
A2A Adenosine Receptor as a Potential Biomarker and a Possible Therapeutic Target in Alzheimer’s Disease


 ,
,  , ,
, ,  ,
, 

Abstract
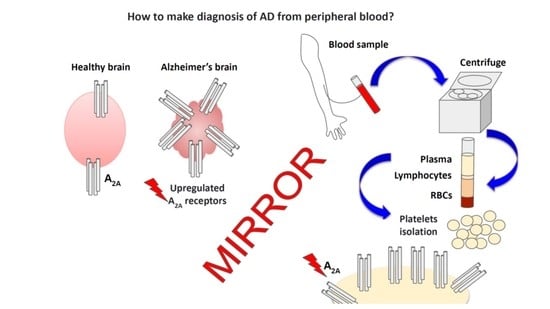
:
1. Alzheimer’s Disease
2. Biomarkers of AD
3. A2A Adenosine Receptors Biology
4. Role of A2A Adenosine Receptors in AD
4.1. Neuronal Injury
4.2. Neuroinflammation
4.2.1. Astrocytes
4.2.2. Microglia
5. A2A Adenosine Receptor as a Novel Peripheral Biomarker in AD
6. A2A Adenosine Receptor as a Possible Therapeutic Target in AD
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Arlington, V.A.; American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Publishing: Washington, DC, USA, 2013. [Google Scholar]
- Vermunt, L.; Sikkes, S.A.M.; van den Hout, A.; Handels, R.; Bos, I.; van der Flier, W.M.; Kern, S.; Ousset, P.J.; Maruff, P.; Skoog, I.; et al. Alzheimer Disease Neuroimaging Initiative; AIBL Research Group; ICTUS/DSA study groups. Duration of preclinical, prodromal, and dementia stages of Alzheimer’s disease in relation to age, sex, and APOE genotype. Alzheimers Dement. 2019, 15, 888–898. [Google Scholar] [CrossRef]
- Williamson, J.; Goldman, J.; Marder, K.S. Genetic aspects of Alzheimer disease. Neurologist 2009, 15, 80–86. [Google Scholar] [CrossRef] [Green Version]
- Livingston, J.M.; McDonald, M.W.; Gagnon, T.; Jeffers, M.S.; Gomez-Smith, M.; Antonescu, S.; Cron, G.O.; Boisvert, C.; Lacoste, B.; Corbett, D. Influence of metabolic syndrome on cerebral perfusion and cognition. Neurobiol. Dis. 2020, 137, 104756. [Google Scholar] [CrossRef] [PubMed]
- Lourida, I.; Hannon, E.; Littlejohns, T.J.; Langa, K.M.; Hyppönen, E.; Kuzma, E.; Llewellyn, D.J. Association of Lifestyle and Genetic Risk With Incidence of Dementia. JAMA 2019, 322, 430–437. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Nortley, R.; Korte, N.; Izquierdo, P.; Hirunpattarasilp, C.; Mishra, A.; Jaunmuktane, Z.; Kyrargyri, V.; Pfeiffer, T.; Khennouf, L.; Madry, C.; et al. Amyloid b oligomers constrict human capillaries in Alzheimer’s disease via signaling to pericytes. Science 2019, 365, eaav9518. [Google Scholar] [CrossRef] [PubMed]
- Perez-Nievas, B.G.; Stein, T.D.; Tai, H.C.; Dols-Icardo, O.; Scotton, T.C.; Barroeta-Espar, I.; Fernandez-Carballo, L.; de Munain, E.L.; Perez, J.; Marquie, M.; et al. Dissecting phenotypic traits linked to human resilience to Alzheimer’s pathology. Brain 2013, 136, 2510–2526. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Wiste, H.J.; Schwarz, C.G.; Lowe, V.J.; Senjem, M.L.; Vemuri, P.; Weigand, S.D.; Therneau, T.M.; Knopman, D.S.; Gunter, J.L.; et al. Longitudinal tau PET in ageing and Alzheimer’s disease. Brain 2018, 141, 1517–1528. [Google Scholar] [CrossRef] [Green Version]
- Mirra, S.S.; Heyman, A.; McKeel, D.; Sumi, S.M.; Crain, B.J.; Brownlee, L.M.; Vogel, F.S.; Hughes, J.P.; Belle, G.V.; Berg, L. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD): Part II. Standardization of the Neuropathologic Assessment of Alzheimer’s Disease. Neurology 1991, 41, 479. [Google Scholar] [CrossRef]
- Thal, D.R.; Rüb, U.; Orantes, M.; Braak, H. Phases of Aβ-deposition in the human brain and its relevance for the development of AD. Neurology 2002, 58, 1791–1800. [Google Scholar] [CrossRef]
- Braak, H.; Alafuzoff, I.; Arzberger, T.; Kretzschmar, H.; Del Tredici, K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006, 112, 389–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montine, T.J.; Phelps, C.H.; Beach, T.G.; Bigio, E.H.; Cairns, N.J.; Dickson, D.W.; Duyckaerts, C.; Frosch, M.P.; Masliah, E.; Mirra, S.S.; et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: A practical approach. Acta Neuropathol. 2012, 123, 1–11. [Google Scholar] [CrossRef] [Green Version]
- McKhann, G.; Drachman, D.; Folstein, M.; Katzman, R.; Price, D.; Stadlan, E.M. Clinical Diagnosis of Alzheimer’s Disease: Report of the NINCDS-ADRDA Work Group under the Auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984, 34, 939–944. [Google Scholar] [CrossRef] [Green Version]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Feldman, H.H.; Frisoni, G.B.; Hampel, H.; Jagust, W.J.; Johnson, K.A.; Knopman, D.S.; et al. A/T/N: An unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology 2016, 87, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Crary, J.F.; Trojanowski, J.Q.; Schneider, J.A.; Abisambra, J.F.; Abner, E.L.; Alafuzoff, I.; Arnold, S.E.; Attems, J.; Beach, T.G.; Bigio, E.H.; et al. Primary age-related tauopathy (PART): A common pathology associated with human aging. Acta Neuropathol. 2014, 128, 755–766. [Google Scholar] [CrossRef] [Green Version]
