
Nogo-A Modulates the Synaptic Excitation of Hippocampal Neurons in a Ca2+-Dependent Manner


 , and
, and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animal Procedures
2.2. Primary Mouse Hippocampal Culture
2.3. Organotypic Hippocampal Slice Culture
2.4. Method Details
2.4.1. Antibody and Peptide Treatment
2.4.2. Patch Clamp Electrophysiology
2.4.3. Live-Cell Labeling and Immunocytochemistry
2.4.4. Widefield Fluorescence Imaging and Analysis
2.4.5. Ca2+-Imaging
3. Results
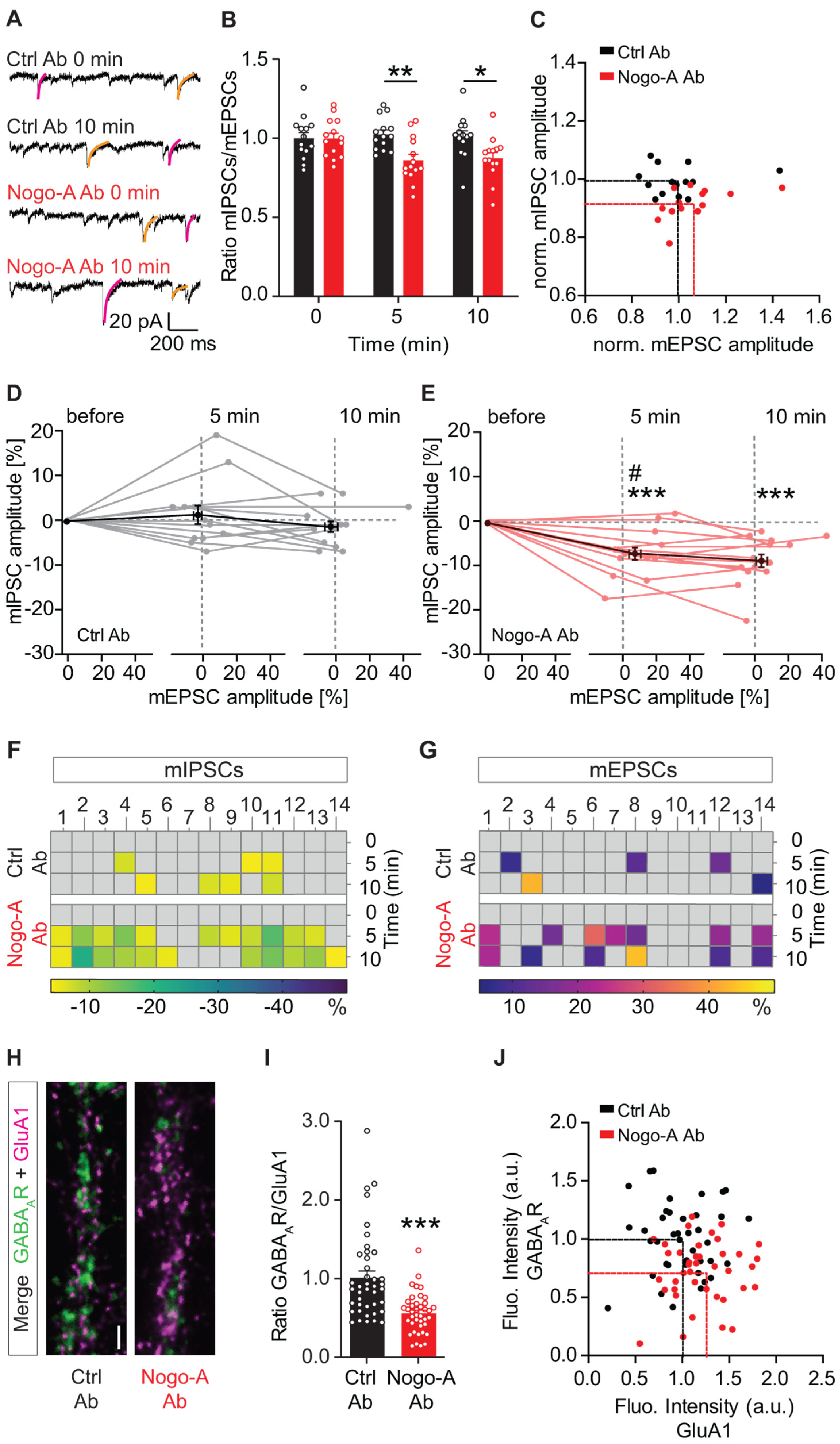
3.1. Nogo-A Loss-of-Function Shifts the Excitation/Inhibition Balance toward a Higher Excitation at a Single Cell Level
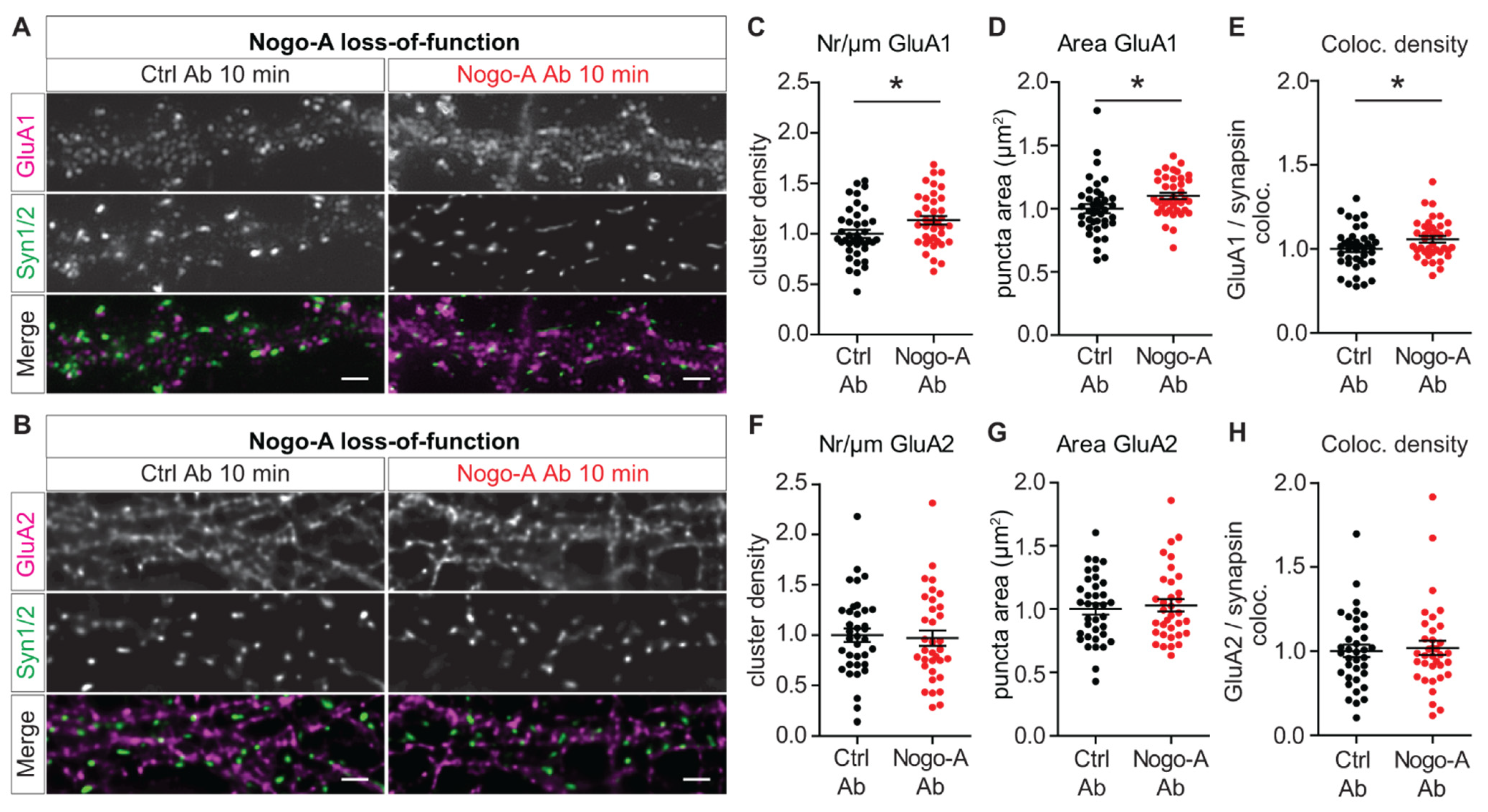
3.2. Nogo-A Regulates the Synaptic Insertion of Calcium Permeable-AMPARs
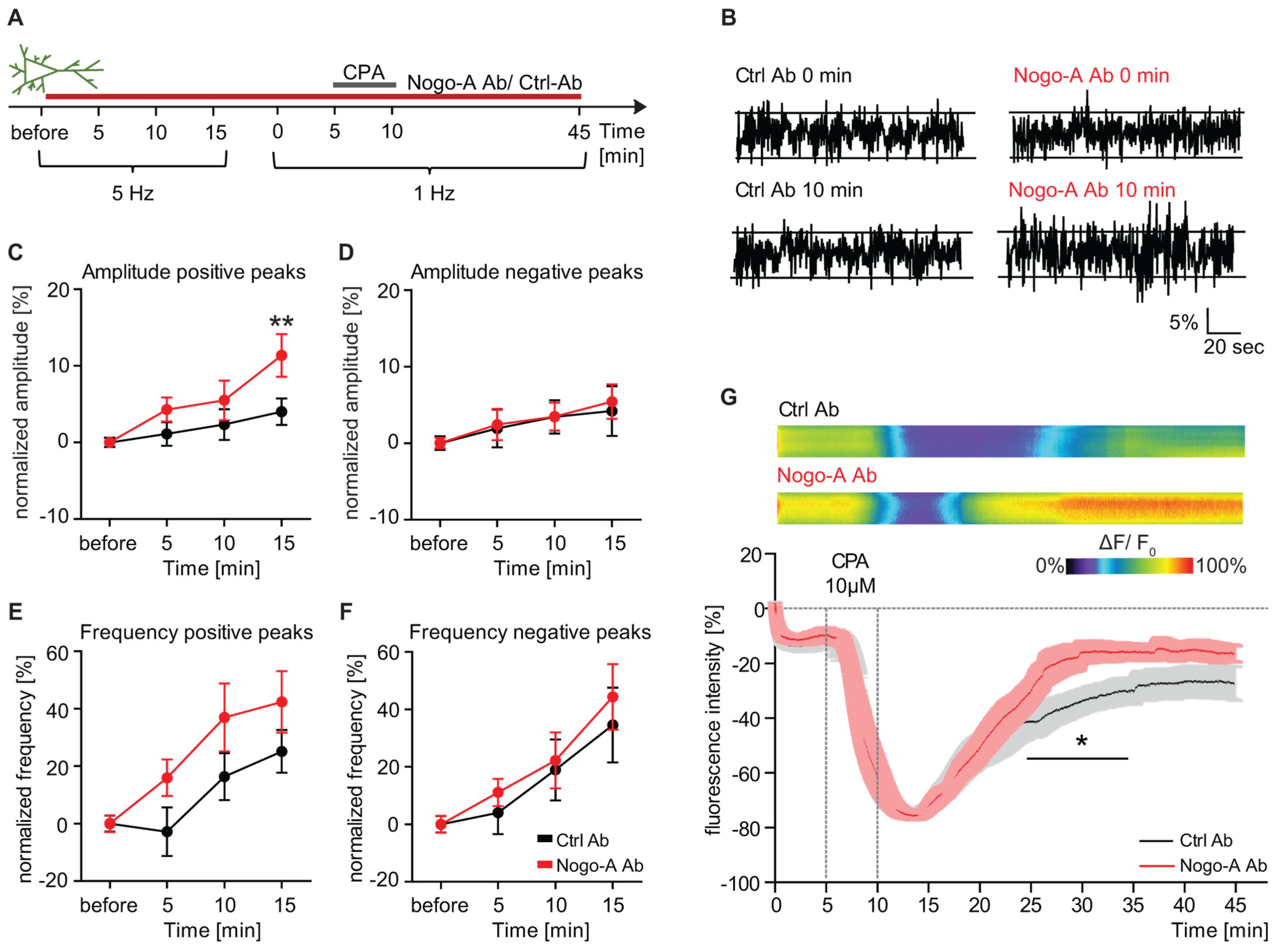
3.3. Nogo-A Neutralization Results in a Calcium Permeable-AMPAR Dependent Increase in Ca2+ Dynamics
3.4. Ca2+ Influx Versus Intracellular Ca2+ Release upon Nogo-A Loss-of-Function
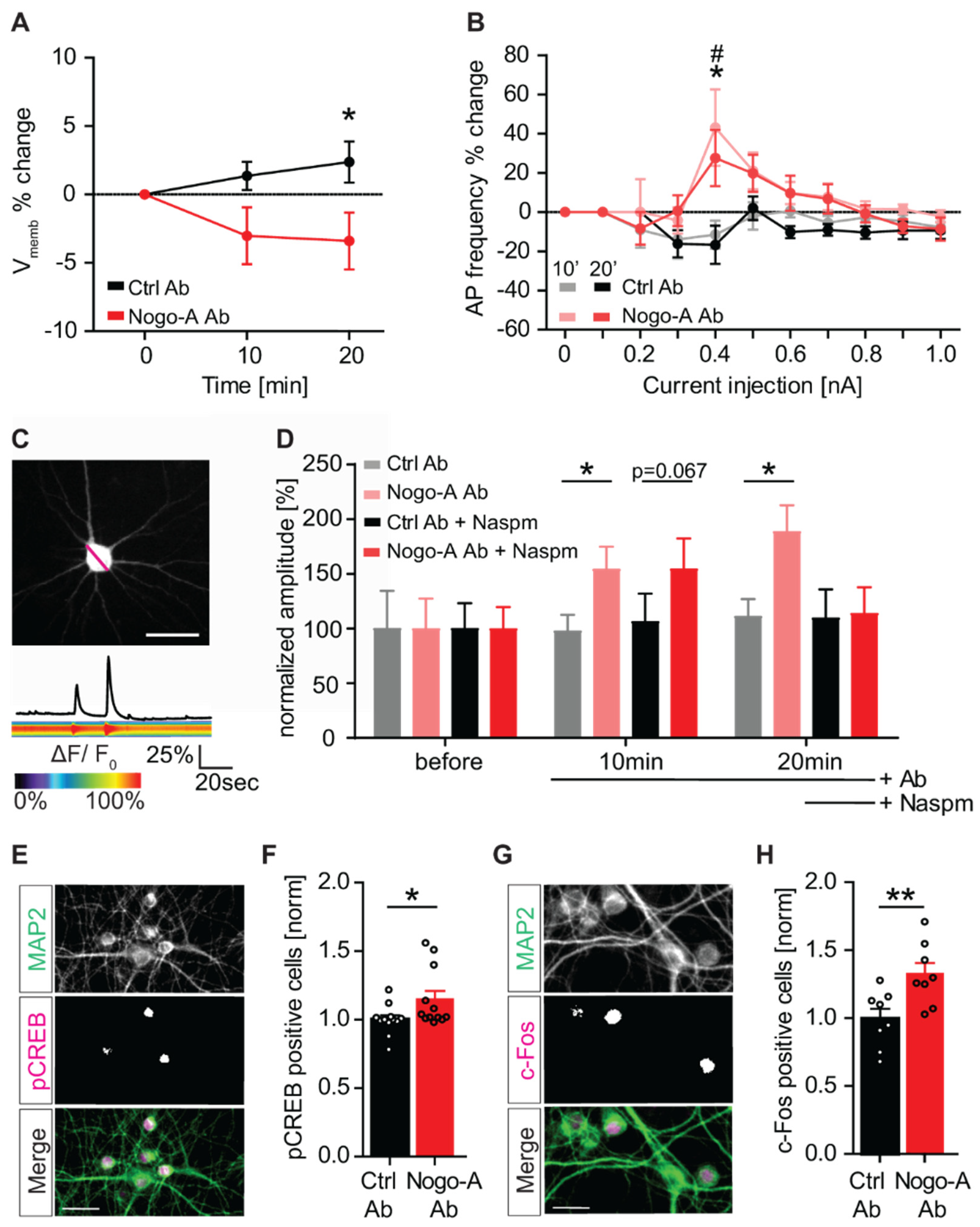
3.5. Nogo-A Loss-of-Function Increases Neuronal Excitability and Neuronal Activation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fagiolini, M.; Fritschy, J.M.; Löw, K.; Möhler, H.; Rudolph, U.; Hensch, T.K. Specific GABAA circuits for visual cortical plasticity. Science 2004, 303, 1681–1683. [Google Scholar] [CrossRef] [Green Version]
- Fagiolini, M.; Hensch, T.K. Inhibitory threshold for critical-period activation in primary visual cortex. Nature 2000, 404, 183–186. [Google Scholar] [CrossRef] [PubMed]
- Sohal, V.S.; Rubenstein, J.L.R. Excitation-inhibition balance as a framework for investigating mechanisms in neuropsychiatric disorders. Mol. Psychiatry 2019, 24, 1248–1257. [Google Scholar] [CrossRef] [PubMed]
- Jacob, T.C.; Moss, S.J.; Jurd, R. GABAA receptor trafficking and its role in the dynamic modulation of neuronal inhibition. Nat. Rev. Neurosci. 2008, 9, 331–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilman, V.; Van Rossum, M.C.W.; Turrigiano, G.G. Activity deprivation reduces miniature IPSC amplitude by decreasing the number of postsynaptic GABAA receptors clustered at neocortical synapses. J. Neurosci. 2002, 22, 1328–1337. [Google Scholar] [CrossRef] [Green Version]
