
Calcium Signaling Regulates Autophagy and Apoptosis

 , , and
, , and
Abstract
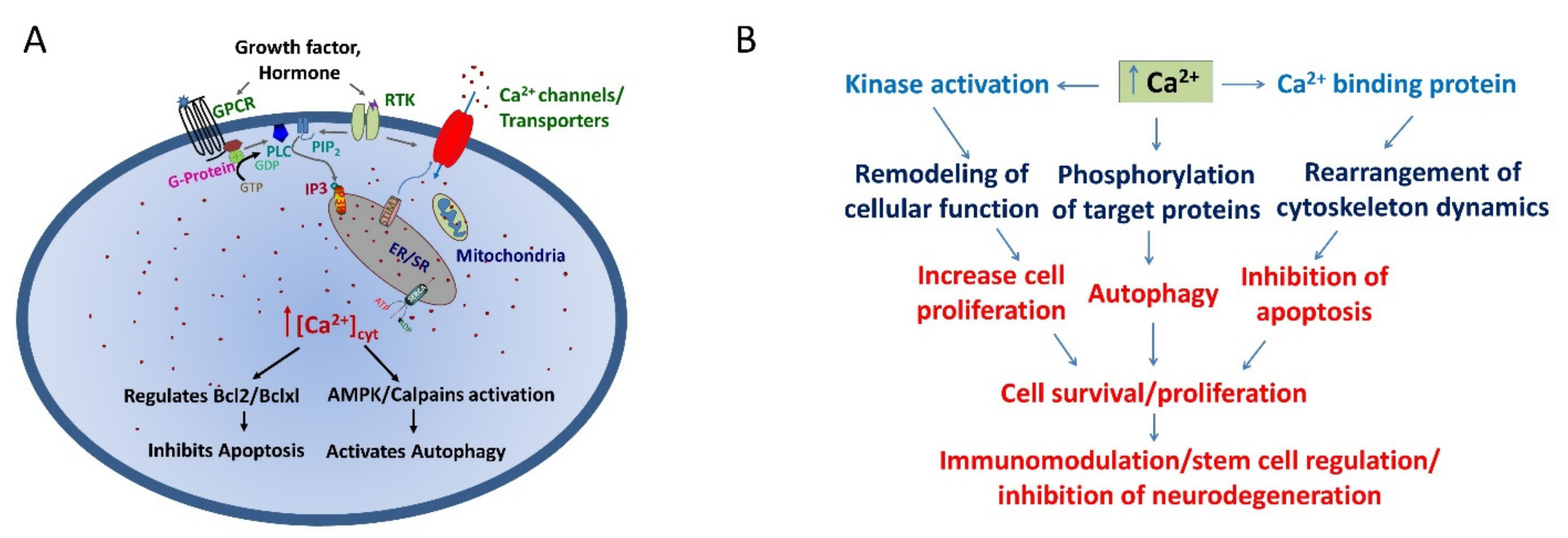
:1. Introduction
2. Autophagy and Apoptosis Crosstalk Is Guided by Ca2+ Influx
3. Calcium as Regulator for Apoptosis
4. Intracellular Ca2+ Stores in Regulating Apoptotic Function
5. Calcium Channels/Receptors and Its Role in Autophagy and Apoptosis
5.1. Inositol Trisphosphate Receptors
5.2. Voltage-Gated Calcium Channels
5.3. Transient Receptor Potential Canonical and Orai1 Channels
5.4. Transient Receptor Potential Melastatin Channels
5.5. Transient Receptor Potential Vanilloid Channels

{kind=link}
| Autophagy/Apoptosis | TRP Channels | Used Materials | Molecular Pathway | Reference |
|---|---|---|---|---|
| Decreased TRPC1 expression subsequently attenuated autophagy along with increased apoptosis. | TRPC1 | Neuronal Cells | ER stress | [67] |
| Depletion of TRPC3 causes activation of autophagy in response to supramaximal CCK8 and to bile acids. | TRPC3 | In mice single pancreatic acinar cells | Intracellular trypsin activation and excessive actin depolymerization | [70] |
| Cannabidiol stimulates autophagy signal transduction via crosstalk between the ERK1/2 and AKT kinases. | TRPV1 | Human neuroblastoma SH-SY5Y and murine astrocyte cell lines. | ERK1/2 and AKT kinases | [82,83] |
| TRPV1 induces autophagy in nitrogen mustard (NM)-caused cutaneous injury. | TRPV1 | The HaCaT cells | Ca2+/calmodulin-dependent kinase β (CaMKβ), AMP-activated protein kinase (AMPK), unc-51-like kinase 1 (ULK1) pathway | [77] |
| TRPV1 activation mitigates hypoxic injury in mouse cardiomyocytes by inducing autophagy. | TRPV1 | Primary cardiomyocytes isolated from C57 mice | AMPK signaling pathway | [84] |
| TRPV2 agonist leads to the activation of autophagy. | TRPV2 | Glioblastoma stem cell (GSC) | Stimulating the expression of several genes involved in the autophagic process and in the unfolded protein response | [80,81] |
| TRPV4 channels promote autophagy. | TRPV4 | Rat Hepatic stellate cell (HSC) | Inhibiting AKT via generating Ca2+ signals | [85] |
| Oxoglaucine protects against cartilage damage by blocking the TRPV5/CAMK-II/calmodulin pathway to inhibit Ca2+ influx and activate autophagy. | TRPV5 | In vitro and in vivo- rat model of osteoarthritis | CAMK-II and calmodulin | [86] |
| Trichostatin A suppresses cervical cancer cell proliferation and induces apoptosis and autophagy through regulation of the PRMT5/STC1/TRPV6/JNK axis. | TRPV6 | HeLa and Caski cervical cancer cell lines | JNK pathway | [87] |
| TRPM2 ion channel has been involved in oxidative stress-mediated induced autophagy. | TRPM2 | TRPM2 KO mice | Via oxidative stress and stimulates NADPH oxidase | [74,75,88,89] |
| TRPM7 channel played a role in regulation of basal autophagy. | TRPM7 | Neuronal cells | Ca(2+)/calmodulin-dependent protein kinase kinase β and AMP-activated protein kinase pathway | [71,90] |
| Silencing TRPM7 trigger autophagy without any detectable increase in free Ca2+. | TRPM7 | hMSC cells isolated from adult human bone marrow | Via Mg2+ channel regulation | [72] |
6. Autophagy/Apoptosis and Stem Cell Function
7. Autophagy and Apoptosis in Immune Cells
7.1. Neutrophils
7.2. Monocytes/Macrophages
7.3. T Cells
7.4. B Cells
7.5. Dendritic Cells
7.6. Nature Killers
8. Autophagy Inhibits Apoptosis in Neuronal Cells
9. Role of Autophagy/Apoptosis in Neurodegenerative Diseases
9.1. Autophagy/Apoptosis in Alzheimer’s Disease (AD)
9.2. Autophagy/Apoptosis in Parkinson’s Disease (PD)
9.3. Autophagy/Apoptosis in Huntington’s Disease (HD)
9.4. Autophagy/Apoptosis in Amyotrophic Lateral Sclerosis (ALS)
10. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Smaili, S.; Pereira, J.S.; Costa, M.; Rocha, K.; Rodrigues, L.; do Carmo, G.; Hsu, T.Y. The role of calcium stores in apoptosis and autophagy. Curr. Mol. Med. 2013, 13, 252–265. [Google Scholar] [CrossRef]
- Sukumaran, P.; Sun, Y.; Schaar, A.; Selvaraj, S.; Singh, B.B. TRPC Channels and Parkinson’s Disease. Adv. Exp. Med. Biol. 2017, 976, 85–94. [Google Scholar] [PubMed] [Green Version]
- Feng, S. TRPC Channel Structure and Properties. Adv. Exp. Med. Biol. 2017, 976, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.X.; Tang, J. TRPC channel interactions with calmodulin and IP3 receptors. In Novartis Foundation Symposium; John Wiley: Chichester, NY, USA, 2004; Volume 258, pp. 44–266, 44–58, discussion 58–62, 98–102, 263–266. [Google Scholar]
- Cárdenas, C.; Miller, R.A.; Smith, I.; Bui, T.; Molgó, J.; Müller, M.; Vais, H.; Cheung, K.-H.; Yang, J.; Parker, I.; et al. Essential Regulation of Cell Bioenergetics by Constitutive InsP3 Receptor Ca2+ Transfer to Mitochondria. Cell 2010, 142, 270–283. [Google Scholar] [CrossRef] [Green Version]
- Høyer-Hansen, M.; Bastholm, L.; Szyniarowski, P.; Campanella, M.; Szabadkai, G.; Farkas, T.; Jäättelä, M. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-β, and Bcl-2. Mol. Cell 2007, 25, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Gordon, J.N.; Shu, W.P.; Schlussel, R.N.; Droller, M.J.; Liu, B.C. Altered extracellular matrices influence cellular processes and nuclear matrix organizations of overlying human bladder urothelial cells. Cancer Res. 1993, 53, 4971–4977. [Google Scholar]
- Decuypere, J.-P.; Bultynck, G.; Parys, J.B. A dual role for Ca2+ in autophagy regulation. Cell Calcium 2011, 50, 242–250. [Google Scholar] [CrossRef] [PubMed]
