. Redox control of microglial function: Molecular mechanisms and functional significance. Antioxid. Redox Signal. 2014, 21, 1766–1801. [Google Scholar] [CrossRef] [Green Version]
- Brune, B.; Dehne, N.; Grossmann, N.; Jung, M.; Namgaladze, D.; Schmid, T.; von Knethen, A.; Weigert, A. Redox control of inflammation in macrophages. Antioxid. Redox Signal. 2013, 19, 595–637. [Google Scholar] [CrossRef] [Green Version]
- Diotallevi, M.; Checconi, P.; Palamara, A.T.; Celestino, I.; Coppo, L.; Holmgren, A.; Abbas, K.; Peyrot, F.; Mengozzi, M.; Ghezzi, P. Glutathione Fine-Tunes the Innate Immune Response toward Antiviral Pathways in a Macrophage Cell Line Independently of Its Antioxidant Properties. Front. Immunol. 2017, 8, 1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soares, C.M.; Baptista, A.M.; Pereira, M.M.; Teixeira, M. Investigation of protonatable residues in Rhodothermus marinus caa3 haem-copper oxygen reductase: Comparison with Paracoccus denitrificans aa3 haem-copper oxygen reductase. J. Biol. Inorg. Chem. 2004, 9, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Sivandzade, F.; Prasad, S.; Bhalerao, A.; Cucullo, L. NRF2 and NF-B interplay in cerebrovascular and neurodegenerative disorders: Molecular mechanisms and possible therapeutic approaches. Redox Biol. 2019, 21, 101059. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, M.; Patil, J.; D’Angelo, B.; Weber, S.G.; Mallard, C. NRF2-regulation in brain health and disease: Implication of cerebral inflammation. Neuropharmacology 2014, 79, 298–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, L.; Chai, Y.; Lin, H.; Chen, C.; Zhao, M.; Xiong, W.; Zhuang, J.; Fan, X. Dihydroquercetin Activates AMPK/Nrf2/HO-1 Signaling in Macrophages and Attenuates Inflammation in LPS-Induced Endotoxemic Mice. Front. Pharmacol. 2020, 11, 662. [Google Scholar] [CrossRef]
- Cheong, S.H.; Lee, D.S. Taurine Chloramine Prevents Neuronal HT22 Cell Damage Through Nrf2-Related Heme Oxygenase-1. Adv. Exp. Med. Biol. 2017, 975 Pt 1, 145–157. [Google Scholar] [CrossRef]
- Lee, D.S.; Kwon, K.H.; Cheong, S.H. Taurine Chloramine Suppresses LPS-Induced Neuroinflammatory Responses through Nrf2-Mediated Heme Oxygenase-1 Expression in Mouse BV2 Microglial Cells. Adv. Exp. Med. Biol. 2017, 975 Pt 1, 131–143. [Google Scholar] [CrossRef]