- Nelson, P.T.; Dickson, D.W.; Trojanowski, J.Q.; Jack, C.R.; Boyle, P.A.; Arfanakis, K.; Rademakers, R.; Alafuzoff, I.; Attems, J.; Brayne, C.; et al. Limbic-predominant age-related TDP-43 encephalopathy (LATE): Consensus working group report. Brain 2019, 142, 1503–1527. [Google Scholar] [CrossRef] [Green Version]
- Smailagic, N.; Vacante, M.; Hyde, C.; Martin, S.; Ukoumunne, O.; Sachpekidis, C. ¹⁸F-FDG PET for the early diagnosis of Alzheimer’s disease dementia and other dementias in people with mild cognitive impairment (MCI). Cochrane Database Syst. Rev. 2015, 1, CD010632. [Google Scholar] [CrossRef]
- Zhang, S.; Smailagic, N.; Hyde, C.; Noel-Storr, A.H.; Takwoingi, Y.; McShane, R.; Feng, J. (11)C-PIB-PET for the early diagnosis of Alzheimer’s disease dementia and other dementias in people with mild cognitive impairment (MCI). Cochrane Database Syst. Rev. 2014, 7, CD010386. [Google Scholar] [CrossRef] [PubMed]
- Counts, S.E.; Ikonomovic, M.D.; Mercado, N.; Vega, I.E.; Mufson, E.J. Biomarkers for the Early Detection and Progression of Alzheimer’s Disease. Neurotherapeutics 2017, 14, 35–53. [Google Scholar] [CrossRef] [Green Version]
- Lewczuk, P.; Ermann, N.; Andreasson, U.; Schultheis, C.; Podhorna, J.; Spitzer, P.; Maler, J.M.; Kornhuber, J.; Blennow, K.; Zetterberg, H. Plasma neurofilament light as a potential biomarker of neurodegeneration in Alzheimer’s disease. Alzheimers Res. Ther. 2018, 10, 71. [Google Scholar] [CrossRef]
- Janelidze, S.; Mattsson, N.; Palmqvist, S.; Smith, R.; Beach, T.G.; Serrano, G.E.; Chai, X.; Proctor, N.K.; Eichenlaub, U.; Zetterberg, H.; et al. Plasma P-tau181 in Alzheimer’s disease: Relationship to other biomarkers, differential diagnosis, neuropathology and longitudinal progression to Alzheimer’s dementia. Nat. Med. 2020, 26, 379–386. [Google Scholar] [CrossRef]
- Wang, M.J.; Yi, S.; Han, J.Y.; Park, S.Y.; Jang, J.W.; Chun, I.K.; Kim, S.E.; Lee, B.S.; Kim, G.J.; Yu, J.S.; et al. Oligomeric forms of amyloid-β protein in plasma as a potential blood-based biomarker for Alzheimer’s disease. Alzheimer’s Res. Ther. 2017, 9, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fandos, N.; Pérez-Grijalba, V.; Pesini, P.; Olmos, S.; Bossa, M.; Villemagne, V.L.; Doecke, J.; Fowler, C.; Masters, C.L.; Sarasa, M. Plasma amyloid β 42/40 ratios as biomarkers for amyloid β cerebral deposition in cognitively normal individuals. Alzheimers Dement. 2017, 8, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Grijalba, V.; Arbizu, J.; Romero, J.; Prieto, E.; Pesini, P.; Sarasa, L.; Guillen, F.; Monleòn, I.; San-Josè, I.; Martìnez-Lage, P.; et al. Plasma Aβ42/40 ratio alone or combined with FDG-PET can accurately predict amyloid-PET positivity: A cross-sectional analysis from the AB255 Study. Alzheimer’s Res. Ther. 2019, 11, 96. [Google Scholar] [CrossRef] [PubMed]
- Mattsson, N.; Cullen, N.C.; Andreasson, U.; Zetterberg, H.; Blennow, K. Association Between Longitudinal Plasma Neurofilament Light and Neurodegeneration in Patients With Alzheimer Disease. JAMA Neurol. 2019, 76, 791–799. [Google Scholar] [CrossRef]
- Mattsson-Carlgren, N.; Janelidze, S.; Palmqvist, S.; Cullen, N.; Svenningsson, A.L.; Strandberg, O.; Mengel, D.; Walsh, D.M.; Stomrud, E.; Dage, J.L.; et al. Longitudinal plasma p-tau217 is increased in early stages of Alzheimer’s disease. Brain 2020, 143, 3234–3241. [Google Scholar] [CrossRef]
- Headley, A.; De Leon-Benedetti, A.; Dong, C.; Levin, B.; Loewenstein, D.; Camargo, C.; Rundek, T.; Zetterberg, H.; Blennow, K.; Wright, C.B.; et al. Alzheimer’s Disease Neuroimaging Initiative. Neurogranin as a predictor of memory and executive function decline in MCI patients. Neurology 2018, 90, e887–e895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrano-Pozo, A.; Betensky, R.A.; Frosch, M.P.; Hyman, B.T. Plaque-Associated Local Toxicity Increases over the Clinical Course of Alzheimer Disease. Am. J. Pathol. 2016, 186, 375–384. [Google Scholar] [CrossRef] [Green Version]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef]
- Poloni, T.E.; Medici, V.; Moretti, M.; Visonà, S.D.; Cirrincione, A.; Carlos, A.F.; Davin, A.; Gagliard, S.; Pansarasa, O.; Cereda, C.; et al. COVID-19-related neuropathology and microglial activation in elderly with and without dementia. Brain Pathol. 2021, 31, e12997. [Google Scholar] [CrossRef] [PubMed]
- Tarkowski, E.; Andreasen, N.; Tarkowski, A.; Blennow, K. Intrathecal inflammation precedes development of Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 2003, 74, 1200–1205. [Google Scholar] [CrossRef] [PubMed]