- East, D.A.; Campanella, M. Ca2+ in quality control: An unresolved riddle critical to autophagy and mitophagy. Autophagy 2013, 9, 1710–1719. [Google Scholar] [CrossRef] [Green Version]
- Su, J.; Zhou, L.; Kong, X.; Yang, X.; Xiang, X.; Zhang, Y.; Li, X.; Sun, L. Endoplasmic Reticulum Is at the Crossroads of Autophagy, Inflammation, and Apoptosis Signaling Pathways and Participates in the Pathogenesis of Diabetes Mellitus. J. Diabetes Res. 2013, 2013, 1–6. [Google Scholar] [CrossRef]
- Gastaldello, A.; Callaghan, H.; Gami-Patel, P.; Campanella, M. Ca2+-dependent autophagy is enhanced by the pharmacological agent PK11195. Autophagy 2010, 6, 607–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Codogno, P.; Mehrpour, M.; Proikas-Cezanne, T. Canonical and non-canonical autophagy: Variations on a common theme of self-eating? Nat. Rev. Mol. Cell Biol. 2012, 13, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Münz, C. Non-canonical Functions of Macroautophagy Proteins During Endocytosis by Myeloid Antigen Presenting Cells. Front. Immunol. 2018, 9, 2765. [Google Scholar] [CrossRef]
- Gui, X.; Yang, H.; Li, T.; Tan, X.; Shi, P.; Li, M.; Du, F.; Chen, Z.J. Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nat. Cell Biol. 2019, 567, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Kania, E.; Pajak, B.; Orzechowski, A. Calcium Homeostasis and ER Stress in Control of Autophagy in Cancer Cells. BioMed Res. Int. 2015, 2015, 352794. [Google Scholar] [CrossRef] [Green Version]
- Valladares, D.; Utreras-Mendoza, Y.; Campos, C.; Morales, C.; Diaz-Vegas, A.; Contreras-Ferrat, A.; Lavandero, S. IP3 receptor blockade restores autophagy and mitochondrial function in skeletal muscle fibers of dystrophic mice. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3685–3695. [Google Scholar] [CrossRef]
- Kondratskyi, A.; Yassine, M.; Kondratska, K.; Skryma, R.; Slomianny, C.; Prevarskaya, N. Calcium-permeable ion channels in control of autophagy and cancer. Front. Physiol. 2013, 4, 272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, W.; Ding, W.-X.; Stolz, N.B.; Yin, X.-M. Induction of macroautophagy by exogenously introduced calcium. Autophagy 2008, 4, 754–761. [Google Scholar] [CrossRef] [Green Version]
- Mirzoeva, O.K.; Hann, B.; Hom, Y.K.; Debnath, J.; Aftab, D.; Shokat, K.; Korn, W.M. Autophagy suppression promotes apoptotic cell death in response to inhibition of the PI3K-mTOR pathway in pancreatic adenocarcinoma. J. Mol. Med. 2011, 89, 877–889. [Google Scholar] [CrossRef]
- Ding, W.-X.; Chen, X.; Yin, X.-M. Tumor cells can evade dependence on autophagy through adaptation. Biochem. Biophys. Res. Commun. 2012, 425, 684–688. [Google Scholar] [CrossRef] [Green Version]
- Selimovic, D.; Porzig, B.B.; El-Khattouti, A.; Badura, H.E.; Ahmad, M.; Ghanjati, F.; Santourlidis, S.; Haikel, Y.; Hassan, M. Bortezomib/proteasome inhibitor triggers both apoptosis and autophagy-dependent pathways in melanoma cells. Cell. Signal. 2013, 25, 308–318. [Google Scholar] [CrossRef]
- Zhao, C.; Yin, S.; Dong, Y.; Guo, X.; Fan, L.; Ye, M.; Hu, H. Autophagy-dependent EIF2AK3 activation compromises ursolic acid-induced apoptosis through upregulation of MCL1 in MCF-7 human breast cancer cells. Autophagy 2013, 9, 196–207. [Google Scholar] [CrossRef] [Green Version]
- Thorburn, A. Crosstalk between autophagy and apoptosis: Mechanisms and therapeutic implications. Prog. Mol. Biol. Transl. Sci. 2020, 172, 55–65. [Google Scholar] [CrossRef]
- Orrenius, S.; Zhivotovsky, B.; Nicotera, P. Regulation of cell death: The calcium–apoptosis link. Nat. Rev. Mol. Cell Biol. 2003, 4, 552–565. [Google Scholar] [CrossRef]
- Martikainen, P.; Kyprianou, N.; Tucker, R.W.; Isaacs, J.T. Programmed death of nonproliferating androgen-independent prostatic cancer cells. Cancer Res. 1991, 51, 4693–4700. [Google Scholar]
- Choi, D.W. Excitotoxic cell death. J. Neurobiol. 1992, 23, 1261–1276. [Google Scholar] [CrossRef] [PubMed]
- Fleckenstein, A.; Kanke, J.; Döring, H.J.; Leder, O. Key role of Ca in the production of noncoronarogenic myocardial necroses. Recent Adv. Stud. Card. Struct. Metab. 1975, 6, 21–32. [Google Scholar]
- McConkey, D.J.; Nicotera, P.; Hartzell, P.; Bellomo, G.; Wyllie, A.H.; Orrenius, S. Glucocorticoids activate a suicide process in thymocytes through an elevation of cytosolic Ca2+ concentration. Arch. Biochem. Biophys. 1989, 269, 365–370. [Google Scholar] [CrossRef]
- Khan, A.A.; Soloski, M.J.; Sharp, A.H.; Schilling, G.; Sabatini, D.M.; Li, S.-H.; Ross, C.A.; Snyder, S.H. Lymphocyte Apoptosis: Mediation by Increased Type 3 Inositol 1,4,5-Trisphosphate Receptor. Science 1996, 273, 503–507. [Google Scholar] [CrossRef] [Green Version]
- Kruman, I.; Guo, Q.; Mattson, M.P. Calcium and reactive oxygen species mediate staurosporine-induced mitochondrial dysfunction and apoptosis in PC12 cells. J. Neurosci. Res. 1998, 51, 293–308. [Google Scholar] [CrossRef]
- Tombal, B.; Denmeade, S.; Isaacs, J. Assessment and validation of a microinjection method for kinetic analysis of [Ca2+]i in individual cells undergoing apoptosis. Cell Calcium 1999, 25, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, R.; Pozzan, T. Microdomains of Intracellular Ca2+: Molecular Determinants and Functional Consequences. Physiol. Rev. 2006, 86, 369–408. [Google Scholar] [CrossRef]
- Kaufman, R.J. Orchestrating the unfolded protein response in health and disease. J. Clin. Investig. 2002, 110, 1389–1398. [Google Scholar] [CrossRef]
