
The Spatiotemporal Expression of Notch1 and Numb and Their Functional Interaction during Cardiac Morphogenesis

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mouse
2.2. Paraffin and Frozen Section Immunofluorescence
2.3. Cardiomyocyte Proliferation Assay via BrdU Pulse Labeling
2.4. mRNA In Situ Hybridization
2.5. Imaging
2.6. Western Blot Analysis
2.7. Trabecular Density and Thickness Quantification and IF Staining
2.8. RNA Isolation and Quantitative PCR (Q-PCR)
2.9. Statistical Analysis
3. Results
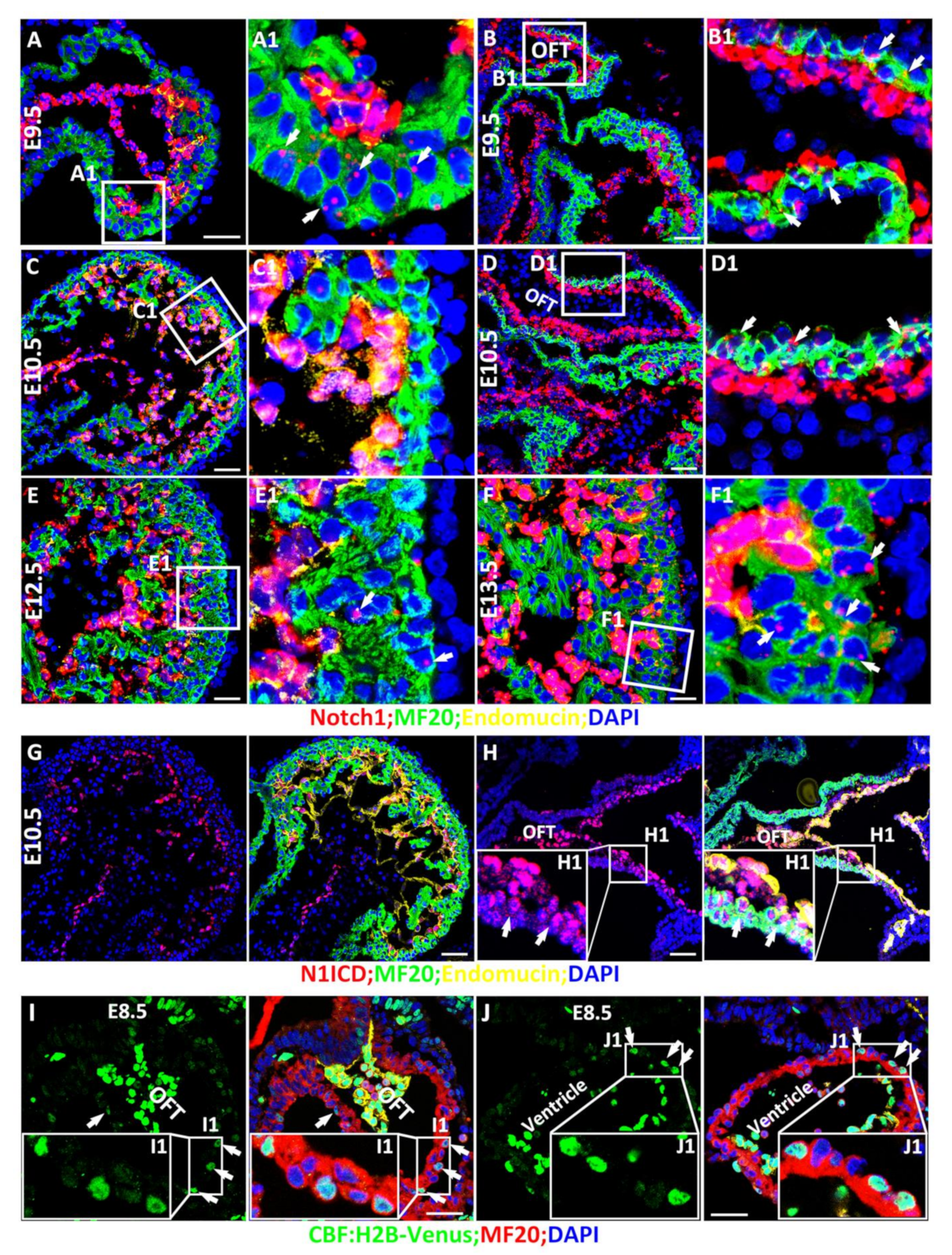
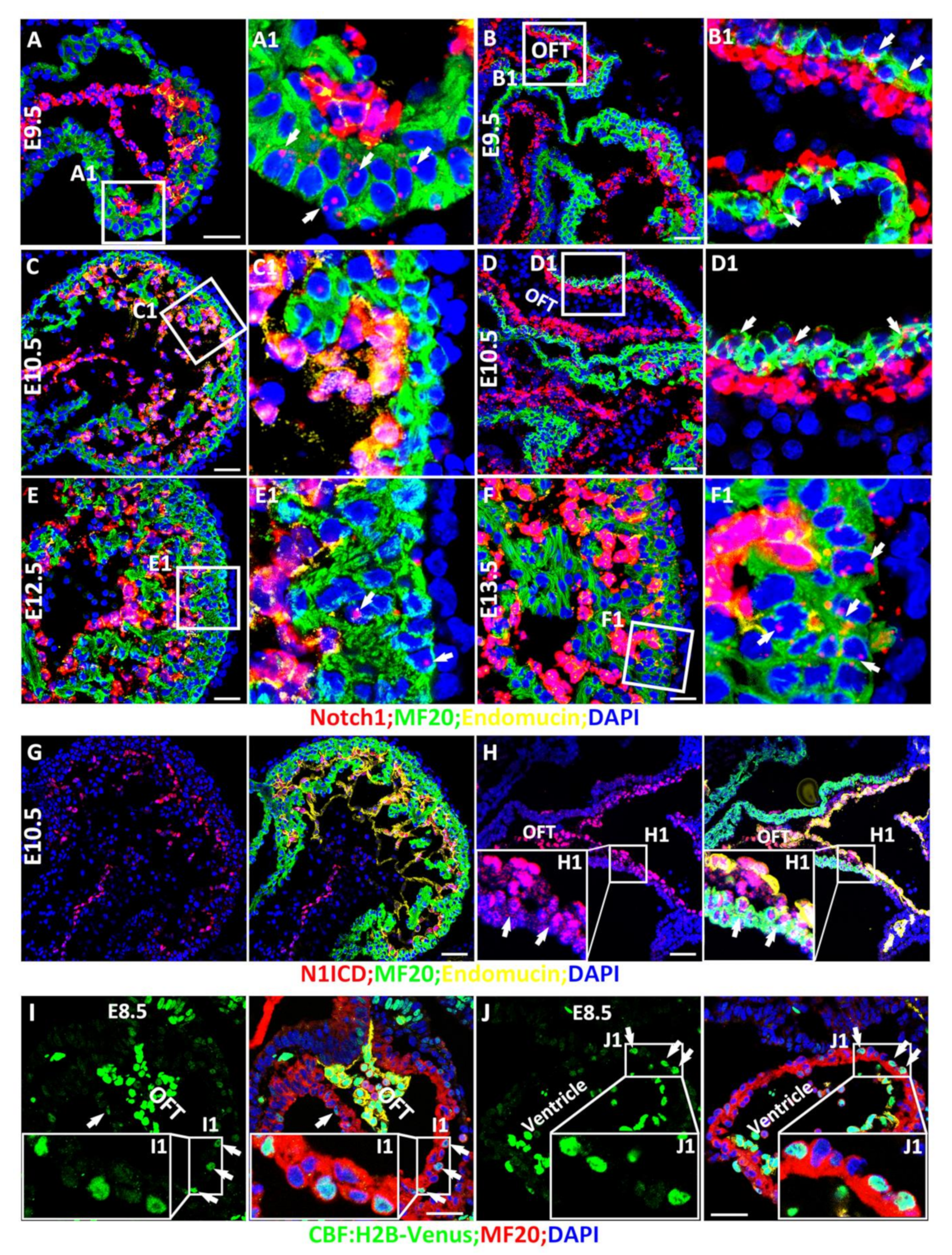
3.1. Spatiotemporal Expression and Activation of Notch1 during Cardiac Morphogenesis
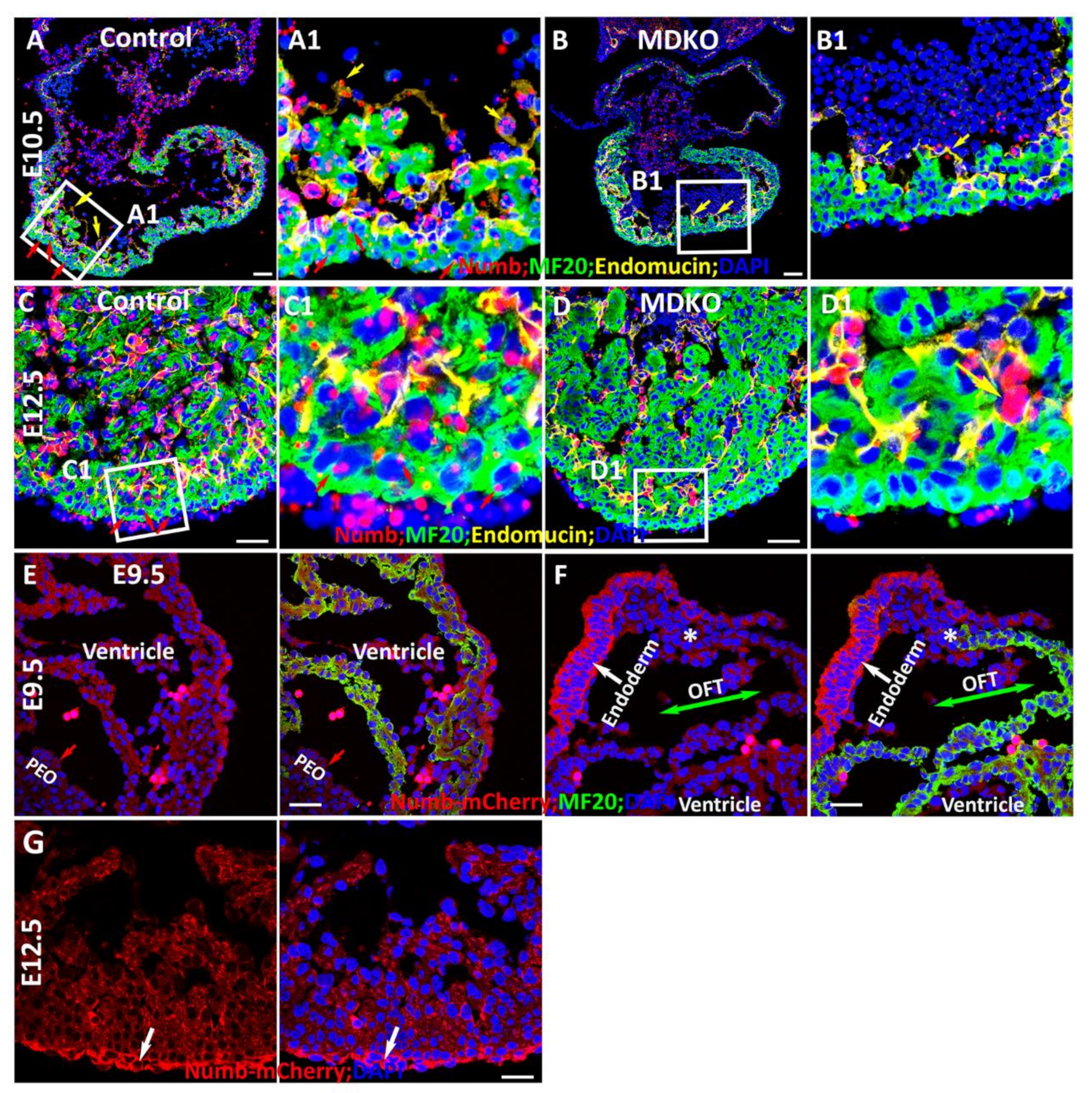
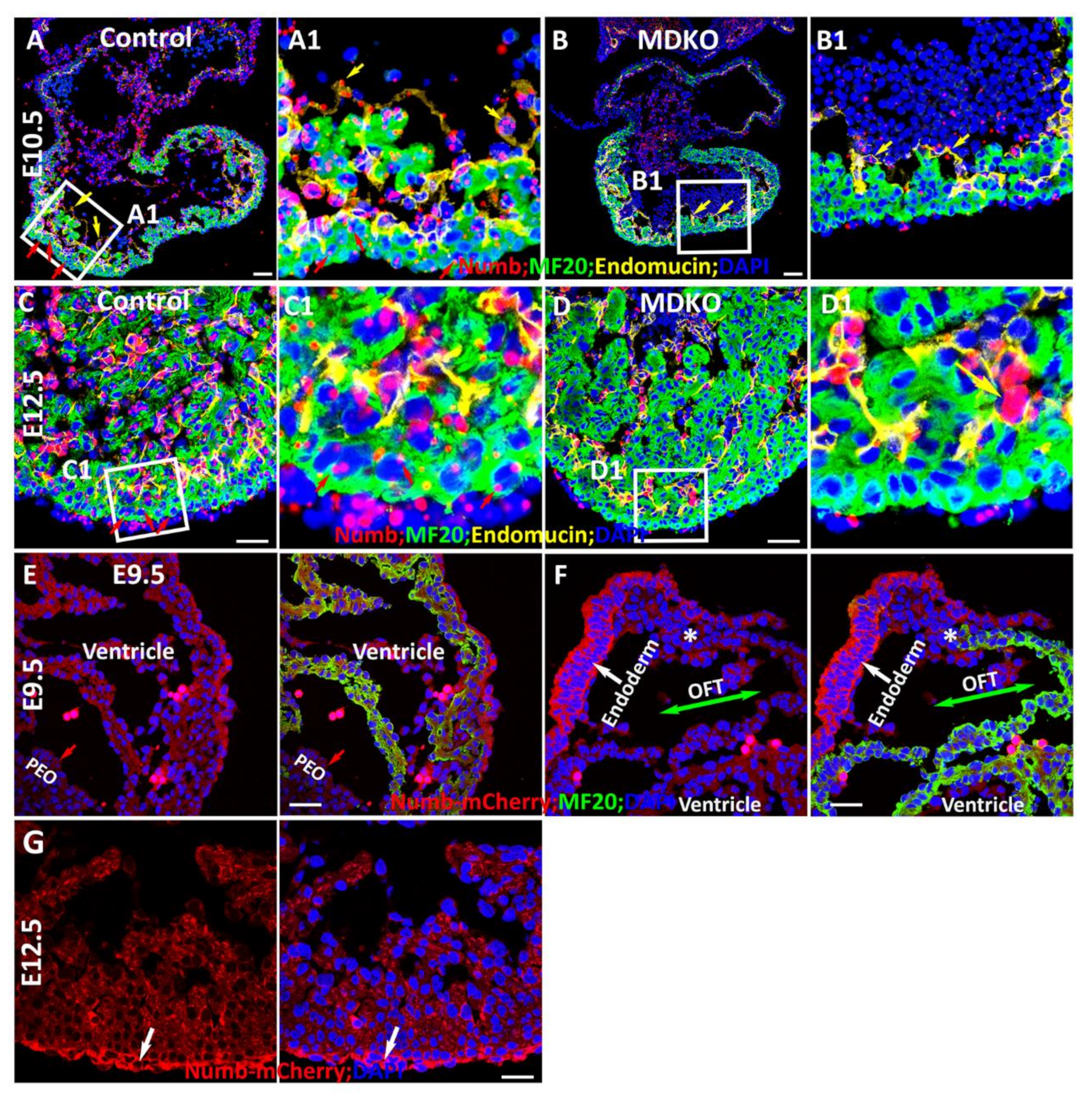
3.2. Spatiotemporal Expression of Numb during Myocardial Morphogenesis
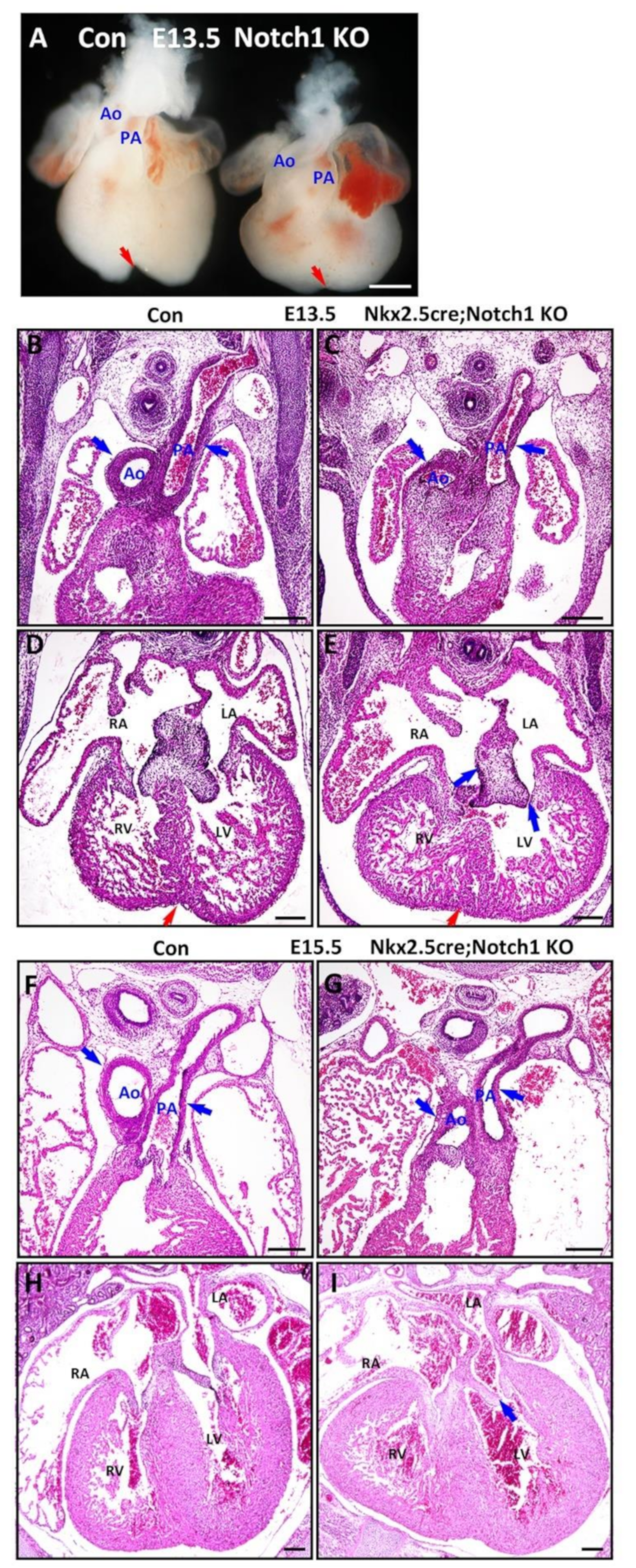
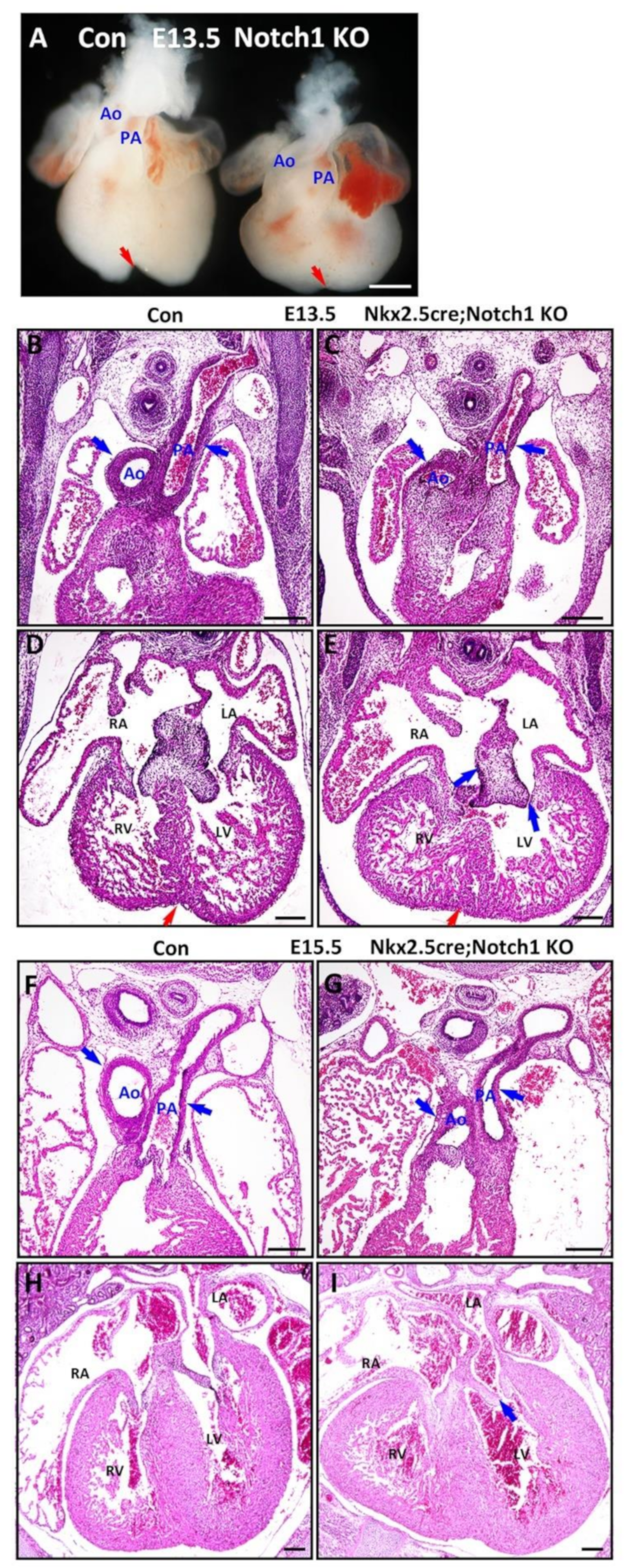
3.3. Notch1 Is Required for Outflow Tract Clockwise Rotation and Septation
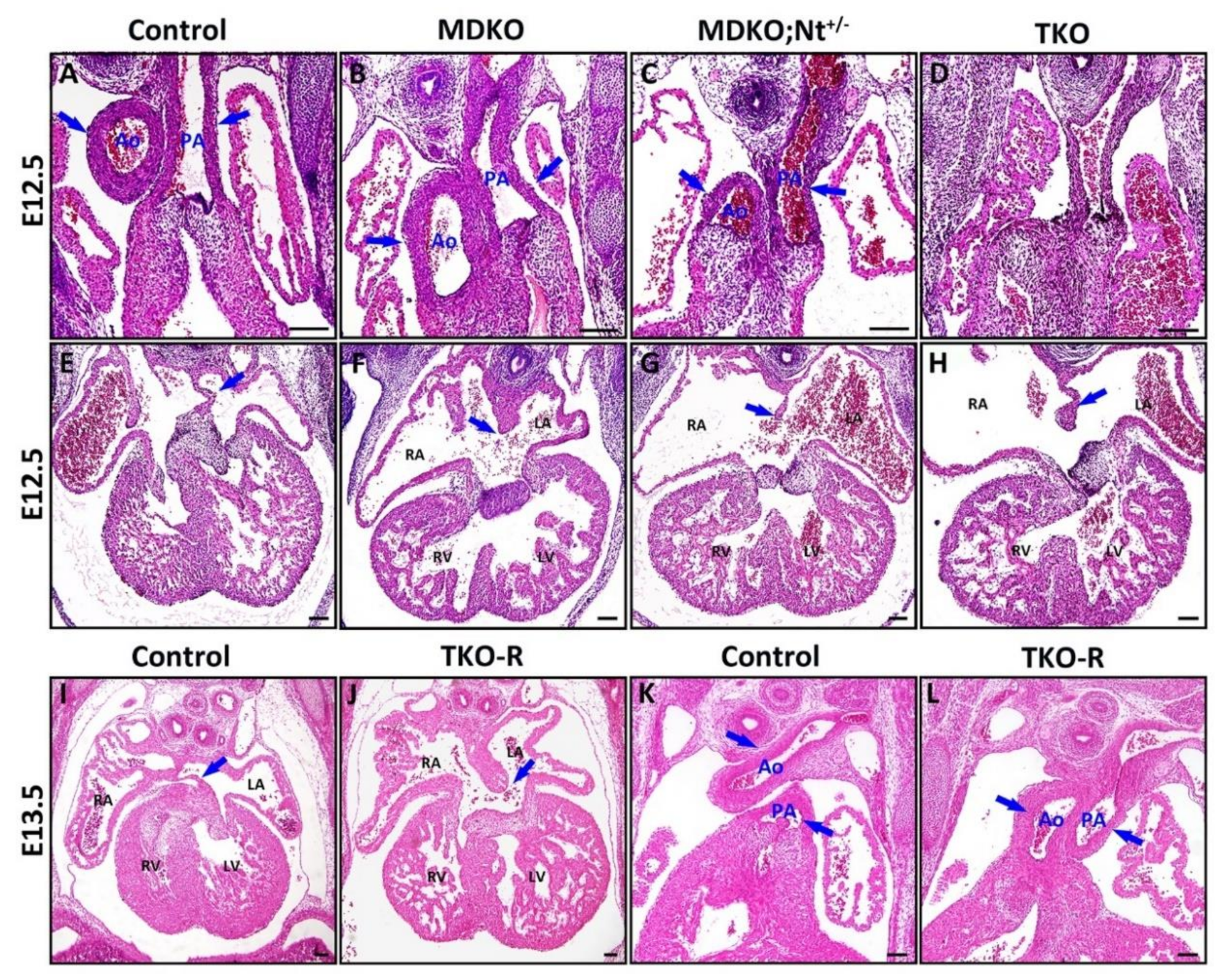
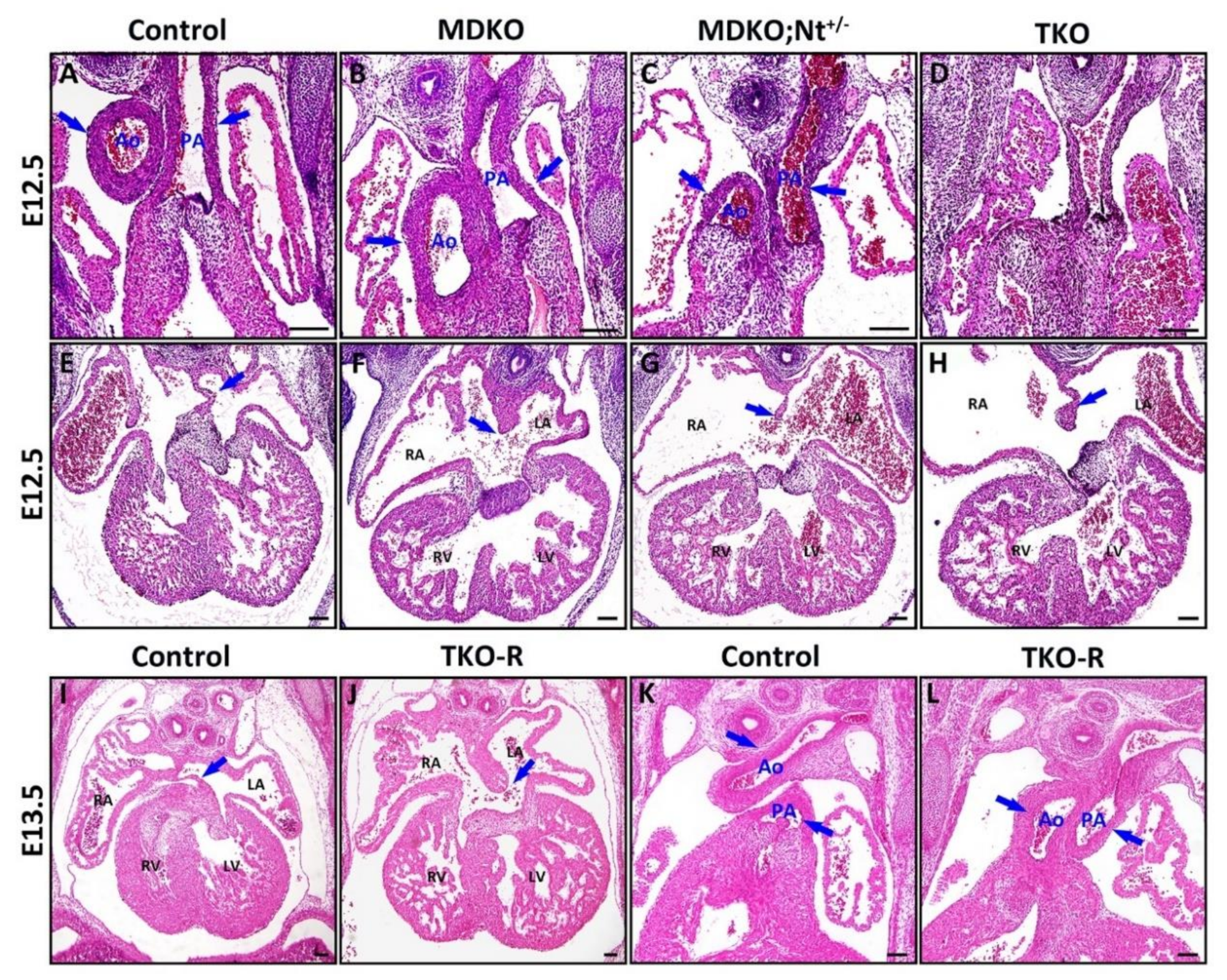
3.4. Notch1 or RBPJk Deletion in MDKO Did Not Rescue the Structural Defects of MDKO
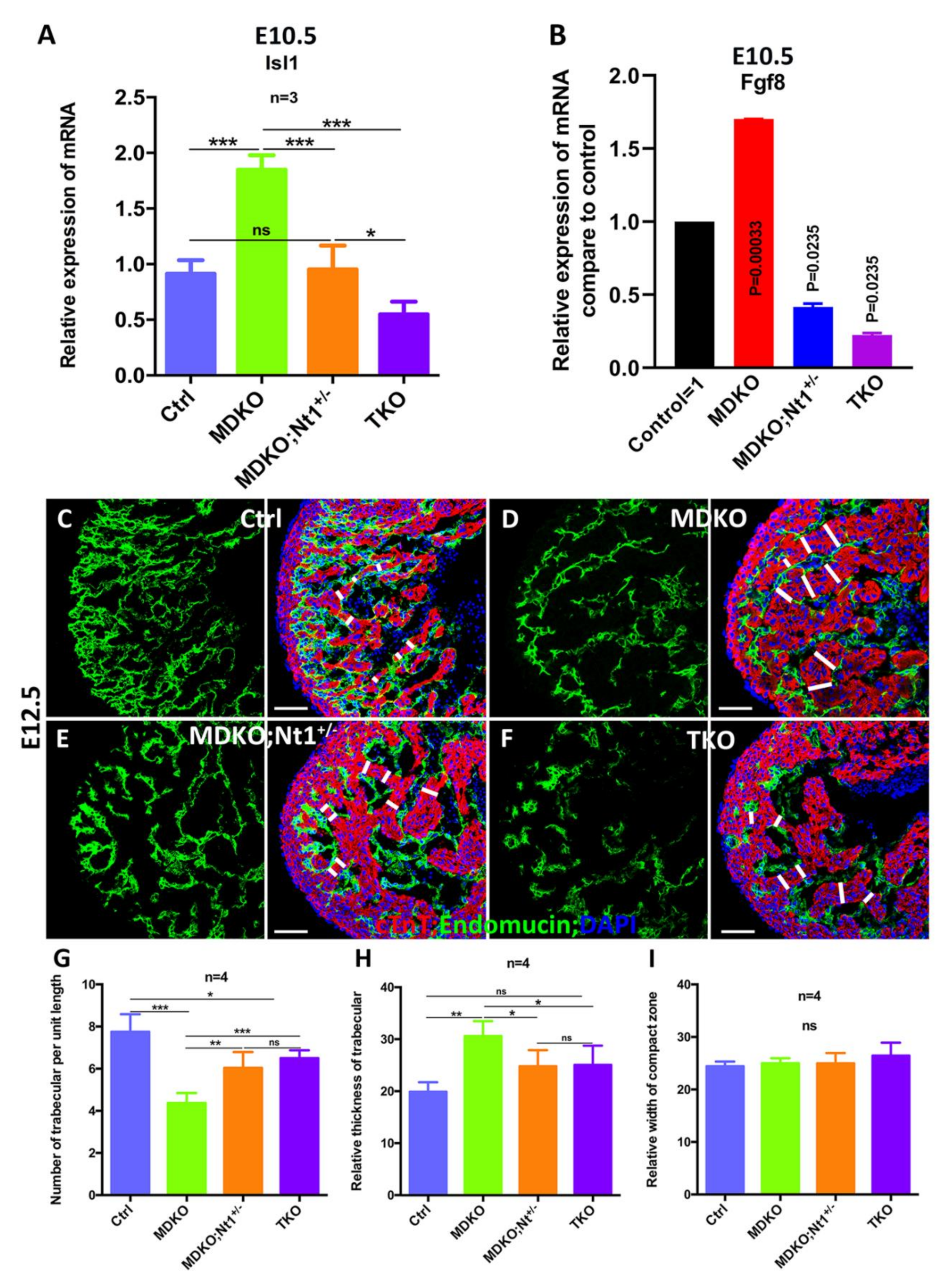
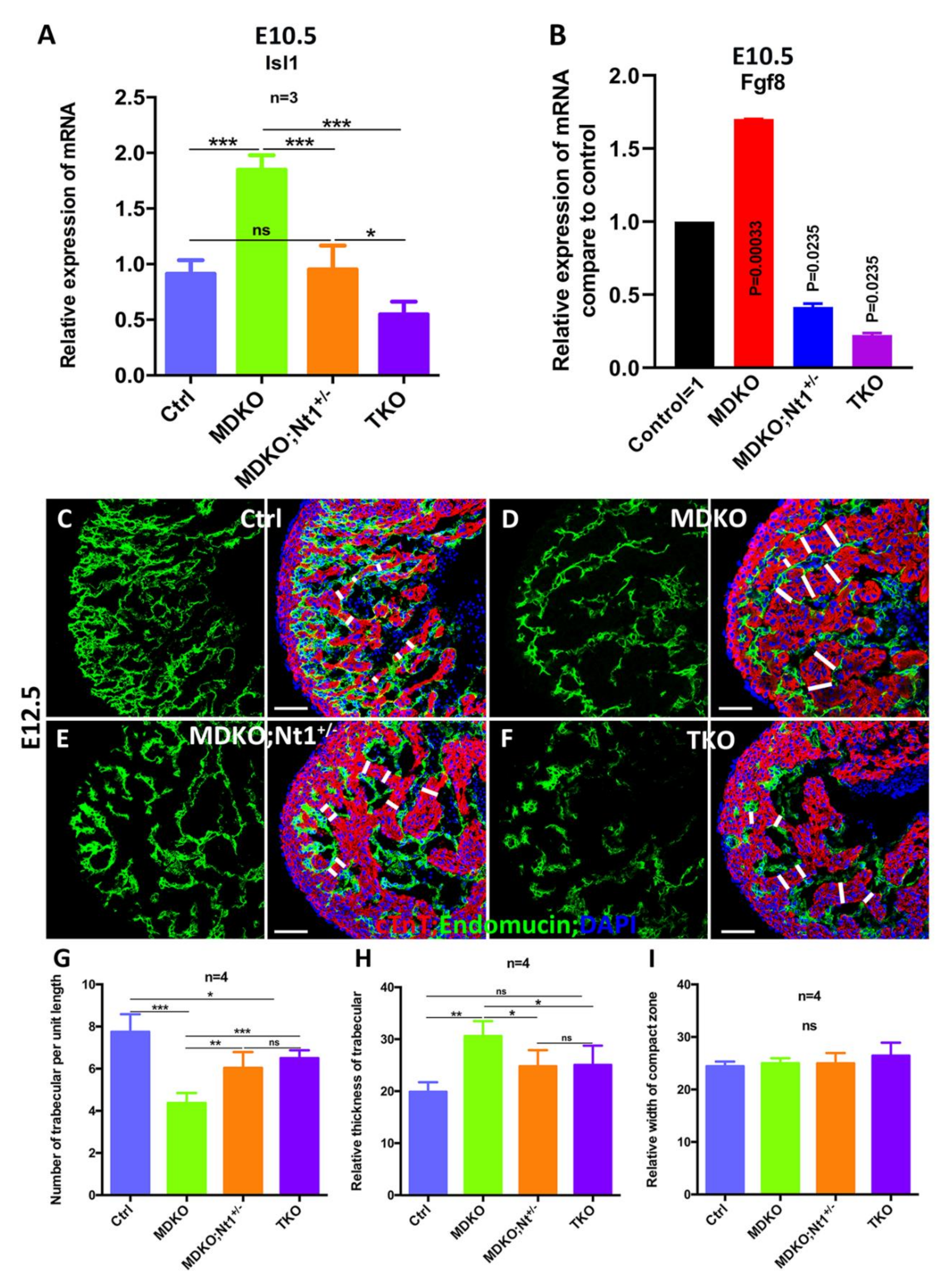
3.5. NFPs Regulate Progenitor Cell Differentiation and Trabecular Morphogenesis Partially through Notch1
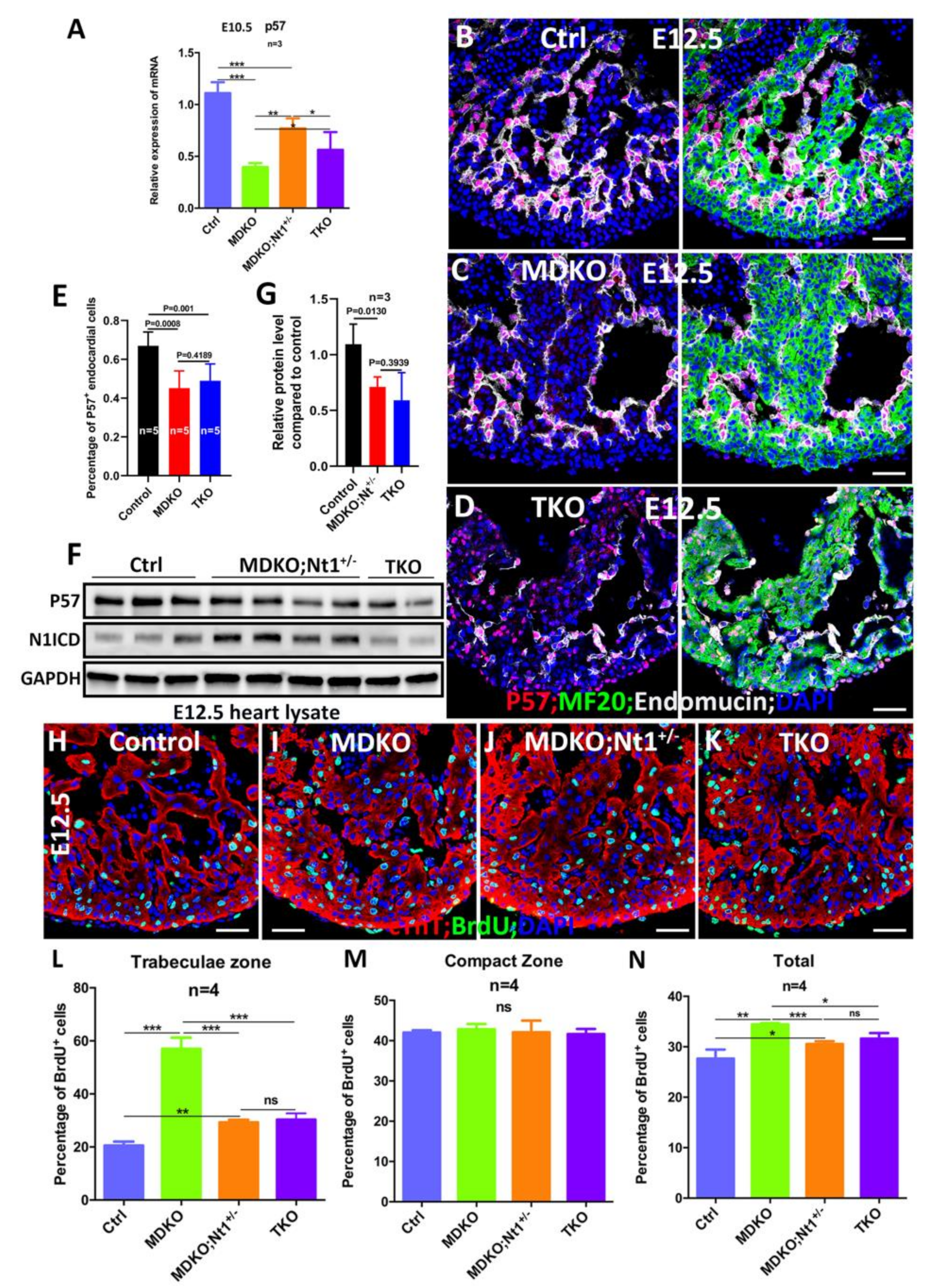
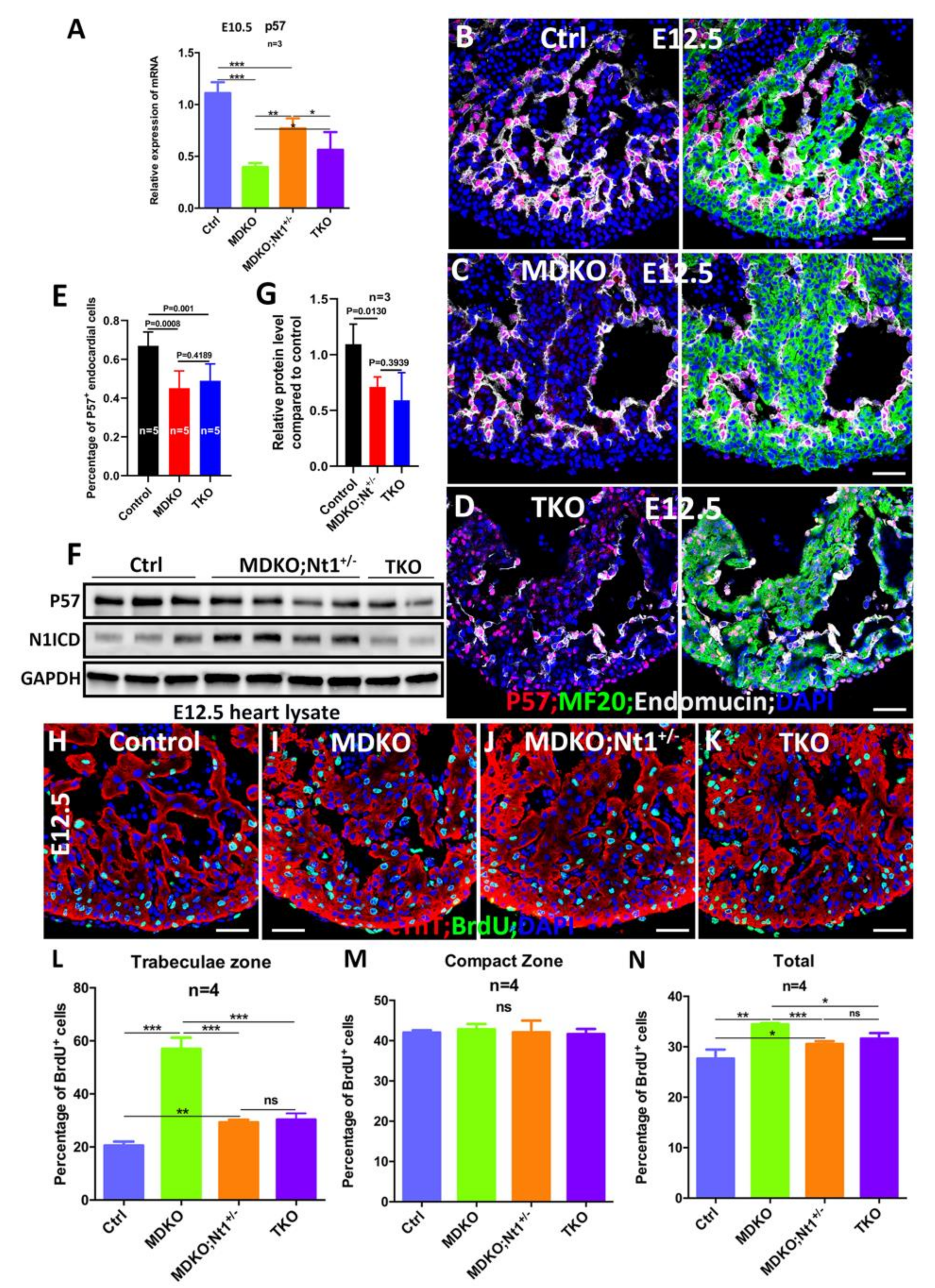
3.6. NFPs Regulate p57 Partially through Notch1
4. Discussion
4.1. NFPs Play Multiple Roles in a Cell Type-Dependent Manner during Cardiac Morphogenesis
4.2. NFPs Regulate Cardiac Morphogenesis via Multiple Molecules/Signaling Pathways
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhong, W.; Feder, J.N.; Jiang, M.-M.; Jan, L.; Jan, Y.N. Asymmetric Localization of a Mammalian Numb Homolog during Mouse Cortical Neurogenesis. Neuron 1996, 17, 43–53. [Google Scholar] [CrossRef] [Green Version]
- Guo, M.; Jan, L.; Jan, Y.N. Control of Daughter Cell Fates during Asymmetric Division: Interaction of Numb and Notch. Neuron 1996, 17, 27–41. [Google Scholar] [CrossRef] [Green Version]
- Spana, E.P.; Doe, C.Q. Numb Antagonizes Notch Signaling to Specify Sibling Neuron Cell Fates. Neuron 1996, 17, 21–26. [Google Scholar] [CrossRef] [Green Version]
- Han, Z.; Bodmer, R. Myogenic cells fates are antagonized by Notch only in asymmetric lineages of the Drosophila heart, with or without cell division. Development 2003, 130, 3039–3051. [Google Scholar] [CrossRef] [Green Version]
- Park, M.; Yaich, L.; Bodmer, R. Mesodermal cell fate decisions in Drosophila are under the control of the lineage genes numb, Notch, and sanpodo. Mech. Dev. 1998, 75, 117–126. [Google Scholar] [CrossRef]
- Niikura, Y.; Tabata, Y.; Tajima, A.; Inoue, I.; Arai, K.-I.; Watanabe, S. Zebrafish Numb homologue: Phylogenetic evolution and involvement in regulation of left–right asymmetry. Mech. Dev. 2006, 123, 407–414. [Google Scholar] [CrossRef]
- Zhong, W.; Jiang, M.; Weinmaster, G.; Jan, L.; Jan, Y.N. Differential expression of mammalian Numb, Numblike and Notch1 suggests distinct roles during mouse cortical neurogenesis. Development 1997, 124, 1887–1897. [Google Scholar] [CrossRef] [PubMed]