- Muszyński, P.; Groblewska, M.; Kulczyńska-Przybik, A.; Kułakowska, A.; Mroczko, B. YKL-40 as a Potential Biomarker and a Possible Target in Therapeutic Strategies of Alzheimer’s Disease. Curr. Neuropharmacol. 2017, 15, 906–917. [Google Scholar] [CrossRef] [Green Version]
- Olsson, B.; Hertze, J.; Lautner, R.; Zetterberg, H.; Nägga, K.; Hoglund, K.; Basun, H.; Annas, P.; Lannfelt, L.; Andreasen, N.; et al. Microglial Markers are Elevated in the Prodromal Phase of Alzheimer’s Disease and Vascular Dementia. J. Alzheimer’s Dis. 2013, 33, 45–53. [Google Scholar] [CrossRef]
- Zetterberg, H.; Bendlin, B.B. Biomarkers for Alzheimer’s disease-preparing for a new era of disease-modifying therapies. Mol. Psychiatry 2021, 26, 296–308. [Google Scholar] [CrossRef]
- Mullard, A. FDA approval for Biogen’s aducanumab sparks Alzheimer disease firestorm. Nat. Rev. Drug Discov. 2021, 20, 496. [Google Scholar]
- Sabbagh, M.N.; Cummings, J. Open Peer Commentary to “Failure to demonstrate efficacy of aducanumab: An analysis of the EMERGE and ENGAGE Trials as reported by Biogen December 2019”. Alzheimers Dement. 2021, 17, 702–703. [Google Scholar] [CrossRef] [PubMed]
- Knopman, D.S.; Jones, D.T.; Greicius, M.D. Failure to demonstrate efficacy of aducanumab: An analysis of the EMERGE and ENGAGE trials as reported by Biogen, December 2019. Alzheimers Dement. 2021, 17, 696–701. [Google Scholar] [CrossRef]
- Borea, P.A.; Gessi, S.; Merighi, S.; Vincenzi, F.; Varani, K. Pharmacology of Adenosine Receptors: The State of the Art. Physiol. Rev. 2018, 98, 1591–1625. [Google Scholar] [CrossRef] [PubMed]
- Fredholm, B.B.; Arslan, G.; Halldner, L.; Kull, B.; Schulte, G.; Wasserman, W. Structure and function of adenosine receptors and their genes. Naunyn Schmiedebergs Arch. Pharmacol. 2000, 362, 364–374. [Google Scholar] [CrossRef]
- Ferré, S.; Navarro, G.; Casadó, V.; Cortés, A.; Mallol, J.; Canela, E.I.; Lluís, C.; Franco, R. G protein-coupled receptor heteromers as new targets for drug development. Prog. Mol. Biol. Transl. Sci. 2010, 91, 41–52. [Google Scholar]
- Antonioli, L.; Fornai, M.; Blandizzi, C.; Pacher, P.; Haskó, G. Adenosine signaling and the immune system: When a lot could be too much. Immunol. Lett. 2019, 205, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Kull, B.; Svenningsson, P.; Fredholm, B.B. Adenosine A(2A) receptors are colocalized with and activate g(olf) in rat striatum. Mol. Pharmacol. 2000, 58, 771–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Preti, D.; Baraldi, P.G.; Moorman, A.R.; Borea, P.A.; Varani, K. History and perspectives of A2A adenosine receptor antagonists as potential therapeutic agents. Med. Res. Rev. 2015, 35, 790–848. [Google Scholar] [CrossRef]
- Baraldi, P.G.; Tabrizi, M.A.; Gessi, S.; Borea, P.A. Adenosine receptor antagonists: Translating medicinal chemistry and pharmacology into clinical utility. Chem. Rev. 2008, 108, 238–263. [Google Scholar] [CrossRef]
- Chen, J.F.; Eltzschig, H.K.; Fredholm, B.B. Adenosine receptors as drug targets–what are the challenges? Nat. Rev. Drug Discov. 2013, 12, 265–286. [Google Scholar] [CrossRef] [Green Version]
- Schulte, G.; Fredholm, B.B. Signalling from adenosine receptors to mitogen-activated protein kinases. Cell Signal 2003, 15, 813–827. [Google Scholar] [CrossRef]
- Terry, R.D.; Masliah, E.; Salmon, D.P.; Butters, N.; DeTeresa, R.; Hill, R.; Hansen, L.A.; Katzman, R. Physical basis of cognitive alterations in Alzheimer’s disease: Synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 1991, 30, 572–580. [Google Scholar] [CrossRef]
- Scheff, S.W.; Price, D.A.; Schmitt, F.A.; DeKosky, S.T.; Mufson, E.J. Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology 2007, 68, 1501–1508. [Google Scholar] [CrossRef] [PubMed]
- Scheff, S.W.; Price, D.A.; Ansari, M.A.; Roberts, K.N.; Schmitt, F.A.; Ikonomovic, M.D.; Mufson, E.J. Synaptic change in the posterior cingulate gyrus in the progression of Alzheimer’s disease. J. Alzheimers Dis. 2015, 43, 1073–1090. [Google Scholar] [CrossRef] [Green Version]
- Selkoe, D.J. Alzheimer’s disease is a synaptic failure. Science 2002, 298, 789–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coleman, P.; Federoff, H.; Kurlan, R. A focus on the synapse for neuroprotection in Alzheimer disease and other dementias. Neurology 2004, 63, 1155–1162. [Google Scholar] [CrossRef]