- Luo, J.F.; Shen, X.Y.; Lio, C.K.; Dai, Y.; Cheng, C.S.; Liu, J.X.; Yao, Y.D.; Yu, Y.; Xie, Y.; Luo, P.; et al. Activation of Nrf2/HO-1 Pathway by Nardochinoid C Inhibits Inflammation and Oxidative Stress in Lipopolysaccharide-Stimulated Macrophages. Front. Pharmacol. 2018, 9, 911. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.L.; Yang, T.Y.; Gowrisankar, Y.V.; Liao, C.H.; Liao, J.W.; Huang, P.J.; Hseu, Y.C. Suppression of LPS-Induced Inflammation by Chalcone Flavokawain A through Activation of Nrf2/ARE-Mediated Antioxidant Genes and Inhibition of ROS/NFkappaB Signaling Pathways in Primary Splenocytes. Oxid. Med. Cell. Longev. 2020, 2020, 3476212. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Meng, P.; Matsumiya, T.; Tanji, K.; Hayakari, R.; Xing, F.; Wang, L.; Tsuruga, K.; Tanaka, H.; Mimura, J.; et al. Carnosic acid suppresses the production of amyloid-beta 1-42 and 1-43 by inducing an alpha-secretase TACE/ADAM17 in U373MG human astrocytoma cells. Neurosci. Res. 2014, 79, 83–93. [Google Scholar] [CrossRef]
- Jo, C.; Gundemir, S.; Pritchard, S.; Jin, Y.N.; Rahman, I.; Johnson, G.V. Nrf2 reduces levels of phosphorylated tau protein by inducing autophagy adaptor protein NDP52. Nat. Commun. 2014, 5, 3496. [Google Scholar] [CrossRef]
- Liddell, J.R. Are Astrocytes the Predominant Cell Type for Activation of Nrf2 in Aging and Neurodegeneration? Antioxidants 2017, 6, 65. [Google Scholar] [CrossRef] [PubMed]
- Rojo, A.I.; Innamorato, N.G.; Martin-Moreno, A.M.; De Ceballos, M.L.; Yamamoto, M.; Cuadrado, A. Nrf2 regulates microglial dynamics and neuroinflammation in experimental Parkinson’s disease. Glia 2010, 58, 588–598. [Google Scholar] [CrossRef]
- Vargas, M.R.; Johnson, D.A.; Sirkis, D.W.; Messing, A.; Johnson, J.A. Nrf2 activation in astrocytes protects against neurodegeneration in mouse models of familial amyotrophic lateral sclerosis. J. Neurosci. 2008, 28, 13574–13581. [Google Scholar] [CrossRef]
- Pan, H.; Wang, H.; Wang, X.; Zhu, L.; Mao, L. The absence of Nrf2 enhances NF-kappaB-dependent inflammation following scratch injury in mouse primary cultured astrocytes. Mediat. Inflamm. 2012, 2012, 217580. [Google Scholar] [CrossRef]
- Frakes, A.E.; Ferraiuolo, L.; Haidet-Phillips, A.M.; Schmelzer, L.; Braun, L.; Miranda, C.J.; Ladner, K.J.; Bevan, A.K.; Foust, K.D.; Godbout, J.P.; et al. Microglia induce motor neuron death via the classical NF-kappaB pathway in amyotrophic lateral sclerosis. Neuron 2014, 81, 1009–1023. [Google Scholar] [CrossRef] [Green Version]