- Hajnóczky, G.; Davies, E.; Madesh, M. Calcium signaling and apoptosis. Biochem. Biophys. Res. Commun. 2003, 304, 445–454. [Google Scholar] [CrossRef]
- Ferri, K.F.; Kroemer, G. Organelle-specific initiation of cell death pathways. Nat. Cell Biol. 2001, 3, E255–E263. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Zhu, H.; Morishima, N.; Li, E.; Xu, J.; Yankner, B.A.; Yuan, J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-β. Nature 2000, 403, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Tu, H.; Nelson, O.; Bezprozvanny, A.; Wang, Z.; Lee, S.F.; Hao, Y.H.; Bezprozvanny, I. Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer’s disease-linked mutations. Cell 2006, 126, 981–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stutzmann, G.E.; Mattson, M.P. Endoplasmic Reticulum Ca2+ Handling in Excitable Cells in Health and Disease. Pharmacol. Rev. 2011, 63, 700–727. [Google Scholar] [CrossRef] [Green Version]
- Jayaraman, S.; Mensi, N.; Webb, D.R.; Dorf, M. Involvement of protein kinase C in competence induction of macrophages to generate T suppressor cells. J. Immunol. 1991, 146, 4085–4091. [Google Scholar]
- Csordás, G.; Várnai, P.; Golenár, T.; Roy, S.; Purkins, G.; Schneider, T.G.; Balla, T.; Hajnóczky, G. Imaging Interorganelle Contacts and Local Calcium Dynamics at the ER-Mitochondrial Interface. Mol. Cell 2010, 39, 121–132. [Google Scholar] [CrossRef]
- Giacomello, M.; Drago, I.; Bortolozzi, M.; Scorzeto, M.; Gianelle, A.; Pizzo, P.; Pozzan, T. Ca2+ Hot Spots on the Mitochondrial Surface Are Generated by Ca2+ Mobilization from Stores, but Not by Activation of Store-Operated Ca2+ Channels. Mol. Cell 2010, 38, 280–290. [Google Scholar] [CrossRef]
- Anderson, N.M.; Mucka, P.; Kern, J.G.; Feng, H. The emerging role and targetability of the TCA cycle in cancer metabolism. Protein Cell 2018, 9, 216–237. [Google Scholar] [CrossRef]
- Korge, P.; Weiss, J.N. Thapsigargin directly induces the mitochondrial permeability transition. JBIC J. Biol. Inorg. Chem. 1999, 265, 273–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akao, Y.; Maruyama, W.; Shimizu, S.; Yi, H.; Nakagawa, Y.; Shamoto-Nagai, M.; Youdim, M.B.H.; Tsujimoto, Y.; Naoi, M. Mitochondrial permeability transition mediates apoptosis induced by N-methyl(R)salsolinol, an endogenous neurotoxin, and is inhibited by Bcl-2 and rasagiline, N-propargyl-1(R)-aminoindan. J. Neurochem. 2002, 82, 913–923. [Google Scholar] [CrossRef] [PubMed]
- Kidd, J.F.; Pilkington, M.F.; Schell, M.J.; Fogarty, K.E.; Skepper, J.N.; Taylor, C.; Thorn, P. Paclitaxel Affects Cytosolic Calcium Signals by Opening the Mitochondrial Permeability Transition Pore. J. Biol. Chem. 2002, 277, 6504–6510. [Google Scholar] [CrossRef] [Green Version]
- Martinou, J.-C.; Desagher, S.; Antonsson, B. Cytochrome c release from mitochondria: All or nothing. Nat. Cell Biol. 2000, 2, E41–E43. [Google Scholar] [CrossRef]
- Joza, N.; Susin, S.A.; Daugas, E.; Stanford, W.L.; Cho, S.K.; Li, C.Y.J.; Sasaki, T.; Elia, A.J.; Cheng, H.-Y.M.; Ravagnan, L.; et al. Essential role of the mitochondrial apoptosis-inducing factor in programmed cell death. Nature 2001, 410, 549–554. [Google Scholar] [CrossRef]
- Verhagen, A.M.; Ekert, P.G.; Pakusch, M.; Silke, J.; Connolly, L.M.; Reid, G.E.; Vaux, D.L. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell 2000, 102, 43–53. [Google Scholar] [CrossRef] [Green Version]
- Parrish, J.; Li, L.; Klotz, K.; Ledwich, D.; Wang, X.; Xue, D. Mitochondrial endonuclease G is important for apoptosis in C. elegans. Nature 2001, 412, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Pinton, P.; Rizzuto, R. Bcl-2 and Ca2+ homeostasis in the endoplasmic reticulum. Cell Death Differ. 2006, 13, 1409–1418. [Google Scholar] [CrossRef]
- Pinton, P.; Ferrari, D.; Magalhães, P.; Schulze-Osthoff, K.; Di Virgilio, F.; Pozzan, T.; Rizzuto, R. Reduced Loading of Intracellular Ca2+ Stores and Downregulation of Capacitative Ca2+Influx in Bcl-2–Overexpressing Cells. J. Cell Biol. 2000, 148, 857–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scorrano, L.; Oakes, S.A.; Opferman, J.T.; Cheng, E.H.; Sorcinelli, M.D.; Pozzan, T.; Korsmeyer, S.J. BAX and BAK regulation of endoplasmic reticulum Ca2+: A control point for apoptosis. Science 2003, 300, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Kondratskyi, A.; Kondratska, K.; Skryma, R.; Klionsky, D.J.; Prevarskaya, N. Ion channels in the regulation of autophagy. Autophagy 2018, 14, 3–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parys, J.B.; Decuypere, J.-P.; Bultynck, G. Role of the inositol 1,4,5-trisphosphate receptor/Ca2+-release channel in autophagy. Cell Commun. Signal. 2012, 10, 17. [Google Scholar] [CrossRef] [Green Version]
- Decuypere, J.-P.; Welkenhuyzen, K.; Luyten, T.; Ponsaerts, R.; Dewaele, M.; Molgó, J.; Agostinis, P.; Missiaen, L.; De Smedt, H.; Parys, J.; et al. Ins(1,4,5)P3receptor-mediated Ca2+signaling and autophagy induction are interrelated. Autophagy 2011, 7, 1472–1489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decuypere, J.-P.; Monaco, G.; Bultynck, G.; Missiaen, L.; De Smedt, H.; Parys, J. The IP3 receptor–mitochondria connection in apoptosis and autophagy. Biochim. Biophys. Acta Bioenerg. 2011, 1813, 1003–1013. [Google Scholar] [CrossRef] [Green Version]
