
Time-Dependent Protective and Pro-Resolving Effects of FPR2 Agonists on Lipopolysaccharide-Exposed Microglia Cells Involve Inhibition of NF-κB and MAPKs Pathways

 , , ,
, , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Chemicals
2.3. Cell Culture
2.4. Cell Treatment
2.5. Lactate Dehydrogenase (LDH) Release Assay
2.6. Mitochondrial Membrane Potential (∆ψm) Assay
2.7. Caspase-3 Activity
2.8. Intracellular ROS Assay
2.9. NO Release Assay (Nitrite Ion in Solution)
2.10. Immunocytochemistry
2.11. Quantitative Analysis of Confocal Fluorescent Images of Microglia
2.12. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
2.13. Enzyme-Linked Immunosorbent Assay (ELISA)
2.14. Western Blot Analyses in Homogenates of Microglial Cells
2.15. Statistical Analysis
3. Results
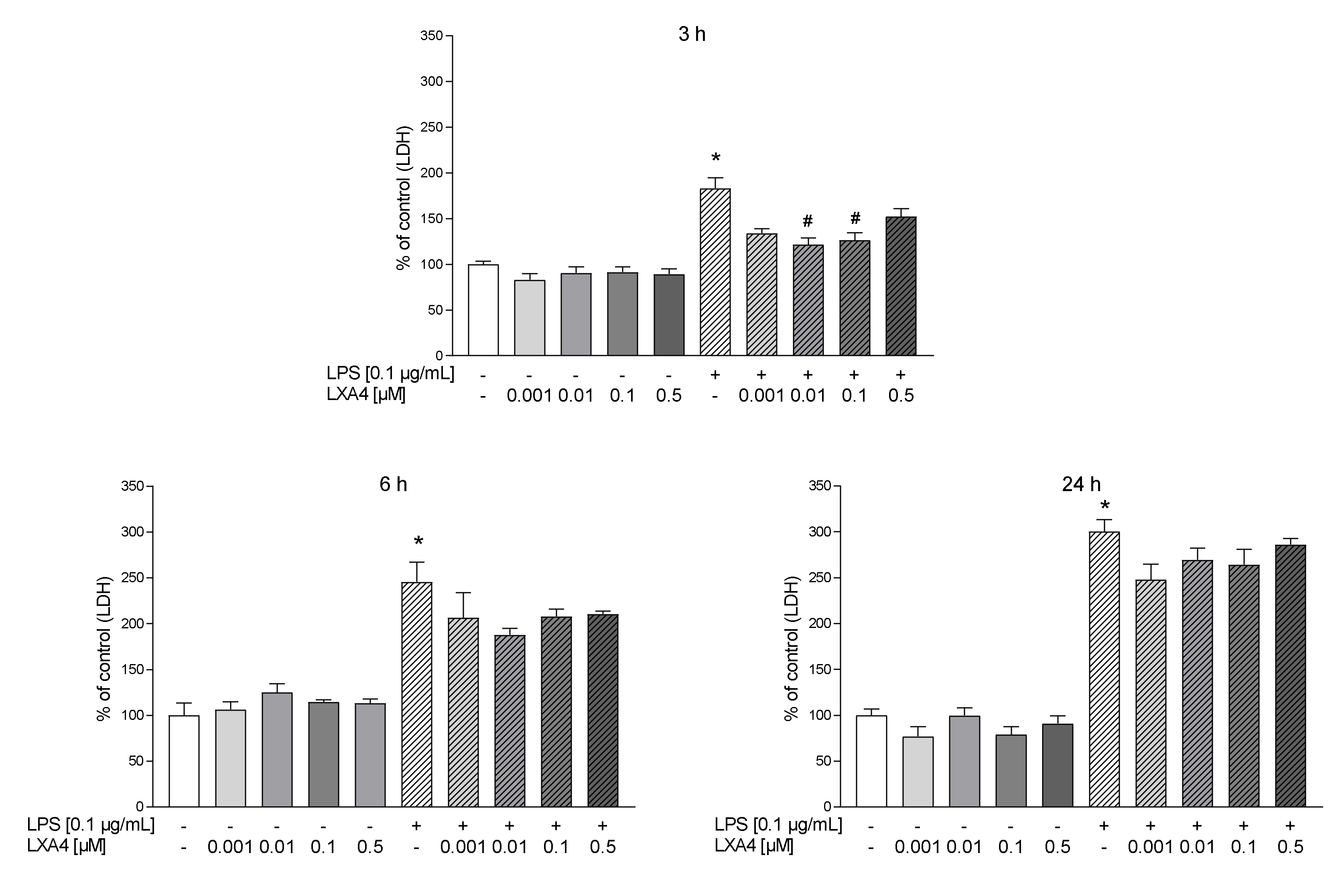
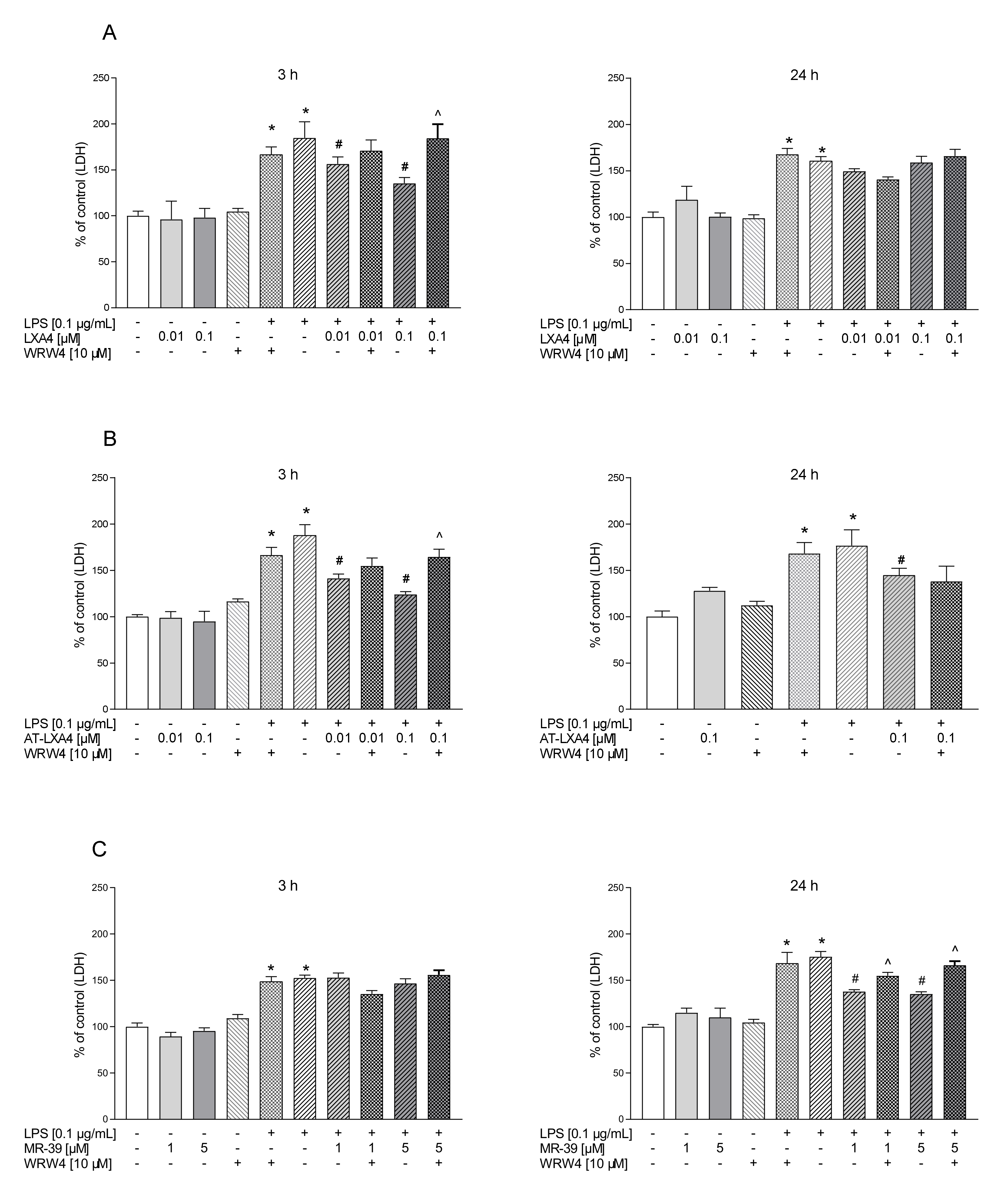
3.1. The Time-Dependent Impact of LXA4, AT-LXA4, and MR-39 on Lactate Dehydrogenase Release in Microglial Cells Stimulated with Lipopolysaccharide
3.2. Visualization of FPR2 Presence in Microglial Cells Stimulated with Lipopolysaccharide
3.3. The Impact of LXA4, AT-LXA4, and MR-39 on the Mitochondrial Membrane Potential in Microglial Cells Stimulated with Lipopolysaccharide
3.4. The Impact of LXA4, AT-LXA4, and MR-39 on Caspase-3 Activity in Microglial Cells Stimulated with Lipopolysaccharide
3.5. The Impact of LXA4, AT-LXA4, and MR-39 on Reactive Oxygen Species (ROS) Production in Microglial Cells Stimulated with Lipopolysaccharide
3.6. The Impact of LXA4, AT-LXA4, and MR-39 on Nitric Oxide Release (NO) in Microglial Cells Stimulated with Lipopolysaccharide
3.7. The Impact of LXA4, AT-LXA4, and MR-39 on Pro- and Anti-Inflammatory Factors’ Expression in Microglial Cells Stimulated with Lipopolysaccharide
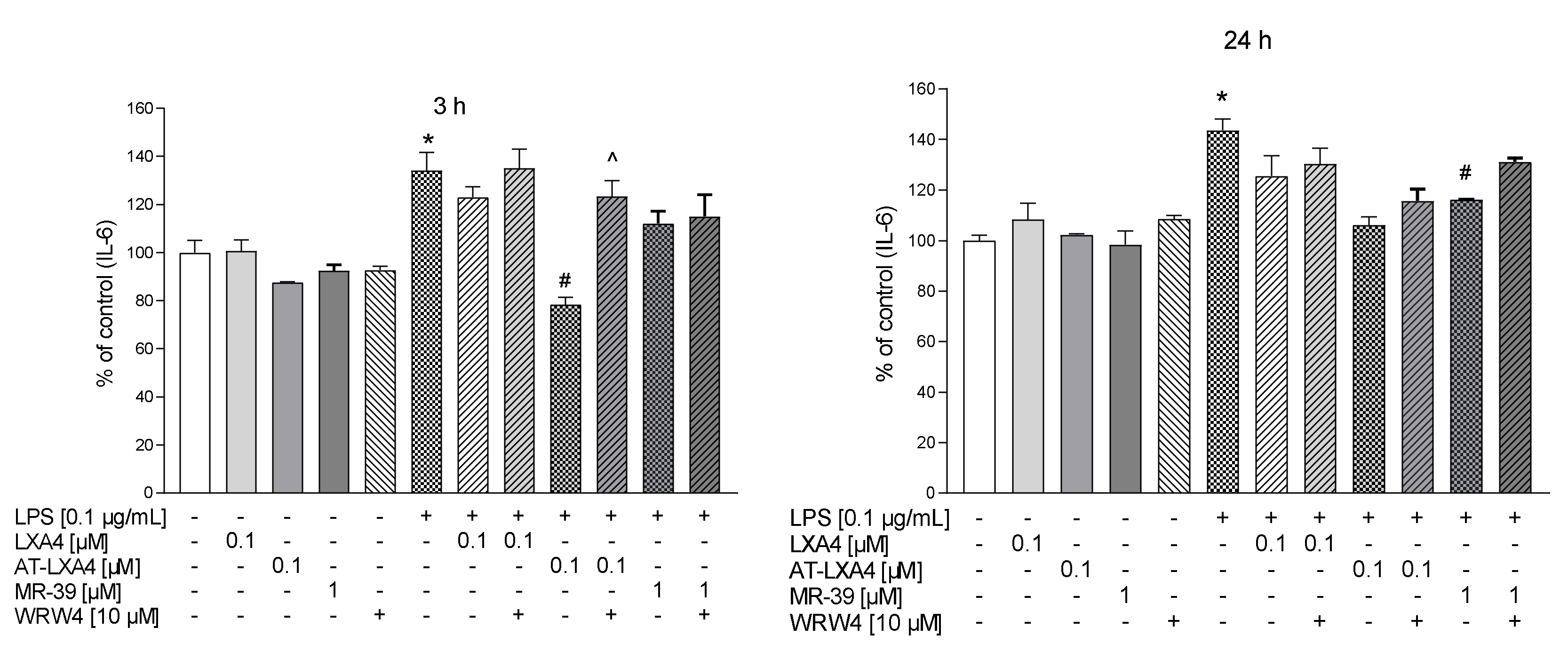
3.8. The Impact of LXA4, AT-LXA4, and MR-39 on Pro- and Anti-Inflammatory Cytokine Production in Microglial Cells Stimulated with Lipopolysaccharide
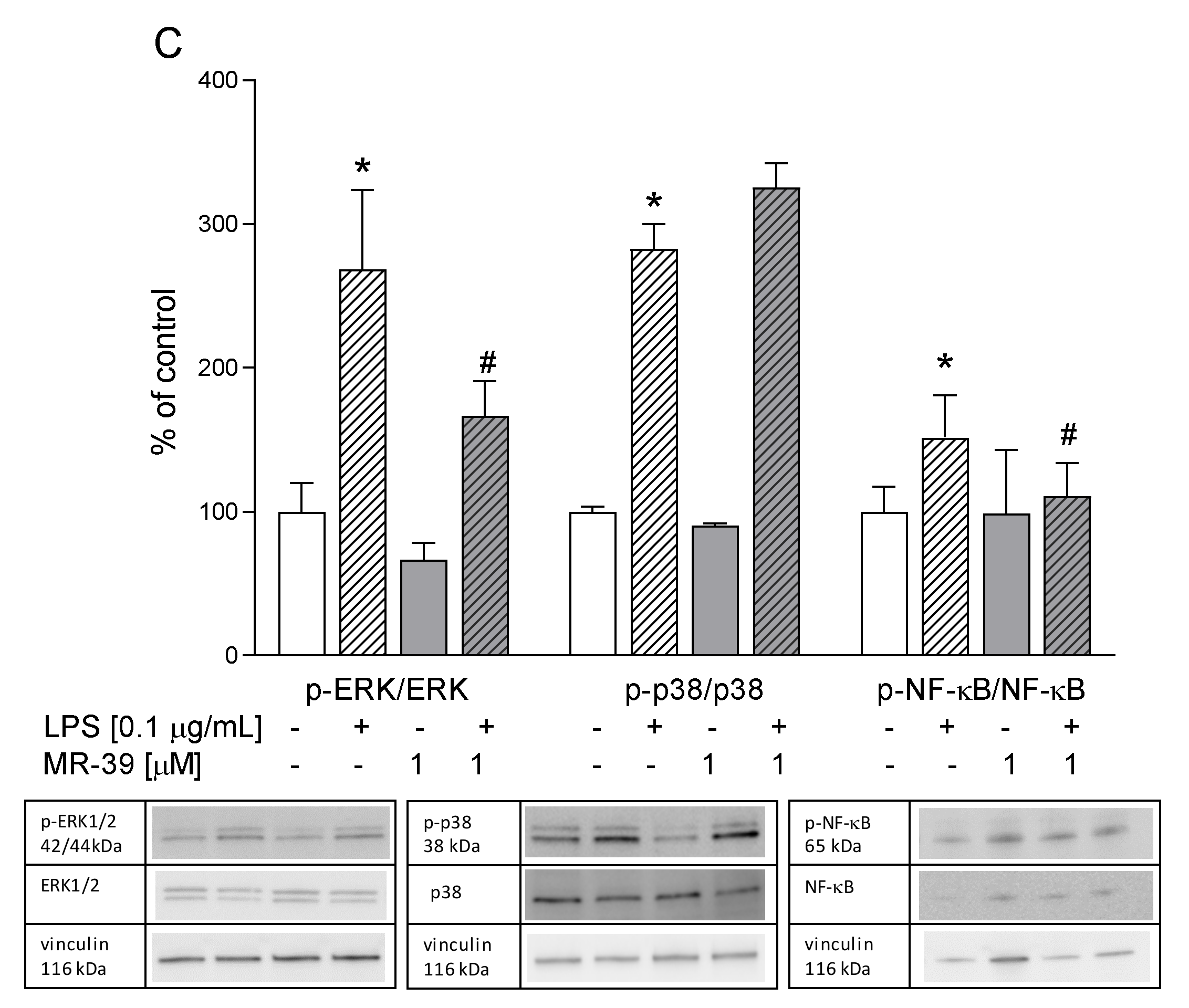
3.9. The Impact of LXA4, AT-LXA4, and MR-39, FPR2 Ligands, on the ERK1/2, p38, and NF-κB Pathways in Microglial Cells Stimulated with Lipopolysaccharide
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Graeber, M.B.; Streit, W.J. Microglia: Biology and pathology. Acta Neuropathol. 2010, 119, 89–105. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Meng, Y.; Li, W.; Yong, Y.; Fan, Z.; Ding, H.; Wei, Y.; Luo, J.; Ke, Z.-J. Neuronal MCP-1 Mediates Microglia Recruitment and Neurodegeneration Induced by the Mild Impairment of Oxidative Metabolism. Brain Pathol. 2011, 21, 279–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polazzi, E.; Monti, B. Microglia and neuroprotection: From in vitro studies to therapeutic applications. Prog. Neurobiol. 2010, 92, 293–315. [Google Scholar] [CrossRef] [PubMed]
- Goldmann, T.; Prinz, M. Role of microglia in CNS autoimmunity. Clin. Dev. Immunol. 2013, 2013, 208093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohman, R.A.; Rhodes, J.S. Neurogenesis, inflammation and behavior. Brain. Behav. Immun. 2013, 27, 22–32. [Google Scholar] [CrossRef] [Green Version]
