
Effects of Extracellular Osteoanabolic Agents on the Endogenous Response of Osteoblastic Cells



 ,
,
Abstract
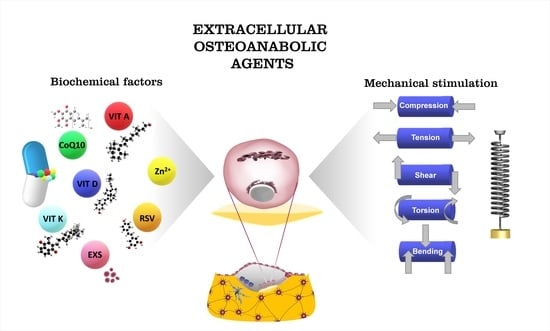
1. Introduction
2. Mechanoresponsive Skeletal Biology
2.1. Cells and Extracellular Matrix Organization in Bone
2.2. Bone Remodeling
2.2.1. Mechanical Properties and Structural Modification of Bone Tissue
2.2.2. Osteoblast Lineage
2.2.3. Osteoblast Functions
2.2.4. The Bone Multicellular Unit
2.2.5. The Bone Biochemical Markers
2.3. Cell Mechanosensing
2.3.1. Molecular Basis of Mechanotransduction in Mechanosensor Cells
Tethers
Focal Adhesion Complexes
2.3.2. Bone Biomechanics
2.3.3. Mechanosignal Transductions: Prominent Pathways for the Biomechanics of Bone Cells
3. Signaling in Bone Differentiation Capacity
3.1. Dedifferentiation and Differentiation of Bone Cells Play a Role in Bone Mineralization
3.1.1. Wnt Signaling
3.1.2. The Effect of Modulation of Wnt Signaling on Bone
3.1.3. Dedifferentiative Capacity of the Osteoblastic Lineage
3.1.4. Regulation of Differentiative Signaling Pathways by Vitamin D
3.2. RANK/RANK Ligand Signaling Pathway
4. Mechanical Stimulation in the Recovery of Bone Loss
4.1. Physical Description of Biomechanics
4.1.1. The Correspondence between Mechanical Stimulus and Strain
4.1.2. Concept and Terms Employed to Describe Mechanical Stimuli Applied to Bone
Stress and Strain Characteristics
Strain Frequency
Strain Rate and Strain Distribution
Strain Volume
4.2. Frost’s Mechanostat Theory
4.3. Bone Adaptation
4.4. Cell Response to Anabolic Mechanical Treatments
5. Ossification Coactivators
5.1. Micronutrients in Bone
5.1.1. Vitamin A
Effects of Retinoids on Osteoblast Cultures
Effect of Retinoids on Bone Health in Humans
5.1.2. Vitamin D
In Vitro Effects of 1α,25(OH)2D3 on Osteoblast Differentiation and Mineralization
Vitamin D Status and Bone Health
5.1.3. Vitamin K Status and Bone Health
5.1.4. Zinc as an Emergent Ossification Stimulus
Cell importers and Cellular Transporters of Zinc
Pro-Osteogenic Action of Zinc
Exogenous Zinc as a Reinforcement for Endogenous Osteogenesis
5.2. Antioxidant Supplements Involved in Bone Metabolism
Effects of Phytochemicals on Bone Health
Resveratrol
Coenzyme Q10
5.3. Exosomes in Bone Metabolism
5.3.1. Exosome Vesicles
5.3.2. Exosome Content
5.3.3. Exosome Biogenesis and Release
5.3.4. Role of Exosomes in Bone Remodeling and Molecular Mechanisms Involved
5.3.5. Possible Applications
6. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Ficai, A.; Marques, C.; Ferreira, J.M.F.; Andronescu, E.; Ficai, D.; Sonmez, M. Multifunctional Materials for Bone Cancer Treatment. Int. J. Nanomed. 2014, 9, 2713. [Google Scholar] [CrossRef]
- Samuel, J.; Park, J.-S.; Almer, J.; Wang, X. Effect of Water on Nanomechanics of Bone Is Different between Tension and Compression. J. Mech. Behav. Biomed. Mater. 2016, 57, 128–138. [Google Scholar] [CrossRef]
- Tang, V.W. Collagen, Stiffness, and Adhesion: The Evolutionary Basis of Vertebrate Mechanobiology. Mol. Biol. Cell 2020, 31, 1823–1834. [Google Scholar] [CrossRef]
- Kenkre, J.; Bassett, J. The Bone Remodelling Cycle. Ann. Clin. Biochem. Int. J. Lab. Med. 2018, 55, 308–327. [Google Scholar] [CrossRef]
- Palmer, L.C.; Newcomb, C.J.; Kaltz, S.R.; Spoerke, E.D.; Stupp, S.I. Biomimetic Systems for Hydroxyapatite Mineralization Inspired By Bone and Enamel. Chem. Rev. 2008, 108, 4754–4783. [Google Scholar] [CrossRef] [PubMed]
- Lambert, L.J.; Challa, A.K.; Niu, A.; Zhou, L.; Tucholski, J.; Johnson, M.S.; Nagy, T.R.; Eberhardt, A.W.; Estep, P.N.; Kesterson, R.A.; et al. Increased Trabecular Bone and Improved Biomechanics in an Osteocalcin Null Rat Model Created by CRISPR/Cas9 Technology. Dis. Model. Mech. 2016, 10, 1169–1179. [Google Scholar] [CrossRef] [PubMed]
- Rosset, E.M.; Bradshaw, A.D. SPARC/Osteonectin in Mineralized Tissue. Matrix Biol. 2016, 52–54, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Gardinier, J.D.; Majumdar, S.; Duncan, R.L.; Wang, L. Cyclic Hydraulic Pressure and Fluid Flow Differentially Modulate Cytoskeleton Re-Organization in MC3T3 Osteoblasts. Cell. Mol. Bioeng. 2009, 2, 133–143. [Google Scholar] [CrossRef]
- Kaigler, D.; Avila, G.; Wisner-Lynch, L.; Nevins, M.L.; Nevins, M.; Rasperini, G.; Lynch, S.E.; Giannobile, W.V. Platelet-Derived Growth Factor Applications in Periodontal and Peri-Implant Bone Regeneration. Expert Opin. Biol. Ther. 2011, 11, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Su, N.; Jin, M.; Chen, L. Role of FGF/FGFR Signaling in Skeletal Development and Homeostasis: Learning from Mouse Models. Bone Res. 2014, 2, 14003. [Google Scholar] [CrossRef]
- Tsiridis, E.; Upadhyay, N.; Giannoudis, P. Molecular Aspects of Fracture Healing:Which Are the Important Molecules? Injury 2007, 38, S11–S25. [Google Scholar] [CrossRef]
- Rosso, F.; Giordano, A.; Barbarisi, M.; Barbarisi, A. From Cell-ECM Interactions to Tissue Engineering. J. Cell. Physiol. 2004, 199, 174–180. [Google Scholar] [CrossRef]
- Shi, M.; Zhu, J.; Wang, R.; Chen, X.; Mi, L.; Walz, T.; Springer, T.A. Latent TGF-β Structure and Activation. Nature 2011, 474, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Kogianni, G.; Mann, V.; Noble, B.S. Apoptotic Bodies Convey Activity Capable of Initiating Osteoclastogenesis and Localized Bone Destruction. J. Bone Miner. Res. 2008, 23, 915–927. [Google Scholar] [CrossRef]
- Thompson, W.R.; Rubin, C.T.; Rubin, J. Mechanical Regulation of Signaling Pathways in Bone. Gene 2012, 503, 179–193. [Google Scholar] [CrossRef] [PubMed]
- O’Conor, C.J.; Ramalingam, S.; Zelenski, N.A.; Benefield, H.C.; Rigo, I.; Little, D.; Wu, C.-L.; Chen, D.; Liedtke, W.; McNulty, A.L.; et al. Cartilage-Specific Knockout of the Mechanosensory Ion Channel TRPV4 Decreases Age-Related Osteoarthritis. Sci. Rep. 2016, 6, 29053. [Google Scholar] [CrossRef] [PubMed]
- Vining, K.H.; Mooney, D.J. Mechanical Forces Direct Stem Cell Behaviour in Development and Regeneration. Nat. Rev. Mol. Cell Biol. 2017, 18, 728–742. [Google Scholar] [CrossRef]
- Bayraktar, H.H.; Morgan, E.F.; Niebur, G.L.; Morris, G.E.; Wong, E.K.; Keaveny, T.M. Comparison of the Elastic and Yield Properties of Human Femoral Trabecular and Cortical Bone Tissue. J. Biomech. 2004, 37, 27–35. [Google Scholar] [CrossRef]
- Taddei, F.; Balestri, M.; Rimondi, E.; Viceconti, M.; Manfrini, M. Tibia Adaptation after Fibula Harvesting: An in Vivo Quantitative Study. Clin. Orthop. 2009, 467, 2149–2158. [Google Scholar] [CrossRef][Green Version]
- Langdahl, B.; Ferrari, S.; Dempster, D.W. Bone Modeling and Remodeling: Potential as Therapeutic Targets for the Treatment of Osteoporosis. Ther. Adv. Musculoskelet. Dis. 2016, 8, 225–235. [Google Scholar] [CrossRef]
- Rutkovskiy, A.; Stensløkken, K.-O.; Vaage, I.J. Osteoblast Differentiation at a Glance. Med. Sci. Monit. Basic Res. 2016, 22, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Maeda, K.; Kobayashi, Y.; Koide, M.; Uehara, S.; Okamoto, M.; Ishihara, A.; Kayama, T.; Saito, M.; Marumo, K. The Regulation of Bone Metabolism and Disorders by Wnt Signaling. Int. J. Mol. Sci. 2019, 20, 5525. [Google Scholar] [CrossRef] [PubMed]
- Komori, T. Regulation of Proliferation, Differentiation and Functions of Osteoblasts by Runx2. Int. J. Mol. Sci. 2019, 20, 1694. [Google Scholar] [CrossRef] [PubMed]
- Marie, P.J. Transcription Factors Controlling Osteoblastogenesis. Arch. Biochem. Biophys. 2008, 473, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, K.; de Crombrugghe, B. Transcriptional Mechanisms in Osteoblast Differentiation and Bone Formation. Trends Genet. 2003, 19, 458–466. [Google Scholar] [CrossRef]
- Bruderer, M.; Richards, R.G.; Alini, M.; Stoddart, M.J. Role and Regulation of RUNX2 in Osteogenesis. Eur. Cell. Mater. 2014, 28. [Google Scholar] [CrossRef]
- Plotkin, L.I.; Mathov, I.; Aguirre, J.I.; Parfitt, A.M.; Manolagas, S.C.; Bellido, T. Mechanical Stimulation Prevents Osteocyte Apoptosis: Requirement of Integrins, Src Kinases, and ERKs. Am. J. Physiol. Cell Physiol. 2005, 289, C633–C643. [Google Scholar] [CrossRef]
- Plotkin, L.I.; Manolagas, S.C.; Bellido, T. Glucocorticoids Induce Osteocyte Apoptosis by Blocking Focal Adhesion Kinase-Mediated Survival. J. Biol. Chem. 2007, 282, 24120–24130. [Google Scholar] [CrossRef] [PubMed]
- van Bezooijen, R.L.; Papapoulos, S.E.; Löwik, C.W.G.M. Bone Morphogenetic Proteins and Their Antagonists: The Sclerostin Paradigm. J. Endocrinol. Investig. 2005, 28, 15–17. [Google Scholar]
- Tabacco, G.; Bilezikian, J.P. Osteoanabolic and Dual Action Drugs. Br. J. Clin. Pharmacol. 2019, 85, 1084–1094. [Google Scholar] [CrossRef]
- Matic, I.; Matthews, B.G.; Wang, X.; Dyment, N.A.; Worthley, D.L.; Rowe, D.W.; Grcevic, D.; Kalajzic, I. Quiescent Bone Lining Cells Are a Major Source of Osteoblasts During Adulthood: Bone Lining Cells and Osteogenesis. Stem Cells 2016, 34, 2930–2942. [Google Scholar] [CrossRef]
- Liu, F.; Malaval, L.; Aubin, J.E. The Mature Osteoblast Phenotype Is Characterized by Extensive Plasticity. Exp. Cell Res. 1997, 232, 97–105. [Google Scholar] [CrossRef]
- Manolagas, S.C. Birth and Death of Bone Cells: Basic Regulatory Mechanisms and Implications for the Pathogenesis and Treatment of Osteoporosis. Endocr. Rev. 2000, 21, 115–137. [Google Scholar] [CrossRef] [PubMed]
- Murshed, M.; Harmey, D.; Millán, J.L.; McKee, M.D.; Karsenty, G. Unique Coexpression in Osteoblasts of Broadly Expressed Genes Accounts for the Spatial Restriction of ECM Mineralization to Bone. Genes Dev. 2005, 19, 1093–1104. [Google Scholar] [CrossRef] [PubMed]
- Daoussis, D.; Andonopoulos, A.P. The Emerging Role of Dickkopf-1 in Bone Biology: Is It the Main Switch Controlling Bone and Joint Remodeling? Semin. Arthritis Rheum. 2011, 41, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, K.; Zhou, X.; Kunkel, G.; Zhang, Z.; Deng, J.M.; Behringer, R.R.; de Crombrugghe, B. The Novel Zinc Finger-Containing Transcription Factor Osterix Is Required for Osteoblast Differentiation and Bone Formation. Cell 2002, 108, 17–29. [Google Scholar] [CrossRef]
- Hartmann, C. A Wnt Canon Orchestrating Osteoblastogenesis. Trends Cell Biol. 2006, 16, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Bonewald, L.F. The Amazing Osteocyte. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2011, 26, 229–238. [Google Scholar] [CrossRef]
- Bonewald, L.F.; Johnson, M.L. Osteocytes, Mechanosensing and Wnt Signaling. Bone 2008, 42, 606–615. [Google Scholar] [CrossRef]
- Dallas, S.L.; Prideaux, M.; Bonewald, L.F. The Osteocyte: An Endocrine Cell and More. Endocr. Rev. 2013, 34, 658–690. [Google Scholar] [CrossRef]
- Chen, H.; Senda, T.; Kubo, K. The Osteocyte Plays Multiple Roles in Bone Remodeling and Mineral Homeostasis. Med. Mol. Morphol. 2015, 48, 61–68. [Google Scholar] [CrossRef]
- Xiong, J.; Onal, M.; Jilka, R.L.; Weinstein, R.S.; Manolagas, S.C.; O’Brien, C.A. Matrix-Embedded Cells Control Osteoclast Formation. Nat. Med. 2011, 17, 1235–1241. [Google Scholar] [CrossRef]
- Nakashima, T.; Hayashi, M.; Fukunaga, T.; Kurata, K.; Oh-Hora, M.; Feng, J.Q.; Bonewald, L.F.; Kodama, T.; Wutz, A.; Wagner, E.F.; et al. Evidence for Osteocyte Regulation of Bone Homeostasis through RANKL Expression. Nat. Med. 2011, 17, 1231–1234. [Google Scholar] [CrossRef]
- Moester, M.J.C.; Papapoulos, S.E.; Löwik, C.W.G.M.; van Bezooijen, R.L. Sclerostin: Current Knowledge and Future Perspectives. Calcif. Tissue Int. 2010, 87, 99–107. [Google Scholar] [CrossRef]
- Boyle, W.J.; Simonet, W.S.; Lacey, D.L. Osteoclast Differentiation and Activation. Nature 2003, 423, 337–342. [Google Scholar] [CrossRef]