- Verdi, J.M.; Schmandt, R.; Bashirullah, A.; Jacob, S.; Salvino, R.; Craig, C.G.; Program, A.E.; Lipshitz, H.D.; McGlade, C. Mammalian NUMB is an evolutionarily conserved signaling adapter protein that specifies cell fate. Curr. Biol. 1996, 6, 1134–1145. [Google Scholar] [CrossRef] [Green Version]
- Petersen, P.H.; Zou, K.; Hwang, J.K.; Jan, Y.N.; Zhong, W. Progenitor cell maintenance requires numb and numblike during mouse neurogenesis. Nature 2002, 419, 929–934. [Google Scholar] [CrossRef]
- Wu, M.; Kwon, H.Y.; Rattis, F.; Blum, J.; Zhao, C.; Ashkenazi, R.; Jackson, T.L.; Gaiano, N.; Oliver, T.; Reya, T. Imaging Hematopoietic Precursor Division in Real Time. Cell Stem Cell 2007, 1, 541–554. [Google Scholar] [CrossRef] [Green Version]
- Conboy, I.M.; Rando, T.A. The Regulation of Notch Signaling Controls Satellite Cell Activation and Cell Fate Determination in Postnatal Myogenesis. Dev. Cell 2002, 3, 397–409. [Google Scholar] [CrossRef] [Green Version]
- Ito, T.; Kwon, H.Y.; Zimdahl, B.; Congdon, K.L.; Blum, J.; Lento, W.E.; Zhao, C.; Lagoo, A.; Gerrard, G.; Foroni, L.; et al. Regulation of myeloid leukaemia by the cell-fate determinant Musashi. Nature 2010, 466, 765–768. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.; Huber, T.L.; Chen, V.C.; Gadue, P.; Keller, G.M. Numb mediates the interaction between Wnt and Notch to modulate primitive erythropoietic specification from the hemangioblast. Development 2008, 135, 3447–3458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dho, S.E.; French, M.B.; Woods, S.A.; McGlade, C.J. Characterization of four mammalian numb protein isoforms. Identification of cytoplasmic and membrane-associated variants of the phosphotyrosine binding domain. J. Biol. Chem. 1999, 274, 33097–33104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGill, M.A.; McGlade, C.J. Mammalian Numb Proteins Promote Notch1 Receptor Ubiquitination and Degradation of the Notch1 Intracellular Domain. J. Biol. Chem. 2003, 278, 23196–23203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Šestan, N.; Artavanis-Tsakonas, S.; Rakic, P. Contact-Dependent Inhibition of Cortical Neurite Growth Mediated by Notch Signaling. Science 1999, 286, 741–746. [Google Scholar] [CrossRef] [Green Version]
- Rhyu, M.S.; Jan, L.; Jan, Y.N. Asymmetric distribution of numb protein during division of the sensory organ precursor cell confers distinct fates to daughter cells. Cell 1994, 76, 477–491. [Google Scholar] [CrossRef]
- Doe, C.Q.; Spana, E.P. A collection of cortical crescents: Asymmetric protein localization in CNS precursor cells. Neuron 1995, 15, 991–995. [Google Scholar] [CrossRef] [Green Version]
- Gómez, M.R.; Bate, M. Segregation of myogenic lineages in Drosophila requires numb. Development 1997, 124, 4857–4866. [Google Scholar]
- Uemura, T.; Shepherd, S.; Ackerman, L.; Jan, L.; Jan, Y.N. numb, a gene required in determination of cell fate during sensory organ formation in Drosophila embryos. Cell 1989, 58, 349–360. [Google Scholar] [CrossRef]
- Cayouette, M.; Whitmore, A.V.; Jeffery, G.; Raff, M. Asymmetric Segregation of Numb in Retinal Development and the Influence of the Pigmented Epithelium. J. Neurosci. 2001, 21, 5643–5651. [Google Scholar] [CrossRef]