- Bannai, H.; Niwa, F.; Sakuragi, S.; Mikoshiba, K. Inhibitory synaptic transmission tuned by Ca2+ and glutamate through the control of GABAAR lateral diffusion dynamics. Dev. Growth Differ. 2020, 62, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Bannai, H.; Niwa, F.; Sherwood, M.W.; Shrivastava, A.N.; Arizono, M.; Miyamoto, A.; Sugiura, K.; Lévi, S.; Triller, A.; Mikoshiba, K. Bidirectional control of synaptic GABAAR clustering by glutamate and calcium. Cell Rep. 2015, 13, 2768–2780. [Google Scholar] [CrossRef]
- Bannai, H.; Lévi, S.; Schweizer, C.; Inoue, T.; Launey, T.; Racine, V.; Sibarita, J.B.; Mikoshiba, K.; Triller, A. Activity-dependent tuning of inhibitory neurotransmission based on GABAAR diffusion dynamics. Neuron 2009, 62, 670–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muir, J.; Arancibia-Carcamo, I.L.; MacAskill, A.F.; Smith, K.R.; Griffin, L.D.; Kittler, J.T. NMDA receptors regulate GABAA receptor lateral mobility and clustering at inhibitory synapses through serine 327 on the γ2 subunit. Proc. Natl. Acad. Sci. USA 2010, 107, 16679–16684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmandke, A.; Schmandke, A.; Schwab, M.E. Nogo-A: Multiple roles in CNS development, maintenance, and disease. Neuroscientist 2014, 20, 372–386. [Google Scholar] [CrossRef]
- Huber, A.B.; Weinmann, O.; Brösamle, C.; Oertle, T.; Schwab, M.E. Patterns of nogo mRNA and protein expression in the developing and adult rat and after CNS lesions. J. Neurosci. 2002, 22, 3553–3567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwab, M.E.; Strittmatter, S.M. Nogo limits neural plasticity and recovery from injury. Curr. Opin. Neurobiol. 2014, 27, 53–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zagrebelsky, M.; Korte, M. Maintaining stable memory engrams: New roles for Nogo-A in the CNS. Neuroscience 2014, 283, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Delekate, A.; Zagrebelsky, M.; Kramer, S.; Schwab, M.E.; Korte, M. NogoA restricts synaptic plasticity in the adult hippocampus on a fast time scale. Proc. Natl. Acad. Sci. USA 2011, 108, 2569–2574. [Google Scholar] [CrossRef] [Green Version]
- Iobbi, C.; Korte, M.; Zagrebelsky, M. Nogo-66 restricts synaptic strengthening via Lingo1 and the ROCK2-cofilin pathway to control actin dynamics. Cereb. Cortex 2017, 27, 2779–2792. [Google Scholar] [CrossRef] [Green Version]
- Kellner, Y.; Fricke, S.; Kramer, S.; Iobbi, C.; Wierenga, C.J.; Schwab, M.E.; Korte, M.; Zagrebelsky, M. Nogo-A controls structural plasticity at dendritic spines by rapidly modulating actin dynamics. Hippocampus 2016, 26, 816–831. [Google Scholar] [CrossRef]
