
Sinapic Acid Controls Inflammation by Suppressing NLRP3 Inflammasome Activation


Abstract
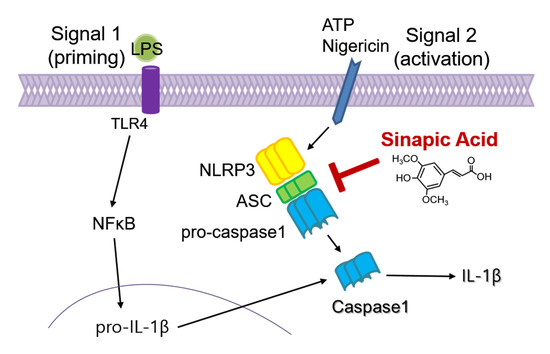
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Mice
2.3. Cell Culture
2.4. MTT Assay
2.5. Western Blot Analysis
2.6. ELISA
2.7. Real-Time PCR
2.8. Reporter Gene Assay Analysis
2.9. Immunofluorescence
2.10. In Vivo LPS Injection
2.11. Statistics
3. Results
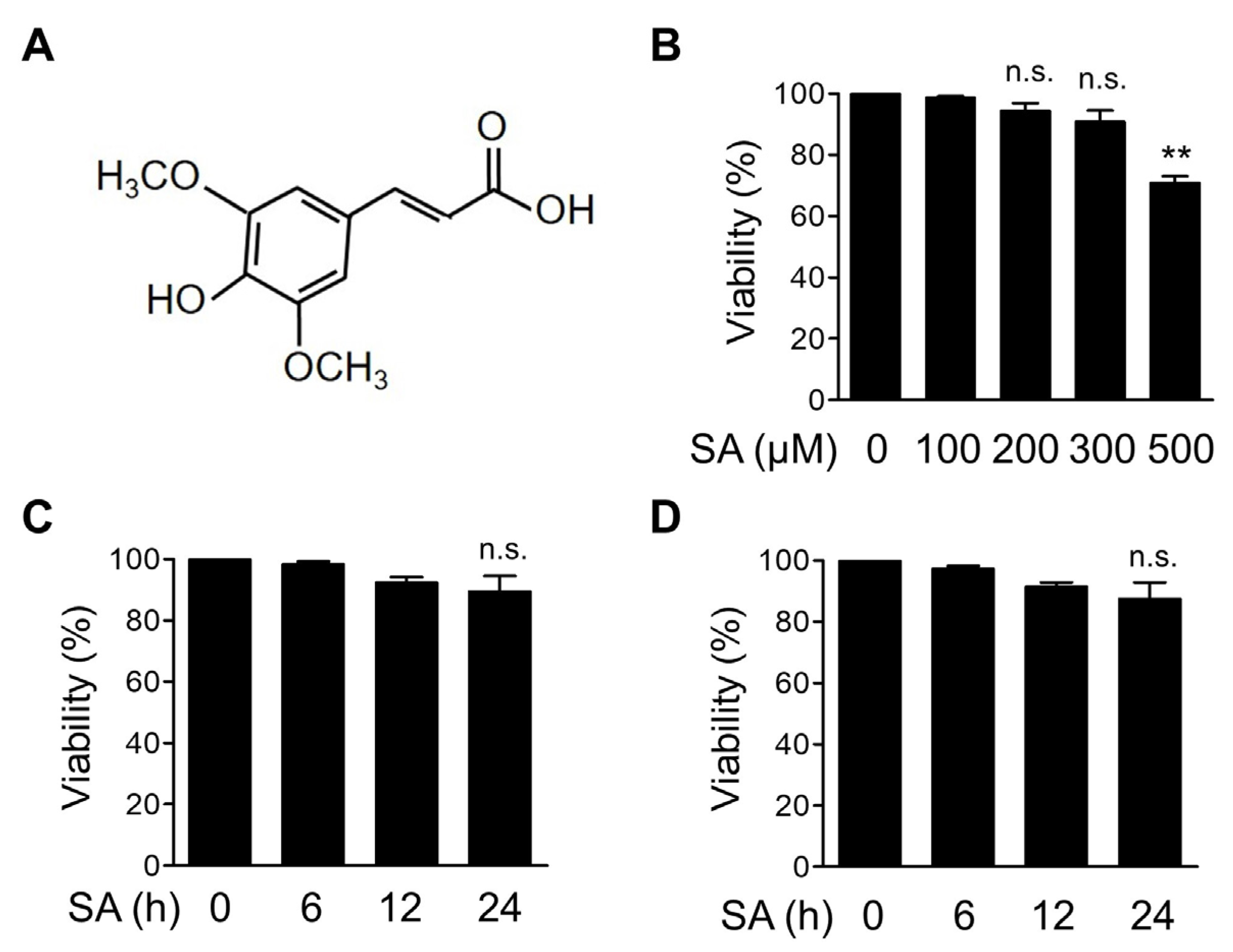
3.1. Dose Optimization of SA in Bone Marrow-Derived Macrophages (BMDMs)
3.2. SA Inhibits NLRP3 Inflammasome Activation
3.3. SA Has No Effect on the Transcription of Proinflammatory Mediators
3.4. SA Has No Effect on the Priming Phase of the NLRP3 Inflammasome
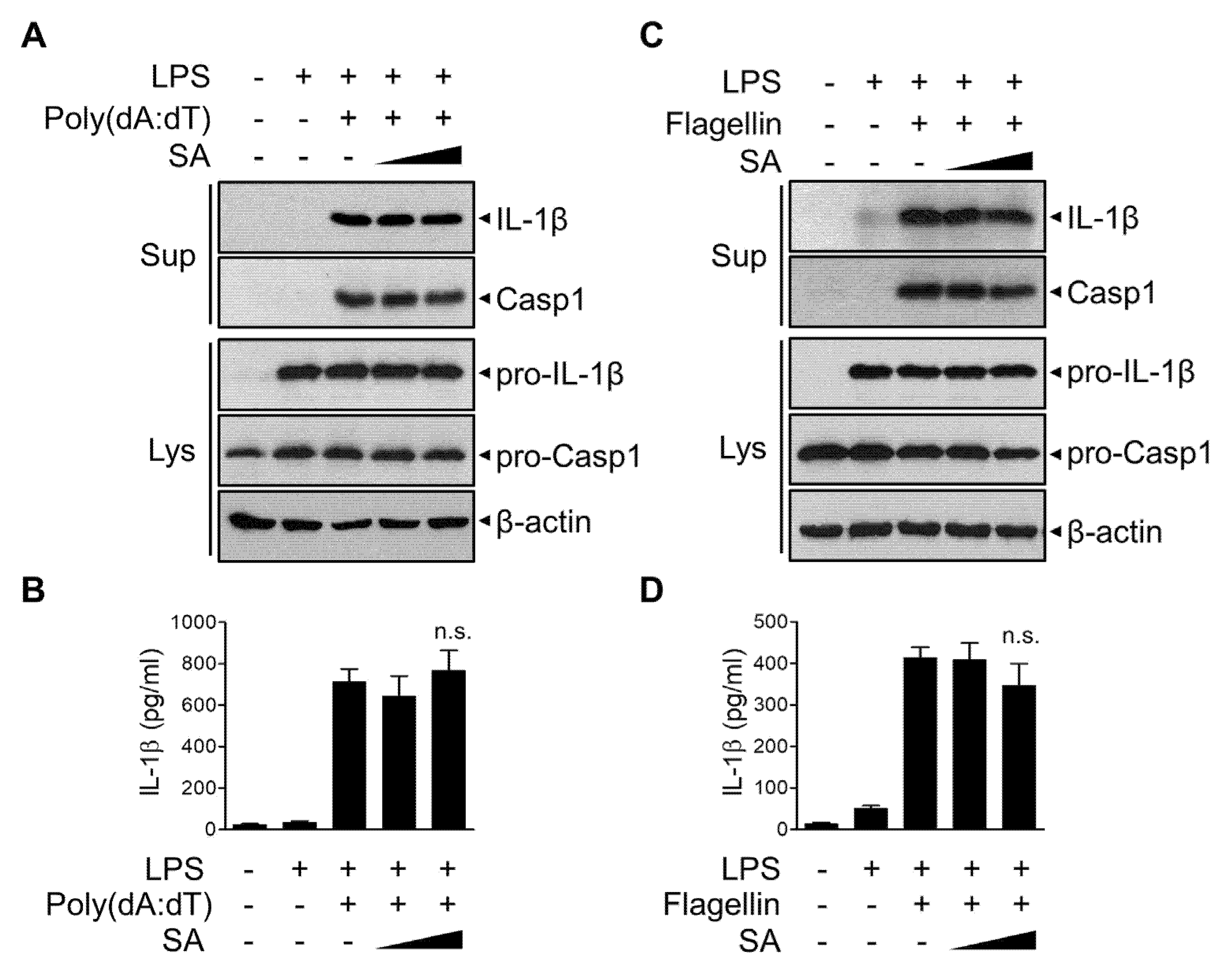
3.5. SA Has No effect on AIM2 or NLRC4 Inflammasomes
3.6. SA Reduces LPS-Induced Systemic Inflammation In Vivo
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, C. Sinapic Acid and Its Derivatives as Medicine in Oxidative Stress-Induced Diseases and Aging. Oxid. Med. Cell. Longev. 2016, 2016, 3571614. [Google Scholar] [CrossRef] [Green Version]
- Menezes, J.C.; Kamat, S.P.; Cavaleiro, J.A.; Gaspar, A.; Garrido, J.; Borges, F. Synthesis and antioxidant activity of long chain alkyl hydroxycinnamates. Eur. J. Med. Chem. 2011, 46, 773–777. [Google Scholar] [CrossRef] [Green Version]
- Lee-Manion, A.M.; Price, R.K.; Strain, J.J.; Dimberg, L.H.; Sunnerheim, K.; Welch, R.W. In vitro antioxidant activity and antigenotoxic effects of avenanthramides and related compounds. J. Agric. Food Chem. 2009, 57, 10619–10624. [Google Scholar] [CrossRef] [PubMed]
- Firuzi, O.; Giansanti, L.; Vento, R.; Seibert, C.; Petrucci, R.; Marrosu, G.; Agostino, R.; Saso, L. Hypochlorite scavenging activity of hydroxycinnamic acids evaluated by a rapid microplate method based on the measurement of chloramines. J. Pharm. Pharmacol. 2003, 55, 1021–1027. [Google Scholar] [CrossRef]
- Niwa, T.; Doi, U.; Kato, Y.; Osawa, T. Inhibitory mechanism of sinapinic acid against peroxynitrite-mediated tyrosine nitration of protein in vitro. FEBS Lett. 1999, 459, 43–46. [Google Scholar] [CrossRef] [Green Version]