- Wakamatsu, Y.; Maynard, T.; Jones, S.U.; A Weston, J. NUMB Localizes in the Basal Cortex of Mitotic Avian Neuroepithelial Cells and Modulates Neuronal Differentiation by Binding to NOTCH-1. Neuron 1999, 23, 71–81. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.; Guo, H.; Li, J.; Myint, T.; Pittman, W.; Yang, L.; Zhong, W.; Schwartz, R.J.; Schwarz, J.; Singer, H.A.; et al. Numb family proteins are essential for cardiac morphogenesis and progenitor differentiation. Development 2014, 141, 281–295. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Li, J. Numb family proteins: Novel players in cardiac morphogenesis and cardiac progenitor cell differentiation. Biomol. Concepts 2015, 6, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Shenje, L.T.; Andersen, P.; Uosaki, H.; Fernandez, L.; Rainer, P.P.; Cho, G.-S.; Lee, D.-I.; Zhong, W.; Harvey, R.; A Kass, D.; et al. Precardiac deletion of Numb and Numblike reveals renewal of cardiac progenitors. eLife 2014, 3, e02164. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Bücker, S.; Jungblut, B.; Böttger, T.; Cinnamon, Y.; Tchorz, J.; Müller, M.; Bettler, B.; Harvey, R.; Sun, Q.-Y.; et al. Inhibition of Notch2 by Numb/Numblike controls myocardial compaction in the heart. Cardiovasc. Res. 2012, 96, 276–285. [Google Scholar] [CrossRef] [Green Version]
- Hirai, M.; Arita, Y.; McGlade, C.J.; Lee, K.-F.; Chen, J.; Evans, S.M. Adaptor proteins NUMB and NUMBL promote cell cycle withdrawal by targeting ERBB2 for degradation. J. Clin. Investig. 2017, 127, 569–582. [Google Scholar] [CrossRef] [Green Version]
- Miao, L.; Li, J.; Li, J.; Lu, Y.; Shieh, D.; Mazurkiewicz, J.E.; Barroso, M.; Schwarz, J.; Xin, H.-B.; Singer, H.A.; et al. Cardiomyocyte orientation modulated by the Numb family proteins–N-cadherin axis is essential for ventricular wall morphogenesis. Proc. Natl. Acad. Sci. USA 2019, 116, 15560–15569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M. Mechanisms of Trabecular Formation and Specification During Cardiogenesis. Pediatr. Cardiol. 2018, 39, 1082–1089. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Smith, C.L.; Hall, J.A.; Lee, I.; Luby-Phelps, K.; Tallquist, M.D. Epicardial Spindle Orientation Controls Cell Entry into the Myocardium. Dev. Cell 2010, 19, 114–125. [Google Scholar] [CrossRef] [Green Version]
- Vincent, S.D.; Buckingham, M.E. How to make a heart: The origin and regulation of cardiac progenitor cells. Curr. Top. Dev. Biol. 2010, 90, 1–41. [Google Scholar]
- Mjaatvedt, C.; Nakaoka, T.; Moreno-Rodriguez, R.; Norris, R.; Kern, M.; Eisenberg, C.; Turner, D.; Markwald, R. The Outflow Tract of the Heart Is Recruited from a Novel Heart-Forming Field. Dev. Biol. 2001, 238, 97–109. [Google Scholar] [CrossRef] [Green Version]
- Kelly, R.G.; Brown, N.A.; Buckingham, M.E. The Arterial Pole of the Mouse Heart Forms from Fgf10-Expressing Cells in Pharyngeal Mesoderm. Dev. Cell 2001, 1, 435–440. [Google Scholar] [CrossRef] [Green Version]
- Waldo, K.L.; Kumiski, D.H.; Wallis, K.T.; Stadt, H.A.; Hutson, M.R.; Platt, D.H.; Kirby, M.L. Conotruncal myocardium arises from a secondary heart field. Development 2001, 128, 3179–3188. [Google Scholar] [CrossRef] [PubMed]
- Zaffran, S.; Kelly, R.; Meilhac, S.; Buckingham, M.E.; Brown, N.A. Right Ventricular Myocardium Derives From the Anterior Heart Field. Circ. Res. 2004, 95, 261–268. [Google Scholar] [CrossRef] [Green Version]