- Szalay, G.; Martinecz, B.; Lénárt, N.; Környei, Z.; Orsolits, B.; Judák, L.; Császár, E.; Fekete, R.; West, B.L.; Katona, G.; et al. Microglia protect against brain injury and their selective elimination dysregulates neuronal network activity after stroke. Nat. Commun. 2016, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Rock, R.B.; Gekker, G.; Hu, S.; Sheng, W.S.; Cheeran, M.; Lokensgard, J.R.; Peterson, P.K. Role of microglia in central nervous system infections. Clin. Microbiol. Rev. 2004, 17, 942–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X. Microglia/macrophage polarization: Fantasy or evidence of functional diversity? J. Cereb. Blood Flow Metab. 2020, 40, S134–S136. [Google Scholar] [CrossRef]
- Masuda, T.; Sankowski, R.; Staszewski, O.; Prinz, M. Microglia Heterogeneity in the Single-Cell Era. Cell Rep. 2020, 30, 1271–1281. [Google Scholar] [CrossRef]
- Ransohoff, R.M. A polarizing question: Do M1 and M2 microglia exist. Nat. Neurosci. 2016, 19, 987–991. [Google Scholar] [CrossRef]
- Bordt, E.A.; Polster, B.M. NADPH oxidase- and mitochondria-derived reactive oxygen species in proinflammatory microglial activation: A Bipartisan affair? Free Radic. Biol. Med. 2014, 76, 34–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orihuela, R.; McPherson, C.A.; Harry, G.J. Microglial M1/M2 polarization and metabolic states. Br. J. Pharmacol. 2016, 173, 649–665. [Google Scholar] [CrossRef] [PubMed]
- Jurga, A.M.; Paleczna, M.; Kuter, K.Z. Overview of General and Discriminating Markers of Differential Microglia Phenotypes. Front. Cell. Neurosci. 2020, 14, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Kierdorf, K.; Prinz, M. Factors regulating microglia activation. Front. Cell. Neurosci. 2013, 7, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Cherry, J.D.; Olschowka, J.A.; O’Banion, M.K. Neuroinflammation and M2 microglia: The good, the bad, and the inflamed. J. Neuroinflamm. 2014, 11, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Kwon, H.S.; Koh, S.H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wickstead, E.S.; Karim, H.A.; Manuel, R.E.; Biggs, C.S.; Getting, S.J.; McArthur, S. Reversal of β-Amyloid-Induced Microglial Toxicity in Vitro by Activation of Fpr2/3. Oxidative Med. Cell. Longev. 2020, 2020, 2139192. [Google Scholar] [CrossRef] [PubMed]
- Feghali, C.A.; Wright, T.M. Cytokines in acute and chronic inflammation. Front. Biosci. 1997, 2, A171. [Google Scholar] [CrossRef] [Green Version]
- Lintermans, L.L.; Stegeman, C.A.; Heeringa, P.; Abdulahad, W.H. T cells in vascular inflammatory diseases. Front. Immunol. 2014, 5, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Ashley, N.T.; Weil, Z.M.; Nelson, R.J. Inflammation: Mechanisms, costs, and natural variation. Annu. Rev. Ecol. Evol. Syst. 2012, 43, 385–406. [Google Scholar] [CrossRef]
- Schett, G.; Neurath, M.F. Resolution of chronic inflammatory disease: Universal and tissue-specific concepts. Nat. Commun. 2018, 9, 1–8. [Google Scholar] [CrossRef]
- Leszek, J.; Barreto, G.E.; Gsiorowski, K.; Koutsouraki, E.; Ávila-Rodrigues, M.; Aliev, G. Inflammatory Mechanisms and Oxidative Stress as Key Factors Responsible for Progression of Neurodegeneration: Role of Brain Innate Immune System. CNS Neurol. Disord. Drug Targets 2016, 15, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N. Novel Pro-Resolving Lipid Mediators in Inflammation Are Leads for Resolution Physiology. Nature 2014, 510, 92–101. [Google Scholar] [CrossRef] [Green Version]
- Trojan, E.; Bryniarska, N.; Leśkiewicz, M.; Regulska, M.; Chamera, K.; Szuster-Głuszczak, M.; Leopoldo, M.; Lacivita, E.; Basta-Kaim, A. The Contribution of Formyl Peptide Receptor Dysfunction to the Course of Neuroinflammation: A Potential Role in the Brain Pathology. Curr. Neuropharmacol. 2019, 18, 229–249. [Google Scholar] [CrossRef] [PubMed]