- Zou, Y.; Kim, A.R.; Kim, J.E.; Choi, J.S.; Chung, H.Y. Peroxynitrite scavenging activity of sinapic acid (3,5-dimethoxy-4-hydroxycinnamic acid) isolated from Brassica juncea. J. Agric. Food Chem. 2002, 50, 5884–5890. [Google Scholar] [CrossRef]
- Kampa, M.; Alexaki, V.I.; Notas, G.; Nifli, A.P.; Nistikaki, A.; Hatzoglou, A.; Bakogeorgou, E.; Kouimtzoglou, E.; Blekas, G.; Boskou, D.; et al. Antiproliferative and apoptotic effects of selective phenolic acids on T47D human breast cancer cells: Potential mechanisms of action. Breast Cancer Res. 2004, 6, R63–R74. [Google Scholar] [CrossRef] [Green Version]
- Yoon, B.H.; Jung, J.W.; Lee, J.J.; Cho, Y.W.; Jang, C.G.; Jin, C.; Oh, T.H.; Ryu, J.H. Anxiolytic-like effects of sinapic acid in mice. Life Sci. 2007, 81, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Yoon, B.H.; Jung, W.Y.; Kim, J.M.; Park, S.J.; Park, D.H.; Huh, Y.; Park, C.; Cheong, J.H.; Lee, K.T.; et al. Sinapic acid attenuates kainic acid-induced hippocampal neuronal damage in mice. Neuropharmacology 2010, 59, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Ferreres, F.; Fernandes, F.; Sousa, C.; Valentao, P.; Pereira, J.A.; Andrade, P.B. Metabolic and bioactivity insights into Brassica oleracea var. acephala. J. Agric. Food Chem. 2009, 57, 8884–8892. [Google Scholar] [CrossRef]
- Lee, J.Y. Anti-inflammatory effects of sinapic acid on 2,4,6-trinitrobenzenesulfonic acid-induced colitis in mice. Arch. Pharm. Res. 2018, 41, 243–250. [Google Scholar] [CrossRef]
- Yun, K.J.; Koh, D.J.; Kim, S.H.; Park, S.J.; Ryu, J.H.; Kim, D.G.; Lee, J.Y.; Lee, K.T. Anti-inflammatory effects of sinapic acid through the suppression of inducible nitric oxide synthase, cyclooxygase-2, and proinflammatory cytokines expressions via nuclear factor-kappaB inactivation. J. Agric. Food Chem. 2008, 56, 10265–10272. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Dixit, V.M. Mechanisms and functions of inflammasomes. Cell 2014, 157, 1013–1022. [Google Scholar] [CrossRef] [Green Version]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Vanaja, S.K.; Rathinam, V.A.; Fitzgerald, K.A. Mechanisms of inflammasome activation: Recent advances and novel insights. Trends Cell Biol. 2015, 25, 308–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vance, R.E. The NAIP/NLRC4 inflammasomes. Curr. Opin. Immunol. 2015, 32, 84–89. [Google Scholar] [CrossRef] [Green Version]