- Horgusluoglu-Moloch, E.; Nho, K.; Risacher, S.L.; Kim, S.; Foroud, T.; Shaw, L.M.; Trojanowski, J.Q.; Aisen, P.S.; Petersen, R.C.; Jack, C.R.; et al. Targeted neurogenesis pathway-based gene analysis identifies ADORA2A associated with hippocampal volume in mild cognitive impairment and Alzheimer’s disease. Neurobiol. Aging 2017, 60, 92–103. [Google Scholar] [CrossRef] [Green Version]
- Rebola, N.; Canas, P.M.; Oliveira, C.R.; Cunha, R.A. Different synaptic and subsynaptic localization of adenosine A2A receptors in the hippocampus and striatum of the rat. Neuroscience 2005, 132, 893–903. [Google Scholar] [CrossRef]
- Rebola, N.; Lujan, R.; Cunha, R.A.; Mulle, C. Adenosine A2A receptors are essential for long-term potentiation of NMDA-EPSCs at hippocampal mossy fiber synapses. Neuron 2008, 57, 121–134. [Google Scholar] [CrossRef] [Green Version]
- Costenla, A.R.; Diógenes, M.J.; Canas, P.M.; Rodrigues, R.J.; Nogueira, C.; Maroco, J.; Agostinho, P.M.; Ribeiro, J.A.; Cunha, R.A.; De Mendonça, A. Enhanced role of adenosine A(2A) receptors in the modulation of LTP in the rat hippocampus upon ageing. Eur. J. Neurosci. 2011, 34, 12–21. [Google Scholar] [CrossRef]
- Tebano, M.T.; Martire, A.; Rebola, N.; Pepponi, R.; Domenici, M.R.; Gro, M.C.; Schwarzschild, M.A.; Chen, J.F.; Cunha, R.A.; PopoliAdenosine, P. A2A receptors and metabotropic glutamate 5 receptors are co-localized and functionally interact in the hippocampus: A possible key mechanism in the modulation of N-methyl-D-aspartate effects. J. Neurochem. 2005, 95, 1188–1200. [Google Scholar] [CrossRef] [Green Version]
- Tebano, M.T.; Martire, A.; Chiodi, V.; Ferrante, A.; Popoli, P. Role of adenosine A(2A) receptors in modulating synaptic functions and brain levels of BDNF: A possible key mechanism in the pathophysiology of Huntington’s disease. Sci. World J. 2010, 10, 1768–1782. [Google Scholar] [CrossRef] [Green Version]
- Temido-Ferreira, M.; Coelho, J.E.; Pousinha, P.A.; Lopes, L.V. Novel players in the aging synapse: Impact on cognition. J. Caffeine Adenosine Res. 2019, 9, 104–127. [Google Scholar] [CrossRef] [PubMed]
- Temido-Ferreira, M.; Ferreira, D.G.; Batalha, V.L.; Marques-Morgado, I.; Coelho, J.E.; Pereira, P.; Gomes, R.; Pinto, A.; Carvalho, S.; Canas, P.M.; et al. Age-related shift in LTD is dependent on neuronal adenosine A 2A receptors interplay with mGluR5 and NMDA receptors. Mol. Psychiatry 2020, 25, 1876–1900. [Google Scholar] [CrossRef] [PubMed]
- Viana da Silva, S.; Haberl, M.G.; Zhang, P.; Bethge, P.; Lemos, C.; Gonçalves, N.; Gorlewicz, A.; Malezieux, M.; Gonçalves, F.Q.; Grosjean, N.; et al. Early synaptic deficits in the APP/PS1 mouse model of Alzheimer’s disease involve neuronal adenosine A2A receptors. Nat. Commun. 2016, 7, 11915. [Google Scholar] [CrossRef]
- Lopes, L.V.; Cunha, R.A.; Ribeiro, J.A. Cross talk between A(1) and A(2A) adenosine receptors in the hippocampus and cortex of young adult and old rats. J. Neurophysiol. 1999, 82, 3196–3203. [Google Scholar] [CrossRef]
- Lopes, L.V.; Cunha, R.A.; Ribeiro, J.A. Increase in the Number, G Protein Coupling, and Efficiency of Facilitatory Adenosine A2A Receptors in the Limbic Cortex, but not Striatum, of Aged Rats. J. Neurochem. 1999, 73, 1733–1738. [Google Scholar] [CrossRef]
- Arendash, G.W.; Schleif, W.; Rezai-Zadeh, K.; Jackson, E.K.; Zacharia, L.C.; Cracchiolo, J.R.; Shippy, D.; Tan, J. Caffeine protects Alzheimer’s mce against cognitive impairment and reduces brain beta-amyloid production. Neuroscience 2006, 142, 941–952. [Google Scholar] [CrossRef]
- Canas, P.M.; Duarte, J.M.; Rodrigues, R.J.; Köfalvi, A.; Cunha, R.A. Modification upon aging of the density of presynaptic modulation systems in the hippocampus. Neurobiol. Aging 2009, 30, 1877–1884. [Google Scholar] [CrossRef] [Green Version]
- Espinosa, J.; Rocha, A.; Nunes, F.; Costa, M.S.; Schein, V.; Kazlauckas, V.; Kalinine, E.; Souza, D.O.; Rodrigo, A.; Cunha, R.A.; et al. Caffeine consumption prevents memory impairment, neuronal damage, and adenosine A2A receptors upregulation in the hippocampus of a rat model of sporadic dementia. J. Alzheimers Dis. 2013, 34, 509–518. [Google Scholar] [CrossRef]
- Orr, A.G.; Hsiao, E.C.; Wang, M.M.; Ho, K.; Kim, D.H.; Wang, X.; Guo, W.; Kang, J.; Yu, G.Q.; Adame, A.; et al. Astrocytic adenosine receptor A2A and Gs-coupled signaling regulate memory. Nat. Neurosci. 2015, 18, 423–434. [Google Scholar] [CrossRef] [Green Version]
- Gonçalves, F.Q.; Lopes, J.P.; Silva, H.B.; Lemos, C.; Silva, A.C.; Gonçalves, N.; Tomé, A.R.; Ferreira, S.G.; Canas, P.M.; Rial, D.; et al. Synaptic and memory dysfunction in a β-amyloid model of early Alzheimer’s disease depends on increased formation of ATP-derived extracellular adenosine. Neurobiol. Dis. 2019, 132, 104570. [Google Scholar] [CrossRef]