- KEMPF, A.; Tews, B.; Arzt, M.E.; Weinmann, O.; Obermair, F.J.; Pernet, V.; Zagrebelsky, M.; Delekate, A.; Iobbi, C.; Zemmar, A.; et al. The sphingolipid receptor S1PR2 is a receptor for nogo-A repressing synaptic plasticity. PLoS Biol. 2014, 12. [Google Scholar] [CrossRef]
- Zagrebelsky, M.; Schweigreiter, R.; Bandtlow, C.E.; Schwab, M.E.; Korte, M. Nogo-A stabilizes the architecture of hippocampal neurons. J. Neurosci. 2010, 30, 13220–13234. [Google Scholar] [CrossRef] [Green Version]
- Zagrebelsky, M.; Lonnemann, N.; Fricke, S.; Kellner, Y.; Preuß, E.; Michaelsen-Preusse, K.; Korte, M. Nogo-A regulates spatial learning as well as memory formation and modulates structural plasticity in the adult mouse hippocampus. Neurobiol. Learn. Mem. 2017, 138, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Akbik, F.V.; Bhagat, S.M.; Patel, P.R.; Cafferty, W.B.J.; Strittmatter, S.M. Anatomical plasticity of adult brain is Titrated by Nogo receptor. Neuron 2013, 77, 859–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zemmar, A.; Weinmann, O.; Kellner, Y.; Yu, X.; Vicente, R.; Gullo, M.; Kasper, H.; Lussi, K.; Ristic, Z.; Luft, A.R.; et al. Neutralization of Nogo-A enhances synaptic plasticity in the rodent motor cortex and improves motor learning in vivo. J. Neurosci. 2014, 34, 8685–8698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jitsuki, S.; Nakajima, W.; Takemoto, K.; Sano, A.; Tada, H.; Takahashi-Jitsuki, A.; Takahashi, T. Nogo Receptor signaling restricts adult neural plasticity by limiting synaptic AMPA receptor delivery. Cereb. Cortex 2016, 26, 427–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fricke, S.; Metzdorf, K.; Ohm, M.; Haak, S.; Heine, M.; Korte, M.; Zagrebelsky, M. Fast regulation of GABAAR diffusion dynamics by Nogo-A signaling. Cell Rep. 2019, 29, 671–684.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, B. Nogo-A and the regulation of neurotransmitter receptors. Neural Regen. Res. 2020, 15, 2037–2038. [Google Scholar] [CrossRef] [PubMed]
- Stoppini, L.; Buchs, P.A.; Muller, D. A simple method for organotypic cultures of nervous tissue. J. Neurosci. Methods 1991, 37, 173–182. [Google Scholar] [CrossRef]
- Michaelsen-Preusse, K.; Kellner, Y.; Korte, M.; Zagrebelsky, M. Analysis of actin turnover and spine dynamics in hippocampal slice cultures. Anim. Models Drug Addict. 2014, 87, 189–217. [Google Scholar]