- Kantarci, A.; Aytan, N.; Palaska, I.; Stephens, D.; Crabtree, L.; Benincasa, C.; Jenkins, B.G.; Carreras, I.; Dedeoglu, A. Combined administration of resolvin E1 and lipoxin A4 resolves inflammation in a murine model of Alzheimer’s disease. Exp. Neurol. 2018, 300, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Chiang, N.; Serhan, C.N.; Dahlén, S.-E.; Drazen, J.M.; Hay, D.W.P.; Rovati, G.E.; Shimizu, T.; Yokomizo, T.; Brink, C. The Lipoxin Receptor ALX: Potent Ligand-Specific and Stereoselective Actions in Vivo. Pharmacol. Rev. 2006, 58, 463–487. [Google Scholar] [CrossRef] [PubMed]
- Regulska, M.; Szuster-Głuszczak, M.; Trojan, E.; Leśkiewicz, M.; Basta-Kaim, A. The Emerging Role of the Double-Edged Impact of Arachidonic Acid- Derived Eicosanoids in the Neuroinflammatory Background of Depression. Curr. Neuropharmacol. 2020, 19, 278–293. [Google Scholar] [CrossRef]
- Serhan, C.N. Lipoxins and aspirin-triggered 15-epi-lipoxins are the first lipid mediators of endogenous anti-inflammation and resolution. Prostaglandins Leukot. Essent. Fat. Acids 2005, 73, 141–162. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Ding, D.H.; Li, Q.Q.; Wang, X.Y.; Sun, Y.Y.; Li, L.J. Lipoxin A4 regulates lipopolysaccharide-induced BV2 microglial activation and differentiation via the notch signaling pathway. Front. Cell. Neurosci. 2019, 13, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.H. Lipoxin A4: A novel anti-inflammatory molecule? Thorax 1995, 50, 111–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Z.; Hu, Q.; Xu, L.; Guo, Z.; Ou, Y.; He, Y.; Yin, C.; Sun, X.; Tang, J.; Zhang, J.H. Lipoxin A4 Reduces Inflammation Through Formyl Peptide Receptor 2/p38 MAPK Signaling Pathway in Subarachnoid Hemorrhage Rats. Stroke 2016, 47, 490–497. [Google Scholar] [CrossRef] [Green Version]
- Vital, S.A.; Becker, F.; Holloway, P.M.; Russell, J.; Perretti, M.; Granger, D.N.; Gavins, F.N.E. Formyl-peptide receptor 2/3/Lipoxin A4 receptor regulates neutrophil-platelet aggregation and attenuates cerebral inflammation: Impact for therapy in cardiovascular disease. Circulation 2016, 133, 2169–2179. [Google Scholar] [CrossRef] [Green Version]
- Martini, A.C.; Berta, T.; Forner, S.; Chen, G.; Bento, A.F.; Ji, R.R.; Rae, G.A. Lipoxin A4 inhibits microglial activation and reduces neuroinflammation and neuropathic pain after spinal cord hemisection. J. Neuroinflamm. 2016, 13, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Tylek, K.; Trojan, E.; Regulska, M.; Lacivita, E.; Leopoldo, M.; Basta-Kaim, A. Formyl peptide receptor 2, as an important target for ligands triggering the inflammatory response regulation: A link to brain pathology. Pharmacol. Rep. 2021, 1–16. [Google Scholar] [CrossRef]
- Lohse, M.J. Dimerization in GPCR mobility and signaling. Curr. Opin. Pharmacol. 2010, 10, 53–58. [Google Scholar] [CrossRef]
- Sodin-Semrl, S.; Spagnolo, A.; Mikus, R.; Barbaro, B.; Varga, J.; Fiore, S. Opposing regulation of interleukin-8 and NF-kB responses by lipoxin A4 and serum amyloid a via the common lipoxin a receptor. Int. J. Immunopathol. Pharmacol. 2004, 17, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, K.E.; DeMars, K.M.; Alexander, J.C.; De Leon, L.G.; Pacheco, S.C.; Graves, C.; Yang, C.; McCrea, A.O.; Frankowski, J.C.; Garrett, T.J.; et al. Targeting resolution of neuroinflammation after ischemic stroke with a lipoxin A4 analog: Protective mechanisms and long-term effects on neurological recovery. Brain Behav. 2017, 7, 1–14. [Google Scholar] [CrossRef] [PubMed]