- Albasanz, J.L.; Perez, S.; Barrachina, M.; Ferrer, I.; Martín, M. Up-regulation of adenosine receptors in the frontal cortex in Alzheimer’s disease. Brain Pathol. 2008, 18, 211–219. [Google Scholar] [CrossRef]
- Merighi, S.; Battistello, E.; Casetta, I.; Gragnaniello, D.; Poloni, T.E.; Medici, V.; Cirrincione, A.; Varani, K.; Vincenzi, F.; Borea, P.A.; et al. Upregulation of Cortical A2A Adenosine Receptors Is Reflected in Platelets of Patients with Alzheimer’s Disease. J. Alzheimers Dis. 2021, 80, 1105–1117. [Google Scholar] [CrossRef]
- Cunha, R.A.; Constantino, M.C.; Sebastião, A.M.; Ribeiro, J.A. Modification of A1 and A2a adenosine receptor binding in aged striatum, hippocampus and cortex of the rat. Neuroreport 1995, 6, 1583–1588. [Google Scholar] [CrossRef]
- Rebola, N.; Sebastião, A.M.; de Mendonca, A.; Oliveira, C.R.; Ribeiro, J.A.; Cunha, R.A. Enhanced adenosine A2A receptor facilitation of synaptic transmission in the hippocampus of aged rats. J. Neurophysiol. 2003, 90, 1295–1303. [Google Scholar] [CrossRef]
- Angulo, E.; Casadó, V.; Mallol, J.; Canela, E.I.; Viñals, F.; Ferrer, I.; Franco, R. A1 adenosine receptors accumulate in neurodegenerative structures in Alzheimer disease and mediate both amyloid precursor protein processing and tau phosphorylation and translocation. Brain Pathol. 2003, 13, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Diógenes, M.J.; Assaife-Lopes, N.; Pinto-Duarte, A.; Ribeiro, J.A.; Sebastião, A.M. Influence of age on BDNF modulation of hippocampal synaptic transmission: Interplay with adenosine A2A receptors. Hippocampus 2007, 17, 577–585. [Google Scholar] [CrossRef]
- Orr, A.G.; Lo, I.; Schumacher, H.; Ho, K.; Gill, M.; Guo, W.; Kim, D.H.; Knox, A.; Saito, T.; Saido, T.C.; et al. Istradefylline reduces memory deficits in aging mice with amyloid pathology. Neurobiol. Dis. 2018, 110, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Reyes-Resina, I.; Aguinaga, D.; Lillo, A.; Jiménez, J.; Raïch, I.; Borroto-Escuela, D.O.; Ferreiro-Vera, C.; Canela, E.I.; Sánchez de Medina, V.; et al. Potentiation of cannabinoid signaling in microglia by adenosine A2A receptor antagonists. Glia 2019, 67, 2410–2423. [Google Scholar] [CrossRef]
- Alonso-Andrés, P.; Albasanz, J.L.; Ferrer, I.; Martín, M. Purine-related metabolites and their converting enzymes are altered in frontal, parietal and temporal cortex at early stages of Alzheimer’s disease pathology. Brain Pathol. 2018, 28, 933–946. [Google Scholar] [CrossRef] [Green Version]
- Arendash, G.W.; Cao, C. Caffeine and coffee as therapeutics against Alzheimer’s disease. J. Alzheimers Dis. 2010, 20 (Suppl. S1), S117–S126. [Google Scholar] [CrossRef] [Green Version]
- Dall’Igna, O.P.; Fett, P.; Gomes, M.W.; Souza, D.O.; Cunha, R.A.; Lara, D.R. Caffeine and adenosine A(2a) receptor antagonists prevent beta-amyloid (25-35)-induced cognitive deficits in mice. Exp. Neurol. 2007, 203, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Canas, P.M.; Porciúncula, L.O.; Cunha, G.M.; Silva, C.G.; Machado, N.J.; Oliveira, J.M.; Oliveira, C.R.; Cunha, R.A. Adenosine A2A receptor blockade prevents synaptotoxicity and memory dysfunction caused by beta-amyloid peptides via p38 mitogen-activated protein kinase pathway. J. Neurosci. 2009, 29, 14741–14751. [Google Scholar] [CrossRef]
- Faivre, E.; Coelho, J.E.; Zornbach, K.; Malik, E.; Baqi, Y.; Schneider, M.; Cellai, L.; Carvalho, K.; Sebda, S.; Figeac, M.; et al. Beneficial Effect of a Selective Adenosine A2A Receptor Antagonist in the APPswe/PS1dE9 Mouse Model of Alzheimer’s Disease. Front. Mol. Neurosci. 2018, 11, 235. [Google Scholar] [CrossRef]
- Jacobson, K.A.; Gao, Z.G.; Matricon, P.; Eddy, M.T.; Carlsson, J. Adenosine A2A receptor antagonists: From caffeine to selective non-xanthines. Br. J. Pharmacol. 2020, 1–16. [Google Scholar] [CrossRef]
- Laurent, C.; Eddarkaoui, S.; Derisbourg, M.; Leboucher, A.; Demeyer, D.; Carrier, S.; Schneider, M.; Hamdane, M.; Müller, C.E.; Buée, L.; et al. Beneficial effects of caffeine in a transgenic model of Alzheimer’s disease-like tau pathology. Neurobiol. Aging 2014, 35, 2079–2090. [Google Scholar] [CrossRef]
- Laurent, C.; Burnouf, S.; Ferry, B.; Batalha, V.L.; Coelho, J.E.; Baqi, Y.; Malik, E.; Mariciniak, E.; Parrot, S.; Van der Jeugd, A.; et al. A2A adenosine receptor deletion is protective in a mouse model of Tauopathy. Mol. Psychiatry 2016, 21, 97–107. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, K.; Faivre, E.; Pietrowski, M.J.; Marques, X.; Gomez-Murcia, V.; Deleau, A.; Huin, V.; Hansen, J.N.; Kozlov, S.; Danis, C.; et al. Exacerbation of C1q dysregulation, synaptic loss and memory deficits in tau pathology linked to neuronal adenosine A2A receptor. Brain 2019, 142, 3636–3654. [Google Scholar] [CrossRef]