- Liebscher, T.; Schnell, L.; Schnell, D.; Scholl, J.; Schneider, R.; Gullo, M.; Fouad, K.; Mir, A.; Rausch, M.; Kindler, D.; et al. Nogo-A antibody improves regeneration and locomotion of spinal cord-injured rats. Ann. Neurol. 2005, 58, 706–719. [Google Scholar] [CrossRef]
- Maier, I.C.; Ichiyama, R.M.; Courtine, G.; Schnell, L.; Lavrov, I.; Edgerton, V.R.; Schwab, M. Differential effects of anti-Nogo-A antibody treatment and treadmill training in rats with incomplete spinal cord injury. Brain 2009, 132, 1426–1440. [Google Scholar] [CrossRef] [Green Version]
- Oertle, T.; Huber, C.; Van der Putten, H.; Schwab, M.E. Genomic structure and functional characterisation of the promoters of human and mouse nogo/Rtnj. Mol. Biol. 2003, 325, 299–323. [Google Scholar] [CrossRef]
- Danielson, E.; Lee, S.H.; Fox, M.A. SynPAnal: Software for rapid quantification of the density and intensity of protein puncta from fluorescence microscopy images of neurons. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [Green Version]
- Akerboom, J.; Chen, T.W.; Wardill, T.J.; Tian, L.; Marvin, J.S.; Mutlu, S.; Calderón, N.C.; Esposti, F.; Borghuis, B.G.; Sun, X.R.; et al. Optimization of a GCaMP calcium indicator for neural activity imaging. J. Neurosci. 2012, 32, 13819–13840. [Google Scholar] [CrossRef] [PubMed]
- De Juan-Sanz, J.; Holt, G.T.; Schreiter, E.R.; de Juan, F.; Kim, D.S.; Ryan, T.A. Axonal endoplasmic reticulum Ca2+ content controls release probability in CNS nerve terminals. Neuron 2017, 93, 867–881.e6. [Google Scholar] [CrossRef] [Green Version]
- Dejanovic, B.; Semtner, M.; Ebert, S.; Lamkemeyer, T.; Neuser, F.; Lüscher, B.; Meier, J.C.; Schwarz, G. Palmitoylation of gephyrin controls receptor clustering and plasticity of GABAergic synapses. PLoS Biol. 2014, 12, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bliss, T.V.P.; Collingridge, G.L. A synaptic model of memory: LTP in the hippocampus. Nature 1993, 361, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Huganir, R.L.; Nicoll, R.A. AMPARs and synaptic plasticity: The last 25 years. Neuron 2013, 80, 704–717. [Google Scholar] [CrossRef] [Green Version]
- Nusser, Z.; Cull-Candy, S.; Farrant, M. Differences in synaptic GABA(A) receptor number underlie variation in GABA mini amplitude. Neuron 1997, 19, 697–709. [Google Scholar] [CrossRef] [Green Version]