- Verzi, M.P.; McCulley, D.J.; De Val, S.; Dodou, E.; Black, B.L. The right ventricle, outflow tract, and ventricular septum comprise a restricted expression domain within the secondary/anterior heart field. Dev. Biol. 2005, 287, 134–145. [Google Scholar] [CrossRef] [Green Version]
- Meilhac, S.; Esner, M.; Kelly, R.; Nicolas, J.-F.; E Buckingham, M. The Clonal Origin of Myocardial Cells in Different Regions of the Embryonic Mouse Heart. Dev. Cell 2004, 6, 685–698. [Google Scholar] [CrossRef] [Green Version]
- Cai, C.-L.; Liang, X.; Shi, Y.; Chu, P.-H.; Pfaff, S.L.; Chen, J.; Evans, S. Isl1 Identifies a Cardiac Progenitor Population that Proliferates Prior to Differentiation and Contributes a Majority of Cells to the Heart. Dev. Cell 2003, 5, 877–889. [Google Scholar] [CrossRef] [Green Version]
- Maeda, J.; Yamagishi, H.; McAnally, J.; Yamagishi, C.; Srivastava, D. Tbx1 is regulated by forkhead proteins in the secondary heart field. Dev. Dyn. 2006, 235, 701–710. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Morishima, M.; Wylie, J.N.; Schwartz, R.J.; Bruneau, B.G.; Lindsay, E.A.; Baldini, A. Tbx1has a dual role in the morphogenesis of the cardiac outflow tract. Development 2004, 131, 3217–3227. [Google Scholar] [CrossRef] [Green Version]
- Ilagan, R.; Abu-Issa, R.; Brown, D.; Yang, Y.-P.; Jiao, K.; Schwartz, R.J.; Klingensmith, J.; Meyers, E.N. Fgf8 is required for anterior heart field development. Development 2006, 133, 2435–2445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, E.J.; Ogden, L.A.; Talbot, A.; Evans, S.; Cai, C.-L.; Black, B.; Frank, D.U.; Moon, A.M. Required, tissue-specific roles for Fgf8 in outflow tract formation and remodeling. Development 2006, 133, 2419–2433. [Google Scholar] [CrossRef] [Green Version]
- Ueno, S.; Weidinger, G.; Osugi, T.; Kohn, A.D.; Golob, J.L.; Pabon, L.; Reinecke, H.; Moon, R.T.; Murry, C.E. Biphasic role for Wnt/beta-catenin signaling in cardiac specification in zebrafish and embryonic stem cells. Proc. Natl. Acad. Sci. USA 2007, 104, 9685–9690. [Google Scholar] [CrossRef] [Green Version]
- Kwon, C.; Arnold, J.; Hsiao, E.C.; Taketo, M.M.; Conklin, B.R.; Srivastava, D. Canonical Wnt signaling is a positive regulator of mammalian cardiac progenitors. Proc. Natl. Acad. Sci. USA 2007, 104, 10894–10899. [Google Scholar] [CrossRef] [Green Version]
- Ai, D.; Fu, X.; Wang, J.; Lu, M.-F.; Chen, L.; Baldini, A.; Klein, W.H.; Martin, J.F. Canonical Wnt signaling functions in second heart field to promote right ventricular growth. Proc. Natl. Acad. Sci. USA 2007, 104, 9319–9324. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.; Cui, L.; Zhou, W.; Dufort, D.; Zhang, X.; Cai, C.-L.; Bu, L.; Yang, L.; Martin, J.; Kemler, R.; et al. beta-Catenin directly regulates Islet1 expression in cardiovascular progenitors and is required for multiple aspects of cardiogenesis. Proc. Natl. Acad. Sci. USA 2007, 104, 9313–9318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smoak, I.W.; Byrd, N.; Abu-Issa, R.; Goddeeris, M.; Anderson, R.; Morris, J.; Yamamura, K.; Klingensmith, J.; Meyers, E. Sonic hedgehog is required for cardiac outflow tract and neural crest cell development. Dev. Biol. 2005, 283, 357–372. [Google Scholar] [CrossRef] [Green Version]