- He, H.Q.; Ye, R.D. The formyl peptide receptors: Diversity of ligands and mechanism for recognition. Molecules 2017, 22, 455. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.J.; Tao, T.; Wang, H.; Zhou, Y.; Gao, X.; Gao, Y.Y.; Hang, C.H.; Li, W. Functions of resolvin D1-ALX/FPR2 receptor interaction in the hemoglobin-induced microglial inflammatory response and neuronal injury. J. Neuroinflamm. 2020, 17, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Duvall, M.G.; Levy, B.D. DHA- and EPA-derived resolvins, protectins, and maresins in airway inflammation. Eur. J. Pharmacol. 2016, 785, 144–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Zhang, L.; Chen, X.; Xue, X.; Guo, Q.; Liu, M.; Zhao, J. Formylpeptide Receptors Promote the Migration and Differentiation of Rat Neural Stem Cells. Sci. Rep. 2016, 6, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Stama, M.L.; Ślusarczyk, J.; Lacivita, E.; Kirpotina, L.N.; Schepetkin, I.A.; Chamera, K.; Riganti, C.; Perrone, R.; Quinn, M.T.; Basta-Kaim, A.; et al. Novel ureidopropanamide based N-formyl peptide receptor 2 (FPR2) agonists with potential application for central nervous system disorders characterized by neuroinflammation. Eur. J. Med. Chem. 2017, 141, 703–720. [Google Scholar] [CrossRef]
- Mastromarino, M.; Lacivita, E.; Colabufo, N.A.; Leopoldo, M. G-protein coupled receptors involved in the resolution of inflammation: Ligands and therapeutic perspectives. Mini Rev. Med. Chem. 2020, 20, 2090–2103. [Google Scholar] [CrossRef] [PubMed]
- Zawadzka, M.; Kaminska, B. A novel mechanism of FK506-mediated neuroprotection: Downregulation of cytokine expression in glial cells. Glia 2005, 49, 36–51. [Google Scholar] [CrossRef] [PubMed]
- Ślusarczyk, J.; Trojan, E.; Głombik, K.; Budziszewska, B.; Kubera, M.; Lasoń, W.; Popiołek-Barczyk, K.; Mika, J.; Wędzony, K.; Basta-Kaim, A. Prenatal stress is a vulnerability factor for altered morphology and biological activity of microglia cells. Front. Cell. Neurosci. 2015, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Ślusarczyk, J.; Trojan, E.; Głombik, K.; Piotrowska, A.; Budziszewska, B.; Kubera, M.; Popiołek-Barczyk, K.; Lasoń, W.; Mika, J.; Basta-Kaim, A. Targeting the NLRP3 inflammasome-related pathways via tianeptine treatment-suppressed microglia polarization to the M1 phenotype in lipopolysaccharide-stimulated cultures. Int. J. Mol. Sci. 2018, 19, 1965. [Google Scholar] [CrossRef] [Green Version]
- Basta-Kaim, A.; Ślusarczyk, J.; Szczepanowicz, K.; Warszyński, P.; Leśkiewicz, M.; Regulska, M.; Trojan, E.; Lasoń, W. Protective effects of polydatin in free and nanocapsulated form on changes caused by lipopolysaccharide in hippocampal organotypic cultures. Pharmacol. Rep. 2019, 71, 603–613. [Google Scholar] [CrossRef]
- Leskiewicz, M.; Regulska, M.; Budziszewska, B.; Jantas, D.; Jaworska-Feil, L.; Basta-Kaim, A.; Kubera, M.; Jagla, G.; Nowak, W.; Lason, W. Effects of neurosteroids on hydrogen peroxide- and staurosporine-induced damage of human neuroblastoma SH-SY5Y cells. J. Neurosci. Res. 2008, 86, 1361–1370. [Google Scholar] [CrossRef] [PubMed]