- Heine, M.; Thoumine, O.; Mondin, M.; Tessier, B.; Giannone, G.; Choquet, D. recruitment of functional AMPA receptors at neurexin/neuroligin contacts. Proc. Natl. Acad. Sci. USA 2008, 105, 20947–20952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blackstone, C.; Murphy, T.H.; Moss, S.J.; Baraban, J.M.; Huganir, R.L. Cyclic AMP and synaptic activity-dependent phosphorylation of AMPA- preferring glutamate receptors. J. Neurosci. 1994, 14, 7585–7593. [Google Scholar] [CrossRef] [Green Version]
- Huganir, R.L.; Song, I. Regulation of AMPA receptors during synaptic plasticity. TRENDS Neurosci. 2002, 25, 578–588. [Google Scholar]
- McGlade-Mcculloh, E.; Yamamoto, H.; Tan, S.E.; Brickey, D.A.; Soderling, T.R. Phosphorylation and regulation of glutamate receptors by calcium/calmodulin-dependent protein kinase II. Nature 1993, 362, 640–642. [Google Scholar] [CrossRef] [PubMed]
- Isaac, J.T.R.; Ashby, M.; McBain, C.J. The role of the GluR2 subunit in AMPA receptor function and synaptic plasticity. Neuron 2007, 54, 859–871. [Google Scholar] [CrossRef] [Green Version]
- Plant, K.; Pelkey, K.A.; Bortolotto, Z.A.; Morita, D.; Terashima, A.; McBain, C.J.; Collingridge, G.L.; Isaac, J.T.R. Transient incorporation of native GluR2-lacking AMPA receptors during hippocampal long-term potentiation. Nat. Neurosci. 2006, 9, 602–604. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Kim, J.; Zhou, Z.; Fink, D.J.; Mata, M. Neuronal Nogo-A regulates glutamate receptor subunit expression in hippocampal neurons. J. Neurochem. 2011, 119, 1183–1193. [Google Scholar] [CrossRef]
- Henderson, M.J.; Baldwin, H.A.; Werley, C.A.; Boccardo, S.; Whitaker, L.R.; Yan, X.; Holt, G.T.; Schreiter, E.R.; Looger, L.L.; Cohen, A.E.; et al. A low affinity GCaMP3 variant (GCaMPer) for imaging the endoplasmic reticulum calcium store. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [Green Version]
- Brunner, J.; Szabadics, J. Analogue modulation of back-propagating action potentials enables dendritic hybrid signalling. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Ziff, E.B. Calcineurin mediates synaptic scaling via synaptic trafficking of Ca2+-permeable AMPA receptors. PLoS Biol. 2014, 12, 1–15. [Google Scholar] [CrossRef]
- Rosen, L.B.; Ginty, D.D.; Greenberg, M.E. Calcium regulation of gene expression. Adv. Second Messenger Phosphoprot. Res. 1995, 30, 225–253. [Google Scholar] [CrossRef]