- Cárdenas, C.; Juretić, N.; Bevilacqua, J.A.; García, I.E.; Figueroa, R.; Hartley, R.; Taratuto, A.L.; Gejman, R.; Riveros, N.; Molgó, J.; et al. Abnormal distribution of inositol 1,4,5-trisphosphate receptors in human muscle can be related to altered calcium signals and gene expression in Duchenne dystrophy-derived cells. FASEB J. 2010, 24, 3210–3221. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.T.; Joseph, S.K. Role of Inositol Trisphosphate Receptors in Autophagy in DT40 Cells. J. Biol. Chem. 2010, 285, 16912–16920. [Google Scholar] [CrossRef] [Green Version]
- Williams, A.; Sarkar, S.; Cuddon, P.; Ttofi, E.K.; Saiki, S.; Siddiqi, F.H.; Rubinsztein, D.C. Novel targets for Huntington’s disease in an mTOR-independent autophagy pathway. Nat. Chem. Biol. 2008, 4, 295–305. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.-A.; Li, L.; Kakhlon, O.; Myerowitz, R.; Raben, N. Defects in calcium homeostasis and mitochondria can be reversed in Pompe disease. Autophagy 2015, 11, 385–402. [Google Scholar] [CrossRef] [Green Version]
- Pushparaj, C.; Das, A.; Purroy, R.; Nàger, M.; Herreros, J.; Pamplona, R.; Cantí, C. Voltage-gated calcium channel blockers deregulate macroautophagy in cardiomyocytes. Int. J. Biochem. Cell Biol. 2015, 68, 166–175. [Google Scholar] [CrossRef]
- Abdelmohsen, K.; Srikantan, S.; Tominaga, K.; Kang, M.-J.; Yaniv, Y.; Martindale, J.L.; Yang, X.; Park, S.-S.; Becker, K.; Subramanian, M.; et al. Growth Inhibition by miR-519 via Multiple p21-Inducing Pathways. Mol. Cell. Biol. 2012, 32, 2530–2548. [Google Scholar] [CrossRef] [Green Version]
- Tang, B.-D.; Xia, X.; Lv, X.-F.; Yu, B.-X.; Yuan, J.-N.; Mai, X.-Y.; Shang, J.-Y.; Zhou, J.-G.; Liang, S.-J.; Pang, R.-P. Inhibition of Orai1-mediated Ca2+entry enhances chemosensitivity of HepG2 hepatocarcinoma cells to 5-fluorouracil. J. Cell. Mol. Med. 2016, 21, 904–915. [Google Scholar] [CrossRef]
- Zheng, C.-B.; Gao, W.-C.; Xie, M.; Li, Z.; Ma, X.; Song, W.; Luo, D.; Huang, Y.; Yang, J.; Zhang, P.; et al. Ang II Promotes Cardiac Autophagy and Hypertrophy via Orai1/STIM1. Front. Pharmacol. 2021, 12, 622774. [Google Scholar] [CrossRef]
- Yang, J.; Yu, J.; Li, D.; Yu, S.; Ke, J.; Wang, L.; Wang, Y.; Qiu, Y.; Gao, X.; Zhang, J.; et al. Store-operated calcium entry-activated autophagy protects EPC proliferation via the CAMKK2-MTOR pathway in ox-LDL exposure. Autophagy 2017, 13, 82–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Z.-D.; Yu, T.; Liu, H.-J.; Jin, J.; He, J. SOCE induced calcium overload regulates autophagy in acute pancreatitis via calcineurin activation. Cell Death Dis. 2018, 9, 50. [Google Scholar] [CrossRef]
- Sukumaran, P.; Sun, Y.; Vyas, M.; Singh, B.B. TRPC1-mediated Ca2+ entry is essential for the regulation of hypoxia and nutrient depletion-dependent autophagy. Cell Death Dis. 2015, 6, e1674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sukumaran, P.; Sun, Y.; Zangbede, F.Q.; da Conceicao, V.N.; Mishra, B.; Singh, B.B. TRPC1 expression and function inhibit ER stress and cell death in salivary gland cells. FASEB BioAdv. 2019, 1, 40–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sukumaran, P.; Sun, Y.; Antonson, N.; Singh, B.B. Dopaminergic neurotoxins induce cell death by attenuating NF-κB-mediated regulation of TRPC1 expression and autophagy. FASEB J. 2018, 32, 1640–1652. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.S.; Hong, J.H.; Li, Q.; Shin, D.M.; Abramowitz, J.; Birnbaumer, L.; Muallem, S. Deletion of TRPC3 in Mice Reduces Store-Operated Ca2+ Influx and the Severity of Acute Pancreatitis. Gastroenterology 2009, 137, 1509–1517. [Google Scholar] [CrossRef] [Green Version]
- Oh, H.G.; Chun, Y.S.; Park, C.-S.; Kim, T.-W.; Park, M.K.; Chung, S. Regulation of basal autophagy by transient receptor potential melastatin 7 (TRPM7) channel. Biochem. Biophys. Res. Commun. 2015, 463, 7–12. [Google Scholar] [CrossRef]
- Castiglioni, S.; Romeo, V.; Locatelli, L.; Cazzaniga, A.; Maier, J.A.M. TRPM7 and MagT1 in the osteogenic differentiation of human mesenchymal stem cells in vitro. Sci. Rep. 2018, 8, 16195. [Google Scholar] [CrossRef]
- Miller, B.A.; Hoffman, N.E.; Merali, S.; Zhang, X.Q.; Wang, J.; Rajan, S.; Cheung, J.Y. TRPM2 channels protect against cardiac ischemia-reperfusion injury: Role of mitochondria. J. Biol. Chem. 2014, 289, 7615–7629. [Google Scholar] [CrossRef] [Green Version]
- Gao, G.; Wang, W.; Tadagavadi, R.K.; Briley, N.E.; Love, M.I.; Miller, B.A.; Reeves, W.B. TRPM2 mediates ischemic kidney injury and oxidant stress through RAC1. J. Clin. Investig. 2014, 124, 4989–5001. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Guo, W.; Hao, B.; Shi, X.; Lu, Y.; Wong, C.W.; Yue, J. Mechanistic study of TRPM2-Ca2+-CAMK2-BECN1 signaling in oxidative stress-induced autophagy inhibition. Autophagy 2016, 12, 1340–1354. [Google Scholar] [CrossRef] [Green Version]
- Farfariello, V.; Amantini, C.; Santoni, G. Transient receptor potential vanilloid 1 activation induces autophagy in thymocytes through ROS-regulated AMPK and Atg4C pathways. J. Leukoc. Biol. 2012, 92, 421–431. [Google Scholar] [CrossRef]