- Kubiak, A.; Chighizola, M.; Schulte, C.; Bryniarska, N.; Wesołowska, J.; Pudełek, M.; Lasota, M.; Ryszawy, D.; Basta-Kaim, A.; Laidler, P.; et al. Stiffening of DU145 prostate cancer cells driven by actin filaments—Microtubule crosstalk conferring resistance to microtubule-targeting drugs. Nanoscale 2021, 13, 6212–6226. [Google Scholar] [CrossRef] [PubMed]
- Prauzner-Bechcicki, S.; Raczkowska, J.; Madej, E.; Pabijan, J.; Lukes, J.; Sepitka, J.; Rysz, J.; Awsiuk, K.; Bernasik, A.; Budkowski, A.; et al. PDMS substrate stiffness affects the morphology and growth profiles of cancerous prostate and melanoma cells. J. Mech. Behav. Biomed. Mater. 2015, 41, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Bollmann, L.; Koser, D.E.; Shahapure, R.; Gautier, H.O.B.; Holzapfel, G.A.; Scarcelli, G.; Gather, M.C.; Ulbricht, E.; Franze, K. Microglia mechanics: Immune activation alters traction forces and durotaxis. Front. Cell. Neurosci. 2015, 9, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Basta-Kaim, A.; Budziszewska, B.; Leśkiewicz, M.; Fijał, K.; Regulska, M.; Kubera, M.; Wȩdzony, K.; Lasoń, W. Hyperactivity of the hypothalamus-pituitary-adrenal axis in lipopolysaccharide-induced neurodevelopmental model of schizophrenia in rats: Effects of antipsychotic drugs. Eur. J. Pharmacol. 2011, 650, 586–595. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Chen, K.; Yoshimura, T.; Liu, Y.; Gong, W.; Le, Y.; Gao, J.L.; Zhao, J.; Wang, J.M.; Wang, A. Formylpeptide receptors mediate rapid neutrophil mobilization to accelerate wound healing. PLoS ONE 2014, 9, e99541. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Qu, C.; Lu, X.; Zhang, S. Activation of microglia by histamine and substance P. Cell. Physiol. Biochem. 2014, 34, 768–780. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Burguillos, M.A.; Joseph, B. Guilt by association, caspase-3 regulates microglia polarization. Cell Cycle 2017, 16, 306–307. [Google Scholar] [CrossRef] [Green Version]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric Oxide and Peroxynitrite in Health and Disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, Y.-H.; Le, Y.; Gong, W.; Proost, P.; Van Damme, J.; Murphy, W.J.; Wang, J.M. Bacterial Lipopolysaccharide Selectively Up-Regulates the Function of the Chemotactic Peptide Receptor Formyl Peptide Receptor 2 in Murine Microglial Cells. J. Immunol. 2002, 168, 434–442. [Google Scholar] [CrossRef] [Green Version]
- Tiefenthaler, M.; Amberger, A.; Bacher, N.; Hartmann, B.L.; Margreiter, R.; Kofler, R.; Konwalinka, G. Increased lactate production follows loss of mitochondrial membrane potential during apoptosis of human leukaemia cells. Br. J. Haematol. 2001, 114, 574–580. [Google Scholar] [CrossRef]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial membrane potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Sivandzade, F.; Bhalerao, A.; Cucullo, L. Analysis of the Mitochondrial Membrane Potential Using the Cationic JC-1 Dye as a Sensitive Fluorescent Probe. Bio Protocol 2019, 9, 1–13. [Google Scholar] [CrossRef]
- Green, D.R.; Reed, J.C. Mitochondria and apoptosis. Science 1998, 281, 1309. [Google Scholar] [CrossRef] [PubMed]
- Burguillos, M.A.; Hajji, N.; Englund, E.; Persson, A.; Cenci, A.M.; Machado, A.; Cano, J.; Joseph, B.; Venero, J.L. Apoptosis-inducing factor mediates dopaminergic cell death in response to LPS-induced inflammatory stimulus. Evidence in Parkinson’s disease patients. Neurobiol. Dis. 2011, 41, 177–188. [Google Scholar] [CrossRef] [Green Version]