- Minatohara, K.; Akiyoshi, M.; Okuno, H. Role of immediate-early genes in synaptic plasticity and neuronal ensembles underlying the memory trace. Front. Mol. Neurosci. 2016, 8, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Sutton, M.A.; Ito, H.T.; Cressy, P.; Kempf, C.; Woo, J.C.; Schuman, E.M. Miniature neurotransmission stabilizes synaptic function via tonic suppression of local dendritic protein synthesis. Cell 2006, 125, 785–799. [Google Scholar] [CrossRef] [Green Version]
- Shepherd, J.D.; Huganir, R.L. The cell biology of synaptic plasticity: AMPA receptor trafficking. Annu. Rev. Cell Dev. Biol. 2007, 23, 613–643. [Google Scholar] [CrossRef] [Green Version]
- Leonoudakis, D.; Zhao, P.; Beattie, E.C. Rapid tumor necrosis factor α-induced exocytosis of glutamate receptor 2-lacking AMPA receptors to extrasynaptic plasma membrane potentiates excitotoxicity. J. Neurosci. 2008, 28, 2119–2130. [Google Scholar] [CrossRef] [Green Version]
- Hou, Q.; Zhang, D.; Jarzylo, L.; Huganir, R.L.; Man, H. Homeostatic regulation of AMPA receptor expression at single hippocampal synapses. Proc. Natl. Acad. Sci. USA 2008, 105, 775–780. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Wang, X.B.; Zhou, Q. Perisynaptic GluR2-lacking AMPA receptors control the reversibility of synaptic and spines modifications. Proc. Natl. Acad. Sci. USA 2010, 107, 11999–12004. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.M.; Mansuy, I.M.; Kandel, E.R.; Roder, J. Calcineurin-mediated LTD of GABAergic inhibition underlies the increased excitability of CA1 neurons associated with LTP. Neuron 2000, 26, 197–205. [Google Scholar] [CrossRef] [Green Version]
- Schwab, M.E. Functions of Nogo proteins and their receptors in the nervous system. Nat. Rev. Neurosci. 2010, 11, 799–811. [Google Scholar] [CrossRef] [PubMed]
- Pulli, I.; Asghar, M.Y.; Kemppainen, K.; Törnquist, K. Sphingolipid-mediated calcium signaling and its pathological effects. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1668–1677. [Google Scholar] [CrossRef] [PubMed]
- Wuytack, F.; Raeymaekers, L.; Missiaen, L. Molecular physiology of the SERCA and SPCA pumps. Cell Calcium 2002, 32, 279–305. [Google Scholar] [CrossRef]
- Samtleben, S.; Wachter, B.; Blum, R. Store-operated calcium entry compensates fast ER calcium loss in resting hippocampal neurons. Cell Calcium 2015, 58, 147–159. [Google Scholar] [CrossRef]
- Ludewig, S.; Herrmann, U.; Michaelsen-Preusse, K.; Metzdorf, K.; Just, J. APPs α rescues impaired Ca2+ homeostasis in APP- and APLP2-deficient hippocampal neurons. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Malenka, R.C.; Kauer, J.A.; Perkel, D.J.; Mauk, M.D.; Kelly, P.T.; Nicoll, R.A.; Waxham, M.N. An essential role for postsynaptic calmodulin and protein kinase activity in long-term potentiation. Nature 1989, 340, 554–557. [Google Scholar] [CrossRef]