- Amantini, C.; Farfariello, V.; Cardinali, C.; Morelli, M.B.; Marinelli, O.; Nabissi, M.; Santoni, M.; Bonfili, L.; Cecarini, V.; Eleuteri, A.M.; et al. The TRPV1 ion channel regulates thymocyte differentiation by modulating autophagy and proteasome activity. Oncotarget 2017, 8, 90766–90780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amantini, C.; Morelli, M.B.; Nabissi, M.; Cardinali, C.; Santoni, M.; Gismondi, A.; Santoni, G. Capsaicin triggers autophagic cell survival which drives epithelial mesenchymal transition and chemoresistance in bladder cancer cells in an Hedgehog-dependent manner. Oncotarget 2016, 7, 50180–50194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, S.; Park, J.; An, I.; Jung, S.J.; Hwang, J. Transient receptor potential cation channel V1 (TRPV1) is degraded by starvation- and glucocorticoid-mediated autophagy. Mol. Cells 2014, 37, 257–263. [Google Scholar] [CrossRef] [Green Version]
- Santoni, G.; Amantini, C. The Transient Receptor Potential Vanilloid Type-2(TRPV2) Ion Channels in Neurogenesis andGliomagenesis: Cross-Talk between TranscriptionFactors and Signaling Molecules. Cancers 2019, 11, 322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nabissi, M.; Morelli, M.B.; Amantini, C.; Liberati, S.; Santoni, M.; Ricci-Vitiani, L.; Santoni, G. Cannabidiol stimulates Aml-1a-dependent glial differentiation and inhibits glioma stem-like cells proliferation by inducing autophagy in a TRPV2-dependent manner. Int. J. Cancer 2015, 137, 1855–1869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhan, L.; Yang, Y.; Ma, T.T.; Huang, C.; Meng, X.M.; Zhang, L.; Li, J. Transient receptor potential vanilloid 4 inhibits rat HSC-T6 apoptosis through induction of autophagy. Mol. Cell Biochem. 2015, 402, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Vrechi, T.A.; Leão, A.H.; Morais, I.B.; Abílio, V.C.; Zuardi, A.W.; Hallak JE, C.; Pereira, G.J. Cannabidiol induces autophagy via ERK1/2 activation in neural cells. Sci. Rep. 2021, 11, 5434. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Lin, J.; Zhang, J.; Tang, D.; Xiang, F.; Cui, L.; Zhang, Q.; Yuan, H.; Song, H.; Lv, Y.; et al. TRPV1 activation mitigates hypoxic injury in mouse cardiomyocytes by inducing autophagy through the AMPK signaling pathway. Am. J. Physiol. Physiol. 2020, 318, C1018–C1029. [Google Scholar] [CrossRef]
- Cao, B.; Dai, X.; Wang, W. Knockdown of TRPV4 suppresses osteoclast differentiation and osteoporosis by inhibiting autophagy through Ca2+–calcineurin–NFATc1 pathway. J. Cell. Physiol. 2019, 234, 6831–6841. [Google Scholar] [CrossRef]
- Zhong, G.; Long, H.; Chen, F.; Yu, Y. Oxoglaucine mediates Ca2+ influx and activates autophagy to alleviate osteoarthritis through the TRPV5/calmodulin/CAMK-II pathway. Br. J. Pharmacol. 2021, 178, 2931–2947. [Google Scholar] [CrossRef]
- Liu, J.-H.; Cao, Y.-M.; Rong, Z.-P.; Ding, J.; Pan, X. Trichostatin A Induces Autophagy in Cervical Cancer Cells by Regulating the PRMT5-STC1-TRPV6-JNK Pathway. Pharmacology 2021, 106, 60–69. [Google Scholar] [CrossRef]
- Wang, M.; Li, J.; Dong, S.; Cai, X.; Simaiti, A.; Yang, X.; Zhu, X.; Luo, J.; Jiang, L.-H.; Du, B.; et al. Silica nanoparticles induce lung inflammation in mice via ROS/PARP/TRPM2 signaling-mediated lysosome impairment and autophagy dysfunction. Part. Fibre Toxicol. 2020, 17, 1–22. [Google Scholar] [CrossRef]
- Huo, Y.; Chen, W.; Zheng, X.; Zhao, J.; Zhang, Q.; Hou, Y.; Jin, X. The protective effect of EGF-activated ROS in human corneal epithelial cells by inducing mitochondrial au-tophagy via activation TRPMJ2. Cell Physiol. 2020, 235, 7018–7029. [Google Scholar] [CrossRef]
- Oh, H.G.; Chung, S. Activation of transient receptor potential melastatin 7 (TRPM7) channel increases basal autophagy and reduces amyloid β-peptide. Biochem. Biophys. Res. Commun. 2017, 493, 494–499. [Google Scholar] [CrossRef]
- Ahamad, N.; Singh, B.B. Calcium channels and their role in regenerative medicine. World J. Stem Cells 2021, 13, 260–280. [Google Scholar] [CrossRef] [PubMed]
- Phadwal, K.; Watson, A.S.; Simon, A.K. Tightrope act: Autophagy in stem cell renewal, differentiation, proliferation, and aging. Cell. Mol. Life Sci. 2013, 70, 89–103. [Google Scholar] [CrossRef] [Green Version]
- Arakawa, S.; Tsujioka, M.; Yoshida, T.; Tajima-Sakurai, H.; Nishida, Y.; Matsuoka, Y.; Yoshino, I.; Tsujimoto, Y.; Shimizu, S. Role of Atg5-dependent cell death in the embryonic development of Bax/Bak double-knockout mice. Cell Death Differ. 2017, 24, 1598–1608. [Google Scholar] [CrossRef]
- Oakes, S.A.; Scorrano, L.; Opferman, J.T.; Bassik, M.C.; Nishino, M.; Pozzan, T.; Korsmeyer, S.J. Proapoptotic BAX and BAK regulate the type 1 inositol trisphosphate receptor and calcium leak from the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 2005, 102, 105–110. [Google Scholar] [CrossRef] [Green Version]
- Vicencio, J.M.; Ortiz, C.; Criollo, A.; Jones, A.W.; Kepp, O.; Galluzzi, L.; Joza, N.; Vitale, I.; Morselli, E.; Tailler, M.; et al. The inositol 1,4,5-trisphosphate receptor regulates autophagy through its interaction with Beclin 1. Cell Death Differ. 2009, 16, 1006–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snoeck, H. Calcium regulation of stem cells. EMBO Rep. 2020, 21, e50028. [Google Scholar] [CrossRef]
- Szabadkai, G.; Simoni, A.M.; Bianchi, K.; De Stefani, D.; Leo, S.; Wieckowski, M.R.; Rizzuto, R. Mitochondrial dynamics and calcium signaling. Biochem. Biophys. Acta 2006, 1763, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Vig, M.; Kinet, J.P. Calcium signaling in immue cells. Nat. Immunol. 2009, 10, 21–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, W.; He, M.-X.; McLeod, L.X.; He, Y.-W. Autophagy, a novel pathway to regulate calcium mobilization in T lymphocytes. Front. Immunol. 2013, 4, 179–182. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Shen, J.; Ran, Z. Emerging views of mitophagy in immunity and autoimmune diseases. Autophagy 2019, 16, 3–17. [Google Scholar] [CrossRef]