- Udagawa, N.; Takahashi, N.; Yasuda, H.; Mizuno, A.; Itoh, K.; Ueno, Y.; Shinki, T.; Gillespie, M.T.; Martin, T.J.; Higashio, K.; et al. Osteoprotegerin Produced by Osteoblasts Is an Important Regulator in Osteoclast Development and Function. Endocrinology 2000, 141, 3478–3484. [Google Scholar] [CrossRef]
- Mitin, N.; Rossman, K.L.; Der, C.J. Signaling Interplay in Ras Superfamily Function. Curr. Biol. CB 2005, 15, R563–R574. [Google Scholar] [CrossRef] [PubMed]
- Ross, F.P. Osteoclast Biology and Bone Resorption. In Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2009; pp. 25–33. ISBN 978-1-118-45392-6. [Google Scholar]
- Reichert, J.C.; Schmalzl, J.; Prager, P.; Gilbert, F.; Quent, V.M.C.; Steinert, A.F.; Rudert, M.; Nöth, U. Synergistic Effect of Indian Hedgehog and Bone Morphogenetic Protein-2 Gene Transfer to Increase the Osteogenic Potential of Human Mesenchymal Stem Cells. Stem Cell Res. Ther. 2013, 4, 105. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Wang, Y.; Wang, W.; Chen, J.; Lai, P.; Liu, Z.; Yan, B.; Xu, S.; Zhang, Z.; Zeng, C.; et al. MTORC1 Prevents Preosteoblast Differentiation through the Notch Signaling Pathway. PLoS Genet. 2015, 11, e1005426. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.W.; Lu, Y.; Williams, E.A.; Lai, F.; Lee, J.Y.; Enishi, T.; Balani, D.H.; Ominsky, M.S.; Ke, H.Z.; Kronenberg, H.M.; et al. Sclerostin Antibody Administration Converts Bone Lining Cells Into Active Osteoblasts. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2017, 32, 892–901. [Google Scholar] [CrossRef]
- Hoffman, B.D.; Grashoff, C.; Schwartz, M.A. Dynamic Molecular Processes Mediate Cellular Mechanotransduction. Nature 2011, 475, 316–323. [Google Scholar] [CrossRef]
- Eriksen, E.F. Cellular Mechanisms of Bone Remodeling. Rev. Endocr. Metab. Disord. 2010, 11, 219–227. [Google Scholar] [CrossRef]
- Papachroni, K.K.; Karatzas, D.N.; Papavassiliou, K.A.; Basdra, E.K.; Papavassiliou, A.G. Mechanotransduction in Osteoblast Regulation and Bone Disease. Trends Mol. Med. 2009, 15, 208–216. [Google Scholar] [CrossRef]
- Raggatt, L.J.; Partridge, N.C. Cellular and Molecular Mechanisms of Bone Remodeling. J. Biol. Chem. 2010, 285, 25103–25108. [Google Scholar] [CrossRef]
- Klein-Nulend, J.; Bacabac, R.; Bakker, A. Mechanical Loading and How It Affects Bone Cells: The Role of the Osteocyte Cytoskeleton in Maintaining Our Skeleton. Eur. Cell. Mater. 2012, 24, 278–291. [Google Scholar] [CrossRef]
- Greenblatt, M.B.; Tsai, J.N.; Wein, M.N. Bone Turnover Markers in the Diagnosis and Monitoring of Metabolic Bone Disease. Clin. Chem. 2017, 63, 464–474. [Google Scholar] [CrossRef]
- Komiya, Y.; Habas, R. Wnt Signal Transduction Pathways. Organogenesis 2008, 4, 68–75. [Google Scholar] [CrossRef]
- Barradas, A.M.C.; Fernandes, H.A.M.; Groen, N.; Chai, Y.C.; Schrooten, J.; van de Peppel, J.; van Leeuwen, J.P.T.M.; van Blitterswijk, C.A.; de Boer, J. A Calcium-Induced Signaling Cascade Leading to Osteogenic Differentiation of Human Bone Marrow-Derived Mesenchymal Stromal Cells. Biomaterials 2012, 33, 3205–3215. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.P.; Albert, C.; Nassar, B.A.; Adachi, J.D.; Cole, D.; Davison, K.S.; Dooley, K.C.; Don-Wauchope, A.; Douville, P.; Hanley, D.A.; et al. Bone Turnover Markers in the Management of Postmenopausal Osteoporosis. Clin. Biochem. 2009, 42, 929–942. [Google Scholar] [CrossRef] [PubMed]
- Twine, N.A.; Chen, L.; Pang, C.N.; Wilkins, M.R.; Kassem, M. Identification of Differentiation-Stage Specific Markers That Define the Ex Vivo Osteoblastic Phenotype. Bone 2014, 67, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Kučukalić-Selimović, E.; Valjevac, A.; Hadžović-Džuvo, A. The Utility of Procollagen Type 1 N-Terminal Propeptide for the Bone Status Assessment in Postmenopausal Women. Bosn. J. Basic Med. Sci. 2013, 13, 259. [Google Scholar] [CrossRef]
- Halleen, J.M.; Tiitinen, S.L.; Ylipahkala, H.; Fagerlund, K.M.; Väänänen, H.K. Tartrate-Resistant Acid Phosphatase 5b (TRACP 5b) as a Marker of Bone Resorption. Clin. Lab. 2006, 52, 499–509. [Google Scholar] [PubMed]
- Everts, V.; Korper, W.; Hoeben, K.A.; Jansen, I.D.; Bromme, D.; Cleutjens, K.B.; Heeneman, S.; Peters, C.; Reinheckel, T.; Saftig, P.; et al. Osteoclastic Bone Degradation and the Role of Different Cysteine Proteinases and Matrix Metalloproteinases: Differences Between Calvaria and Long Bone. J. Bone Miner. Res. 2006, 21, 1399–1408. [Google Scholar] [CrossRef] [PubMed]
- Warden, S.J.; Thompson, W.R. Become One with the Force: Optimising Mechanotherapy through an Understanding of Mechanobiology. Br. J. Sports Med. 2017, 51, 989–990. [Google Scholar] [CrossRef] [PubMed]
- Na, S.; Collin, O.; Chowdhury, F.; Tay, B.; Ouyang, M.; Wang, Y.; Wang, N. Rapid Signal Transduction in Living Cells Is a Unique Feature of Mechanotransduction. Proc. Natl. Acad. Sci. USA 2008, 105, 6626–6631. [Google Scholar] [CrossRef]
- Holle, A.W.; Engler, A.J. More than a Feeling: Discovering, Understanding, and Influencing Mechanosensing Pathways. Curr. Opin. Biotechnol. 2011, 22, 648–654. [Google Scholar] [CrossRef] [PubMed]
- Gardel, M.L.; Sabass, B.; Ji, L.; Danuser, G.; Schwarz, U.S.; Waterman, C.M. Traction Stress in Focal Adhesions Correlates Biphasically with Actin Retrograde Flow Speed. J. Cell Biol. 2008, 183, 999–1005. [Google Scholar] [CrossRef]
- Bose, S.; Dasbiswas, K.; Gopinath, A. Matrix Stiffness Modulates Mechanical Interactions and Promotes Contact between Motile Cells. Biomedicines 2021, 9, 428. [Google Scholar] [CrossRef] [PubMed]
- Olsen, B. Nearly all cells in vertebrates and many cells in invertebrates contain primary cilia. Matrix Biol. 2005, 24, 449–450. [Google Scholar] [CrossRef]
- Yavropoulou, M.P.; Yovos, J.G. The Molecular Basis of Bone Mechanotransduction. J. Musculoskelet. Neuronal Interact. 2016, 16, 221–236. [Google Scholar]
- Peng, A.W.; Salles, F.T.; Pan, B.; Ricci, A.J. Integrating the Biophysical and Molecular Mechanisms of Auditory Hair Cell Mechanotransduction. Nat. Commun. 2011, 2, 523. [Google Scholar] [CrossRef]
- Han, Y.; Cowin, S.C.; Schaffler, M.B.; Weinbaum, S. Mechanotransduction and Strain Amplification in Osteocyte Cell Processes. Proc. Natl. Acad. Sci. USA 2004, 101, 16689–16694. [Google Scholar] [CrossRef]
- Mohammed, D.; Versaevel, M.; Bruyère, C.; Alaimo, L.; Luciano, M.; Vercruysse, E.; Procès, A.; Gabriele, S. Innovative Tools for Mechanobiology: Unraveling Outside-In and Inside-Out Mechanotransduction. Front. Bioeng. Biotechnol. 2019, 7, 162. [Google Scholar] [CrossRef]
- Benito-Jardón, M.; Strohmeyer, N.; Ortega-Sanchís, S.; Bharadwaj, M.; Moser, M.; Müller, D.J.; Fässler, R.; Costell, M. Av-Class Integrin Binding to Fibronectin Is Solely Mediated by RGD and Unaffected by an RGE Mutation. J. Cell Biol. 2020, 219, e202004198. [Google Scholar] [CrossRef]
- Selig, M.; Lauer, J.C.; Hart, M.L.; Rolauffs, B. Mechanotransduction and Stiffness-Sensing: Mechanisms and Opportunities to Control Multiple Molecular Aspects of Cell Phenotype as a Design Cornerstone of Cell-Instructive Biomaterials for Articular Cartilage Repair. Int. J. Mol. Sci. 2020, 21, 5399. [Google Scholar] [CrossRef]
- Roca-Cusachs, P.; del Rio, A.; Puklin-Faucher, E.; Gauthier, N.C.; Biais, N.; Sheetz, M.P. Integrin-Dependent Force Transmission to the Extracellular Matrix by α-Actinin Triggers Adhesion Maturation. Proc. Natl. Acad. Sci. USA 2013, 110, E1361–E1370. [Google Scholar] [CrossRef] [PubMed]
- Horwitz, A.; Duggan, K.; Buck, C.; Beckerle, M.C.; Burridge, K. Interaction of Plasma Membrane Fibronectin Receptor with Talin—a Transmembrane Linkage. Nature 1986, 320, 531–533. [Google Scholar] [CrossRef] [PubMed]
- Subauste, M.C.; Pertz, O.; Adamson, E.D.; Turner, C.E.; Junger, S.; Hahn, K.M. Vinculin Modulation of Paxillin–FAK Interactions Regulates ERK to Control Survival and Motility. J. Cell Biol. 2004, 165, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Schlaepfer, D.D.; Hauck, C.R.; Sieg, D.J. Signaling through Focal Adhesion Kinase. Prog. Biophys. Mol. Biol. 1999, 71, 435–478. [Google Scholar] [CrossRef]
- Mullender, M.G.; Huiskes, R. Osteocytes and Bone Lining Cells: Which Are the Best Candidates for Mechano-Sensors in Cancellous Bone? Bone 1997, 20, 527–532. [Google Scholar] [CrossRef]
- Rubin, J.; Murphy, T.C.; Fan, X.; Goldschmidt, M.; Taylor, W.R. Activation of Extracellular Signal-Regulated Kinase Is Involved in Mechanical Strain Inhibition of RANKL Expression in Bone Stromal Cells. J. Bone Miner. Res. 2002, 17, 1452–1460. [Google Scholar] [CrossRef]
- Lieben, L.; Carmeliet, G. The Involvement of TRP Channels in Bone Homeostasis. Front. Endocrinol. 2012, 3, 99. [Google Scholar] [CrossRef]
- Guilak, F.; Leddy, H.A.; Liedtke, W. Transient Receptor Potential Vanilloid 4: The Sixth Sense of the Musculoskeletal System? Ann. N. Y. Acad. Sci. 2010, 1192, 404–409. [Google Scholar] [CrossRef]
- Kärki, T.; Tojkander, S. TRPV Protein Family-From Mechanosensing to Cancer Invasion. Biomolecules 2021, 11, 1019. [Google Scholar] [CrossRef]
- Du, G.; Li, L.; Zhang, X.; Liu, J.; Hao, J.; Zhu, J.; Wu, H.; Chen, W.; Zhang, Q. Roles of TRPV4 and Piezo Channels in Stretch-Evoked Ca2+ Response in Chondrocytes. Exp. Biol. Med. Maywood NJ 2020, 245, 180–189. [Google Scholar] [CrossRef]
- Zhou, T.; Gao, B.; Fan, Y.; Liu, Y.; Feng, S.; Cong, Q.; Zhang, X.; Zhou, Y.; Yadav, P.S.; Lin, J.; et al. Piezo1/2 Mediate Mechanotransduction Essential for Bone Formation through Concerted Activation of NFAT-YAP1-ß-Catenin. eLife 2020, 9, e52779. [Google Scholar] [CrossRef] [PubMed]
- Liedert, A.; Wagner, L.; Seefried, L.; Ebert, R.; Jakob, F.; Ignatius, A. Estrogen Receptor and Wnt Signaling Interact to Regulate Early Gene Expression in Response to Mechanical Strain in Osteoblastic Cells. Biochem. Biophys. Res. Commun. 2010, 394, 755–759. [Google Scholar] [CrossRef] [PubMed]
- Liedert, A.; Kaspar, D.; Blakytny, R.; Claes, L.; Ignatius, A. Signal Transduction Pathways Involved in Mechanotransduction in Bone Cells. Biochem. Biophys. Res. Commun. 2006, 349, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Huang, D.; Lu, X.; Feng, G.; Xing, J.; Lu, J.; Xu, K.; Xia, W.; Meng, Y.; Tao, T.; et al. Insulin-like Growth Factor 1 Can Promote Proliferation and Osteogenic Differentiation of Human Dental Pulp Stem Cells via MTOR Pathway. Dev. Growth Differ. 2014, 56, 615–624. [Google Scholar] [CrossRef]
- Liu, D.; Genetos, D.C.; Shao, Y.; Geist, D.J.; Li, J.; Ke, H.Z.; Turner, C.H.; Duncan, R.L. Activation of Extracellular-Signal Regulated Kinase (ERK1/2) by Fluid Shear Is Ca2+- and ATP-Dependent in MC3T3-E1 Osteoblasts. Bone 2008, 42, 644–652. [Google Scholar] [CrossRef]
- Rosa, N.; Simoes, R.; Magalhães, F.D.; Marques, A.T. From Mechanical Stimulus to Bone Formation: A Review. Med. Eng. Phys. 2015, 37, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Robling, A.G.; Niziolek, P.J.; Baldridge, L.A.; Condon, K.W.; Allen, M.R.; Alam, I.; Mantila, S.M.; Gluhak-Heinrich, J.; Bellido, T.M.; Harris, S.E.; et al. Mechanical Stimulation of Bone in Vivo Reduces Osteocyte Expression of Sost/Sclerostin. J. Biol. Chem. 2008, 283, 5866–5875. [Google Scholar] [CrossRef] [PubMed]
- Michaletti, A.; Gioia, M.; Tarantino, U.; Zolla, L. Effects of Microgravity on Osteoblast Mitochondria: A Proteomic and Metabolomics Profile. Sci. Rep. 2017, 7, 15376. [Google Scholar] [CrossRef] [PubMed]
- Gioia, M.; Michaletti, A.; Scimeca, M.; Marini, M.; Tarantino, U.; Zolla, L.; Coletta, M. Simulated Microgravity Induces a Cellular Regression of the Mature Phenotype in Human Primary Osteoblasts. Cell Death Discov. 2018, 4, 59. [Google Scholar] [CrossRef] [PubMed]