- Burckstummer, T.; Baumann, C.; Bluml, S.; Dixit, E.; Durnberger, G.; Jahn, H.; Planyavsky, M.; Bilban, M.; Colinge, J.; Bennett, K.L.; et al. An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat. Immunol. 2009, 10, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Hagar, J.A.; Powell, D.A.; Aachoui, Y.; Ernst, R.K.; Miao, E.A. Cytoplasmic LPS activates caspase-11: Implications in TLR4-independent endotoxic shock. Science 2013, 341, 1250–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Huang, H.; Liu, B.; Zhang, Y.; Pan, X.; Yu, X.Y.; Shen, Z.; Song, Y.H. Inflammasomes as therapeutic targets in human diseases. Signal. Transduct. Target. Ther. 2021, 6, 247. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Callaway, J.B.; Ting, J.P. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauernfeind, F.G.; Horvath, G.; Stutz, A.; Alnemri, E.S.; MacDonald, K.; Speert, D.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.G.; Fitzgerald, K.A.; et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 2009, 183, 787–791. [Google Scholar] [CrossRef]
- Mariathasan, S.; Weiss, D.S.; Newton, K.; McBride, J.; O’Rourke, K.; Roose-Girma, M.; Lee, W.P.; Weinrauch, Y.; Monack, D.M.; Dixit, V.M. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006, 440, 228–232. [Google Scholar] [CrossRef]
- Coll, R.C.; Robertson, A.A.; Chae, J.J.; Higgins, S.C.; Munoz-Planillo, R.; Inserra, M.C.; Vetter, I.; Dungan, L.S.; Monks, B.G.; Stutz, A.; et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015, 21, 248–255. [Google Scholar] [CrossRef] [Green Version]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nunez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef] [Green Version]
- Youm, Y.H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D’Agostino, D.; Planavsky, N.; Lupfer, C.; Kanneganti, T.D.; et al. The ketone metabolite beta-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat. Med. 2015, 21, 263–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.H.; Shin, J.H.; Kim, S.S.; Lee, H.; Yang, S.R.; Seo, S.R. Laurus nobilis leaf extract controls inflammation by suppressing NLRP3 inflammasome activation. J. Cell. Physiol. 2019, 234, 6854–6864. [Google Scholar] [CrossRef]