- Mayadas, T.N.; Cullere, X.; Lowell, C.A. The Multifaceted Functions of Neutrophils. Annu. Rev. Pathol. Mech. Dis. 2014, 9, 181–218. [Google Scholar] [CrossRef] [Green Version]
- Mitroulis, I.; Kourtzelis, I.; Kambas, K.; Rafail, S.; Chrysanthopoulou, A.; Speletas, M.; Ritis, K. Regulation of the autophagic machinery in human neutrophils. Eur. J. Immunol. 2010, 40, 1461–1472. [Google Scholar] [CrossRef]
- Yu, Y.; Sun, B. Autophagy-mediated regulation of neutrophils and clinical applications. Burn. Trauma 2020, 8, tkz001. [Google Scholar] [CrossRef] [Green Version]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef]
- Narni-Mancinelli, E.; Soudja, S.M.; Crozat, K.; Dalod, M.; Gounon, P.; Geissmann, F.; Lauvau, G. Inflammatory Monocytes and Neutrophils Are Licensed to Kill during Memory Responses In Vivo. PLoS Pathog. 2011, 7, e1002457. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Galluzzi, L.; Zitvogel, L.; Kroemer, G. Autophagy and Cellular Immune Responses. Immunity 2013, 39, 211–227. [Google Scholar] [CrossRef] [Green Version]
- Chauhan, A.; Sun, Y.; Sukumaran, P.; Zangbede FO, Q.; Jondle, C.N.; Sharma, A.; Mishra, B.B. M1 polarization is depedent on TRPC1-mediated calcium entry. iScience 2018, 8, 85–102. [Google Scholar] [CrossRef] [Green Version]
- Xia, H.; Green, D.R.; Zou, W. Autophagy in tumour immunity and therapy. Nat. Rev. Cancer 2021, 21, 281–297. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.S.; Kehrl, J.H. TRAF6 and A20 regulate lysine 63-linked ubiquitination of Beclin-1 to control TLR4-induced autophagy. Sci. Signal. 2010, 3, ra42. [Google Scholar] [CrossRef] [PubMed]
- Gong, T.; Liu, L.; Jiang, W.; Zhou, R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 2020, 20, 95–112. [Google Scholar] [CrossRef] [PubMed]
- Jia, W.; Pua, H.H.; Li, Q.-J.; He, Y.-W. Autophagy Regulates Endoplasmic Reticulum Homeostasis and Calcium Mobilization in T Lymphocytes. J. Immunol. 2010, 186, 1564–1574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parekh, V.V.; Wu, L.; Boyd, K.L.; Williams, J.A.; Gaddy, J.A.; Olivares-Villagómez, D.; Cover, T.; Zong, W.-X.; Zhang, J.; Van Kaer, L. Impaired Autophagy, Defective T Cell Homeostasis, and a Wasting Syndrome in Mice with a T Cell–Specific Deletion of Vps34. J. Immunol. 2013, 190, 5086–5101. [Google Scholar] [CrossRef]
- Miller, B.; Zhao, Z.; Stephenson, L.M.; Cadwell, K.; Pua, H.H.; Lee, H.K.; Mizushima, N.; Iwasaki, A.; He, Y.-W.; Swat, W.; et al. The autophagy geneATG5plays an essential role in B lymphocyte development. Autophagy 2008, 4, 309–314. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.J. Immunological function of Blimp-1 in dendritic cells and relevance to autoimmune diseases. Immunol. Res. 2015, 63, 113–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Martin, N.; Maldonado, P.; Gasparrini, F.; Frederico, B.; Aggarwal, S.; Gaya, M.; Tsui, C.; Burbage, M.; Keppler, S.J.; Montaner, B.; et al. A switch from canonical to noncanonical autophagy shapes B cell responses. Science 2017, 355, 641–647. [Google Scholar] [CrossRef]
- Ghislat, G.; Lawrence, T. Autophagy in dendritic cells. Cell. Mol. Immunol. 2018, 15, 944–952. [Google Scholar] [CrossRef] [PubMed]
- Baghdadi, M.; Yoneda, A.; Yamashina, T.; Nagao, H.; Komohara, Y.; Nagai, S.; Akiba, H.; Foretz, M.; Yoshiyama, H.; Kinoshita, I.; et al. TIM-4 Glycoprotein-Mediated Degradation of Dying Tumor Cells by Autophagy Leads to Reduced Antigen Presentation and Increased Immune Tolerance. Immunity 2013, 39, 1070–1081. [Google Scholar] [CrossRef] [Green Version]
- Alissafi, T.; Banos, A.; Boon, L.; Sparwasser, T.; Ghigo, A.; Wing, K.; Vassilopoulos, D.; Boumpas, D.; Chavakis, T.; Cadwell, K.; et al. Tregs restrain dendritic cell autophagy to ameliorate autoimmunity. J. Clin. Investig. 2017, 127, 2789–2804. [Google Scholar] [CrossRef] [Green Version]
- Vivier, E.; Tomasello, E.; Baratin, M.; Walzer, T.; Ugolini, S. Functions of natural killer cells. Nat. Immunol. 2008, 9, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Xia, P.; Huang, G.; Zhu, P.; Liu, J.; Ye, B.; Du, Y.; Fan, Z. FoxO1-mediated autophagy is required for NK cell development and innate immunity. Nat. Commun. 2016, 7, 11023. [Google Scholar] [CrossRef] [PubMed]
- Germic, N.; Frangež, Ž.; Yousefi, S.; Simon, H.-U. Regulation of the innate immune system by autophagy: Neutrophils, eosinophils, mast cells, NK cells. Cell Death Differ. 2019, 26, 703–714. [Google Scholar] [CrossRef]
- Frake, R.; Ricketts, T.; Menzies, F.M.; Rubinsztein, D.C. Autophagy and neurodegeneration. J. Clin. Investig. 2015, 125, 65–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Z.; Yang, B.; Mo, X.; Xiao, H. Mechanism and Regulation of Autophagy and Its Role in Neuronal Diseases. Mol. Neurobiol. 2014, 52, 1190–1209. [Google Scholar] [CrossRef] [PubMed]
- Schaeffer, V.; Lavenir, I.; Ozcelik, S.; Tolnay, M.; Winkler, D.T.; Goedert, M. Stimulation of autophagy reduces neurodegeneration in a mouse model of human tauopathy. Brain 2012, 135, 2169–2177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barmada, S.J.; Serio, A.; Arjun, A.; Bilican, B.; Daub, A.; Ando, D.; Tsvetkov, A.S.; Pleiss, M.; Li, X.; Peisach, D.; et al. Autophagy induction enhances TDP43 turnover and survival in neuronal ALS models. Nat. Chem. Biol. 2014, 10, 677–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikoletopoulou, V.; Papandreou, M.-E.; Tavernarakis, N. Autophagy in the physiology and pathology of the central nervous system. Cell Death Differ. 2015, 22, 398–407. [Google Scholar] [CrossRef] [Green Version]