- Venero, J.L.; Burguillos, M.A.; Brundin, P.; Joseph, B. The executioners sing a new song: Killer caspases activate microglia. Cell Death Differ. 2011, 18, 1679–1691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, J.W.; Leigh, N.J.; Mellas, R.E.; McCall, A.D.; Aguirre, A.; Baker, O.J. ALX/FPR2 receptor for RvD1 is expressed and functional in salivary glands. Am. J. Physiol. Cell Physiol. 2014, 306, 178–185. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Min, J.S.; Kim, B.; Chae, U.-B.; Yun, J.W.; Choi, M.S.; Kong, I.K.; Chang, K.T.; Lee, D.S. Mitochondrial ROS govern the LPS-induced pro-inflammatory response in microglia cells by regulating MAPK and NF-κB pathways. Neurosci. Lett. 2015, 584, 191–196. [Google Scholar] [CrossRef]
- Block, M.L.; Zecca, L.; Hong, J.S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Allen, R.G.; Tresini, M. Oxidative stress and gene regulation. Free Radic. Biol. Med. 2000, 28, 463–499. [Google Scholar] [CrossRef]
- Brown, G.C.; Neher, J.J. Inflammatory neurodegeneration and mechanisms of microglial killing of neurons. Mol. Neurobiol. 2010, 41, 242–247. [Google Scholar] [CrossRef]
- Wang, Y.P.; Wu, Y.; Li, L.Y.; Zheng, J.; Liu, R.G.; Zhou, J.P.; Yuan, S.Y.; Shang, Y.; Yao, S.L. Aspirin-triggered lipoxin A4attenuates LPS-induced pro-inflammatory responses by inhibiting activation of NF-κB and MAPKs in BV-2 microglial cells. J. Neuroinflamm. 2011, 8, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araújo, I.M.; Carvalho, C.M. Role of nitric oxide and calpain activation in neuronal death and survival. Curr. Drug Targets CNS Neurol. Disord. 2005, 4, 319–324. [Google Scholar] [CrossRef]
- Romano, M. Lipoxin and aspirin-triggered lipoxins. Sci. World J. 2010, 10, 1048–1064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, T.; Xu, G.; Newton, P.T.; Chagin, A.S.; Mkrtchian, S.; Carlström, M.; Zhang, X.M.; Harris, R.A.; Cooter, M.; Berger, M.; et al. Maresin 1 attenuates neuroinflammation in a mouse model of perioperative neurocognitive disorders. Br. J. Anaesth. 2019, 122, 350–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serhan, C.N.; Yacoubian, S.; Yang, R. Anti-Inflammatory and Proresolving Lipid Mediators. Annu. Rev. Pathol. Mech. Dis. 2008, 3, 279–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Ye, X.H.; Guo, P.P.; Xu, S.P.; Wang, J.; Yuan, S.Y.; Yao, S.L.; Shang, Y. Neuroprotective effect of lipoxin a4 methyl ester in a rat model of permanent focal cerebral ischemia. J. Mol. Neurosci. 2010, 42, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Wada, K.; Arita, M.; Nakajima, A.; Katayama, K.; Kudo, C.; Kamisaki, Y.; Serhan, C.N. Leukotriene B 4 and lipoxin A 4 are regulatory signals for neural stem cell proliferation and differentiation. FASEB J. 2006, 20, 1785–1792. [Google Scholar] [CrossRef]
- Wu, Y.; Zhai, H.; Wang, Y.; Li, L.; Wu, J.; Wang, F.; Sun, S.; Yao, S.; Shang, Y. Aspirin-triggered lipoxin A 4 attenuates lipopolysaccharide- induced intracellular ROS in BV2 microglia cells by inhibiting the function of NADPH oxidase. Neurochem. Res. 2012, 37, 1690–1696. [Google Scholar] [CrossRef]
- Wu, J.; Ding, D.; Wang, X.; Li, Q.; Sun, Y.; Li, L.; Wang, Y. Regulation of aquaporin 4 expression by lipoxin A4 in astrocytes stimulated by lipopolysaccharide. Cell. Immunol. 2019, 344, 103959. [Google Scholar] [CrossRef]