- Martí Navia, A.; Dal Ben, D.; Lambertucci, C.; Spinaci, A.; Volpini, R.; Marques-Morgado, I.; Coelho, J.E.; Lopes, L.V.; Marucci, G.; Buccioni, M. Adenosine Receptors as Neuroinflammation Modulators: Role of A(1) Agonists and A(2A) Antagonists. Cells 2020, 9, 1739. [Google Scholar] [CrossRef]
- Chambers, E.S.; Akbar, A.N. Can blocking inflammation enhance immunity during aging? J. Allergy Clin. Immunol. 2020, 145, 1323–1331. [Google Scholar] [CrossRef]
- Illes, P.; Rubini, P.; Ulrich, H.; Zhao, Y.; Tang, Y. Regulation of Microglial Functions by Purinergic Mechanisms in the Healthy and Diseased CNS. Cells 2020, 9, 1108. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Nedergaard, M. Physiology of Astroglia. Physiol. Rev. 2018, 98, 239–389. [Google Scholar] [CrossRef] [PubMed]
- Matos, M.; Augusto, E.; Machado, N.J.; Dos Santos-Rodrigues, A.; Cunha, R.A.; Agostinho, P. Astrocytic adenosine A2A receptors control the amyloid-beta peptide-induced decrease of glutamate uptake. J. Alzheimers Dis. 2012, 31, 555–567. [Google Scholar] [CrossRef]
- Lopes, C.R.; Cunha, R.A.; Agostinho, P. Astrocytes and Adenosine A(2A) Receptors: Active Players in Alzheimer’s Disease. Front. Neurosci. 2021, 15, 666710. [Google Scholar] [CrossRef]
- Lee, C.C.; Chang, C.P.; Lin, C.J.; Lai, H.L.; Kao, Y.H.; Cheng, S.J.; Chen, H.M.; Liao, Y.P.; Faivre, E.; Buée, L.; et al. Adenosine Augmentation Evoked by an ENT1 Inhibitor Improves Memory Impairment and Neuronal Plasticity in the APP/PS1 Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2018, 55, 8936–8952. [Google Scholar] [CrossRef]
- Paiva, I.; Carvalho, K.; Santos, P.; Cellai, L.; Pavlou, M.A.S.; Jain, G.; Gnad, T.; Pfeifer, A.; Vieau, D.; Fischer, A.; et al. A2AR-induced transcriptional deregulation in astrocytes: An in vitro study. Glia 2019, 67, 2329–2342. [Google Scholar] [CrossRef]
- Badimon, A.; Strasburger, H.J.; Ayata, P.; Chen, X.; Nair, A.; Ikegami, A.; Hwang, P.; Chan, A.T.; Graves, S.M.; Uweru, J.O.; et al. Negative feedback control of neuronal activity by microglia. Nature 2020, 586, 417–423. [Google Scholar] [CrossRef]
- Monje, M.L.; Toda, H.; Palmer, T.D. Inflammatory blockade restores adult hippocampal neurogenesis. Science 2003, 302, 1760–1765. [Google Scholar] [CrossRef]
- Franco, R.; Rivas-Santisteban, R.; Casanovas, M.; Lillo, A.; Saura, C.A.; Navarro, G. Adenosine A(2A) receptor antagonists affects NMDA glutamate receptor function. Potential to address neurodegeneration in Alzheimer’s disease. Cells 2020, 9, 1075. [Google Scholar] [CrossRef]
- Franco, R.; Lillo, A.; Rivas-Santisteban, R.; Reyes-Resina, I.; Navarro, G. Microglial Adenosine Receptors: From Preconditioning to Modulating the M1/M2 Balance in Activated Cells. Cells 2021, 10, 1124. [Google Scholar] [CrossRef] [PubMed]
- Colella, M.; Zinni, M.; Pansiot, J.; Cassanello, M.; Mairesse, J.; Ramenghi, L.; Baud, O. Modulation of Microglial Activation by Adenosine A2a Receptor in Animal Models of Perinatal Brain Injury. Front. Neurol. 2018, 9, 605. [Google Scholar] [CrossRef] [PubMed]
- Borea, P.A.; Gessi, S.; Merighi, S.; Varani, K. Adenosine as a multisignalling guardian angel in human diseases: When, where and how does it exert its protective effects? Trends Pharmacol. Sci. 2016, 37, 419–434. [Google Scholar] [CrossRef]
- Borea, P.A.; Gessi, S.; Merighi, S.; Vincenzi, F.; Varani, K. Pathological overproduction: The bad side of adenosine. Br. J. Pharmacol. 2017, 174, 1945–1960. [Google Scholar] [CrossRef] [Green Version]
- Brodde, O.E.; Beckering, J.J.; Michel, M.C. Human heart β-adrenoceptors: A fair comparison with lymphocyte β-adrenoceptors? Trends Pharmacol. Sci. 1987, 8, 403–407. [Google Scholar] [CrossRef]
- Nijhuis, E.W.; Oostervink, F.; Hinloopen, B.; Rozing, J.; Nagelkerken, L. Differences in dexamethasone-sensitivity between lymphocytes from patients with Alzheimer’s disease and patients with multi-infarct dementia. Brain Behav. Immun. 1996, 10, 115–125. [Google Scholar] [CrossRef]
- Varani, K.; Caramori, G.; Vincenzi, F.; Adcock, I.; Casolari, P.; Leung, E.; Maclennan, S.; Gessi, S.; Morello, S.; Barnes, P.J.; et al. Alteration of adenosine receptors in patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2006, 173, 398–406. [Google Scholar] [CrossRef]