- Thimmulappa, R.K.; Lee, H.; Rangasamy, T.; Reddy, S.P.; Yamamoto, M.; Kensler, T.W.; Biswal, S. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J. Clin. Investig. 2006, 116, 984–995. [Google Scholar] [CrossRef] [Green Version]
- Thimmulappa, R.K.; Scollick, C.; Traore, K.; Yates, M.; Trush, M.A.; Liby, K.T.; Sporn, M.B.; Yamamoto, M.; Kensler, T.W.; Biswal, S. Nrf2-dependent protection from LPS induced inflammatory response and mortality by CDDO-Imidazolide. Biochem. Biophys. Res. Commun. 2006, 351, 883–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jian, Z.; Li, K.; Liu, L.; Zhang, Y.; Zhou, Z.; Li, C.; Gao, T. Heme oxygenase-1 protects human melanocytes from H2O2-induced oxidative stress via the Nrf2-ARE pathway. J. Investig. Dermatol. 2011, 131, 1420–1427. [Google Scholar] [CrossRef] [Green Version]
- Habtemariam, S. The Nrf2/HO-1 Axis as Targets for Flavanones: Neuroprotection by Pinocembrin, Naringenin, and Eriodictyol. Oxid. Med. Cell. Longev. 2019, 2019, 4724920. [Google Scholar] [CrossRef] [PubMed]
- Jeong, W.S.; Kim, I.W.; Hu, R.; Kong, A.N. Modulatory properties of various natural chemopreventive agents on the activation of NF-kappaB signaling pathway. Pharm. Res. 2004, 21, 661–670. [Google Scholar] [CrossRef]
- Xu, C.; Shen, G.; Chen, C.; Gelinas, C.; Kong, A.N. Suppression of NF-kappaB and NF-kappaB-regulated gene expression by sulforaphane and PEITC through IkappaBα, IKK pathway in human prostate cancer PC-3 cells. Oncogene 2005, 24, 4486–4495. [Google Scholar] [CrossRef] [Green Version]
- Jang, M.; Cho, I.H. Sulforaphane Ameliorates 3-Nitropropionic Acid-Induced Striatal Toxicity by Activating the Keap1-Nrf2-ARE Pathway and Inhibiting the MAPKs and NF-kappaB Pathways. Mol. Neurobiol. 2016, 53, 2619–2635. [Google Scholar] [CrossRef]
- Takaya, K.; Suzuki, T.; Motohashi, H.; Onodera, K.; Satomi, S.; Kensler, T.W.; Yamamoto, M. Validation of the multiple sensor mechanism of the Keap1-Nrf2 system. Free Radic. Biol. Med. 2012, 53, 817–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Oliveira, M.R.; de Bittencourt Brasil, F.; Furstenau, C.R. Sulforaphane Promotes Mitochondrial Protection in SH-SY5Y Cells Exposed to Hydrogen Peroxide by an Nrf2-Dependent Mechanism. Mol. Neurobiol. 2018, 55, 4777–4787. [Google Scholar] [CrossRef] [PubMed]