- Miller, Y.I.; Viriyakosol, S.; Worrall, D.S.; Boullier, A.; Butler, S.; Witztum, J.L. Toll-like receptor 4-dependent and -independent cytokine secretion induced by minimally oxidized low-density lipoprotein in macrophages. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1213–1219. [Google Scholar] [CrossRef] [Green Version]
- Koistinaho, M.; Koistinaho, J. Interactions between Alzheimer’s disease and cerebral ischemia—Focus on inflammation. Brain Res. Rev. 2005, 48, 240–250. [Google Scholar] [CrossRef]
- Bachstetter, A.D.; Van Eldik, L.J. The p38 map kinase family as regulators of proinflammatory cytokine production in degenerative diseases of the CNS. Aging Dis. 2010, 1, 199–211. [Google Scholar]
- Falcicchia, C.; Tozzi, F.; Arancio, O.; Watterson, D.M.; Origlia, N. Involvement of p38 mapk in synaptic function and dysfunction. Int. J. Mol. Sci. 2020, 21, 5624. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.C.; Dickson, D.W.; Liu, W.; Brosnan, C.F. Induction of nitric oxide synthase activity in human astrocytes by interleukin-1β and interferon-γ. J. Neuroimmunol. 1993, 46, 19–24. [Google Scholar] [CrossRef]
- Hwang, J.; Zheng, L.T.; Ock, J.; Lee, M.G.; Suk, K. Anti-inflammatory effects of m-chlorophenylpiperazine in brain glia cells. Int. Immunopharmacol. 2008, 8, 1686–1694. [Google Scholar] [CrossRef] [PubMed]
- Qin, C.X.; May, L.T.; Li, R.; Cao, N.; Rosli, S.; Deo, M.; Alexander, A.E.; Horlock, D.; Bourke, J.E.; Yang, Y.H.; et al. Small-molecule-biased formyl peptide receptor agonist compound 17b protects against myocardial ischaemia-reperfusion injury in mice. Nat. Commun. 2017, 8, 1–13. [Google Scholar] [CrossRef]
- Moynagh, P.N. The NF-κB pathway. J. Cell Sci. 2005, 118, 4589–4592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, J.Y.; Crews, F.T. TNFα potentiates glutamate neurotoxicity by inhibiting glutamate uptake in organotypic brain slice cultures: Neuroprotection by NFκB inhibition. Brain Res. 2005, 1034, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Majumder, S.; Zhou, L.Z.; Chaturvedi, P.; Babcock, G.; Aras, S.; Ransohoff, R.M. p48/STAT-1alpha-containing complexes play a predominant role in induction of IFN-gamma-inducible protein, 10 kDa (IP-10) by IFN-gamma alone or in synergy with TNF-alpha. J. Immunol. 1998, 161, 4736–4744. [Google Scholar] [PubMed]
- Hyam, S.R.; Lee, I.A.; Gu, W.; Kim, K.A.; Jeong, J.J.; Jang, S.E.; Han, M.J.; Kim, D.H. Arctigenin ameliorates inflammation in vitro and in vivo by inhibiting the PI3K/AKT pathway and polarizing M1 macrophages to M2-like macrophages. Eur. J. Pharmacol. 2013, 708, 21–29. [Google Scholar] [CrossRef]
- Le, Y.; Murphy, P.M.; Wang, J.M. Formyl-peptide receptors revisited. Trends Immunol. 2002, 23, 541–548. [Google Scholar] [CrossRef]
- De Gaetano, M.; Butler, E.; Gahan, K.; Zanetti, A.; Marai, M.; Chen, J.; Cacace, A.; Hams, E.; Maingot, C.; McLoughlin, A.; et al. Asymmetric synthesis and biological evaluation of imidazole- and oxazole-containing synthetic lipoxin A4 mimetics (sLXms). Eur. J. Med. Chem. 2019, 162, 80–108. [Google Scholar] [CrossRef]