- Chrzanowska-Wodnicka, M.; Burridge, K. Rho-Stimulated Contractility Drives the Formation of Stress Fibers and Focal Adhesions. J. Cell Biol. 1996, 133, 1403–1415. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.H.-C.; Thampatty, B.P. An Introductory Review of Cell Mechanobiology. Biomech. Model. Mechanobiol. 2006, 5, 1–16. [Google Scholar] [CrossRef]
- Chen, N.X.; Ryder, K.D.; Pavalko, F.M.; Turner, C.H.; Burr, D.B.; Qiu, J.; Duncan, R.L. Ca2+ Regulates Fluid Shear-Induced Cytoskeletal Reorganization and Gene Expression in Osteoblasts. Am. J. Physiol.-Cell Physiol. 2000, 278, C989–C997. [Google Scholar] [CrossRef]
- Case, N.; Sen, B.; Thomas, J.A.; Styner, M.; Xie, Z.; Jacobs, C.R.; Rubin, J. Steady and Oscillatory Fluid Flows Produce a Similar Osteogenic Phenotype. Calcif. Tissue Int. 2011, 88, 189–197. [Google Scholar] [CrossRef]
- Manokawinchoke, J.; Pavasant, P.; Sawangmake, C.; Limjeerajarus, N.; Limjeerajarus, C.N.; Egusa, H.; Osathanon, T. Intermittent Compressive Force Promotes Osteogenic Differentiation in Human Periodontal Ligament Cells by Regulating the Transforming Growth Factor-β Pathway. Cell Death Dis. 2019, 10, 761. [Google Scholar] [CrossRef]
- Searby, N.D.; Steele, C.R.; Globus, R.K. Influence of Increased Mechanical Loading by Hypergravity on the Microtubule Cytoskeleton and Prostaglandin E 2 Release in Primary Osteoblasts. Am. J. Physiol.-Cell Physiol. 2005, 289, C148–C158. [Google Scholar] [CrossRef]
- McAllister, T.N.; Frangos, J.A. Steady and Transient Fluid Shear Stress Stimulate NO Release in Osteoblasts Through Distinct Biochemical Pathways. J. Bone Miner. Res. 1999, 14, 930–936. [Google Scholar] [CrossRef] [PubMed]
- McGarry, J.G.; Klein-Nulend, J.; Prendergast, P.J. The Effect of Cytoskeletal Disruption on Pulsatile Fluid Flow-Induced Nitric Oxide and Prostaglandin E2 Release in Osteocytes and Osteoblasts. Biochem. Biophys. Res. Commun. 2005, 330, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Foster, A.D. The Impact of Bipedal Mechanical Loading History on Longitudinal Long Bone Growth. PLoS ONE 2019, 14, e0211692. [Google Scholar] [CrossRef] [PubMed]
- Hutchings, G.; Moncrieff, L.; Dompe, C.; Janowicz, K.; Sibiak, R.; Bryja, A.; Jankowski, M.; Mozdziak, P.; Bukowska, D.; Antosik, P.; et al. Bone Regeneration, Reconstruction and Use of Osteogenic Cells; from Basic Knowledge, Animal Models to Clinical Trials. J. Clin. Med. 2020, 9, 139. [Google Scholar] [CrossRef] [PubMed]
- Rubin, J.; Murphy, T.C.; Zhu, L.; Roy, E.; Nanes, M.S.; Fan, X. Mechanical Strain Differentially Regulates Endothelial Nitric-Oxide Synthase and Receptor Activator of Nuclear ΚB Ligand Expression via ERK1/2 MAPK. J. Biol. Chem. 2003, 278, 34018–34025. [Google Scholar] [CrossRef]
- Wang, B.; Du, T.; Wang, Y.; Yang, C.; Zhang, S.; Cao, X. Focal Adhesion Kinase Signaling Pathway Is Involved in Mechanotransduction in MG-63 Cells. Biochem. Biophys. Res. Commun. 2011, 410, 671–676. [Google Scholar] [CrossRef]
- Hung, C.T.; Pollack, S.R.; Reilly, T.M.; Brighton, C.T. Real-Time Calcium Response of Cultured Bone Cells to Fluid Flow. Clin. Orthop. 1995, 313, 256–269. [Google Scholar]
- Donahue, S.W.; Donahue, H.J.; Jacobs, C.R. Osteoblastic Cells Have Refractory Periods for Fluid-Flow-Induced Intracellular Calcium Oscillations for Short Bouts of Flow and Display Multiple Low-Magnitude Oscillations during Long-Term Flow. J. Biomech. 2003, 36, 35–43. [Google Scholar] [CrossRef]
- Kwon, R.Y.; Temiyasathit, S.; Tummala, P.; Quah, C.C.; Jacobs, C.R. Primary Cilium-Dependent Mechanosensing Is Mediated by Adenylyl Cyclase 6 and Cyclic AMP in Bone Cells. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2010, 24, 2859–2868. [Google Scholar] [CrossRef]
- Ryder, K.D.; Duncan, R.L. Parathyroid Hormone Enhances Fluid Shear-Induced [Ca2+]i Signaling in Osteoblastic Cells through Activation of Mechanosensitive and Voltage-Sensitive Ca2+ Channels. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2001, 16, 240–248. [Google Scholar] [CrossRef]
- Lessey, E.C.; Guilluy, C.; Burridge, K. From Mechanical Force to RhoA Activation. Biochemistry 2012, 51, 7420–7432. [Google Scholar] [CrossRef] [PubMed]
- Case, N.; Rubin, J. Beta-Catenin--a Supporting Role in the Skeleton. J. Cell. Biochem. 2010, 110, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Boyden, L.M.; Mao, J.; Belsky, J.; Mitzner, L.; Farhi, A.; Mitnick, M.A.; Wu, D.; Insogna, K.; Lifton, R.P. High Bone Density Due to a Mutation in LDL-Receptor-Related Protein 5. N. Engl. J. Med. 2002, 346, 1513–1521. [Google Scholar] [CrossRef] [PubMed]
- Norvell, S.M.; Alvarez, M.; Bidwell, J.P.; Pavalko, F.M. Fluid Shear Stress Induces Beta-Catenin Signaling in Osteoblasts. Calcif. Tissue Int. 2004, 75, 396–404. [Google Scholar] [CrossRef] [PubMed]
- Bidwell, J.P.; Pavalko, F.M. The Load-Bearing Mechanosome Revisited. Clin. Rev. Bone Miner. Metab. 2010, 8, 213–223. [Google Scholar] [CrossRef]
- Pitsillides, A.A.; Rawlinson, S.C.; Suswillo, R.F.; Bourrin, S.; Zaman, G.; Lanyon, L.E. Mechanical Strain-Induced NO Production by Bone Cells: A Possible Role in Adaptive Bone (Re)Modeling? FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1995, 9, 1614–1622. [Google Scholar] [CrossRef]
- Ajubi, N.E.; Klein-Nulend, J.; Alblas, M.J.; Burger, E.H.; Nijweide, P.J. Signal Transduction Pathways Involved in Fluid Flow-Induced PGE2 Production by Cultured Osteocytes. Am. J. Physiol. 1999, 276, E171–E178. [Google Scholar] [CrossRef]
- Klein-Nulend, J.; Semeins, C.M.; Ajubi, N.E.; Nijweide, P.J.; Burger, E.H. Pulsating Fluid Flow Increases Nitric Oxide (NO) Synthesis by Osteocytes but Not Periosteal Fibroblasts--Correlation with Prostaglandin Upregulation. Biochem. Biophys. Res. Commun. 1995, 217, 640–648. [Google Scholar] [CrossRef]
- Bakker, A.D.; Joldersma, M.; Klein-Nulend, J.; Burger, E.H. Interactive Effects of PTH and Mechanical Stress on Nitric Oxide and PGE2 Production by Primary Mouse Osteoblastic Cells. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E608–E613. [Google Scholar] [CrossRef]
- Zhu, W.; Boachie-Adjei, O.; Rawlins, B.A.; Frenkel, B.; Boskey, A.L.; Ivashkiv, L.B.; Blobel, C.P. A Novel Regulatory Role for Stromal-Derived Factor-1 Signaling in Bone Morphogenic Protein-2 Osteogenic Differentiation of Mesenchymal C2C12 Cells. J. Biol. Chem. 2007, 282, 18676–18685. [Google Scholar] [CrossRef]
- Jung, Y.; Wang, J.; Schneider, A.; Sun, Y.-X.; Koh-Paige, A.J.; Osman, N.I.; McCauley, L.K.; Taichman, R.S. Regulation of SDF-1 (CXCL12) Production by Osteoblasts; a Possible Mechanism for Stem Cell Homing. Bone 2006, 38, 497–508. [Google Scholar] [CrossRef]
- You, J.; Jacobs, C.R.; Steinberg, T.H.; Donahue, H.J. P2Y Purinoceptors Are Responsible for Oscillatory Fluid Flow-Induced Intracellular Calcium Mobilization in Osteoblastic Cells. J. Biol. Chem. 2002, 277, 48724–48729. [Google Scholar] [CrossRef]
- Zaman, G.; Saxon, L.K.; Sunters, A.; Hilton, H.; Underhill, P.; Williams, D.; Price, J.S.; Lanyon, L.E. Loading-Related Regulation of Gene Expression in Bone in the Contexts of Estrogen Deficiency, Lack of Estrogen Receptor α and Disuse. Bone 2010, 46, 628–642. [Google Scholar] [CrossRef]
- Galea, G.L.; Meakin, L.B.; Sugiyama, T.; Zebda, N.; Sunters, A.; Taipaleenmaki, H.; Stein, G.S.; van Wijnen, A.J.; Lanyon, L.E.; Price, J.S. Estrogen Receptor α Mediates Proliferation of Osteoblastic Cells Stimulated by Estrogen and Mechanical Strain, but Their Acute down-Regulation of the Wnt Antagonist Sost Is Mediated by Estrogen Receptor β. J. Biol. Chem. 2013, 288, 9035–9048. [Google Scholar] [CrossRef]
- Windahl, S.H.; Saxon, L.; Börjesson, A.E.; Lagerquist, M.K.; Frenkel, B.; Henning, P.; Lerner, U.H.; Galea, G.L.; Meakin, L.B.; Engdahl, C.; et al. Estrogen Receptor-α Is Required for the Osteogenic Response to Mechanical Loading in a Ligand-independent Manner Involving Its Activation Function 1 but Not 2. J. Bone Miner. Res. 2013, 28, 291–301. [Google Scholar] [CrossRef]
- Saxon, L.K.; Galea, G.; Meakin, L.; Price, J.; Lanyon, L.E. Estrogen Receptors α and β Have Different Gender-Dependent Effects on the Adaptive Responses to Load Bearing in Cancellous and Cortical Bone. Endocrinology 2012, 153, 2254–2266. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Uehara, S.; Udagawa, N.; Takahashi, N. Regulation of Bone Metabolism by Wnt Signals. J. Biochem. 2016, 159, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Visweswaran, M.; Pohl, S.; Arfuso, F.; Newsholme, P.; Dilley, R.; Pervaiz, S.; Dharmarajan, A. Multi-Lineage Differentiation of Mesenchymal Stem Cells—To Wnt, or Not Wnt. Int. J. Biochem. Cell Biol. 2015, 68, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Grumolato, L.; Liu, G.; Mong, P.; Mudbhary, R.; Biswas, R.; Arroyave, R.; Vijayakumar, S.; Economides, A.N.; Aaronson, S.A. Canonical and Noncanonical Wnts Use a Common Mechanism to Activate Completely Unrelated Coreceptors. Genes Dev. 2010, 24, 2517–2530. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.S.; Robling, A.G. New Insights into Wnt-Lrp5/6-β-Catenin Signaling in Mechanotransduction. Front. Endocrinol. 2015, 5, 246. [Google Scholar] [CrossRef]
- Spencer, G.J.; Utting, J.C.; Etheridge, S.L.; Arnett, T.R.; Genever, P.G. Wnt Signalling in Osteoblasts Regulates Expression of the Receptor Activator of NFκB Ligand and Inhibits Osteoclastogenesis in Vitro. J. Cell Sci. 2006, 119, 1283–1296. [Google Scholar] [CrossRef]
- Bodine, P.V.N.; Komm, B.S. Wnt Signaling and Osteoblastogenesis. Rev. Endocr. Metab. Disord. 2007, 7, 33–39. [Google Scholar] [CrossRef]
- Holguin, N.; Brodt, M.D.; Silva, M.J. Activation of Wnt Signaling by Mechanical Loading Is Impaired in the Bone of Old Mice: Load-induced activation of wnt signaling impaired in bone of old mice. J. Bone Miner. Res. 2016, 31, 2215–2226. [Google Scholar] [CrossRef]
- Robinson, J.A.; Chatterjee-Kishore, M.; Yaworsky, P.J.; Cullen, D.M.; Zhao, W.; Li, C.; Kharode, Y.; Sauter, L.; Babij, P.; Brown, E.L.; et al. Wnt/β-Catenin Signaling Is a Normal Physiological Response to Mechanical Loading in Bone. J. Biol. Chem. 2006, 281, 31720–31728. [Google Scholar] [CrossRef]
- Hou, W.W.; Zhu, Z.L.; Zhou, Y.; Zhang, C.X.; Yu, H.Y. Involvement of Wnt Activation in the Micromechanical Vibration-Enhanced Osteogenic Response of Osteoblasts. J. Orthop. Sci. 2011, 16, 598–605. [Google Scholar] [CrossRef]
- Torreggiani, E.; Matthews, B.G.; Pejda, S.; Matic, I.; Horowitz, M.C.; Grcevic, D.; Kalajzic, I. Preosteocytes/Osteocytes Have the Potential to Dedifferentiate Becoming a Source of Osteoblasts. PLoS ONE 2013, 8, e75204. [Google Scholar] [CrossRef]
- DeLuca, H.F. Overview of General Physiologic Features and Functions of Vitamin D. Am. J. Clin. Nutr. 2004, 80, 1689S–1696S. [Google Scholar] [CrossRef] [PubMed]
- Holick, M.F. Resurrection of Vitamin D Deficiency and Rickets. J. Clin. Investig. 2006, 116, 2062–2072. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, T.A.; Horst, R.L. Parathyroid Hormone Down-Regulates 1,25-Dihydroxyvitamin D Receptors (VDR) and VDR Messenger Ribonucleic Acid in Vitro and Blocks Homologous up-Regulation of VDR in Vivo. Endocrinology 1990, 127, 942–948. [Google Scholar] [CrossRef]
- Godschalk, M.; Levy, J.R.; Downs, R.W. Glucocorticoids Decrease Vitamin D Receptor Number and Gene Expression in Human Osteosarcoma Cells. J. Bone Miner. Res. 2009, 7, 21–27. [Google Scholar] [CrossRef] [PubMed]