- Moon, D.O.; Kim, M.O.; Kang, S.H.; Choi, Y.H.; Kim, G.Y. Sulforaphane suppresses TNF-alpha-mediated activation of NF-kappaB and induces apoptosis through activation of reactive oxygen species-dependent caspase-3. Cancer Lett. 2009, 274, 132–142. [Google Scholar] [CrossRef]
- Zhao, H.; Zhao, X.; Liu, L.; Zhang, H.; Xuan, M.; Guo, Z.; Wang, H.; Liu, C. Neurochemical effects of the R form of alpha-lipoic acid and its neuroprotective mechanism in cellular models of Parkinson’s disease. Int. J. Biochem. Cell Biol. 2017, 87, 86–94. [Google Scholar] [CrossRef]
- Kim, H.V.; Kim, H.Y.; Ehrlich, H.Y.; Choi, S.Y.; Kim, D.J.; Kim, Y. Amelioration of Alzheimer’s disease by neuroprotective effect of sulforaphane in animal model. Amyloid 2013, 20, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Yoo, I.H.; Kim, M.J.; Kim, J.; Sung, J.J.; Park, S.T.; Ahn, S.W. The Anti-Inflammatory Effect of Sulforaphane in Mice with Experimental Autoimmune Encephalomyelitis. J. Korean Med. Sci. 2019, 34, e197. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Alfieri, A.; Siow, R.C.; Mann, G.E.; Fraser, P.A. Temporal and spatial distribution of Nrf2 in rat brain following stroke: Quantification of nuclear to cytoplasmic Nrf2 content using a novel immunohistochemical technique. J. Physiol. 2013, 591, 3525–3538. [Google Scholar] [CrossRef] [PubMed]
- Kwak, M.K.; Cho, J.M.; Huang, B.; Shin, S.; Kensler, T.W. Role of increased expression of the proteasome in the protective effects of sulforaphane against hydrogen peroxide-mediated cytotoxicity in murine neuroblastoma cells. Free Radic. Biol. Med. 2007, 43, 809–817. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.; Zhan, P.; Wang, Q.; Wang, C.; Liu, Y.; Yu, Z.; Zhang, S. Curcumin upregulates the Nrf2 system by repressing inflammatory signaling-mediated Keap1 expression in insulin-resistant conditions. Biochem. Biophys. Res. Commun. 2019, 514, 691–698. [Google Scholar] [CrossRef] [PubMed]
- Zheng, K.; Dai, X.; Xiao, N.; Wu, X.; Wei, Z.; Fang, W.; Zhu, Y.; Zhang, J.; Chen, X. Curcumin Ameliorates Memory Decline via Inhibiting BACE1 Expression and beta-Amyloid Pathology in 5× FAD Transgenic Mice. Mol. Neurobiol. 2017, 54, 1967–1977. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wu, C.; Zhao, S.; Yuan, D.; Lian, G.; Wang, X.; Wang, L.; Yang, J. Demethoxycurcumin, a natural derivative of curcumin attenuates LPS-induced pro-inflammatory responses through down-regulation of intracellular ROS-related MAPK/NF-kappaB signaling pathways in N9 microglia induced by lipopolysaccharide. Int. Immunopharmacol. 2010, 10, 331–338. [Google Scholar] [CrossRef]