- Boland, B.; Kumar, A.; Lee, S.; Platt, F.M.; Wegiel, J.; Yu, W.H.; Nixon, R.A. Autophagy induction and autophagosome clearance in neurons: Relationship to autophagic pathology in Alzheimer’s disease. J. Neurosci. 2008, 28, 6926–6937. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.-I.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef]
- Yoshii, S.; Kuma, A.; Akashi, T.; Hara, T.; Yamamoto, A.; Kurikawa, Y.; Itakura, E.; Tsukamoto, S.; Shitara, H.; Eishi, Y.; et al. Systemic Analysis of Atg5-Null Mice Rescued from Neonatal Lethality by Transgenic ATG5 Expression in Neurons. Dev. Cell 2016, 39, 116–130. [Google Scholar] [CrossRef] [Green Version]
- Saitsu, H.; Nishimura, T.; Muramatsu, K.; Kodera, H.; Kumada, S.; Sugai, K.; Matsumoto, N. De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neuro-degeneration in adulthood. Nat. Genet. 2013, 45, 445–449. [Google Scholar] [CrossRef]
- Kim, M.; Sandford, E.; Gatica, D.; Qiu, Y.; Liu, X.; Zheng, Y.; Schulman, B.; Xu, J.; Semple, I.; Ro, S.-H.; et al. Mutation in ATG5 reduces autophagy and leads to ataxia with developmental delay. eLife 2016, 5, 5. [Google Scholar] [CrossRef] [Green Version]
- Moran, M.M.; McAlexander, M.A.; Bíró, T.; Szallasi, A. Tranisent receptor potrential channels as therapeutics tragets. Nat. Rev. Drug. Discov. 2012, 10, 601–620. [Google Scholar] [CrossRef]
- Descloux, C.; Ginet, V.; Clarke, P.G.H.; Puyal, J.; Truttmann, A.C. Neuronal death after perinatal cerebral hypoxia-ischemia: Focus on autophagy—mediated cell death. Int. J. Dev. Neurosci. 2015, 45, 75–85. [Google Scholar] [CrossRef]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef]
- Chu, C.T. Mechanisms of selective autophagy and mitophagy: Implications for neurodegenerative diseases. Neurobiol. Dis. 2019, 122, 23–34. [Google Scholar] [CrossRef]
- Boland, B.; Yu, W.H.; Corti, O.; Mollereau, B.; Henriques, A.; Bezard, E.; Pastores, G.M.; Rubinsztein, D.C.; Nixon, R.A.; Duchen, M.; et al. Promoting the clearance of neurotoxic proteins in neurodegenerative disorders of ageing. Nat. Rev. Drug Discov. 2018, 17, 660–688. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A.; Wegiel, J.; Kumar, A.; Yu, W.H.; Peterhoff, C.; Cataldo, A.; Cuervo, A.M. Extensive Involvement of Autophagy in Alzheimer Disease: An Immuno-Electron Microscopy Study. J. Neuropathol. Exp. Neurol. 2005, 64, 113–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilsson, P.; Loganathan, K.; Sekiguchi, M.; Matsuba, Y.; Hui, K.; Tsubuki, S.; Saido, T.C. Aβ secretion and plaque formation depend on autophagy. Cell Rep. 2013, 5, 61–69. [Google Scholar] [CrossRef] [Green Version]
- Djajadikerta, A.; Keshri, S.; Pavel, M.; Prestil, R.; Ryan, L.; Rubinsztein, D.C. Autophagy Induction as a Therapeutic Strategy for Neurodegenerative Diseases. J. Mol. Biol. 2020, 432, 2799–2821. [Google Scholar] [CrossRef] [PubMed]
- François, A.; Bilan, A.R.; Quellard, N.; Fernandez, B.; Janet, T.; Chassaing, D.; Paccalin, M.; Terro, F.; Page, G. Longitudinal follow-up of autophagy and inflammation in brain of APPswePS1dE9 transgenic mice. J. Neuroinflamm. 2014, 11, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreau, K.; Fleming, A.; Imarisio, S.; Ramirez, A.L.; Mercer, J.L.; Jimenez-Sanchez, M.; Bento, C.F.; Puri, C.; Zavodszky, E.; Siddiqi, F.H.; et al. PICALM modulates autophagy activity and tau accumulation. Nat. Commun. 2014, 5, 4998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ando, K.; Brion, J.-P.; Stygelbout, V.; Suain, V.; Authelet, M.; Dedecker, R.; Chanut, A.; Lacor, P.; Lavaur, J.; Sazdovitch, V.; et al. Clathrin adaptor CALM/PICALM is associated with neurofibrillary tangles and is cleaved in Alzheimer’s brains. Acta Neuropathol. 2013, 125, 861–878. [Google Scholar] [CrossRef]
- Lee, M.; Slunt, H.H.; Martin, L.J.; Thinakaran, G.; Kim, G.; Gandy, S.E.; Seeger, M.; Koo, E.; Price, D.L.; Sisodia, S.S. Expression of Presenilin 1 and 2 (PS1 and PS2) in Human and Murine Tissues. J. Neurosci. 1996, 16, 7513–7525. [Google Scholar] [CrossRef] [Green Version]
- Cao, L.-L.; Guan, P.-P.; Liang, Y.-Y.; Huang, X.-S.; Wang, P. Calcium Ions Stimulate the Hyperphosphorylation of Tau by Activating Microsomal Prostaglandin E Synthase 1. Front. Aging Neurosci. 2019, 11, 108. [Google Scholar] [CrossRef] [Green Version]
- Anglade, P.; Vyas, S.; Javoy-Agid, F.; Herrero, M.T.; Michel, P.P.; Marquez, J.; Agid, Y. Apoptosis and autophagy in nigral neurons of patients with Parkinson’s disease. Histol. Histopathol. 1997, 12, 25–31. [Google Scholar]
- Lachenmayer, M.L.; Yue, Z. Genetic animal models for evaluating the role of autophagy in etiopathogenesis of Parkinson disease. Autophagy 2012, 8, 1837–1838. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhang, H.; Selvaraj, S.; Sukumaran, P.; Lei, S.; Birnbaumer, L.; Singh, B.B. Inhibition of Cav1.3 channels by STIM1 in dopaminergic neurons is regulated by TRPC1. J. Neurosci. 2017, 37, 3364–3377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bollimuntha, S.; Singh, B.B.; Shavali, S.; Sharma, S.K.; Ebadi, M. TRPC1-mediated inhibition of MPP+ neurotoxicity in human SH-SY5Y neuroblastoma cells. J. Biol. Chem. 2005, 280, 2132–2140. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Xie, Z.; Turkson, S.; Zhuang, X. A53T human α-synuclein overexpression in transgenic mice induces pervasive mitochondria macroau-tophagy defects preceding dopamine neuron degeneration. J. Neurosci. 2015, 35, 890–905. [Google Scholar] [CrossRef] [PubMed]