- Pouille, F.; Marin-Burgin, A.; Adesnik, H.; Atallah, B.V.; Scanziani, M. Input normalization by global feedforward inhibition expands cortical dynamic range. Nat. Neurosci. 2009, 12, 1577–1585. [Google Scholar] [CrossRef]
- Kohonen, T. Physiological interpretation of the self-organizing map algorithm. Neural Netw. 1993, 6, 895–905. [Google Scholar] [CrossRef]
- Kaski, S.; Kohonen, T. Winner-take-all networks for physiological models of competitive learning. Neural Netw. 1994, 7, 973–984. [Google Scholar] [CrossRef]
- Wang, J.H.; Stelzer, A. Shared calcium signaling pathways in the induction of long-term potentiation and synaptic disinhibition in CA1 pyramidal cell dendrites. J. Neurophysiol. 1996, 75, 1687–1702. [Google Scholar] [CrossRef]
- McGee, A.W.; Yang, Y.; Fischer, Q.S.; Daw, N.W.; Strittmatter, S.H. Neuroscience: Experience-driven plasticity of visual cortex limited by myelin and nogo receptor. Science 2005, 309, 2222–2226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodson, W.; Nitecka, L.; Ben-Ari, Y. Organization of the GABAergic system in the rat hippocampal formation: A quantitative immunocytochemical study. J. Comp. Neurol. 1989, 280, 254–271. [Google Scholar] [CrossRef]
- Bezare, M.J.; Soltész, I. Quantitative assessment of CA1 local circuits: Knowledge base for interneuron-pyramidal cell connectivity. Hippocampus 2013, 23, 751–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenzie, S. Inhibition shapes the organization of hippocampal representations. Hippocampus 2018, 28, 659–671. [Google Scholar] [CrossRef]
- Stephany, C.É.; Ikrar, T.; Nguyen, C.; Xu, X.; McGee, A.W. Nogo receptor 1 confines a disinhibitory microcircuit to the critical period in visual cortex. J. Neurosci. 2016, 36, 11006–11012. [Google Scholar] [CrossRef] [Green Version]
- Bhagat, S.M.; Butler, S.S.; Taylor, J.R.; McEwen, B.S.; Strittmatter, S.M. Erasure of fear memories is prevented by nogo receptor 1 in adulthood. Mol. Psychiatry 2016, 21, 1281–1289. [Google Scholar] [CrossRef] [Green Version]