- Harry, G.J.; Kraft, A.D. Neuroinflammation and microglia: Considerations and approaches for neurotoxicity assessment. Expert Opin. Drug Metab. Toxicol. 2008, 4, 1265–1277. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wu, Y.; Wang, Y.; Wu, J.; Song, L.; Xian, W.; Yuan, S.; Pei, L.; Shang, Y. Resolvin D1 promotes the interleukin-4-induced alternative activation in BV-2 microglial cells. J. Neuroinflamm. 2014, 11, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obuchowicz, E.; Bielecka, A.M.; Paul-Samojedny, M.; Pudełko, A.; Kowalski, J. Imipramine and fluoxetine inhibit LPS-induced activation and affect morphology of microglial cells in the rat glial culture. Pharmacol. Rep. 2014, 66, 34–43. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A | |||||
|---|---|---|---|---|---|
| Control | LPS | MR + LPS | LXA4 + LPS | AT-LXA4 + LPS | |
| Pro-inflammatory markers | |||||
| Cd40 Il-1β Tnf-α Cd68 | 1.04 ± 0.27 1.06 ± 0.19 1.22 ± 0.70 1.00 ± 0.00 | 5.44 ± 1.67 19.57 ± 1.57 * 19.13 ± 0.68 * 0.22 ± 0.1 * | 1.42 ± 0.68 13.84 ± 2.04 # 4.10 ± 0.10 # 0.10 ± 0.04 | 3.40 ± 1.99 42.34 ± 9.95 # 9.17 ± 3.12 # 0.20 ± 0.10 | 4.16 ± 3.23 35.61 ± 2.83 # 9.55 ± 1.89 # 0.17 ± 0.03 |
| Anti-inflammatory markers | |||||
| Cd206 Arg1 Igf-1 Il-10 | 1.07 ± 0.38 1.00 ± 0.07 1.02 ± 0.19 1.05 ± 0.17 | 0.21 ± 0.09 1.81 ± 0.02 * 0.13 ± 0.05 * 1.69 ± 0.38 | 0.07 ± 0.03 0.27 ± 0.03 # 0.03 ± 0.01 15.60 ± 7.95 # | 0.16 ± 0.03 1.02 ± 0.65 0.13 ± 0.07 18.88 ± 9.67 | 0.11 ± 0.01 1.40 ± 0.88 0.09 ± 0.01 29.58 ± 14.80 # |
| B | |||||
| Pro-inflammatory markers | |||||
| Cd40 Il-1β Tnf-α Cd68 | 1.01 ± 0.07 1.07 ± 0.13 1.02 ± 0.10 1.01 ± 0.07 | 0.46 ± 0.14 * 9.06 ± 1.79 * 0.33 ± 0.07 * 0.04 ± 0.01 | 0.62 ± 0.18 13.24 ± 1.42 0.88 ± 0.32 0.07 ± 0.02 | 0.39 ± 0.04 35.19 ± 7.12 # 0.56 ± 0.12 0.10 ± 0.02 | 0.68 ± 0.25 52.54 ± 13.15# 0.88 ± 0.35 0.03 ± 0.02 |
| Anti-inflammatory markers | |||||
| Cd206 Arg1 Igf-1 Il-10 | 1.01 ± 0.06 1.04 ± 0.16 1.05 ± 0.19 1.04 ± 0.09 | 0.01 ± 0.00 * 0.38 ± 0.10 * 0.01 ± 0.00 * 4.46 ± 0.72 | 0.01 ± 0.00 0.22 ± 0.06 0.01 ± 0.00 8.45 ± 1.61 # | 0.02 ± 0.01 0.48 ± 0.07 0.04 ± 0.01 23.78 ± 2.58 # | 0.03 ± 0.01 0.67 ± 0.19 0.03 ± 0.01 31.56 ± 5.77 # |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tylek, K.; Trojan, E.; Leśkiewicz, M.; Regulska, M.; Bryniarska, N.; Curzytek, K.; Lacivita, E.; Leopoldo, M.; Basta-Kaim, A. Time-Dependent Protective and Pro-Resolving Effects of FPR2 Agonists on Lipopolysaccharide-Exposed Microglia Cells Involve Inhibition of NF-κB and MAPKs Pathways. Cells 2021, 10, 2373. https://doi.org/10.3390/cells10092373
Tylek K, Trojan E, Leśkiewicz M, Regulska M, Bryniarska N, Curzytek K, Lacivita E, Leopoldo M, Basta-Kaim A. Time-Dependent Protective and Pro-Resolving Effects of FPR2 Agonists on Lipopolysaccharide-Exposed Microglia Cells Involve Inhibition of NF-κB and MAPKs Pathways. Cells. 2021; 10(9):2373. https://doi.org/10.3390/cells10092373
Chicago/Turabian StyleTylek, Kinga, Ewa Trojan, Monika Leśkiewicz, Magdalena Regulska, Natalia Bryniarska, Katarzyna Curzytek, Enza Lacivita, Marcello Leopoldo, and Agnieszka Basta-Kaim. 2021. "Time-Dependent Protective and Pro-Resolving Effects of FPR2 Agonists on Lipopolysaccharide-Exposed Microglia Cells Involve Inhibition of NF-κB and MAPKs Pathways" Cells 10, no. 9: 2373. https://doi.org/10.3390/cells10092373
APA StyleTylek, K., Trojan, E., Leśkiewicz, M., Regulska, M., Bryniarska, N., Curzytek, K., Lacivita, E., Leopoldo, M., & Basta-Kaim, A. (2021). Time-Dependent Protective and Pro-Resolving Effects of FPR2 Agonists on Lipopolysaccharide-Exposed Microglia Cells Involve Inhibition of NF-κB and MAPKs Pathways. Cells, 10(9), 2373. https://doi.org/10.3390/cells10092373