- Dyer, L.A.; Kirby, M.L. Sonic hedgehog maintains proliferation in secondary heart field progenitors and is required for normal arterial pole formation. Dev. Biol. 2009, 330, 305–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitelli, F.; Morishima, M.; Taddei, I.; Lindsay, E.A.; Baldini, A. Tbx1 mutation causes multiple cardiovascular defects and disrupts neural crest and cranial nerve migratory pathways. Hum. Mol. Genet. 2002, 11, 915–922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- High, F.A.; Jain, R.; Stoller, J.; Antonucci, N.B.; Lu, M.M.; Loomes, K.M.; Kaestner, K.H.; Pear, W.S.; Epstein, J.A. Murine Jagged1/Notch signaling in the second heart field orchestrates Fgf8 expression and tissue-tissue interactions during outflow tract development. J. Clin. Investig. 2009, 119, 1986–1996. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Selever, J.; Wang, D.; Lu, M.-F.; Moses, K.A.; Schwartz, R.J.; Martin, J.F. Bmp4 signaling is required for outflow-tract septation and branchial-arch artery remodeling. Proc. Natl. Acad. Sci. USA 2004, 101, 4489–4494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryckebusch, L.; Wang, Z.; Bertrand, N.; Lin, S.-C.; Chi, X.; Schwartz, R.; Zaffran, S.; Niederreither, K. Retinoic acid deficiency alters second heart field formation. Proc. Natl. Acad. Sci. USA 2008, 105, 2913–2918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Black, B.L. Transcriptional pathways in second heart field development. Semin. Cell Dev. Biol. 2007, 18, 67–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rochais, F.; Mesbah, K.; Kelly, R.G. Signaling Pathways Controlling Second Heart Field Development. Circ. Res. 2009, 104, 933–942. [Google Scholar] [CrossRef] [Green Version]
- Buckingham, M.; Meilhac, S.; Zaffran, S. Building the mammalian heart from two sources of myocardial cells. Nat. Rev. Genet. 2005, 6, 826–835. [Google Scholar] [CrossRef]
- Nowotschin, S.; Xenopoulos, P.; Schrode, N.; Hadjantonakis, A.-K. A bright single-cell resolution live imaging reporter of Notch signaling in the mouse. BMC Dev. Biol. 2013, 13, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Klein, R.; Tian, X.; Cheng, H.-T.; Kopan, R.; Shen, J. Notch activation induces apoptosis in neural progenitor cells through a p53-dependent pathway. Dev. Biol. 2004, 269, 81–94. [Google Scholar] [CrossRef] [Green Version]
- Han, H.; Tanigaki, K.; Yamamoto, N.; Kuroda, K.; Yoshimoto, M.; Nakahata, T.; Ikuta, K.; Honjo, T. Inducible gene knockout of transcription factor recombination signal binding protein-J reveals its essential role in T versus B lineage decision. Int. Immunol. 2002, 14, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Zilian, O.; Saner, C.; Hagedorn, L.; Lee, H.-Y.; Säuberli, E.; Suter, U.; Sommer, L.; Aguet, M. Multiple roles of mouse Numb in tuning developmental cell fates. Curr. Biol. 2001, 11, 494–501. [Google Scholar] [CrossRef] [Green Version]
- Wilson, A.; Ardiet, D.-L.; Saner, C.; Vilain, N.; Beermann, F.; Aguet, M.; Macdonald, H.R.; Zilian, O. Normal Hemopoiesis and Lymphopoiesis in the Combined Absence of Numb and Numblike. J. Immunol. 2007, 178, 6746–6751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moses, K.A.; DeMayo, F.; Braun, R.M.; Reecy, J.; Schwartz, R.J. Embryonic expression of anNkx2-5/Cre gene usingROSA26 reporter mice. Genesis 2001, 31, 176–180. [Google Scholar] [CrossRef]