- Varani, K.; Laghi-Pasini, F.; Camurri, A.; Capecchi, P.L.; Maccherini, M.; Diciolla, F.; Ceccatelli, L.; Lazzerini, P.E.; Ulouglu, C.; Cattabeni, F.; et al. Changes of peripheral A2A adenosine receptors in chronic heart failure and cardiac transplantation. FASEB J. 2003, 17, 280–282. [Google Scholar] [CrossRef] [Green Version]
- Varani, K.; Vincenzi, F.; Tosi, A.; Gessi, S.; Casetta, I.; Granieri, G.; Fazio, P.; Leung, E.; MacLennan, S.; Granieri, E.; et al. A2A adenosine receptor overexpression and functionality, as well as TNF-alpha levels, correlate with motor symptoms in Parkinson’s disease. FASEB J. 2010, 24, 587–598. [Google Scholar] [CrossRef]
- Gessi, S.; Cattabriga, E.; Avitabile, A.; Gafa’, R.; Lanza, G.; Cavazzini, L.; Bianchi, N.; Gambari, R.; Feo, C.; Liboni, A.; et al. Elevated expression of A3 adenosine receptors in human colorectal cancer is reflected in peripheral blood cells. Clin. Cancer Res. 2004, 10, 5895–5901. [Google Scholar] [CrossRef] [Green Version]
- Pluta, R.; Ułamek-Kozioł, M.; Januszewski, S.; Czuczwar, S.J. Platelets, Lymphocytes and erythrocytes from Alzheimer’s disease patients: The quest for blood cell-based biomarkers. Folia Neuropathol. 2018, 56, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Arosio, B.; Mastronardi, L.; As, C.; Nicolini, P.; Casè, A.; Ziglioli, E.; Bergamaschini, L. Adenosine A(2A) Receptor and IL-10 in Peripheral Blood Mononuclear Cells of Patients with Mild Cognitive Impairment. Int. J. Alzheimers Dis. 2011, 2011, 484021. [Google Scholar]
- Arosio, B.; Viazzoli, C.; Mastronardi, L.; Bilotta, C.; Vergani, C.; Bergamaschini, L. Adenosine A2A receptor expression in peripheral blood mononuclear cells of patients with mild cognitive impairment. J. Alzheimers.Dis. 2010, 20, 991–996. [Google Scholar] [CrossRef] [PubMed]
- Gussago, C.; Arosio, B.; Casati, M.; Ferri, E.; Gualandris, F.; Tedone, E.; Nicolini, P.; Rossi, P.D.; Abbate, C.; Mari, D. Different adenosine A2A receptor expression in peripheral cells from elderly patients with vascular dementia and Alzheimer’s disease. J. Alzheimer’s Dis. 2014, 40, 45–49. [Google Scholar] [CrossRef]
- Pasquini, S.; Vincenzi, F.; Casetta, I.; Laudisi, M.; Merighi, S.; Gessi, S.; Borea, P.A.; Varani, K. Adenosinergic System Involvement in Ischemic Stroke Patients’ Lymphocytes. Cells 2020, 9, 1072. [Google Scholar] [CrossRef]
- Ravani, A.; Vincenzi, F.; Bortoluzzi, A.; Padovan, M.; Pasquini, S.; Gessi, S.; Merighi, S.; Borea, P.A.; Govoni, M.; Varani, K. Role and Function of A(2A) and A3 Adenosine Receptors in Patients with Ankylosing Spondylitis, Psoriatic Arthritis and Rheumatoid Arthritis. Int. J. Mol. Sci. 2017, 18, 697. [Google Scholar] [CrossRef] [PubMed]
- Bortoluzzi, A.; Vincenzi, F.; Govoni, M.; Padovan, M.; Ravani, A.; Borea, P.A.; Varani, K. A2A adenosine receptor upregulation correlates with disease activity in patients with systemic lupus erythematosus. Arthritis Res. Ther. 2016, 18, 192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vincenzi, F.; Corciulo, C.; Targa, M.; Casetta, I.; Gentile, M.; Granieri, E.; Borea, P.A.; Popoli, P.; Varani, K. A2A adenosine receptors are up-regulated in lymphocytes from amyotrophic lateral sclerosis patients. Amyotroph. Lateral Scler. Front. Degener. 2013, 14, 406–413. [Google Scholar] [CrossRef]
- Vincenzi, F.; Corciulo, C.; Targa, M.; Merighi, S.; Gessi, S.; Casetta, I.; Gentile, M.; Granieri, E.; Borea, P.A.; Varani, K. Multiple sclerosis lymphocytes upregulate A2A adenosine receptors that are antiinflammatory when stimulated. Eur. J. Immunol. 2013, 43, 2206–2216. [Google Scholar] [CrossRef]
- Varani, K.; Abbracchio, M.P.; Cannella, M.; Cislaghi, G.; Giallonardo, P.; Mariotti, C.; Cattabriga, E.; Cattabeni, F.; Borea, P.A.; Squitieri, F.; et al. Aberrant A2A receptor function in peripheral blood cells in Huntington’s disease. FASEB J. 2003, 17, 2148–2150. [Google Scholar] [CrossRef]
- Godoy-Marín, H.; Duroux, R.; Jacobson, K.A.; Soler, C.; Colino-Lage, H.; Jiménez-Sábado, V.; Montiel, J.; Hove-Madsen, L.; Ciruela, F. Adenosine A2A Receptors Are Upregulated in Peripheral Blood Mononuclear Cells from Atrial Fibrillation Patients. Int. J. Mol. Sci. 2021, 22, 3467. [Google Scholar] [CrossRef]
- Yang, K.; Yang, Z.; Chen, X.; Li, W. The Significance of Sialylation on the Pathogenesis of Alzheimer’s Disease. Brain Res. Bull. 2021, 173, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Merighi, S.; Poloni, T.E.; Pelloni, L.; Varani, K.; Vincenzi, F.; Borea, P.A.; Gessi, S. An open question: Is the A2A adenosine receptor a novel target for Alzheimer’s disease treatment? Front. Pharmacol. 2021, 12, 652455. [Google Scholar] [CrossRef] [PubMed]