- van Leeuwen, J.P.T.M.; Birkenhäger, J.C.; Vink-van Wijngaarden, T.; van den Bemd, G.J.C.M.; Pols, H.A.P. Regulation of 1,25-Dihydroxyvitamin D3 Receptor Gene Expression by Parathyroid Hormone and CAMP-Agonists. Biochem. Biophys. Res. Commun. 1992, 185, 881–886. [Google Scholar] [CrossRef]
- Breen, E.C.; van Wijnen, A.J.; Lian, J.B.; Stein, G.S.; Stein, J.L. In Vivo Occupancy of the Vitamin D Responsive Element in the Osteocalcin Gene Supports Vitamin D-Dependent Transcriptional Upregulation in Intact Cells. Proc. Natl. Acad. Sci. USA 1994, 91, 12902–12906. [Google Scholar] [CrossRef]
- Boyan, B.D.; Wang, L.; Wong, K.L.; Jo, H.; Schwartz, Z. Plasma Membrane Requirements for 1α,25(OH)2D3 Dependent PKC Signaling in Chondrocytes and Osteoblasts. Steroids 2006, 71, 286–290. [Google Scholar] [CrossRef]
- Zanello, L.P.; Norman, A. 1α,25(OH)2 Vitamin D3 Actions on Ion Channels in Osteoblasts. Steroids 2006, 71, 291–297. [Google Scholar] [CrossRef]
- Khanal, R.; Nemere, I. The ERp57/GRp58/1,25D3-MARRS Receptor: Multiple Functional Roles in Diverse Cell Systems. Curr. Med. Chem. 2007, 14, 1087–1093. [Google Scholar] [CrossRef]
- van de Peppel, J.; van Leeuwen, J.P.T.M. Vitamin D and Gene Networks in Human Osteoblasts. Front. Physiol. 2014, 5, 137. [Google Scholar] [CrossRef]
- Narayanan, R.; Sepulveda, V.A.T.; Falzon, M.; Weigel, N.L. The Functional Consequences of Cross-Talk between the Vitamin D Receptor and ERK Signaling Pathways Are Cell-Specific. J. Biol. Chem. 2004, 279, 47298–47310. [Google Scholar] [CrossRef]
- Corral, D.A.; Amling, M.; Priemel, M.; Loyer, E.; Fuchs, S.; Ducy, P.; Baron, R.; Karsenty, G. Dissociation between Bone Resorption and Bone Formation in Osteopenic Transgenic Mice. Proc. Natl. Acad. Sci. USA 1998, 95, 13835–13840. [Google Scholar] [CrossRef]
- Galli, C.; Fu, Q.; Wang, W.; Olsen, B.R.; Manolagas, S.C.; Jilka, R.L.; O’Brien, C.A. Commitment to the Osteoblast Lineage Is Not Required for RANKL Gene Expression. J. Biol. Chem. 2009, 284, 12654–12662. [Google Scholar] [CrossRef]
- Xiong, J.; Piemontese, M.; Onal, M.; Campbell, J.; Goellner, J.J.; Dusevich, V.; Bonewald, L.; Manolagas, S.C.; O’Brien, C.A. Osteocytes, Not Osteoblasts or Lining Cells, Are the Main Source of the RANKL Required for Osteoclast Formation in Remodeling Bone. PLoS ONE 2015, 10, e0138189. [Google Scholar] [CrossRef]
- Feng, J.Q.; Ye, L.; Schiavi, S. Do Osteocytes Contribute to Phosphate Homeostasis? : Curr. Opin. Nephrol. Hypertens. 2009, 18, 285–291. [Google Scholar] [CrossRef]
- Drissi, H. 1,25-(OH)2-Vitamin D3 Suppresses the Bone-Related Runx2/Cbfa1 Gene Promoter. Exp. Cell Res. 2002, 274, 323–333. [Google Scholar] [CrossRef]
- Nemere, I.; Farach-Carson, M.C.; Rohe, B.; Sterling, T.M.; Norman, A.W.; Boyan, B.D.; Safford, S.E. Ribozyme Knockdown Functionally Links a 1,25(OH)2D3 Membrane Binding Protein (1,25D3-MARRS) and Phosphate Uptake in Intestinal Cells. Proc. Natl. Acad. Sci. USA 2004, 101, 7392–7397. [Google Scholar] [CrossRef] [PubMed]
- Nemere, I.; Safford, S.E.; Rohe, B.; DeSouza, M.M.; Farach-Carson, M.C. Identification and Characterization of 1,25D3-Membrane-Associated Rapid Response, Steroid (1,25D3-MARRS) Binding Protein. J. Steroid Biochem. Mol. Biol. 2004, 89–90, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Olivares-Navarrete, R.; Wang, Y.; Herman, T.R.; Boyan, B.D.; Schwartz, Z. Protein-Disulfide Isomerase-Associated 3 (Pdia3) Mediates the Membrane Response to 1,25-Dihydroxyvitamin D3 in Osteoblasts. J. Biol. Chem. 2010, 285, 37041–37050. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Dosier, C.R.; Park, J.H.; De, S.; Guldberg, R.E.; Boyan, B.D.; Schwartz, Z. Mineralization of Three-Dimensional Osteoblast Cultures Is Enhanced by the Interaction of 1α,25-Dihydroxyvitamin D3 and BMP2 via Two Specific Vitamin D Receptors. J. Tissue Eng. Regen. Med. 2016, 10, 40–51. [Google Scholar] [CrossRef]
- Willems, H.M.E.; van den Heuvel, E.G.H.M.; Carmeliet, G.; Schaafsma, A.; Klein-Nulend, J.; Bakker, A.D. VDR Dependent and Independent Effects of 1,25-Dihydroxyvitamin D3 on Nitric Oxide Production by Osteoblasts. Steroids 2012, 77, 126–131. [Google Scholar] [CrossRef]
- Fretz, J.A.; Zella, L.A.; Kim, S.; Shevde, N.K.; Pike, J.W. 1,25-Dihydroxyvitamin D3 Induces Expression of the Wnt Signaling Co-Regulator LRP5 via Regulatory Elements Located Significantly Downstream of the Gene’s Transcriptional Start Site. J. Steroid Biochem. Mol. Biol. 2007, 103, 440–445. [Google Scholar] [CrossRef]
- Cianferotti, L.; Demay, M.B. VDR-Mediated Inhibition of DKK1 and SFRP2 Suppresses Adipogenic Differentiation of Murine Bone Marrow Stromal Cells. J. Cell. Biochem. 2007, 101, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Boyce, B.F.; Xing, L. Biology of RANK, RANKL, and Osteoprotegerin. Arthritis Res. Ther. 2007, 9, S1. [Google Scholar] [CrossRef]
- Dougall, W.C. Molecular Pathways: Osteoclast-Dependent and Osteoclast-Independent Roles of the RANKL/RANK/OPG Pathway in Tumorigenesis and Metastasis. Clin. Cancer Res. 2012, 18, 326–335. [Google Scholar] [CrossRef]
- Kusumi, A.; Sakaki, H.; Kusumi, T.; Oda, M.; Narita, K.; Nakagawa, H.; Kubota, K.; Satoh, H.; Kimura, H. Regulation of Synthesis of Osteoprotegerin and Soluble Receptor Activator of Nuclear Factor-ΚB Ligand in Normal Human Osteoblasts via the P38 Mitogen-Activated Protein Kinase Pathway by the Application of Cyclic Tensile Strain. J. Bone Miner. Metab. 2005, 23, 373–381. [Google Scholar] [CrossRef]
- Bartel, L.; Mosabbir, A. Possible Mechanisms for the Effects of Sound Vibration on Human Health. Healthcare 2021, 9, 597. [Google Scholar] [CrossRef]
- Narasimhan, B.N.; Ting, M.S.; Kollmetz, T.; Horrocks, M.S.; Chalard, A.E.; Malmström, J. Mechanical Characterization for Cellular Mechanobiology: Current Trends and Future Prospects. Front. Bioeng. Biotechnol. 2020, 8, 595978. [Google Scholar] [CrossRef]
- Nagaraja, M.; Jo, H. The Role of Mechanical Stimulation in Recovery of Bone Loss—High versus Low Magnitude and Frequency of Force. Life 2014, 4, 117–130. [Google Scholar] [CrossRef]
- Hart, N.H.; Nimphius, S.; Rantalainen, T.; Ireland, A.; Siafarikas, A.; Newton, R.U. Mechanical Basis of Bone Strength: Influence of Bone Material, Bone Structure and Muscle Action. J. Musculoskelet. Neuronal Interact. 2017, 17, 114–139. [Google Scholar]
- Hattner, R.; Epker, B.N.; Frost, H.M. Suggested Sequential Mode of Control of Changes in Cell Behaviour in Adult Bone Remodelling. Nature 1965, 206, 489–490. [Google Scholar] [CrossRef]
- Tyrovola, J.B.; Odont, X. The “Mechanostat Theory” of Frost and the OPG/RANKL/RANK System. J. Cell. Biochem. 2015, 116, 2724–2729. [Google Scholar] [CrossRef]
- Uzer, G.; Pongkitwitoon, S.; Ete Chan, M.; Judex, S. Vibration Induced Osteogenic Commitment of Mesenchymal Stem Cells Is Enhanced by Cytoskeletal Remodeling but Not Fluid Shear. J. Biomech. 2013, 46, 2296–2302. [Google Scholar] [CrossRef]
- Kim, I.S.; Song, Y.M.; Lee, B.; Hwang, S.J. Human Mesenchymal Stromal Cells Are Mechanosensitive to Vibration Stimuli. J. Dent. Res. 2012, 91, 1135–1140. [Google Scholar] [CrossRef]
- Tanaka, S.M.; Li, J.; Duncan, R.L.; Yokota, H.; Burr, D.B.; Turner, C.H. Effects of Broad Frequency Vibration on Cultured Osteoblasts. J. Biomech. 2003, 36, 73–80. [Google Scholar] [CrossRef]
- Robling, A.G.; Hinant, F.M.; Burr, D.B.; Turner, C.H. Improved Bone Structure and Strength After Long-Term Mechanical Loading Is Greatest If Loading Is Separated Into Short Bouts. J. Bone Miner. Res. 2002, 17, 1545–1554. [Google Scholar] [CrossRef]
- Bhandari, M.; Mundi, R.; Petis, S.; Kaloty, R.; Shetty, V. Low-Intensity Pulsed Ultrasound: Fracture Healing. Indian J. Orthop. 2009, 43, 132. [Google Scholar] [CrossRef] [PubMed]
- Rubin, C.; Recker, R.; Cullen, D.; Ryaby, J.; McCabe, J.; McLeod, K. Prevention of Postmenopausal Bone Loss by a Low-Magnitude, High-Frequency Mechanical Stimuli: A Clinical Trial Assessing Compliance, Efficacy, and Safety. J. Bone Miner. Res. 2003, 19, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Callewaert, F.; Bakker, A.; Schrooten, J.; Meerbeek, B.V.; Verhoeven, G.; Boonen, S.; Vanderschueren, D. Androgen Receptor Disruption Increases the Osteogenic Response to Mechanical Loading in Male Mice. J. Bone Miner. Res. 2010, 25, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Castillo, A.B.; Triplett, J.W.; Pavalko, F.M.; Turner, C.H. Estrogen Receptor-β Regulates Mechanical Signaling in Primary Osteoblasts. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E937–E944. [Google Scholar] [CrossRef]
- Steppe, L.; Liedert, A.; Ignatius, A.; Haffner-Luntzer, M. Influence of Low-Magnitude High-Frequency Vibration on Bone Cells and Bone Regeneration. Front. Bioeng. Biotechnol. 2020, 8, 595139. [Google Scholar] [CrossRef]
- Haffner-Luntzer, M.; Lackner, I.; Liedert, A.; Fischer, V.; Ignatius, A. Effects of Low-Magnitude High-Frequency Vibration on Osteoblasts Are Dependent on Estrogen Receptor α Signaling and Cytoskeletal Remodeling. Biochem. Biophys. Res. Commun. 2018, 503, 2678–2684. [Google Scholar] [CrossRef]
- Lombardi, G.; Di Somma, C.; Rubino, M.; Faggiano, A.; Vuolo, L.; Guerra, E.; Contaldi, P.; Savastano, S.; Colao, A. The Roles of Parathyroid Hormone in Bone Remodeling: Prospects for Novel Therapeutics. J. Endocrinol. Investig. 2011, 34, 18–22. [Google Scholar]
- Fuchs, R.K.; Warden, S.J. Combination Therapy Using Exercise and Pharmaceutical Agents to Optimize Bone Health. Clin. Rev. Bone Miner. Metab. 2008, 6, 37–45. [Google Scholar] [CrossRef]
- Peters, B.S.E.; Martini, L.A. Nutritional Aspects of the Prevention and Treatment of Osteoporosis. Arq. Bras. Endocrinol. Metabol. 2010, 54, 179–185. [Google Scholar] [CrossRef][Green Version]
- Gaffney-Stomberg, E. The Impact of Trace Minerals on Bone Metabolism. Biol. Trace Elem. Res. 2019, 188, 26–34. [Google Scholar] [CrossRef]
- Zofková, I.; Nemcikova, P.; Matucha, P. Trace Elements and Bone Health. Clin. Chem. Lab. Med. 2013, 51, 1–7. [Google Scholar] [CrossRef]
- Kim, G.S.; Kim, C.-H.; Park, J.Y.; Lee, K.-U.; Park, C.S. Effects of Vitamin B12 on Cell Proliferation and Cellular Alkaline Phosphatase Activity in Human Bone Marrow Stromal Osteoprogenitor Cells and UMR106 Osteoblastic Cells. Metabolism 1996, 45, 1443–1446. [Google Scholar] [CrossRef]
- Yazdanpanah, N.; Zillikens, M.C.; Rivadeneira, F.; de Jong, R.; Lindemans, J.; Uitterlinden, A.G.; Pols, H.A.P.; van Meurs, J.B.J. Effect of Dietary B Vitamins on BMD and Risk of Fracture in Elderly Men and Women: The Rotterdam Study. Bone 2007, 41, 987–994. [Google Scholar] [CrossRef]
- Ahmadieh, H.; Arabi, A. Vitamins and Bone Health: Beyond Calcium and Vitamin D: Nutrition Reviews. Nutr. Rev. 2011, 69, 584–598. [Google Scholar] [CrossRef]
- Rodríguez-Olleros Rodríguez, C.; Díaz Curiel, M. Vitamin K and Bone Health: A Review on the Effects of Vitamin K Deficiency and Supplementation and the Effect of Non-Vitamin K Antagonist Oral Anticoagulants on Different Bone Parameters. J. Osteoporos. 2019, 2019, 1–8. [Google Scholar] [CrossRef]
- Zhu, K.; Beilby, J.; Dick, I.M.; Devine, A.; Soós, M.; Prince, R.L. The Effects of Homocysteine and MTHFR Genotype on Hip Bone Loss and Fracture Risk in Elderly Women. Osteoporos. Int. 2009, 20, 1183–1191. [Google Scholar] [CrossRef] [PubMed]
- Toti, E.; Chen, C.-Y.O.; Palmery, M.; Villaño Valencia, D.; Peluso, I. Non-Provitamin A and Provitamin A Carotenoids as Immunomodulators: Recommended Dietary Allowance, Therapeutic Index, or Personalized Nutrition? Oxid. Med. Cell. Longev. 2018, 2018, 1–20. [Google Scholar] [CrossRef]