- Stephany, C.É.; Chan, L.L.H.; Parivash, S.N.; Dorton, H.M.; Piechowicz, M.; Qiu, S.; McGee, A.W. Plasticity of binocularity and visual acuity are differentially limited by nogo receptor. J. Neurosci. 2014, 34, 11631–11640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandolfi, D.; Cerri, S.; Mapelli, J.; Polimeni, M.; Tritto, S.; Fuzzati-Armentero, M.T.; Bigiani, A.; Blandini, F.; Mapelli, L.; D’Angelo, E. Activation of the CREB/c-Fos pathway during long-term synaptic plasticity in the cerebellum granular layer. Front. Cell. Neurosci. 2017, 11, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, M.; McFadden, G.; Greenberg, M.E. Membrane depolarization and calcium induce c-fos transcription via phosphorylation of transcription factor CREB. Neuron 1990, 4, 571–582. [Google Scholar] [CrossRef]
- Joset, A.; Dodd, D.A.; Halegoua, S.; Schwab, M.E. Pincher-generated Nogo-A endosomes mediate growth cone collapse and retrograde signaling. J. Cell Biol. 2010, 188, 271–285. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Deng, K.; Hou, J.; Bryson, J.B.; Barco, A.; Nikulina, E.; Spencer, T.; Mellado, W.; Kandel, E.R.; Filbin, M.T. Activated CREB is sufficient to overcome inhibitors in myelin and promote spinal axon regeneration in vivo. Neuron 2004, 44, 609–621. [Google Scholar] [CrossRef] [Green Version]
- Bito, H.; Deisseroth, K.; Tsien, R.W. CREB phosphorylation and dephosphorylation: A Ca2+- and stimulus duration-dependent switch for hippocampal gene expression. Cell 1996, 87, 1203–1214. [Google Scholar] [CrossRef] [Green Version]
- Holtmaat, A.; Caroni, P. Functional and structural underpinnings of neuronal assembly formation in learning. Nat. Neurosci. 2016, 19, 1553–1562. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Metzdorf, K.; Fricke, S.; Balia, M.T.; Korte, M.; Zagrebelsky, M. Nogo-A Modulates the Synaptic Excitation of Hippocampal Neurons in a Ca2+-Dependent Manner. Cells 2021, 10, 2299. https://doi.org/10.3390/cells10092299
Metzdorf K, Fricke S, Balia MT, Korte M, Zagrebelsky M. Nogo-A Modulates the Synaptic Excitation of Hippocampal Neurons in a Ca2+-Dependent Manner. Cells. 2021; 10(9):2299. https://doi.org/10.3390/cells10092299
Chicago/Turabian StyleMetzdorf, Kristin, Steffen Fricke, Maria Teresa Balia, Martin Korte, and Marta Zagrebelsky. 2021. "Nogo-A Modulates the Synaptic Excitation of Hippocampal Neurons in a Ca2+-Dependent Manner" Cells 10, no. 9: 2299. https://doi.org/10.3390/cells10092299
APA StyleMetzdorf, K., Fricke, S., Balia, M. T., Korte, M., & Zagrebelsky, M. (2021). Nogo-A Modulates the Synaptic Excitation of Hippocampal Neurons in a Ca2+-Dependent Manner. Cells, 10(9), 2299. https://doi.org/10.3390/cells10092299