- Merighi, S.; Poloni, T.E.; Terrazzan, A.; Moretti, E.; Gessi, S.; Ferrari, D. Alzheimer and Purinergic Signaling: Just a matter of inflammation? Cells 2021, 10, 1267. [Google Scholar] [CrossRef]
- Atif, M.; Alsrhani, A.; Naz, F.; Imran, M.; Imran, M.; Ullah, M.I.; Alameen, A.A.M.; Gondal, T.A.; Raza, Q. Targeting Adenosine Receptors in Neurological Diseases. Cell Reprogram 2021, 23, 57–72. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.F.; Cunha, R. The belated US FDA approval of the adenosine A2A receptor antagonist istradefylline for treatment of Parkinson’s disease. Purinergic Signal. 2020, 16, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Je, Y.; Giovannucci, E. Coffee consumption and all-cause and cause-specific mortality: A meta-analysis by potential modifiers. Eur. J. Epidemiol. 2019, 34, 731–752. [Google Scholar] [CrossRef] [PubMed]
- Charvin, D.; Medori, R.; Hauser, R.A.; Rascol, O. Therapeutic strategies for Parkinson disease: Beyond dopaminergic drugs. Nat. Rev. Drug Discov. 2018, 17, 804–822. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| ATN System | |
|---|---|
| A-T-N- | No biomarkers of degenerative brain pathology |
| A + T-N- | Amyloid deposition (Alzheimer’s continuum) |
| A + T-N+ | Amyloid deposition and non-tau degeneration (Alzheimer’s continuum; suspected non-AD pathology) |
| A + T + N- | Early Alzheimer’s Disease (Alzheimer’s continuum) |
| A + T + N+ | Alzheimer’s Disease (Alzheimer’s continuum) |
| A-T + N- A-T-N+ A-T + N+ | Non-Alzheimer neurodegenerative Diseases (e.g., primary TAUpahies, Fronto-Temporal Dementia due to TDP-43, LATE, other rare forms) |
| Biomarkers of Alzheimer’s Disease | |
|---|---|
| ATN System [15,16] | |
| Biomarkers of β-amyloid plaques (A) | Cortical amyloid PET |
| Low CSF β-amyloid 42 | |
| Biomarkers of tau (T) | Cortical tau PET |
| Elevated CSF phospho-tau | |
| Biomarkers of neurodegeneration and neuronal injury (N) | [(18)F]-fluorodeoxyglucose PET hypometabolism |
| Atrophy on MRI | |
| Elevated CSF total-tau | |
| Fluid biomarkers | |
| Increased levels of plasma Neurofilament light (NfL) [22,27] | |
| Increased levels of plasma tau phosphorylated at threonine 181 (P-tau181) [23] | |
| Increased levels of plasma tau phosphorylated at threonine-217 (P-tau217) [28] | |
| Increased levels of plasma Aβ-amyloid oligomers [24] | |
| Lower levels of plasma Aβ42/Aβ40 ratio (TP42/40) [25,26] | |
| Increased levels of CSF Neurogranin (Ng) [29] | |
| Increased levels of CSF YKL-40 [34,35] | |
| Aged/Animal Models of AD/AD Patients | Tissue/Cell | References |
|---|---|---|
| aged rats | cortex and hippocampus | [72] |
| aged vs. young rats | cortical membranes | [64] |
| aged vs. young rats | hippocampal neurons | [57,73] |
| AD patients | microglia | [74] |
| APPsw tg mice | hyppocampus | [65] |
| adult and aged rats | hyppocampus | [75] |
| AD patients | frontal cortex | [70] |
| Rats with different ages | nerve terminals purified from the hippocampus | [66] |
| AD rat model | hippocampus | [67] |
| AD patients/aged mice | astrocytes | [68,76] |
| APP/PS1 mice | CA3 synaptic membranes | [62] |
| Early AD mice model | hippocampal synaptosomes | [69] |
| APPSw/Ind AD transgenic mice model | microglia | [77] |
| Aged subjects and AD patients | hippocampal neurons | [60] |
| AD patients | cortex, hippocampus, platelets | [71] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gessi, S.; Poloni, T.E.; Negro, G.; Varani, K.; Pasquini, S.; Vincenzi, F.; Borea, P.A.; Merighi, S. A2A Adenosine Receptor as a Potential Biomarker and a Possible Therapeutic Target in Alzheimer’s Disease. Cells 2021, 10, 2344. https://doi.org/10.3390/cells10092344
Gessi S, Poloni TE, Negro G, Varani K, Pasquini S, Vincenzi F, Borea PA, Merighi S. A2A Adenosine Receptor as a Potential Biomarker and a Possible Therapeutic Target in Alzheimer’s Disease. Cells. 2021; 10(9):2344. https://doi.org/10.3390/cells10092344
Chicago/Turabian StyleGessi, Stefania, Tino Emanuele Poloni, Giulia Negro, Katia Varani, Silvia Pasquini, Fabrizio Vincenzi, Pier Andrea Borea, and Stefania Merighi. 2021. "A2A Adenosine Receptor as a Potential Biomarker and a Possible Therapeutic Target in Alzheimer’s Disease" Cells 10, no. 9: 2344. https://doi.org/10.3390/cells10092344
APA StyleGessi, S., Poloni, T. E., Negro, G., Varani, K., Pasquini, S., Vincenzi, F., Borea, P. A., & Merighi, S. (2021). A2A Adenosine Receptor as a Potential Biomarker and a Possible Therapeutic Target in Alzheimer’s Disease. Cells, 10(9), 2344. https://doi.org/10.3390/cells10092344