- Englen, M.D.; Valdez, Y.E.; Lehnert, N.M.; Lehnert, B.E. Granulocyte/macrophage colony-stimulating factor is expressed and secreted in cultures of murine L929 cells. J. Immunol. Methods 1995, 184, 281–283. [Google Scholar] [CrossRef]
- Boltz-Nitulescu, G.; Wiltschke, C.; Holzinger, C.; Fellinger, A.; Scheiner, O.; Gessl, A.; Forster, O. Differentiation of rat bone marrow cells into macrophages under the influence of mouse L929 cell supernatant. J. Leukoc. Biol. 1987, 41, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Bode, J.G.; Ehlting, C.; Haussinger, D. The macrophage response towards LPS and its control through the p38(MAPK)-STAT3 axis. Cell. Signal. 2012, 24, 1185–1194. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Franchi, L.; Nunez, G. TLR agonists stimulate Nlrp3-dependent IL-1beta production independently of the purinergic P2X7 receptor in dendritic cells and in vivo. J. Immunol. 2013, 190, 334–339. [Google Scholar] [CrossRef] [Green Version]
- Mao, K.; Chen, S.; Chen, M.; Ma, Y.; Wang, Y.; Huang, B.; He, Z.; Zeng, Y.; Hu, Y.; Sun, S.; et al. Nitric oxide suppresses NLRP3 inflammasome activation and protects against LPS-induced septic shock. Cell Res. 2013, 23, 201–212. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Wang, H.; Zheng, M.; Xu, W.; Yang, Y.; Shi, F. Ginsenoside Rg3 suppresses the NLRP3 inflammasome activation through inhibition of its assembly. FASEB J. 2020, 34, 208–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinarello, C.A.; Simon, A.; van der Meer, J.W. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat. Rev. Drug Discov. 