- D’Ambrosio, D.N.; Clugston, R.D.; Blaner, W.S. Vitamin A Metabolism: An Update. Nutrients 2011, 3, 63–103. [Google Scholar] [CrossRef] [PubMed]
- Henning, P.; Conaway, H.H.; Lerner, U.H. Retinoid Receptors in Bone and Their Role in Bone Remodeling. Front. Endocrinol. 2015, 6, 31. [Google Scholar] [CrossRef] [PubMed]
- Laue, K.; Pogoda, H.-M.; Daniel, P.B.; van Haeringen, A.; Alanay, Y.; von Ameln, S.; Rachwalski, M.; Morgan, T.; Gray, M.J.; Breuning, M.H.; et al. Craniosynostosis and Multiple Skeletal Anomalies in Humans and Zebrafish Result from a Defect in the Localized Degradation of Retinoic Acid. Am. J. Hum. Genet. 2011, 89, 595–606. [Google Scholar] [CrossRef]
- Weng, Z.; Wang, C.; Zhang, C.; Xu, J.; Chai, Y.; Jia, Y.; Han, P.; Wen, G. All-Trans Retinoic Acid Promotes Osteogenic Differentiation and Bone Consolidation in a Rat Distraction Osteogenesis Model. Calcif. Tissue Int. 2019, 104, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Chaudhary, S.C.; Atigadda, V.R.; Belyaeva, O.V.; Harville, S.R.; Elmets, C.A.; Muccio, D.D.; Athar, M.; Kedishvili, N.Y. Retinoid X Receptor Agonists Upregulate Genes Responsible for the Biosynthesis of All-Trans-Retinoic Acid in Human Epidermis. PLoS ONE 2016, 11, e0153556. [Google Scholar] [CrossRef]
- Jacobson, A.; Johansson, S.; Branting, M.; Melhus, H. Vitamin A Differentially Regulates RANKL and OPG Expression in Human Osteoblasts. Biochem. Biophys. Res. Commun. 2004, 322, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Song, H.M.; Nacamuli, R.P.; Xia, W.; Bari, A.S.; Shi, Y.-Y.; Fang, T.D.; Longaker, M.T. High-Dose Retinoic Acid Modulates Rat Calvarial Osteoblast Biology. J. Cell. Physiol. 2005, 202, 255–262. [Google Scholar] [CrossRef]
- Hisada, K.; Hata, K.; Ichida, F.; Matsubara, T.; Orimo, H.; Nakano, T.; Yatani, H.; Nishimura, R.; Yoneda, T. Retinoic Acid Regulates Commitment of Undifferentiated Mesenchymal Stem Cells into Osteoblasts and Adipocytes. J. Bone Miner. Metab. 2013, 31, 53–63. [Google Scholar] [CrossRef]
- Yee, M.M.F.; Chin, K.-Y.; Ima-Nirwana, S.; Wong, S.K. Vitamin A and Bone Health: A Review on Current Evidence. Molecules 2021, 26, 1757. [Google Scholar] [CrossRef]
- Zhang, L.; Zhou, Q.; Zhang, N.; Li, W.; Ying, M.; Ding, W.-J.; Yang, B.; He, Q. E2F1 Impairs All-Trans Retinoic Acid-Induced Osteogenic Differentiation of Osteosarcoma via Promoting Ubiquitination-Mediated Degradation of RARα. Cell Cycle 2014, 13, 1277–1287. [Google Scholar] [CrossRef]
- Malladi, P.; Xu, Y.; Yang, G.P.; Longaker, M.T. Functions of Vitamin D, Retinoic Acid, and Dexamethasone in Mouse Adipose-Derived Mesenchymal Cells. Tissue Eng. 2006, 12, 2031–2040. [Google Scholar] [CrossRef]
- Wan, D.C.; Shi, Y.-Y.; Nacamuli, R.P.; Quarto, N.; Lyons, K.M.; Longaker, M.T. Osteogenic Differentiation of Mouse Adipose-Derived Adult Stromal Cells Requires Retinoic Acid and Bone Morphogenetic Protein Receptor Type IB Signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 12335–12340. [Google Scholar] [CrossRef] [PubMed]
- Wan, D.C.; Siedhoff, M.T.; Kwan, M.D.; Nacamuli, R.P.; Wu, B.M.; Longaker, M.T. Refining Retinoic Acid Stimulation for Osteogenic Differentiation of Murine Adipose-Derived Adult Stromal Cells. Tissue Eng. 2007, 13, 1623–1631. [Google Scholar] [CrossRef] [PubMed]
- Conaway, H.H.; Pirhayati, A.; Persson, E.; Pettersson, U.; Svensson, O.; Lindholm, C.; Henning, P.; Tuckermann, J.; Lerner, U.H. Retinoids Stimulate Periosteal Bone Resorption by Enhancing the Protein RANKL, a Response Inhibited by Monomeric Glucocorticoid Receptor. J. Biol. Chem. 2011, 286, 31425–31436. [Google Scholar] [CrossRef]
- Iba, K.; Chiba, H.; Yamashita, T.; Ishii, S.; Sawada, N. Phase-Independent Inhibition by Retinoic Acid of Mineralization Correlated with Loss of Tetranectin Expression in a Human Osteoblastic Cell Line. Cell Struct. Funct. 2001, 26, 227–233. [Google Scholar] [CrossRef]
- Kneissel, M.; Studer, A.; Cortesi, R.; Šuša, M. Retinoid-Induced Bone Thinning Is Caused by Subperiosteal Osteoclast Activity in Adult Rodents. Bone 2005, 36, 202–214. [Google Scholar] [CrossRef]
- Lind, T.; Sundqvist, A.; Hu, L.; Pejler, G.; Andersson, G.; Jacobson, A.; Melhus, H. Vitamin A Is a Negative Regulator of Osteoblast Mineralization. PLoS ONE 2013, 8, e82388. [Google Scholar] [CrossRef]
- Wiper-Bergeron, N.; St-Louis, C.; Lee, J.M. CCAAT/Enhancer Binding Protein β Abrogates Retinoic Acid-Induced Osteoblast Differentiation via Repression of Runx2 Transcription. Mol. Endocrinol. 2007, 21, 2124–2135. [Google Scholar] [CrossRef] [PubMed]
- Skillington, J.; Choy, L.; Derynck, R. Bone Morphogenetic Protein and Retinoic Acid Signaling Cooperate to Induce Osteoblast Differentiation of Preadipocytes. J. Cell Biol. 2002, 159, 135–146. [Google Scholar] [CrossRef]
- Mattinzoli, D.; Messa, P.; Corbelli, A.; Ikehata, M.; Zennaro, C.; Armelloni, S.; Li, M.; Giardino, L.; Rastaldi, M.P. A Novel Model of in Vitro Osteocytogenesis Induced by Retinoic Acid Treatment. Eur. Cell. Mater. 2012, 24, 403–425. [Google Scholar] [CrossRef]
- Roa, L.A.; Bloemen, M.; Carels, C.E.L.; Wagener, F.A.D.T.G.; Von den Hoff, J.W. Retinoic Acid Disrupts Osteogenesis in Pre-Osteoblasts by down-Regulating WNT Signaling. Int. J. Biochem. Cell Biol. 2019, 116, 105597. [Google Scholar] [CrossRef]
- Li, X.; Zhang, L.; Yin, X.; Gao, Z.; Zhang, H.; Liu, X.; Pan, X.; Li, N.; Yu, Z. Retinoic Acid Remodels Extracellular Matrix (ECM) of Cultured Human Fetal Palate Mesenchymal Cells (HFPMCs) through down-Regulation of TGF-β/Smad Signaling. Toxicol. Lett. 2014, 225, 208–215. [Google Scholar] [CrossRef]
- Nuka, S.; Sawada, N.; Iba, K.; Chiba, H.; Ishii, S.; Mori, M. All-Trans Retinoic Acid Inhibits Dexamethasone-Induced ALP Activity and Mineralization in Human Osteoblastic Cell Line SV HFO. Cell Struct. Funct. 1997, 22, 27–32. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Maggio, D.; Barabani, M.; Pierandrei, M.; Polidori, M.C.; Catani, M.; Mecocci, P.; Senin, U.; Pacifici, R.; Cherubini, A. Marked Decrease in Plasma Antioxidants in Aged Osteoporotic Women: Results of a Cross-Sectional Study. J. Clin. Endocrinol. Metab. 2003, 88, 1523–1527. [Google Scholar] [CrossRef] [PubMed]
- Barker, M.E.; McCloskey, E.; Saha, S.; Gossiel, F.; Charlesworth, D.; Powers, H.J.; Blumsohn, A. Serum Retinoids and β-Carotene as Predictors of Hip and Other Fractures in Elderly Women. J. Bone Miner. Res. 2005, 20, 913–920. [Google Scholar] [CrossRef]
- de Jonge, E.A.L.; Kiefte-de Jong, J.C.; Campos-Obando, N.; Booij, L.; Franco, O.H.; Hofman, A.; Uitterlinden, A.G.; Rivadeneira, F.; Zillikens, M.C. Dietary Vitamin A Intake and Bone Health in the Elderly: The Rotterdam Study. Eur. J. Clin. Nutr. 2015, 69, 1360–1368. [Google Scholar] [CrossRef] [PubMed]
- Feskanich, D. Vitamin A Intake and Hip Fractures Among Postmenopausal Women. JAMA 2002, 287, 47. [Google Scholar] [CrossRef]
- Lim, L.S.; Harnack, L.J.; Lazovich, D.; Folsom, A.R. Vitamin A Intake and the Risk of Hip Fracture in Postmenopausal Women: The Iowa Women?S Health Study. Osteoporos. Int. 2004, 15, 552–559. [Google Scholar] [CrossRef] [PubMed]
- Wolf, R.L.; Cauley, J.A.; Pettinger, M.; Jackson, R.; Lacroix, A.; Leboff, M.S.; Lewis, C.E.; Nevitt, M.C.; Simon, J.A.; Stone, K.L.; et al. Lack of a Relation between Vitamin and Mineral Antioxidants and Bone Mineral Density: Results from the Women’s Health Initiative. Am. J. Clin. Nutr. 2005, 82, 581–588. [Google Scholar] [CrossRef]
- Penniston, K.L.; Weng, N.; Binkley, N.; Tanumihardjo, S.A. Serum Retinyl Esters Are Not Elevated in Postmenopausal Women with and without Osteoporosis Whose Preformed Vitamin A Intakes Are High. Am. J. Clin. Nutr. 2006, 84, 1350–1356. [Google Scholar] [CrossRef]
- Maggio, D.; Polidori, M.C.; Barabani, M.; Tufi, A.; Ruggiero, C.; Cecchetti, R.; Aisa, M.C.; Stahl, W.; Cherubini, A. Low Levels of Carotenoids and Retinol in Involutional Osteoporosis. Bone 2006, 38, 244–248. [Google Scholar] [CrossRef]
- Navarro-Valverde, C.; Caballero-Villarraso, J.; Mata-Granados, J.M.; Casado-Díaz, A.; Sosa-Henríquez, M.; Malouf-Sierra, J.; Nogués-Solán, X.; Rodríguez-Mañas, L.; Cortés-Gil, X.; Delgadillo-Duarte, J.; et al. High Serum Retinol as a Relevant Contributor to Low Bone Mineral Density in Postmenopausal Osteoporotic Women. Calcif. Tissue Int. 2018, 102, 651–656. [Google Scholar] [CrossRef]
- Pinheiro, M.M.; Ciconelli, R.M.; Chaves, G.V.; Aquino, L.; Juzwiak, C.R.; de Souza Genaro, P.; Ferraz, M.B. Antioxidant Intake among Brazilian Adults—The Brazilian Osteoporosis Study (BRAZOS): A Cross-Sectional Study. Nutr. J. 2011, 10, 39. [Google Scholar] [CrossRef]
- Wu, A.-M.; Huang, C.-Q.; Lin, Z.-K.; Tian, N.-F.; Ni, W.-F.; Wang, X.-Y.; Xu, H.-Z.; Chi, Y.-L. The Relationship Between Vitamin A and Risk of Fracture: Meta-Analysis of Prospective Studies: Vitamin a and fracture risk. J. Bone Miner. Res. 2014, 29, 2032–2039. [Google Scholar] [CrossRef]
- Caire-Juvera, G.; Ritenbaugh, C.; Wactawski-Wende, J.; Snetselaar, L.G.; Chen, Z. Vitamin A and Retinol Intakes and the Risk of Fractures among Participants of the Women’s Health Initiative Observational Study. Am. J. Clin. Nutr. 2009, 89, 323–330. [Google Scholar] [CrossRef]
- Mata-Granados, J.M.; Cuenca-Acevedo, J.R.; de Castro, M.D.L.; Holick, M.F.; Quesada-Gómez, J.M. Vitamin D Insufficiency Together with High Serum Levels of Vitamin A Increases the Risk for Osteoporosis in Postmenopausal Women. Arch. Osteoporos. 2013, 8, 124. [Google Scholar] [CrossRef] [PubMed]
- Mata-Granados, J.M.; Cuenca-Acevedo, R.; Luque de Castro, M.D.; Sosa, M.; Quesada-Gómez, J.M. Vitamin D Deficiency and High Serum Levels of Vitamin A Increase the Risk of Osteoporosis Evaluated by Quantitative Ultrasound Measurements (QUS) in Postmenopausal Spanish Women. Clin. Biochem. 2010, 43, 1064–1068. [Google Scholar] [CrossRef]
- Johansson, S.; Melhus, H. Vitamin A Antagonizes Calcium Response to Vitamin D in Man. J. Bone Miner. Res. 2001, 16, 1899–1905. [Google Scholar] [CrossRef] [PubMed]
- van Driel, M.; Pols, H.; van Leeuwen, J.P. Osteoblast Differentiation and Control by Vitamin D and Vitamin D Metabolites. Curr. Pharm. Des. 2004, 10, 2535–2555. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Glowacki, J.; Kim, S.W.; Hahne, J.; Geng, S.; Mueller, S.M.; Shen, L.; Bleiberg, I.; LeBoff, M.S. Clinical Characteristics Influence in Vitro Action of 1,25-Dihydroxyvitamin D 3 in Human Marrow Stromal Cells. J. Bone Miner. Res. 2012, 27, 1992–2000. [Google Scholar] [CrossRef] [PubMed]
- Noda, M.; Vogel, R.L.; Craig, A.M.; Prahl, J.; DeLuca, H.F.; Denhardt, D.T. Identification of a DNA Sequence Responsible for Binding of the 1,25-Dihydroxyvitamin D3 Receptor and 1,25-Dihydroxyvitamin D3 Enhancement of Mouse Secreted Phosphoprotein 1 (SPP-1 or Osteopontin) Gene Expression. Proc. Natl. Acad. Sci. USA 1990, 87, 9995–9999. [Google Scholar] [CrossRef] [PubMed]
- Paredes, R.; Arriagada, G.; Cruzat, F.; Villagra, A.; Olate, J.; Zaidi, K.; van Wijnen, A.; Lian, J.B.; Stein, G.S.; Stein, J.L.; et al. Bone-Specific Transcription Factor Runx2 Interacts with the 1α,25-Dihydroxyvitamin D3 Receptor To Up-Regulate Rat Osteocalcin Gene Expression in Osteoblastic Cells. Mol. Cell. Biol. 2004, 24, 8847–8861. [Google Scholar] [CrossRef]
- Li, Y.; Xiao, Z. Advances in Runx2 Regulation and Its Isoforms. Med. Hypotheses 2007, 68, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Woeckel, V.J.; Bruedigam, C.; Koedam, M.; Chiba, H.; van der Eerden, B.C.J.; van Leeuwen, J.P.T.M. 1α,25-Dihydroxyvitamin D3 and Rosiglitazone Synergistically Enhance Osteoblast-Mediated Mineralization. Gene 2013, 512, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Kveiborg, M.; Rattan, S.I.; Clark, B.F.; Eriksen, E.F.; Kassem, M. Treatment with 1,25-Dihydroxyvitamin D3 Reduces Impairment of Human Osteoblast Functions during Cellular Aging in Culture. J. Cell. Physiol. 2001, 186, 298–306. [Google Scholar] [CrossRef]