- Song, J.X.; Lu, J.H.; Liu, L.F.; Chen, L.L.; Durairajan, S.S.K.; Yue, Z.; Li, M. HMGB1 is involved in autophagy inhibition caused by SNCA/α-synuclein overexpression: A process modulated by the natural autophagy inducer corynoxine B. Autophagy 2014, 10, 144–154. [Google Scholar] [CrossRef] [Green Version]
- Dehay, B.; Ramirez, A.; Martinez-Vicente, M.; Perier, C.; Canron, M.-H.; Doudnikoff, E.; Vital, A.; Vila, M.; Klein, C.; Bezard, E. Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration. Proc. Natl. Acad. Sci. USA 2012, 109, 9611–9616. [Google Scholar] [CrossRef] [Green Version]
- Rideout, H.J.; Stefanis, L. The Neurobiology of LRRK2 and its Role in the Pathogenesis of Parkinson’s Disease. Neurochem. Res. 2014, 39, 576–592. [Google Scholar] [CrossRef] [PubMed]
- Cherra, S.J., III; Steer, E.; Gusdon, A.M.; Kiselyov, K.; Chu, C.T. Mutant LRRK2 Elicits Calcium Imbalance and Depletion of Dendritic Mitochondria in Neurons. Am. J. Pathol. 2013, 182, 474–484. [Google Scholar] [CrossRef] [Green Version]
- Gandhi, S.; Wood-Kaczmar, A.; Yao, Z.; Plun-Favreau, H.; Deas, E.; Klupsch, K.; Abramov, A.Y. PINK1-Associated Parkinson’s Disease Is Caused by Neuronal Vulnerability to Calcium-Induced Cell Death. Mol. Cell 2009, 33, 627–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, P.; Dawson, V.L.; Dawson, T.M. PINK1 and Parkin mitochondrial quality control: A source of regional vulnerability in Parkinson’s disease. Mol. Neurodegener. 2020, 15, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geisler, S.; Holmström, K.; Treis, A.; Skujat, D.; Weber, S.S.; Fiesel, F.; Kahle, P.J.; Springer, W. The PINK1/Parkin-mediated mitophagy is compromised by PD-associated mutations. Autophagy 2010, 6, 871–878. [Google Scholar] [CrossRef] [Green Version]
- Tellez-Nagel, I.; Johnson, A.B.; Terry, R.D. Studies on brain biopsies of patients with Huntington’s chorea. J. Neuropathol. Exp. Neurol. 1974, 33, 308–332. [Google Scholar] [CrossRef]
- Ravikumar, B.; Vacher, C.; Berger, Z.; Davies, J.E.; Luo, S.; Oroz, L.G.; Scaravilli, F.; Easton, D.F.; Duden, R.; O’Kane, C.J.; et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 2004, 36, 585–595. [Google Scholar] [CrossRef] [Green Version]
- Menzies, F.M.; Garcia-Arencibia, M.; Imarisio, S.; O’Sullivan, N.C.; Ricketts, T.; Kent, B.A.; Rao, M.V.; Lam, W.; Green-Thompson, Z.W.; Nixon, R.A.; et al. Calpain inhibition mediates autophagy-dependent protection against polyglutamine toxicity. Cell Death Differ. 2015, 22, 433–444. [Google Scholar] [CrossRef] [Green Version]
- Mealer, R.G.; Murray, A.J.; Shahani, N.; Subramaniam, S.; Snyder, S.H. Rhes, a Striatal-selective Protein Implicated in Huntington Disease, Binds Beclin-1 and Activates Autophagy. J. Biol. Chem. 2014, 289, 3547–3554. [Google Scholar] [CrossRef] [Green Version]
- Tedeschi, V.; Petrozziello, T.; Secondo, A. Calcium Dyshomeostasis and Lysosomal Ca2+ Dysfunction in Amyotrophic Lateral Sclerosis. Cells 2019, 8, 1216. [Google Scholar] [CrossRef] [Green Version]
- Turner, M.; Hardiman, O.; Benatar, M.; Brooks, B.R.; Chio, A.; de Carvalho, M.; Ince, P.G.; Lin, C.S.-Y.; Miller, R.G.; Mitsumoto, H.; et al. Controversies and priorities in amyotrophic lateral sclerosis. Lancet Neurol. 2013, 12, 310–322. [Google Scholar] [CrossRef] [Green Version]
- Li, S.H.; Li, X.J. Huntingtin-protein interactions and the pathogenesis of Huntington’s disease. Trends Genet. 2004, 20, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Gal, J.; Ström, A.-L.; Kilty, R.; Zhang, F.; Zhu, H. p62 Accumulates and Enhances Aggregate Formation in Model Systems of Familial Amyotrophic Lateral Sclerosis. J. Biol. Chem. 2007, 282, 11068–11077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, A.S.; Holzbaur, E.L.F. Dynamic recruitment and activation of ALS-associated TBK1 with its target optineurin are required for efficient mitophagy. Proc. Natl. Acad. Sci. USA 2016, 113, E3349–E3358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Rademakers, R. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromo-some 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sukumaran, P.; Nascimento Da Conceicao, V.; Sun, Y.; Ahamad, N.; Saraiva, L.R.; Selvaraj, S.; Singh, B.B. Calcium Signaling Regulates Autophagy and Apoptosis. Cells 2021, 10, 2125. https://doi.org/10.3390/cells10082125
Sukumaran P, Nascimento Da Conceicao V, Sun Y, Ahamad N, Saraiva LR, Selvaraj S, Singh BB. Calcium Signaling Regulates Autophagy and Apoptosis. Cells. 2021; 10(8):2125. https://doi.org/10.3390/cells10082125
Chicago/Turabian StyleSukumaran, Pramod, Viviane Nascimento Da Conceicao, Yuyang Sun, Naseem Ahamad, Luis R Saraiva, Senthil Selvaraj, and Brij B Singh. 2021. "Calcium Signaling Regulates Autophagy and Apoptosis" Cells 10, no. 8: 2125. https://doi.org/10.3390/cells10082125
APA StyleSukumaran, P., Nascimento Da Conceicao, V., Sun, Y., Ahamad, N., Saraiva, L. R., Selvaraj, S., & Singh, B. B. (2021). Calcium Signaling Regulates Autophagy and Apoptosis. Cells, 10(8), 2125. https://doi.org/10.3390/cells10082125