- Miao, L.; Li, J.; Li, J.; Tian, X.; Lu, Y.; Hu, S.; Shieh, D.; Kanai, R.; Zhou, B.-Y.; Zhou, B.; et al. Notch signaling regulates Hey2 expression in a spatiotemporal dependent manner during cardiac morphogenesis and trabecular specification. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGill, M.A.; Dho, S.E.; Weinmaster, G.; McGlade, C. Numb Regulates Post-endocytic Trafficking and Degradation of Notch1. J. Biol. Chem. 2009, 284, 26427–26438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de la Pompa, J.L. Notch signaling in cardiac development and disease. Pediatr. Cardiol. 2009, 30, 643–650. [Google Scholar] [CrossRef]
- McCright, B.; Gao, X.; Shen, L.; Lozier, J.; Lan, Y.; Maguire, M.; Herzlinger, D.; Weinmaster, G.; Jiang, R.; Gridley, T. Defects in development of the kidney, heart and eye vasculature in mice homozygous for a hypomorphic Notch2 mutation. Development 2001, 128, 491–502. [Google Scholar] [CrossRef]
- Li, J.; Miao, L.; Shieh, D.; Spiotto, E.; Li, J.; Zhou, B.; Paul, A.; Schwartz, R.J.; Firulli, A.B.; Singer, H.A.; et al. Single-Cell Lineage Tracing Reveals that Oriented Cell Division Contributes to Trabecular Morphogenesis and Regional Specification. Cell Rep. 2016, 15, 158–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Strooper, B.; Annaert, W.; Cupers, P.; Saftig, P.; Craessaerts, K.; Mumm, J.S.; Schroeter, E.H.; Schrijvers, V.; Wolfe, M.S.; Ray, W.J.; et al. A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain. Nature 1999, 398, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Grego-Bessa, J.; Zurita, L.L.; del Monte, G.; Bolós, V.; Melgar, P.; Arandilla, A.; Garratt, A.; Zang, H.; Mukouyama, Y.-S.; Chen, H.; et al. Notch Signaling Is Essential for Ventricular Chamber Development. Dev. Cell 2007, 12, 415–429. [Google Scholar] [CrossRef] [Green Version]
- Kratsios, P.; Catela, C.; Salimova, E.; Huth, M.; Berno, V.; Rosenthal, N.; Mourkioti, F. Distinct Roles for Cell-Autonomous Notch Signaling in Cardiomyocytes of the Embryonic and Adult Heart. Circ. Res. 2010, 106, 559–572. [Google Scholar] [CrossRef]
- Han, P.; Bloomekatz, J.; Ren, J.; Zhang, R.; Grinstein, J.D.; Zhao, L.; Burns, C.G.; Burns, C.E.; Anderson, R.M.; Chi, N.C. Coordinating cardiomyocyte interactions to direct ventricular chamber morphogenesis. Nature 2016, 534, 700–704. [Google Scholar] [CrossRef] [Green Version]
- Salguero-Jiménez, A.; Grego-Bessa, J.; D’Amato, G.; Jiménez-Borreguero, L.J.; De La Pompa, J.L. Myocardial Notch1-Rbpj deletion does not affect NOTCH signaling, heart development or function. PLoS ONE 2018, 13, e0203100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castel, D.; Mourikis, P.; Bartels, S.J.; Brinkman, A.; Tajbakhsh, S.; Stunnenberg, H.G. Dynamic binding of RBPJ is determined by Notch signaling status. Genes Dev. 2013, 27, 1059–1071. [Google Scholar] [CrossRef] [Green Version]
- Nemir, M.; Croquelois, A.; Pedrazzini, T.; Radtke, F. Induction of Cardiogenesis in Embryonic Stem Cells via Downregulation of Notch1 Signaling. Circ. Res. 2006, 98, 1471–1478. [Google Scholar] [CrossRef] [Green Version]
- De Zoysa, P.; Liu, J.; Toubat, O.; Choi, J.; Moon, A.; Gill, P.S.; Duarte, A.; Sucov, H.M.; Kumar, S.R. Delta-like ligand-4 mediated Notch signaling controls proliferation of second heart field progenitor cells by regulating Fgf8 expression. Development 2020. [Google Scholar] [CrossRef]