- Lisse, T.S.; Chun, R.F.; Rieger, S.; Adams, J.S.; Hewison, M. Vitamin D Activation of Functionally Distinct Regulatory MiRNAs in Primary Human Osteoblasts: VITAMIN D AND MIRNAS IN OSTEOBLASTS. J. Bone Miner. Res. 2013, 28, 1478–1488. [Google Scholar] [CrossRef] [PubMed]
- Owen, T.A.; Aronow, M.S.; Barone, L.M.; Bettencourt, B.; Stein, G.S.; Lian, J.B. Pleiotropic Effects of Vitamin D on Osteoblast Gene Expression Are Related to the Proliferative and Differentiated State of the Bone Cell Phenotype: Dependency upon Basal Levels of Gene Expression, Duration of Exposure, and Bone Matrix Competency in Normal Rat Osteoblast Cultures. Endocrinology 1991, 128, 1496–1504. [Google Scholar] [CrossRef]
- Bellows, C.G.; Reimers, S.M.; Heersche, J.N.M. Expression of MRNAs for Type-I Collagen, Bone Sialoprotein, Osteocalcin, and Osteopontin at Different Stages of Osteoblastic Differentiation and Their Regulation by 1,25 Dihydroxyvitamin D. Cell Tissue Res. 1999, 297, 249. [Google Scholar] [CrossRef] [PubMed]
- Eelen, G.; Verlinden, L.; Van Camp, M.; Van Hummelen, P.; Marchal, K.; De Moor, B.; Mathieu, C.; Carmeliet, G.; Bouillon, R.; Verstuyf, A. The Effects of 1α,25-Dihydroxyvitamin D3 on the Expression of DNA Replication Genes. J. Bone Miner. Res. 2003, 19, 133–146. [Google Scholar] [CrossRef]
- Tarroni, P.; Villa, I.; Mrak, E.; Zolezzi, F.; Mattioli, M.; Gattuso, C.; Rubinacci, A. Microarray Analysis of 1,25(OH)₂D₃ Regulated Gene Expression in Human Primary Osteoblasts. J. Cell. Biochem. 2012, 113, 640–649. [Google Scholar] [CrossRef] [PubMed]
- Verlinden, L.; Kriebitzsch, C.; Eelen, G.; Van Camp, M.; Leyssens, C.; Tan, B.K.; Beullens, I.; Verstuyf, A. The Odd-Skipped Related Genes Osr1 and Osr2 Are Induced by 1,25-Dihydroxyvitamin D3. J. Steroid Biochem. Mol. Biol. 2013, 136, 94–97. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Goto, M.; Uchiyama, S.; Nakagawa, T. Effect of Zinc on Gene Expression in Osteoblastic MC3T3-E1 Cells: Enhancement of Runx2, OPG, and Regucalcin MRNA Expressions. Mol. Cell. Biochem. 2008, 312, 157–166. [Google Scholar] [CrossRef]
- van Driel, M.; van Leeuwen, J.P.T.M. Vitamin D Endocrine System and Osteoblasts. BoneKEy Rep. 2014, 3, 493. [Google Scholar] [CrossRef] [PubMed]
- Viereck, V.; Siggelkow, H.; Tauber, S.; Raddatz, D.; Schutze, N.; Hüfner, M. Differential Regulation of Cbfa1/Runx2 and Osteocalcin Gene Expression by Vitamin-D3, Dexamethasone, and Local Growth Factors in Primary Human Osteoblasts: Regulation of Cbfa1 During Osteoblast Differentiation. J. Cell. Biochem. 2002, 86, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Maehata, Y.; Takamizawa, S.; Ozawa, S.; Kato, Y.; Sato, S.; Kubota, E.; Hata, R.-I. Both Direct and Collagen-Mediated Signals Are Required for Active Vitamin D3-Elicited Differentiation of Human Osteoblastic Cells: Roles of Osterix, an Osteoblast-Related Transcription Factor. Matrix Biol. 2006, 25, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Thomas, G.P.; Bourne, A.; Eisman, J.A.; Gardiner, E.M. Species-Divergent Regulation of Human and Mouse Osteocalcin Genes by Calciotropic Hormones. Exp. Cell Res. 2000, 258, 395–402. [Google Scholar] [CrossRef]
- Zhang, R.; Ducy, P.; Karsenty, G. 1,25-Dihydroxyvitamin D3 Inhibits Osteocalcin Expression in Mouse through an Indirect Mechanism. J. Biol. Chem. 1997, 272, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Lian, J.B.; Shalhoub, V.; Aslam, F.; Frenkel, B.; Green, J.; Hamrah, M.; Stein, G.S.; Stein, J.L. Species-Specific Glucocorticoid and 1,25-Dihydroxyvitamin D Responsiveness in Mouse MC3T3-E1 Osteoblasts: Dexamethasone Inhibits Osteoblast Differentiation and Vitamin D Down-Regulates Osteocalcin Gene Expression. Endocrinology 1997, 138, 2117–2127. [Google Scholar] [CrossRef] [PubMed]
- Haussler, M.R.; Whitfield, G.K.; Kaneko, I.; Haussler, C.A.; Hsieh, D.; Hsieh, J.-C.; Jurutka, P.W. Molecular Mechanisms of Vitamin D Action. Calcif. Tissue Int. 2013, 92, 77–98. [Google Scholar] [CrossRef]
- Wang, Y.; Zhu, J.; DeLuca, H.F. Identification of the Vitamin D Receptor in Osteoblasts and Chondrocytes But Not Osteoclasts in Mouse Bone: Vdr in osteoblasts & chondrocytes but not osteoclasts in mouse bone. J. Bone Miner. Res. 2014, 29, 685–692. [Google Scholar] [CrossRef]
- Haussler, M.R.; Haussler, C.A.; Whitfield, G.K.; Hsieh, J.-C.; Thompson, P.D.; Barthel, T.K.; Bartik, L.; Egan, J.B.; Wu, Y.; Kubicek, J.L.; et al. The Nuclear Vitamin D Receptor Controls the Expression of Genes Encoding Factors Which Feed the “Fountain of Youth” to Mediate Healthful Aging. J. Steroid Biochem. Mol. Biol. 2010, 121, 88–97. [Google Scholar] [CrossRef]
- Chen, G.; Deng, C.; Li, Y.-P. TGF-β and BMP Signaling in Osteoblast Differentiation and Bone Formation. Int. J. Biol. Sci. 2012, 8, 272–288. [Google Scholar] [CrossRef]
- Weitzmann, M.N. High Dose 1,25(OH)2D3 Inhibits Osteoblast Mineralization in Vitro. Int. J. Mol. Med. 2012, 29, 934–938. [Google Scholar] [CrossRef][Green Version]
- Katzburg, S.; Hendel, D.; Waisman, A.; Posner, G.H.; Kaye, A.M.; Somjen, D. Treatment with Non-Hypercalcemic Analogs of 1,25-Dihydroxyvitamin D3 Increases Responsiveness to 17β-Estradiol, Dihydrotestosterone or Raloxifene in Primary Human Osteoblasts. J. Steroid Biochem. Mol. Biol. 2004, 88, 213–219. [Google Scholar] [CrossRef] [PubMed]
- van der Eerden, B.C.J.; Hoenderop, J.G.J.; de Vries, T.J.; Schoenmaker, T.; Buurman, C.J.; Uitterlinden, A.G.; Pols, H.A.P.; Bindels, R.J.M.; van Leeuwen, J.P.T.M. The Epithelial Ca2+ Channel TRPV5 Is Essential for Proper Osteoclastic Bone Resorption. Proc. Natl. Acad. Sci. USA 2005, 102, 17507–17512. [Google Scholar] [CrossRef]
- Lieben, L.; Masuyama, R.; Torrekens, S.; Van Looveren, R.; Schrooten, J.; Baatsen, P.; Lafage-Proust, M.-H.; Dresselaers, T.; Feng, J.Q.; Bonewald, L.F.; et al. Normocalcemia Is Maintained in Mice under Conditions of Calcium Malabsorption by Vitamin D–Induced Inhibition of Bone Mineralization. J. Clin. Investig. 2012, 122, 1803–1815. [Google Scholar] [CrossRef] [PubMed]
- Lips, P. Vitamin D Deficiency and Secondary Hyperparathyroidism in the Elderly: Consequences for Bone Loss and Fractures and Therapeutic Implications. Endocr. Rev. 2001, 22, 477–501. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Naughton, D.P. Vitamin D in Health and Disease: Current Perspectives. Nutr. J. 2010, 9, 65. [Google Scholar] [CrossRef]
- Weinstein, R.S.; Underwood, J.L.; Hutson, M.S.; DeLuca, H.F. Bone Histomorphometry in Vitamin D-Deficient Rats Infused with Calcium and Phosphorus. Am. J. Physiol.-Endocrinol. Metab. 1984, 246, E499–E505. [Google Scholar] [CrossRef]
- Dardenne, O.; Prud’homme, J.; Hacking, S.A.; Glorieux, F.H.; St-Arnaud, R. Correction of the Abnormal Mineral Ion Homeostasis with a High-Calcium, High-Phosphorus, High-Lactose Diet Rescues the PDDR Phenotype of Mice Deficient for the 25-Hydroxyvitamin D-1α-Hydroxylase (CYP27B1). Bone 2003, 32, 332–340. [Google Scholar] [CrossRef]
- Reid, I.R.; Bolland, M.J.; Grey, A. Effects of Vitamin D Supplements on Bone Mineral Density: A Systematic Review and Meta-Analysis. Lancet 2014, 383, 146–155. [Google Scholar] [CrossRef]
- Balsan, S.; Garabédian, M.; Larchet, M.; Gorski, A.M.; Cournot, G.; Tau, C.; Bourdeau, A.; Silve, C.; Ricour, C. Long-Term Nocturnal Calcium Infusions Can Cure Rickets and Promote Normal Mineralization in Hereditary Resistance to 1,25-Dihydroxyvitamin D. J. Clin. Investig. 1986, 77, 1661–1667. [Google Scholar] [CrossRef]
- Laird, E.; Ward, M.; McSorley, E.; Strain, J.J.; Wallace, J. Vitamin D and Bone Health; Potential Mechanisms. Nutrients 2010, 2, 693–724. [Google Scholar] [CrossRef]
- Bikle, D.D. Vitamin D and Bone. Curr. Osteoporos. Rep. 2012, 10, 151–159. [Google Scholar] [CrossRef]
- Bikle, D.D. Extra Renal Synthesis of 1,25-Dihydroxyvitamin D and Its Health Implications. Clin. Rev. Bone Miner. Metab. 2009, 7, 114–125. [Google Scholar] [CrossRef]
- Morales, O.; Faulds, M.H.; Lindgren, U.J.; Haldosén, L.-A. 1α,25-Dihydroxyvitamin D3 Inhibits GH-Induced Expression of SOCS-3 and CIS and Prolongs Growth Hormone Signaling via the Janus Kinase (JAK2)/Signal Transducers and Activators of Transcription (STAT5) System in Osteoblast-like Cells. J. Biol. Chem. 2002, 277, 34879–34884. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Ono, T.; Tuan, R.S. 1,25-Dihydroxy Vitamin D3 Stimulation of TGF-β Expression in Chick Embryonic Calvarial Bone. Differentiation 1993, 52, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.S.; Yamazaki, K.; Nohtomi, K.; Shizume, K.; Ohsumi, K.; Shibuya, M.; Demura, H.; Sato, K. Increase of Vascular Endothelial Growth Factor MRNA Expression by 1,25-Dihydroxyvitamin D3 in Human Osteoblast-like Cells. J. Bone Miner. Res. 2009, 11, 472–479. [Google Scholar] [CrossRef] [PubMed]
- Lacey, D.L.; Grosso, L.E.; Moser, S.A.; Erdmann, J.; Tan, H.L.; Pacifici, R.; Villareal, D.T. IL-1-Induced Murine Osteoblast IL-6 Production Is Mediated by the Type 1 IL-1 Receptor and Is Increased by 1,25 Dihydroxyvitamin D3. J. Clin. Investig. 1993, 91, 1731–1742. [Google Scholar] [CrossRef]
- Nambi, P.; Wu, H.L.; Lipshutz, D.; Prabhakar, U. Identification and Characterization of Endothelin Receptors on Rat Osteoblastic Osteosarcoma Cells: Down-Regulation by 1,25-Dihydroxy-Vitamin D3. Mol. Pharmacol. 1995, 47, 266–271. [Google Scholar]
- Ross, A.C.; Manson, J.E.; Abrams, S.A.; Aloia, J.F.; Brannon, P.M.; Clinton, S.K.; Durazo-Arvizu, R.A.; Gallagher, J.C.; Gallo, R.L.; Jones, G.; et al. The 2011 Report on Dietary Reference Intakes for Calcium and Vitamin D from the Institute of Medicine: What Clinicians Need to Know. J. Clin. Endocrinol. Metab. 2011, 96, 53–58. [Google Scholar] [CrossRef]
- Gillespie, W.; Avenell, A.; Henry, D.; O’Connell, D.; Robertson, J. Vitamin D and vitamin D analogues for preventing fractures associated with involutional and post-menopausal osteoporosis. In Cochrane Database of Systematic Reviews; The Cochrane Collaboration, Ed.; John Wiley & Sons, Ltd.: Chichester, UK, 2001; p. CD000227. [Google Scholar]
- Trivedi, D.P. Effect of Four Monthly Oral Vitamin D3 (Cholecalciferol) Supplementation on Fractures and Mortality in Men and Women Living in the Community: Randomised Double Blind Controlled Trial. BMJ 2003, 326, 469. [Google Scholar] [CrossRef]
- Heaney, R.P.; Dowell, M.S.; Hale, C.A.; Bendich, A. Calcium Absorption Varies within the Reference Range for Serum 25-Hydroxyvitamin D. J. Am. Coll. Nutr. 2003, 22, 142–146. [Google Scholar] [CrossRef] [PubMed]
- Bischoff-Ferrari, H.A.; Dawson-Hughes, B.; Willett, W.C.; Staehelin, H.B.; Bazemore, M.G.; Zee, R.Y.; Wong, J.B. Effect of Vitamin D on Falls: A Meta-Analysis. JAMA 2004, 291, 1999. [Google Scholar] [CrossRef] [PubMed]
- Shearer, M.J.; Newman, P. Metabolism and Cell Biology of Vitamin K. Thromb. Haemost. 2008, 100, 530–547. [Google Scholar] [CrossRef] [PubMed]
- Fusaro, M. Vitamin K and Bone. Clin. Cases Miner. Bone Metab. 2017, 14, 200. [Google Scholar] [CrossRef] [PubMed]
- Beulens, J.W.J.; Booth, S.L.; van den Heuvel, E.G.H.M.; Stoecklin, E.; Baka, A.; Vermeer, C. The Role of Menaquinones (Vitamin K2) in Human Health. Br. J. Nutr. 2013, 110, 1357–1368. [Google Scholar] [CrossRef] [PubMed]
- Stafford, D.W. The Vitamin K Cycle. J. Thromb. Haemost. 2005, 3, 1873–1878. [Google Scholar] [CrossRef] [PubMed]