2012, 11, 633–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, H.; He, H.; Chen, Y.; Huang, W.; Cheng, J.; Ye, J.; Wang, A.; Tao, J.; Wang, C.; Liu, Q.; et al. Identification of a selective and direct NLRP3 inhibitor to treat inflammatory disorders. J. Exp. Med. 2017, 214, 3219–3238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinon, F.; Petrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006, 440, 237–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Dong, Y.; Ye, M.; Jin, S.; Yang, J.; Joosse, M.E.; Sun, Y.; Zhang, J.; Lazarev, M.; Brant, S.R.; et al. The Pathogenic Role of NLRP3 Inflammasome Activation in Inflammatory Bowel Diseases of Both Mice and Humans. J. Crohn’s Colitis 2017, 11, 737–750. [Google Scholar] [CrossRef]
- Mangan, M.S.J.; Olhava, E.J.; Roush, W.R.; Seidel, H.M.; Glick, G.D.; Latz, E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat. Rev. Drug Discov. 2018, 17, 688. [Google Scholar] [CrossRef] [Green Version]
- de Torre-Minguela, C.; Mesa Del Castillo, P.; Pelegrin, P. The NLRP3 and Pyrin Inflammasomes: Implications in the Pathophysiology of Autoinflammatory Diseases. Front. Immunol. 2017, 8, 43. [Google Scholar] [CrossRef] [Green Version]
- Marchetti, C.; Swartzwelter, B.; Gamboni, F.; Neff, C.P.; Richter, K.; Azam, T.; Carta, S.; Tengesdal, I.; Nemkov, T.; D’Alessandro, A.; et al. OLT1177, a beta-sulfonyl nitrile compound, safe in humans, inhibits the NLRP3 inflammasome and reverses the metabolic cost of inflammation. Proc. Natl. Acad. Sci. USA 2018, 115, E1530–E1539. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Jiang, H.; Chen, Y.; Wang, X.; Yang, Y.; Tao, J.; Deng, X.; Liang, G.; Zhang, H.; Jiang, W.; et al. Tranilast directly targets NLRP3 to treat inflammasome-driven diseases. EMBO Mol. Med. 2018, 10, e8689. [Google Scholar] [CrossRef]
- He, H.; Jiang, H.; Chen, Y.; Ye, J.; Wang, A.; Wang, C.; Liu, Q.; Liang, G.; Deng, X.; Jiang, W.; et al. Oridonin is a covalent NLRP3 inhibitor with strong anti-inflammasome activity. Nat. Commun. 2018, 9, 2550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fautrel, B. Economic benefits of optimizing anchor therapy for rheumatoid arthritis. Rheumatology 2012, 51, iv21–iv26. [Google Scholar] [CrossRef] [Green Version]
- Andreasen, M.F.; Landbo, A.K.; Christensen, L.P.; Hansen, A.; Meyer, A.S. Antioxidant effects of phenolic rye (Secale cereale L.) extracts, monomeric hydroxycinnamates, and ferulic acid dehydrodimers on human low-density lipoproteins. J. Agric. Food Chem. 2001, 49, 4090–4096. [Google Scholar] [CrossRef]