- Li, W.; Suwanwela, N.C.; Patumraj, S. Curcumin by down-regulating NF-kB and elevating Nrf2, reduces brain edema and neurological dysfunction after cerebral I/R. Microvasc. Res. 2016, 106, 117–127. [Google Scholar] [CrossRef]
- Lee, J.W.; Lee, Y.K.; Ban, J.O.; Ha, T.Y.; Yun, Y.P.; Han, S.B.; Oh, K.W.; Hong, J.T. Green tea (-)-epigallocatechin-3-gallate inhibits beta-amyloid-induced cognitive dysfunction through modification of secretase activity via inhibition of ERK and NF-kappaB pathways in mice. J. Nutr. 2009, 139, 1987–1993. [Google Scholar] [CrossRef]
- Ma, L.; Cao, T.T.; Kandpal, G.; Warren, L.; Fred Hess, J.; Seabrook, G.R.; Ray, W.J. Genome-wide microarray analysis of the differential neuroprotective effects of antioxidants in neuroblastoma cells overexpressing the familial Parkinson’s disease alpha-synuclein A53T mutation. Neurochem. Res. 2010, 35, 130–142. [Google Scholar] [CrossRef]
- Itoh, T.; Tabuchi, M.; Mizuguchi, N.; Imano, M.; Tsubaki, M.; Nishida, S.; Hashimoto, S.; Matsuo, K.; Nakayama, T.; Ito, A.; et al. Neuroprotective effect of (−)-epigallocatechin-3-gallate in rats when administered pre- or post-traumatic brain injury. J. Neural Transm. 2013, 120, 767–783. [Google Scholar] [CrossRef]
- Semnani, M.; Mashayekhi, F.; Azarnia, M.; Salehi, Z. Effects of green tea epigallocatechin-3-gallate on the proteolipid protein and oligodendrocyte transcription factor 1 messenger RNA gene expression in a mouse model of multiple sclerosis. Folia Neuropathol. 2017, 55, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Jayachandran, M.; Wu, Z.; Ganesan, K.; Khalid, S.; Chung, S.M.; Xu, B. Isoquercetin upregulates antioxidant genes, suppresses inflammatory cytokines and regulates AMPK pathway in streptozotocin-induced diabetic rats. Chem. Biol. Interact. 2019, 303, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Zhang, H.; Zhang, J.; Yan, M. Isoquercetin attenuates oxidative stress and neuronal apoptosis after ischemia/reperfusion injury via Nrf2-mediated inhibition of the NOX4/ROS/NF-kappaB pathway. Chem. Biol. Interact. 2018, 284, 32–40. [Google Scholar] [CrossRef]