- Marles, R.J.; Roe, A.L.; Oketch-Rabah, H.A. US Pharmacopeial Convention Safety Evaluation of Menaquinone-7, a Form of Vitamin K. Nutr. Rev. 2017, 75, 553–578. [Google Scholar] [CrossRef]
- Shearer, M.J. The Roles of Vitamins D and K in Bone Health and Osteoporosis Prevention. Proc. Nutr. Soc. 1997, 56, 915–937. [Google Scholar] [CrossRef]
- Weber, P. Vitamin K and Bone Health. Nutrition 2001, 17, 880–887. [Google Scholar] [CrossRef]
- Vermeer, C.V. Vitamin K: The Effect on Health beyond Coagulation—An Overview. Food Nutr. Res. 2012, 56, 5329. [Google Scholar] [CrossRef]
- Oldenburg, J.; Bevans, C.G.; Müller, C.R.; Watzka, M. Vitamin K Epoxide Reductase Complex Subunit 1 (VKORC1): The Key Protein of the Vitamin K Cycle. Antioxid. Redox Signal. 2006, 8, 347–353. [Google Scholar] [CrossRef]
- Akbari, S.; Rasouli-Ghahroudi, A.A. Vitamin K and Bone Metabolism: A Review of the Latest Evidence in Preclinical Studies. BioMed Res. Int. 2018, 2018, 1–8. [Google Scholar] [CrossRef]
- Booth, S.L.; Tucker, K.L.; Chen, H.; Hannan, M.T.; Gagnon, D.R.; Cupples, L.A.; Wilson, P.W.; Ordovas, J.; Schaefer, E.J.; Dawson-Hughes, B.; et al. Dietary Vitamin K Intakes Are Associated with Hip Fracture but Not with Bone Mineral Density in Elderly Men and Women. Am. J. Clin. Nutr. 2000, 71, 1201–1208. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-M.; Kim, K.-M.; Kim, B.-T.; Joo, N.-S.; Kim, K.-N.; Lee, D.-J. Correlation of Undercarboxylated Osteocalcin (UcOC) Concentration and Bone Density with Age in Healthy Korean Women. J. Korean Med. Sci. 2010, 25, 1171. [Google Scholar] [CrossRef] [PubMed]
- Jaghsi, S.; Hammoud, T.; Haddad, S. Relation Between Circulating Vitamin K1 and Osteoporosis in the Lumbar Spine in Syrian Post-Menopausal Women. Open Rheumatol. J. 2018, 12, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Buitenhuis, H.; Soute, B.; Vermeer, C. Comparison of the Vitamins K1, K2 and K3 as Cofactors for the Hepatic Vitamin K-Dependent Carboxylase. Biochim. Biophys. Acta BBA Gen. Subj. 1990, 1034, 170–175. [Google Scholar] [CrossRef]
- Schurgers, L.J.; Teunissen, K.J.F.; Hamulyák, K.; Knapen, M.H.J.; Vik, H.; Vermeer, C. Vitamin K–Containing Dietary Supplements: Comparison of Synthetic Vitamin K1 and Natto-Derived Menaquinone-7. Blood 2007, 109, 3279–3283. [Google Scholar] [CrossRef]
- Yao, J.; Guihard, P.J.; Blazquez-Medela, A.M.; Guo, Y.; Liu, T.; Boström, K.I.; Yao, Y. Matrix Gla Protein Regulates Differentiation of Endothelial Cells Derived from Mouse Embryonic Stem Cells. Angiogenesis 2016, 19, 1–7. [Google Scholar] [CrossRef]
- Azuma, K.; Ouchi, Y.; Inoue, S. Vitamin K: Novel Molecular Mechanisms of Action and Its Roles in Osteoporosis: Molecular Mechanism of Vitamin K. Geriatr. Gerontol. Int. 2014, 14, 1–7. [Google Scholar] [CrossRef]
- Azuma, K.; Casey, S.C.; Ito, M.; Urano, T.; Horie, K.; Ouchi, Y.; Kirchner, S.; Blumberg, B.; Inoue, S. Pregnane X Receptor Knockout Mice Display Osteopenia with Reduced Bone Formation and Enhanced Bone Resorption. J. Endocrinol. 2010, 207, 257–263. [Google Scholar] [CrossRef]
- Wu, W.-J.; Kim, M.S.; Ahn, B.-Y. The Inhibitory Effect of Vitamin K on RANKL-Induced Osteoclast Differentiation and Bone Resorption. Food Funct. 2015, 6, 3351–3358. [Google Scholar] [CrossRef]
- Myneni, V.; Mezey, E. Immunomodulatory Effect of Vitamin K2: Implications for Bone Health. Oral Dis. 2018, 24, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Katsuyama, H.; Otsuki, T.; Tomita, M.; Fukunaga, M.; Fukunaga, T.; Suzuki, N.; Saijoh, K.; Fushimi, S.; Sunami, S. Menaquinone-7 Regulates the Expressions of Osteocalcin, OPG, RANKL and RANK in Osteoblastic MC3T3E1 Cells. Int. J. Mol. Med. 2005, 15, 231–236. [Google Scholar] [CrossRef]
- Gigante, A.; Brugè, F.; Cecconi, S.; Manzotti, S.; Littarru, G.P.; Tiano, L. Vitamin MK-7 Enhances Vitamin D3-Induced Osteogenesis in HMSCs: Modulation of Key Effectors in Mineralization and Vascularization: Vitamin MK-7 Enhances Vitamin D3 Effects on HMSCs. J. Tissue Eng. Regen. Med. 2015, 9, 691–701. [Google Scholar] [CrossRef] [PubMed]
- Koshihara, Y.; Hoshi, K. Vitamin K2 Enhances Osteocalcin Accumulation in the Extracellular Matrix of Human Osteoblasts In Vitro. J. Bone Miner. Res. 1997, 12, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, J.; Sato, Y.; Takeda, T.; Matsumoto, H. High-Dose Vitamin K Supplementation Reduces Fracture Incidence in Postmenopausal Women: A Review of the Literature. Nutr. Res. 2009, 29, 221–228. [Google Scholar] [CrossRef]
- Apalset, E.M.; Gjesdal, C.G.; Eide, G.E.; Tell, G.S. Intake of Vitamin K1 and K2 and Risk of Hip Fractures: The Hordaland Health Study. Bone 2011, 49, 990–995. [Google Scholar] [CrossRef]
- Hao, G.; Zhang, B.; Gu, M.; Chen, C.; Zhang, Q.; Zhang, G.; Cao, X. Vitamin K Intake and the Risk of Fractures: A Meta-Analysis. Medicine 2017, 96, e6725. [Google Scholar] [CrossRef]
- Nakano, M.; Nagaishi, K.; Konari, N.; Saito, Y.; Chikenji, T.; Mizue, Y.; Fujimiya, M. Bone Marrow-Derived Mesenchymal Stem Cells Improve Diabetes-Induced Cognitive Impairment by Exosome Transfer into Damaged Neurons and Astrocytes. Sci. Rep. 2016, 6, 24805. [Google Scholar] [CrossRef]
- Khaliq, H.; Juming, Z.; Ke-Mei, P. The Physiological Role of Boron on Health. Biol. Trace Elem. Res. 2018, 186, 31–51. [Google Scholar] [CrossRef]
- Hakki, S.S.; Bozkurt, B.S.; Hakki, E.E. Boron Regulates Mineralized Tissue-Associated Proteins in Osteoblasts (MC3T3-E1). J. Trace Elem. Med. Biol. Organ Soc. Miner. Trace Elem. GMS 2010, 24, 243–250. [Google Scholar] [CrossRef]
- Wu, C.; Miron, R.; Sculean, A.; Kaskel, S.; Doert, T.; Schulze, R.; Zhang, Y. Proliferation, Differentiation and Gene Expression of Osteoblasts in Boron-Containing Associated with Dexamethasone Deliver from Mesoporous Bioactive Glass Scaffolds. Biomaterials 2011, 32, 7068–7078. [Google Scholar] [CrossRef]
- Gümüşderelioğlu, M.; Tunçay, E.Ö.; Kaynak, G.; Demirtaş, T.T.; Aydın, S.T.; Hakkı, S.S. Encapsulated Boron as an Osteoinductive Agent for Bone Scaffolds. J. Trace Elem. Med. Biol. Organ Soc. Miner. Trace Elem. GMS 2015, 31, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Lynch, R.J.M.; Duckworth, R.M. Chapter 4: Microelements: Part I: Zn, Sn, Cu, Fe and I. Monogr. Oral Sci. 2020, 28, 32–47. [Google Scholar] [CrossRef] [PubMed]
- Rucker, R.B.; Kosonen, T.; Clegg, M.S.; Mitchell, A.E.; Rucker, B.R.; Uriu-Hare, J.Y.; Keen, C.L. Copper, Lysyl Oxidase, and Extracellular Matrix Protein Cross-Linking. Am. J. Clin. Nutr. 1998, 67, 996S–1002S. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, J.P.; Ríos, S.; González, M. Modulation of the Proliferation and Differentiation of Human Mesenchymal Stem Cells by Copper. J. Cell. Biochem. 2002, 85, 92–100. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, W.; Yao, Q. Copper-Based Biomaterials for Bone and Cartilage Tissue Engineering. J. Orthop. Transl. 2021, 29, 60–71. [Google Scholar] [CrossRef]
- Balogh, E.; Paragh, G.; Jeney, V. Influence of Iron on Bone Homeostasis. Pharmaceuticals 2018, 11, 107. [Google Scholar] [CrossRef]
- Everett, E.T. Fluoride’s Effects on the Formation of Teeth and Bones, and the Influence of Genetics. J. Dent. Res. 2011, 90, 552–560. [Google Scholar] [CrossRef]
- Vestergaard, P.; Jorgensen, N.R.; Schwarz, P.; Mosekilde, L. Effects of Treatment with Fluoride on Bone Mineral Density and Fracture Risk--a Meta-Analysis. Osteoporos. Int. J. Establ. Result Coop. Eur. Found. Osteoporos. Natl. Osteoporos. Found. USA 2008, 19, 257–268. [Google Scholar] [CrossRef]
- Pan, L.; Shi, X.; Liu, S.; Guo, X.; Zhao, M.; Cai, R.; Sun, G. Fluoride Promotes Osteoblastic Differentiation through Canonical Wnt/β-Catenin Signaling Pathway. Toxicol. Lett. 2014, 225, 34–42. [Google Scholar] [CrossRef]
- Ciosek, Ż.; Kot, K.; Kosik-Bogacka, D.; Łanocha-Arendarczyk, N.; Rotter, I. The Effects of Calcium, Magnesium, Phosphorus, Fluoride, and Lead on Bone Tissue. Biomolecules 2021, 11, 506. [Google Scholar] [CrossRef]
- Bodnar, M.; Szczyglowska, M.; Konieczka, P.; Namiesnik, J. Methods of Selenium Supplementation: Bioavailability and Determination of Selenium Compounds. Crit. Rev. Food Sci. Nutr. 2016, 56, 36–55. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, J.; Xiao, J. Selenoproteins and Selenium Status in Bone Physiology and Pathology. Biochim. Biophys. Acta 2014, 1840, 3246–3256. [Google Scholar] [CrossRef]
- Vescini, F.; Chiodini, I.; Palermo, A.; Cesareo, R.; De Geronimo, V.; Scillitani, A.; Gennari, L.; Falchetti, A. Selenium: A Trace Element for a Healthy Skeleton—A Narrative Review. Endocr. Metab. Immune Disord. Drug Targets 2021, 21, 577–585. [Google Scholar] [CrossRef]
- Shahid, M.; Shamshad, S.; Rafiq, M.; Khalid, S.; Bibi, I.; Niazi, N.K.; Dumat, C.; Rashid, M.I. Chromium Speciation, Bioavailability, Uptake, Toxicity and Detoxification in Soil-Plant System: A Review. Chemosphere 2017, 178, 513–533. [Google Scholar] [CrossRef]
- Rodríguez, J.; Mandalunis, P.M. A Review of Metal Exposure and Its Effects on Bone Health. J. Toxicol. 2018, 2018, 4854152. [Google Scholar] [CrossRef]
- Zijlstra, W.P.; Bulstra, S.K.; van Raay, J.J.A.M.; van Leeuwen, B.M.; Kuijer, R. Cobalt and Chromium Ions Reduce Human Osteoblast-like Cell Activity in Vitro, Reduce the OPG to RANKL Ratio, and Induce Oxidative Stress. J. Orthop. Res. Off. Publ. Orthop. Res. Soc. 2012, 30, 740–747. [Google Scholar] [CrossRef]
- Simonsen, L.O.; Harbak, H.; Bennekou, P. Cobalt Metabolism and Toxicology—A Brief Update. Sci. Total Environ. 2012, 432, 210–215. [Google Scholar] [CrossRef]
- Czarnek, K.; Terpiłowska, S.; Siwicki, A.K. Selected Aspects of the Action of Cobalt Ions in the Human Body. Cent. Eur. J. Immunol. 2015, 40, 236–242. [Google Scholar] [CrossRef]
- Chen, X.; Zhu, G.; Gu, S.; Jin, T.; Shao, C. Effects of Cadmium on Osteoblasts and Osteoclasts in Vitro. Environ. Toxicol. Pharmacol. 2009, 28, 232–236. [Google Scholar] [CrossRef]
- Kazantzis, G. Cadmium, Osteoporosis and Calcium Metabolism. Biometals Int. J. Role Met. Ions Biol. Biochem. Med. 2004, 17, 493–498. [Google Scholar] [CrossRef]
- O’Connor, J.P.; Kanjilal, D.; Teitelbaum, M.; Lin, S.S.; Cottrell, J.A. Zinc as a Therapeutic Agent in Bone Regeneration. Materials 2020, 13, 2211. [Google Scholar] [CrossRef]
- Bhattacharya, P.T.; Misra, S.R.; Hussain, M. Nutritional Aspects of Essential Trace Elements in Oral Health and Disease: An Extensive Review. Scientifica 2016, 2016, 1–12. [Google Scholar] [CrossRef]
- King, J.C.; Shames, D.M.; Woodhouse, L.R. Zinc Homeostasis in Humans. J. Nutr. 2000, 130, 1360S–1366S. [Google Scholar] [CrossRef]
- Vallee, B.L.; Falchuk, K.H. The Biochemical Basis of Zinc Physiology. Physiol. Rev. 1993, 73, 79–118. [Google Scholar] [CrossRef]
- Jackson, M.J.; Jones, D.A.; Edwards, R.H.T. Tissue Zinc Levels as an Index of Body Zinc Status. Clin. Physiol. 1982, 2, 333–343. [Google Scholar] [CrossRef]
- Haumont, S. Distribution of zinc in bone tissue. J. Histochem. Cytochem. 1961, 9, 141–145. [Google Scholar] [CrossRef]
- Huang, T.; Yan, G.; Guan, M. Zinc Homeostasis in Bone: Zinc Transporters and Bone Diseases. Int. J. Mol. Sci. 2020, 21, 1236. [Google Scholar] [CrossRef]
- Meshkini, A. A Correlation Between Intracellular Zinc Content and Osteosarcoma. Biol. Trace Elem. Res. 2021, 199, 3222–3231. [Google Scholar] [CrossRef]
- Fukada, T.; Hojyo, S.; Furuichi, T. Zinc Signal: A New Player in Osteobiology. J. Bone Miner. Metab. 2013, 31, 129–135. [Google Scholar] [CrossRef]
- Klug, A. The Discovery of Zinc Fingers and Their Applications in Gene Regulation and Genome Manipulation. Annu. Rev. Biochem. 2010, 79, 213–231. [Google Scholar] [CrossRef]