- Riccio, O.; E Van Gijn, M.; Bezdek, A.C.; Pellegrinet, L.; Van Es, J.H.; Zimber-Strobl, U.; Strobl, L.J.; Honjo, T.; Clevers, H.; Radtke, F. Loss of intestinal crypt progenitor cells owing to inactivation of both Notch1 and Notch2 is accompanied by derepression of CDK inhibitors p27 Kip1 and p57 Kip2. EMBO Rep. 2008, 9, 377–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Shi, S.; Acosta, L.; Li, W.; Lu, J.; Bao, S.; Chen, Z.; Yang, Z.; Schneider, M.; Chien, K.R.; et al. BMP10 is essential for maintaining cardiac growth during murine cardiogenesis. Development 2004, 131, 2219–2231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Zhang, W.; Sun, X.; Yoshimoto, M.; Chen, Z.; Zhu, W.; Liu, J.; Shen, Y.; Yong, W.; Li, D.; et al. Fkbp1a controls ventricular myocardium trabeculation and compaction by regulating endocardial Notch1 activity. Development 2013, 140, 1946–1957. [Google Scholar] [CrossRef] [Green Version]
- Luxán, G.; Casanova, J.C.; Poveda, B.M.; Prados, B.; D’Amato, G.; MacGrogan, D.; Gonzalez-Rajal, A.; Dobarro, D.; Torroja, C.; Martinez, F.; et al. Mutations in the NOTCH pathway regulator MIB1 cause left ventricular noncompaction cardiomyopathy. Nat. Med. 2013, 19, 193–201. [Google Scholar] [CrossRef]
- van Lessen, M.; Nakayama, M.; Kato, K.; Kim, J.M.; Kaibuchi, K.; Adams, R.H. Regulation of vascular endothelial growth factor receptor function in angiogenesis by numb and numb-like. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1815–1825. [Google Scholar] [CrossRef] [Green Version]
- Koenig, S.; Lahaye, S.; Feller, J.; Rowland, P.; Hor, K.N.; Trask, A.J.; Janssen, P.M.; Radtke, F.; Lilly, B.; Garg, V. Notch1 haploinsufficiency causes ascending aortic aneurysms in mice. JCI Insight 2017, 2. [Google Scholar] [CrossRef]
- Garg, V.; Muth, A.N.; Ransom, J.F.; Schluterman, M.K.; Barnes, R.; King, I.N.; Grossfeld, P.D.; Srivastava, D. Mutations in NOTCH1 cause aortic valve disease. Nature 2005, 437, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Cherian, A.V.; Fukuda, R.; Augustine, S.M.; Maischein, H.-M.; Stainier, D.Y.R. N-cadherin relocalization during cardiac trabeculation. Proc. Natl. Acad. Sci. USA 2016, 113, 7569–7574. [Google Scholar] [CrossRef] [Green Version]
- Varadkar, P.; Kraman, M.; Despres, D.; Ma, G.; Lozier, J.; McCright, B. Notch2 is required for the proliferation of cardiac neural crest-derived smooth muscle cells. Dev. Dyn. 2008, 237, 1144–1152. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhang, W.; Li, D.; Cordes, T.M.; Payne, R.M.; Shou, W. Analysis of Ventricular Hypertrabeculation and Noncompaction Using Genetically Engineered Mouse Models. Pediatr. Cardiol. 2009, 30, 626–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frise, E.; Knoblich, J.; Younger-Shepherd, S.; Jan, L.; Jan, Y.N. The Drosophila Numb protein inhibits signaling of the Notch receptor during cell-cell interaction in sensory organ lineage. Proc. Natl. Acad. Sci. USA 1996, 93, 11925–11932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, P.H.; Zou, K.; Zhong, W.; Tang, H. The Enigma of the Numb-Notch Relationship during Mammalian Embryogenesis. Dev. Neurosci. 2006, 28, 156–168. [Google Scholar] [CrossRef]