- Lv, H.; Yu, Z.; Zheng, Y.; Wang, L.; Qin, X.; Cheng, G.; Ci, X. Isovitexin Exerts Anti-Inflammatory and Anti-Oxidant Activities on Lipopolysaccharide-Induced Acute Lung Injury by Inhibiting MAPK and NF-kappaB and Activating HO-1/Nrf2 Pathways. Int. J. Biol. Sci. 2016, 12, 72–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Choi, S.Y.; Choo, Y.Y.; Kim, O.; Tran, P.T.; Dao, C.T.; Min, B.S.; Lee, J.H. Sappanone A exhibits anti-inflammatory effects via modulation of Nrf2 and NF-kappaB. Int. Immunopharmacol. 2015, 28, 328–336. [Google Scholar] [CrossRef]
- Li, C.Z.; Jin, H.H.; Sun, H.X.; Zhang, Z.Z.; Zheng, J.X.; Li, S.H.; Han, S.H. Eriodictyol attenuates cisplatin-induced kidney injury by inhibiting oxidative stress and inflammation. Eur. J. Pharmacol. 2016, 772, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Negi, G.; Kumar, A.; Sharma, S.S. Melatonin modulates neuroinflammation and oxidative stress in experimental diabetic neuropathy: Effects on NF-kappaB and Nrf2 cascades. J. Pineal Res. 2011, 50, 124–131. [Google Scholar] [CrossRef]
- Kumar, A.; Sharma, S.S. NF-kappaB inhibitory action of resveratrol: A probable mechanism of neuroprotection in experimental diabetic neuropathy. Biochem. Biophys. Res. Commun. 2010, 394, 360–365. [Google Scholar] [CrossRef]
- Palazon, A.; Goldrath, A.W.; Nizet, V.; Johnson, R.S. HIF transcription factors, inflammation, and immunity. Immunity 2014, 41, 518–528. [Google Scholar] [CrossRef] [Green Version]
- Toth, R.K.; Warfel, N.A. Strange Bedfellows: Nuclear Factor, Erythroid 2-Like 2 (Nrf2) and Hypoxia-Inducible Factor 1 (HIF-1) in Tumor Hypoxia. Antioxidants 2017, 6, 27. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.H.; Hur, E.G.; Kang, S.J.; Kim, J.A.; Thapa, D.; Lee, Y.M.; Ku, S.K.; Jung, Y.; Kwak, M.K. NRF2 blockade suppresses colon tumor angiogenesis by inhibiting hypoxia-induced activation of HIF-1alpha. Cancer Res. 2011, 71, 2260–2275. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Wang, B.; Shi, Q.; Wang, X.; Wang, D.; Zhu, L. Brusatol inhibits HIF-1 signaling pathway and suppresses glucose uptake under hypoxic conditions in HCT116 cells. Sci. Rep. 2016, 6, 39123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, E.T.; Kim, J.W.; Kim, J.M.; Kim, S.J.; Lee, J.S.; Hong, S.S.; Goodwin, J.; Ruthenborg, R.J.; Jung, M.G.; Lee, H.J.; et al. NQO1 inhibits proteasome-mediated degradation of HIF-1alpha. Nat. Commun. 2016, 7, 13593. [Google Scholar] [CrossRef] [Green Version]