- Zhu, M.J.; Kang, Y.; Xue, Y.; Liang, X.; Garcia, M.P.G.; Rodgers, D.; Kagel, D.R.; Du, M. Red raspberries suppress NLRP3 inflammasome and attenuate metabolic abnormalities in diet-induced obese mice. J. Nutr. Biochem. 2018, 53, 96–103. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, G.; Gurley, E.C.; Zhou, H. Flavonoid apigenin inhibits lipopolysaccharide-induced inflammatory response through multiple mechanisms in macrophages. PLoS ONE 2014, 9, e107072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, F.; Ye, B.; Cao, J.; Cai, X.; Lin, L.; Huang, S.; Huang, W.; Huang, Z. Curcumin Represses NLRP3 Inflammasome Activation via TLR4/MyD88/NF-kappaB and P2X7R Signaling in PMA-Induced Macrophages. Front. Pharmacol. 2016, 7, 369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nyandwi, J.B.; Ko, Y.S.; Jin, H.; Yun, S.P.; Park, S.W.; Kim, H.J. Rosmarinic acid inhibits oxLDL-induced inflammasome activation under high-glucose conditions through downregulating the p38-FOXO1-TXNIP pathway. Biochem. Pharmacol. 2020, 182, 114246. [Google Scholar] [CrossRef]
- Hameed, H.; Aydin, S.; Başaran, A.A.; Başaran, N. Assessment of cytotoxic properties of sinapic acid in vitro. Turk. J. Pharm. Sci. 2016, 13, 225–232. [Google Scholar] [CrossRef]
- Kim, Y.O.; Lee, S.W.; Oh, M.S.; Lee, H.J. Effects of sinapic Acid of 4 vessel occlusion model-induced ischemia and cognitive impairments in the rat. Clin. Psychopharmacol. Neurosci. 2011, 9, 86–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, E.H.; Shin, J.H.; Kim, S.S.; Seo, S.R. Sinapic Acid Controls Inflammation by Suppressing NLRP3 Inflammasome Activation. Cells 2021, 10, 2327. https://doi.org/10.3390/cells10092327
Lee EH, Shin JH, Kim SS, Seo SR. Sinapic Acid Controls Inflammation by Suppressing NLRP3 Inflammasome Activation. Cells. 2021; 10(9):2327. https://doi.org/10.3390/cells10092327
Chicago/Turabian StyleLee, Eun Hye, Jin Hak Shin, Seon Sook Kim, and Su Ryeon Seo. 2021. "Sinapic Acid Controls Inflammation by Suppressing NLRP3 Inflammasome Activation" Cells 10, no. 9: 2327. https://doi.org/10.3390/cells10092327