- Chen, X.; Liu, Z.; Shan, Z.; Yao, W.; Gu, A.; Wen, W. Structural determinants controlling 14-3-3 recruitment to the endocytic adaptor Numb and dissociation of the Numb·α-adaptin complex. J. Biol. Chem. 2018, 293, 4149–4158. [Google Scholar] [CrossRef] [Green Version]
- Shao, X.; Liu, Y.; Yu, Q.; Ding, Z.; Qian, W.; Zhang, L.; Zhang, J.; Jiang, N.; Gui, L.; Xu, Z.; et al. Numb regulates vesicular docking for homotypic fusion of early endosomes via membrane recruitment of Mon1b. Cell Res. 2016, 26, 593–612. [Google Scholar] [CrossRef] [Green Version]
- Santolini, E.; Puri, C.; Salcini, A.E.; Gagliani, M.C.; Pelicci, P.G.; Tacchetti, C.; Di Fiore, P.P. Numb Is an Endocytic Protein. J. Cell Biol. 2000, 151, 1345–1352. [Google Scholar] [CrossRef] [PubMed]
- Shao, X.; Ding, Z.; Zhao, M.; Liu, K.; Sun, H.; Chen, J.; Liu, X.; Zhang, Y.; Hong, Y.; Li, H.; et al. Mammalian Numb protein antagonizes Notch by controlling postendocytic trafficking of the Notch ligand Delta-like 4. J. Biol. Chem. 2017, 292, 20628–20643. [Google Scholar] [CrossRef] [Green Version]
- Sato, K.; Watanabe, T.; Wang, S.; Kakeno, M.; Matsuzawa, K.; Matsui, T.; Yokoi, K.; Murase, K.; Sugiyama, I.; Ozawa, M.; et al. Numb controls E-cadherin endocytosis through p120 catenin with aPKC. Mol. Biol. Cell 2011, 22, 3103–3119. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Age | Total Embryos | KO | Harvested/Expected Percentage of KO |
|---|---|---|---|
| E9.5 | 75 | 17 | 90.67 |
| E10.5 | 52 | 24 | 184.62 |
| E11.5 | 7 | 2 | 114.29 |
| E12.5 | 35 | 6 | 68.57 |
| E13.5 | 59 | 15 | 101.69 |
| E14.5 | 26 | 5 | 76.92 |
| E15.5 | 26 | 5 | 76.92 |
| E16.5 | 41 | 6 | 58.54 |
| E17.5 | 2 | 0 | 0.00 |
| Postnatal | 42 | 0 | 0.00 |
| Age | Total Embryos | MDKO;Nt1+/− | TKO | Harvested/Expected Percentage of MDKO;Nt1+/− | Harvested/Expected Percentage of TKO | Harvested/Expected Percentage of MDKO |
|---|---|---|---|---|---|---|
| E9.5 | 204 | 25 | 18 | 98.04 | 70.59 | 100.00 |
| E10.5 | 439 | 53 | 32 | 96.58 | 58.31 | 100.00 |
| E11.5 | 149 | 15 | 11 | 80.54 | 59.06 | 100.00 |
| E12.5 | 577 | 59 | 31 | 81.80 | 42.98 | 76.00 |
| E13.5 | 173 | 17 | 8 | 78.61 | 36.99 | 68.00 |
| E14.5 | 44 | 3 | 0 | 54.54 | 0.00 | 40.00 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miao, L.; Lu, Y.; Nusrat, A.; Abdelnasser, H.Y.; Datta, S.; Zhou, B.; Schwartz, R.J.; Wu, M. The Spatiotemporal Expression of Notch1 and Numb and Their Functional Interaction during Cardiac Morphogenesis. Cells 2021, 10, 2192. https://doi.org/10.3390/cells10092192
Miao L, Lu Y, Nusrat A, Abdelnasser HY, Datta S, Zhou B, Schwartz RJ, Wu M. The Spatiotemporal Expression of Notch1 and Numb and Their Functional Interaction during Cardiac Morphogenesis. Cells. 2021; 10(9):2192. https://doi.org/10.3390/cells10092192
Chicago/Turabian StyleMiao, Lianjie, Yangyang Lu, Anika Nusrat, Hala Y. Abdelnasser, Sayantap Datta, Bin Zhou, Robert J. Schwartz, and Mingfu Wu. 2021. "The Spatiotemporal Expression of Notch1 and Numb and Their Functional Interaction during Cardiac Morphogenesis" Cells 10, no. 9: 2192. https://doi.org/10.3390/cells10092192
APA StyleMiao, L., Lu, Y., Nusrat, A., Abdelnasser, H. Y., Datta, S., Zhou, B., Schwartz, R. J., & Wu, M. (2021). The Spatiotemporal Expression of Notch1 and Numb and Their Functional Interaction during Cardiac Morphogenesis. Cells, 10(9), 2192. https://doi.org/10.3390/cells10092192