- Cummins, E.P.; Berra, E.; Comerford, K.M.; Ginouves, A.; Fitzgerald, K.T.; Seeballuck, F.; Godson, C.; Nielsen, J.E.; Moynagh, P.; Pouyssegur, J.; et al. Prolyl hydroxylase-1 negatively regulates IkappaB kinase-beta, giving insight into hypoxia-induced NFkappaB activity. Proc. Natl. Acad. Sci. USA 2006, 103, 18154–18159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzpatrick, S.F.; Tambuwala, M.M.; Bruning, U.; Schaible, B.; Scholz, C.C.; Byrne, A.; O’Connor, A.; Gallagher, W.M.; Lenihan, C.R.; Garvey, J.F.; et al. An intact canonical NF-kappaB pathway is required for inflammatory gene expression in response to hypoxia. J. Immunol. 2011, 186, 1091–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rius, J.; Guma, M.; Schachtrup, C.; Akassoglou, K.; Zinkernagel, A.S.; Nizet, V.; Johnson, R.S.; Haddad, G.G.; Karin, M. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature 2008, 453, 807–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walmsley, S.R.; Print, C.; Farahi, N.; Peyssonnaux, C.; Johnson, R.S.; Cramer, T.; Sobolewski, A.; Condliffe, A.M.; Cowburn, A.S.; Johnson, N.; et al. Hypoxia-induced neutrophil survival is mediated by HIF-1alpha-dependent NF-kappaB activity. J. Exp. Med. 2005, 201, 105–115. [Google Scholar] [CrossRef]
- Ransone, L.J.; Verma, I.M. Nuclear proto-oncogenes Fos and Jun. Annu. Rev. Cell Biol. 1990, 6, 539–557. [Google Scholar] [CrossRef]
- Shaulian, E.; Karin, M. AP-1 as a regulator of cell life and death. Nat. Cell Biol. 2002, 4, E131–E136. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.; Barve, A.; Khor, T.O.; Shen, G.X.; Lin, W.; Chan, J.Y.; Cai, L.; Kong, A.N. Regulation of Nrf2- and AP-1-mediated gene expression by epigallocatechin-3-gallate and sulforaphane in prostate of Nrf2-knockout or C57BL/6J mice and PC-3 AP-1 human prostate cancer cells. Acta Pharmacol. Sin. 2010, 31, 1223–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujioka, S.; Niu, J.; Schmidt, C.; Sclabas, G.M.; Peng, B.; Uwagawa, T.; Li, Z.; Evans, D.B.; Abbruzzese, J.L.; Chiao, P.J. NF-kappaB and AP-1 connection: Mechanism of NF-kappaB-dependent regulation of AP-1 activity. Mol. Cell. Biol. 2004, 24, 7806–7819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, I.M.; Stevenson, J.K.; Schwarz, E.M.; Van Antwerp, D.; Miyamoto, S. Rel/NF-kappa B/I kappa B family: Intimate tales of association and dissociation. Genes Dev. 1995, 9, 2723–2735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, H.; Tai, H.; Huang, N.; Xiao, P.; Mo, C.; Wang, X.; Han, X.; Zhou, J.; Chen, H.; Tang, X.; et al. Nrf2-SHP Cascade-Mediated STAT3 Inactivation Contributes to AMPK-Driven Protection Against Endotoxic Inflammation. Front. Immunol. 2020, 11, 414. [Google Scholar] [CrossRef] [Green Version]
- Snyder, M.; Huang, J.; Huang, X.Y.; Zhang, J.J. A signal transducer and activator of transcription 3 Nuclear Factor kappaB (Stat3.NFkappaB) complex is necessary for the expression of fascin in metastatic breast cancer cells in response to interleukin (IL)-6 and tumor necrosis factor (TNF)-alpha. J. Biol. Chem. 2014, 289, 30082–30089. [Google Scholar] [CrossRef] [Green Version]
- Arlt, A.; Schafer, H.; Kalthoff, H. The ‘N-factors’ in pancreatic cancer: Functional relevance of NF-kappaB, NFAT and Nrf2 in pancreatic cancer. Oncogenesis 2012, 1, e35. [Google Scholar] [CrossRef] [Green Version]
- Serfling, E.; Berberich-Siebelt, F.; Avots, A.; Chuvpilo, S.; Klein-Hessling, S.; Jha, M.K.; Kondo, E.; Pagel, P.; Schulze-Luehrmann, J.; Palmetshofer, A. NFAT and NF-kappaB factors-the distant relatives. Int. J. Biochem. Cell Biol. 2004, 36, 1166–1170. [Google Scholar] [CrossRef]
- Klotz, L.O.; Steinbrenner, H. Cellular adaptation to xenobiotics: Interplay between xenosensors, reactive oxygen species and FOXO transcription factors. Redox Biol. 2017, 13, 646–654. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Hron, J.D.; Peng, S.L. Regulation of NF-kappaB, Th activation, and autoinflammation by the forkhead transcription factor Foxo3a. Immunity 2004, 21, 203–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
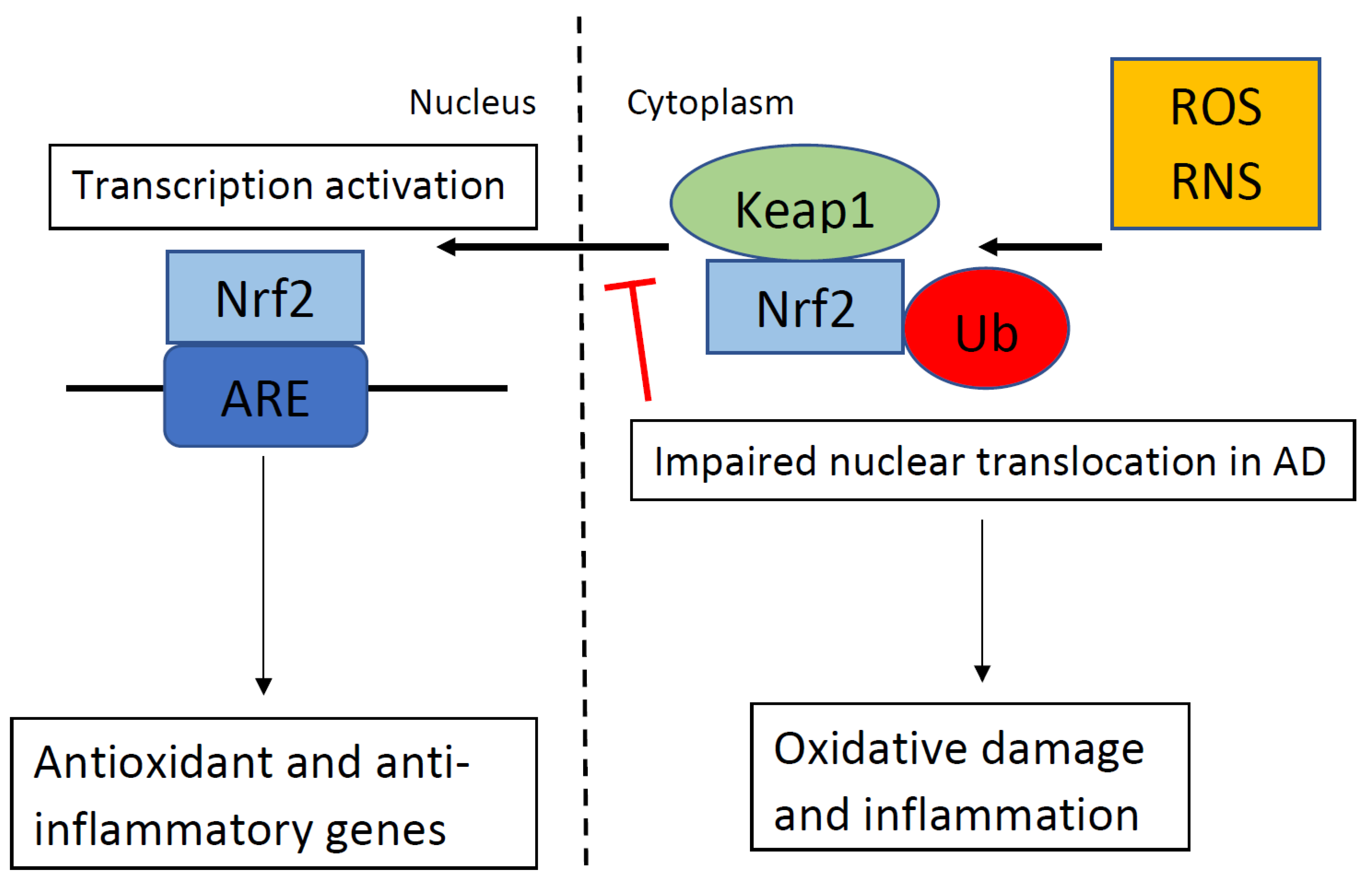
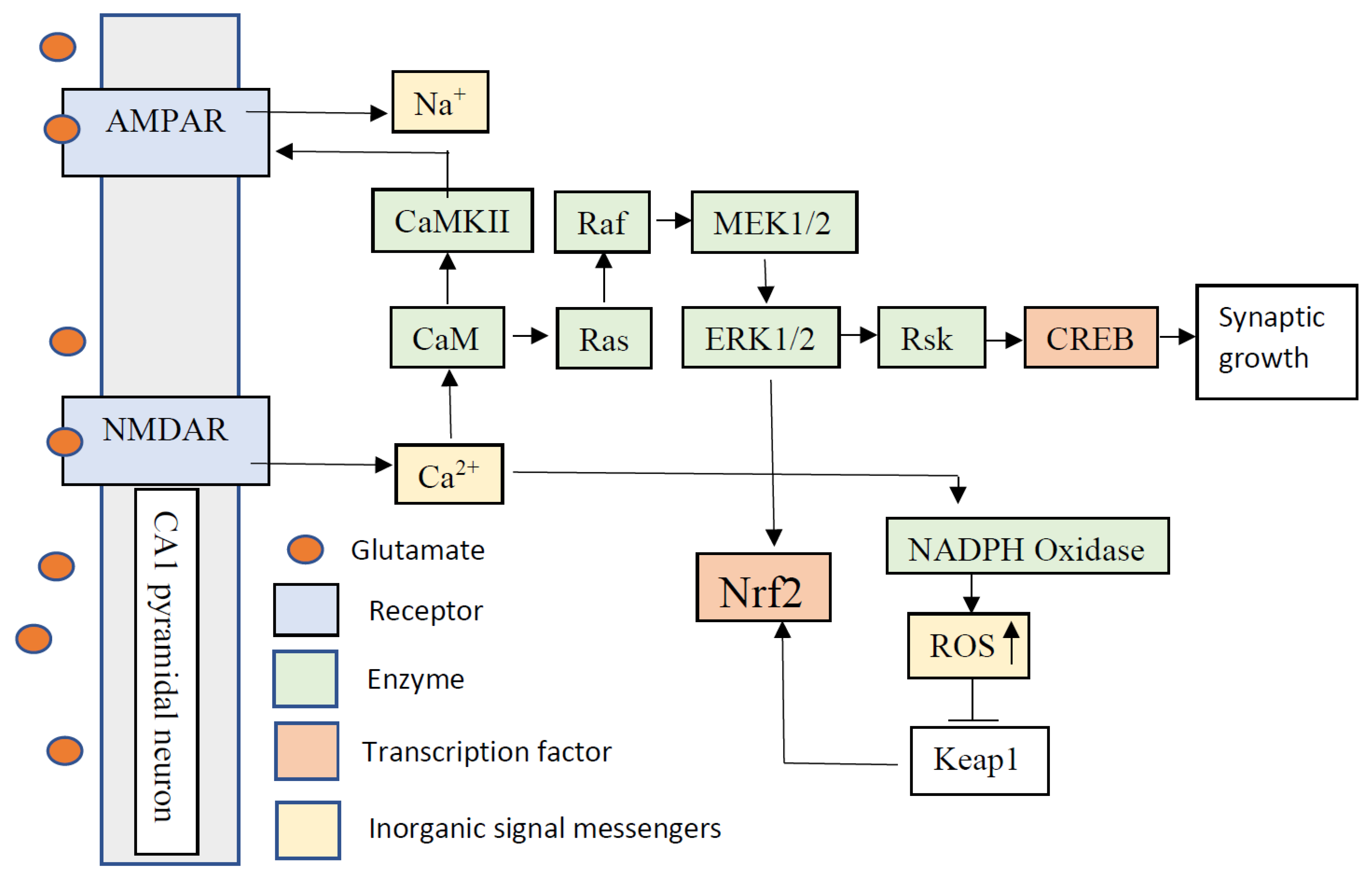
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Davies, D.A.; Adlimoghaddam, A.; Albensi, B.C. Role of Nrf2 in Synaptic Plasticity and Memory in Alzheimer’s Disease. Cells 2021, 10, 1884. https://doi.org/10.3390/cells10081884
Davies DA, Adlimoghaddam A, Albensi BC. Role of Nrf2 in Synaptic Plasticity and Memory in Alzheimer’s Disease. Cells. 2021; 10(8):1884. https://doi.org/10.3390/cells10081884
Chicago/Turabian StyleDavies, Don A., Aida Adlimoghaddam, and Benedict C. Albensi. 2021. "Role of Nrf2 in Synaptic Plasticity and Memory in Alzheimer’s Disease" Cells 10, no. 8: 1884. https://doi.org/10.3390/cells10081884
APA StyleDavies, D. A., Adlimoghaddam, A., & Albensi, B. C. (2021). Role of Nrf2 in Synaptic Plasticity and Memory in Alzheimer’s Disease. Cells, 10(8), 1884. https://doi.org/10.3390/cells10081884