- Jensen, E.D.; Gopalakrishnan, R.; Westendorf, J.J. Regulation of Gene Expression in Osteoblasts. BioFactors 2010, 36, 25–32. [Google Scholar] [CrossRef]
- Kambe, T.; Tsuji, T.; Hashimoto, A.; Itsumura, N. The Physiological, Biochemical, and Molecular Roles of Zinc Transporters in Zinc Homeostasis and Metabolism. Physiol. Rev. 2015, 95, 749–784. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Kambe, T. The Functions of Metallothionein and ZIP and ZnT Transporters: An Overview and Perspective. Int. J. Mol. Sci. 2016, 17, 336. [Google Scholar] [CrossRef]
- Levaot, N.; Hershfinkel, M. How Cellular Zn2+ Signaling Drives Physiological Functions. Cell Calcium 2018, 75, 53–63. [Google Scholar] [CrossRef]
- Fu, X.; Li, Y.; Huang, T.; Yu, Z.; Ma, K.; Yang, M.; Liu, Q.; Pan, H.; Wang, H.; Wang, J.; et al. Runx2/Osterix and Zinc Uptake Synergize to Orchestrate Osteogenic Differentiation and Citrate Containing Bone Apatite Formation. Adv. Sci. 2018, 5, 1700755. [Google Scholar] [CrossRef]
- Tang, Z.; Sahu, S.N.; Khadeer, M.A.; Bai, G.; Franklin, R.B.; Gupta, A. Overexpression of the ZIP1 Zinc Transporter Induces an Osteogenic Phenotype in Mesenchymal Stem Cells. Bone 2006, 38, 181–198. [Google Scholar] [CrossRef]
- Khadeer, M.A.; Sahu, S.N.; Bai, G.; Abdulla, S.; Gupta, A. Expression of the Zinc Transporter ZIP1 in Osteoclasts. Bone 2005, 37, 296–304. [Google Scholar] [CrossRef]
- Aydemir, T.B.; Cousins, R.J. The Multiple Faces of the Metal Transporter ZIP14 (SLC39A14). J. Nutr. 2018, 148, 174–184. [Google Scholar] [CrossRef]
- Kronenberg, H.M. Developmental Regulation of the Growth Plate. Nature 2003, 423, 332–336. [Google Scholar] [CrossRef]
- Sasaki, S.; Tsukamoto, M.; Saito, M.; Hojyo, S.; Fukada, T.; Takami, M.; Furuichi, T. Disruption of the Mouse Slc39a14 Gene Encoding Zinc Transporter ZIP 14 Is Associated with Decreased Bone Mass, Likely Caused by Enhanced Bone Resorption. FEBS Open Bio 2018, 8, 655–663. [Google Scholar] [CrossRef]
- Long, F.; Schipani, E.; Asahara, H.; Kronenberg, H.; Montminy, M. The CREB Family of Activators Is Required for Endochondral Bone Development. Dev. Camb. Engl. 2001, 128, 541–550. [Google Scholar]
- Hendrickx, G.; Borra, V.M.; Steenackers, E.; Yorgan, T.A.; Hermans, C.; Boudin, E.; Waterval, J.J.; Jansen, I.D.C.; Aydemir, T.B.; Kamerling, N.; et al. Conditional Mouse Models Support the Role of SLC39A14 (ZIP14) in Hyperostosis Cranialis Interna and in Bone Homeostasis. PLoS Genet. 2018, 14, e1007321. [Google Scholar] [CrossRef]
- Kim, J.-H.; Jeon, J.; Shin, M.; Won, Y.; Lee, M.; Kwak, J.-S.; Lee, G.; Rhee, J.; Ryu, J.-H.; Chun, C.-H.; et al. Regulation of the Catabolic Cascade in Osteoarthritis by the Zinc-ZIP8-MTF1 Axis. Cell 2014, 156, 730–743. [Google Scholar] [CrossRef]
- Bin, B.-H.; Fukada, T.; Hosaka, T.; Yamasaki, S.; Ohashi, W.; Hojyo, S.; Miyai, T.; Nishida, K.; Yokoyama, S.; Hirano, T. Biochemical Characterization of Human ZIP13 Protein: A Homo-Dimerized Zinc Transporter Involved in the Spondylocheiro Dysplastic Ehlers-Danlos Syndrome. J. Biol. Chem. 2011, 286, 40255–40265. [Google Scholar] [CrossRef]
- Hata, A.; Chen, Y.-G. TGF-β Signaling from Receptors to Smads. Cold Spring Harb. Perspect. Biol. 2016, 8, a022061. [Google Scholar] [CrossRef]
- Zhang, Y.E. Non-Smad Pathways in TGF-β Signaling. Cell Res. 2009, 19, 128–139. [Google Scholar] [CrossRef]
- Giunta, C.; Elçioglu, N.H.; Albrecht, B.; Eich, G.; Chambaz, C.; Janecke, A.R.; Yeowell, H.; Weis, M.; Eyre, D.R.; Kraenzlin, M.; et al. Spondylocheiro Dysplastic Form of the Ehlers-Danlos Syndrome—An Autosomal-Recessive Entity Caused by Mutations in the Zinc Transporter Gene SLC39A13. Am. J. Hum. Genet. 2008, 82, 1290–1305. [Google Scholar] [CrossRef]
- Inoue, K.; Matsuda, K.; Itoh, M.; Kawaguchi, H.; Tomoike, H.; Aoyagi, T.; Nagai, R.; Hori, M.; Nakamura, Y.; Tanaka, T. Osteopenia and Male-Specific Sudden Cardiac Death in Mice Lacking a Zinc Transporter Gene, Znt5. Hum. Mol. Genet. 2002, 11, 1775–1784. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.; Xiang, L.; Yang, M.; Zhang, X.; Guo, B.; Chen, Y.; Yang, L.; Cao, J. ZnT7 Can Protect MC3T3-E1 Cells from Oxidative Stress-Induced Apoptosis via PI3K/Akt and MAPK/ERK Signaling Pathways. Cell. Signal. 2013, 25, 1126–1135. [Google Scholar] [CrossRef] [PubMed]
- Seo, H.-J.; Cho, Y.-E.; Kim, T.; Shin, H.-I.; Kwun, I.-S. Zinc May Increase Bone Formation through Stimulating Cell Proliferation, Alkaline Phosphatase Activity and Collagen Synthesis in Osteoblastic MC3T3-E1 Cells. Nutr. Res. Pract. 2010, 4, 356. [Google Scholar] [CrossRef] [PubMed]
- Kwun, I.-S.; Cho, Y.-E.; Lomeda, R.-A.R.; Shin, H.-I.; Choi, J.-Y.; Kang, Y.-H.; Beattie, J.H. Zinc Deficiency Suppresses Matrix Mineralization and Retards Osteogenesis Transiently with Catch-up Possibly through Runx 2 Modulation. Bone 2010, 46, 732–741. [Google Scholar] [CrossRef]
- Komori, T. Regulation of Osteoblast Differentiation by Transcription Factors. J. Cell. Biochem. 2006, 99, 1233–1239. [Google Scholar] [CrossRef] [PubMed]
- Pajarinen, J.; Lin, T.; Gibon, E.; Kohno, Y.; Maruyama, M.; Nathan, K.; Lu, L.; Yao, Z.; Goodman, S.B. Mesenchymal Stem Cell-Macrophage Crosstalk and Bone Healing. Biomaterials 2019, 196, 80–89. [Google Scholar] [CrossRef]
- Alcantara, E.H.; Lomeda, R.-A.R.; Feldmann, J.; Nixon, G.F.; Beattie, J.H.; Kwun, I.-S. Zinc Deprivation Inhibits Extracellular Matrix Calcification through Decreased Synthesis of Matrix Proteins in Osteoblasts. Mol. Nutr. Food Res. 2011, 55, 1552–1560. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Yang, M.; Liang, D.; Yang, L.; Cao, J.; Zhang, L. Cell Apoptosis Induced by Zinc Deficiency in Osteoblastic MC3T3-E1 Cells via a Mitochondrial-Mediated Pathway. Mol. Cell. Biochem. 2012, 361, 209–216. [Google Scholar] [CrossRef]
- Park, K.H.; Choi, Y.; Yoon, D.S.; Lee, K.-M.; Kim, D.; Lee, J.W. Zinc Promotes Osteoblast Differentiation in Human Mesenchymal Stem Cells Via Activation of the CAMP-PKA-CREB Signaling Pathway. Stem Cells Dev. 2018, 27, 1125–1135. [Google Scholar] [CrossRef]
- Cerovic, A.; Miletic, I.; Sobajic, S.; Blagojevic, D.; Radusinovic, M.; El-Sohemy, A. Effects of Zinc on the Mineralization of Bone Nodules from Human Osteoblast-like Cells. Biol. Trace Elem. Res. 2007, 116, 61–71. [Google Scholar] [CrossRef]
- Kanno, S.; Anuradha, C.D.; Hirano, S. Chemotactic Responses of Osteoblastic MC3T3-E1 Cells Toward Zinc Chloride. Biol. Trace Elem. Res. 2001, 83, 49–55. [Google Scholar] [CrossRef]
- Srivastava, S.; Kumar, N.; Thakur, R.S.; Roy, P. Role of Vanadium (V) in the Differentiation of C3H10t1/2 Cells Towards Osteoblast Lineage: A Comparative Analysis with Other Trace Elements. Biol. Trace Elem. Res. 2013, 152, 135–142. [Google Scholar] [CrossRef]
- Ovesen, J.; Møller-Madsen, B.; Nielsen, P.T.; Christensen, P.H.; Simonsen, O.; Hoeck, H.C.; Laursen, M.B.; Thomsen, J.S. Differences in Zinc Status between Patients with Osteoarthritis and Osteoporosis. J. Trace Elem. Med. Biol. 2009, 23, 1–8. [Google Scholar] [CrossRef]
- Prasad, A.S. Discovery of Human Zinc Deficiency: Its Impact on Human Health and Disease. Adv. Nutr. 2013, 4, 176–190. [Google Scholar] [CrossRef]
- Hyun, T.H.; Barrett-Connor, E.; Milne, D.B. Zinc Intakes and Plasma Concentrations in Men with Osteoporosis: The Rancho Bernardo Study. Am. J. Clin. Nutr. 2004, 80, 715–721. [Google Scholar] [CrossRef]
- Elmståhl, S.; Gullberg, B.; Janzon, L.; Johnell, O.; Elmståhl, B. Increased Incidence of Fractures in Middle-Aged and Elderly Men with Low Intakes of Phosphorus and Zinc. Osteoporos. Int. 1998, 8, 333–340. [Google Scholar] [CrossRef]
- Strause, L.; Saltman, P.; Smith, K.T.; Bracker, M.; Andon, M.B. Spinal Bone Loss in Postmenopausal Women Supplemented with Calcium and Trace Minerals. J. Nutr. 1994, 124, 1060–1064. [Google Scholar] [CrossRef]
- Houschyar, K.S.; Tapking, C.; Borrelli, M.R.; Popp, D.; Duscher, D.; Maan, Z.N.; Chelliah, M.P.; Li, J.; Harati, K.; Wallner, C.; et al. Wnt Pathway in Bone Repair and Regeneration—What Do We Know So Far. Front. Cell Dev. Biol. 2019, 6, 170. [Google Scholar] [CrossRef]
- Amin, N.; Clark, C.C.T.; Taghizadeh, M.; Djafarnejad, S. Zinc Supplements and Bone Health: The Role of the RANKL-RANK Axis as a Therapeutic Target. J. Trace Elem. Med. Biol. 2020, 57, 126417. [Google Scholar] [CrossRef]
- Hie, M.; Iitsuka, N.; Otsuka, T.; Nakanishi, A.; Tsukamoto, I. Zinc Deficiency Decreases Osteoblasts and Osteoclasts Associated with the Reduced Expression of Runx2 and RANK. Bone 2011, 49, 1152–1159. [Google Scholar] [CrossRef]
- Hadley, K.B.; Newman, S.M.; Hunt, J.R. Dietary Zinc Reduces Osteoclast Resorption Activities and Increases Markers of Osteoblast Differentiation, Matrix Maturation, and Mineralization in the Long Bones of Growing Rats. J. Nutr. Biochem. 2010, 21, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Hosea, H.J.; Taylor, C.G.; Wood, T.; Mollard, R.; Weiler, H.A. Zinc-Deficient Rats Have More Limited Bone Recovery During Repletion Than Diet-Restricted Rats. Exp. Biol. Med. 2004, 229, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.-E.; Lomeda, R.-A.R.; Ryu, S.-H.; Sohn, H.-Y.; Shin, H.-I.; Beattie, J.H.; Kwun, I.-S. Zinc Deficiency Negatively Affects Alkaline Phosphatase and the Concentration of Ca, Mg and P in Rats. Nutr. Res. Pract. 2007, 1, 113. [Google Scholar] [CrossRef] [PubMed]
- Trzeciakiewicz, A.; Habauzit, V.; Mercier, S.; Lebecque, P.; Davicco, M.-J.; Coxam, V.; Demigne, C.; Horcajada, M.-N. Hesperetin Stimulates Differentiation of Primary Rat Osteoblasts Involving the BMP Signalling Pathway. J. Nutr. Biochem. 2010, 21, 424–431. [Google Scholar] [CrossRef]
- Yamashita, M.; Otsuka, F.; Mukai, T.; Yamanaka, R.; Otani, H.; Matsumoto, Y.; Nakamura, E.; Takano, M.; Sada, K.; Makino, H. Simvastatin Inhibits Osteoclast Differentiation Induced by Bone Morphogenetic Protein-2 and RANKL through Regulating MAPK, AKT and Src Signaling. Regul. Pept. 2010, 162, 99–108. [Google Scholar] [CrossRef]
- Tsuji, M.; Yamamoto, H.; Sato, T.; Mizuha, Y.; Kawai, Y.; Taketani, Y.; Kato, S.; Terao, J.; Inakuma, T.; Takeda, E. Dietary Quercetin Inhibits Bone Loss without Effect on the Uterus in Ovariectomized Mice. J. Bone Miner. Metab. 2009, 27, 673–681. [Google Scholar] [CrossRef]
- Liang, W.; Luo, Z.; Ge, S.; Li, M.; Du, J.; Yang, M.; Yan, M.; Ye, Z.; Luo, Z. Oral Administration of Quercetin Inhibits Bone Loss in Rat Model of Diabetic Osteopenia. Eur. J. Pharmacol. 2011, 670, 317–324. [Google Scholar] [CrossRef]
- Robaszkiewicz, A.; Balcerczyk, A.; Bartosz, G. Antioxidative and Prooxidative Effects of Quercetin on A549 Cells. Cell Biol. Int. 2007, 31, 1245–1250. [Google Scholar] [CrossRef]
- Lo, Y.-C.; Chang, Y.-H.; Wei, B.-L.; Huang, Y.-L.; Chiou, W.-F. Betulinic Acid Stimulates the Differentiation and Mineralization of Osteoblastic MC3T3-E1 Cells: Involvement of BMP/Runx2 and β-Catenin Signals. J. Agric. Food Chem. 2010, 58, 6643–6649. [Google Scholar] [CrossRef]
- Lee, Y.S.; Choi, E.M. Apocynin Stimulates Osteoblast Differentiation and Inhibits Bone-Resorbing Mediators in MC3T3-E1 Cells. Cell. Immunol. 2011, 270, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Chang, E.-J.; Kim, H.-M.; Lee, S.B.; Kim, H.-D.; Su Kim, G.; Kim, H.-H. Antioxidant α-Lipoic Acid Inhibits Osteoclast Differentiation by Reducing Nuclear Factor-ΚB DNA Binding and Prevents in Vivo Bone Resorption Induced by Receptor Activator of Nuclear Factor-ΚB Ligand and Tumor Necrosis Factor-α. Free Radic. Biol. Med. 2006, 40, 1483–1493. [Google Scholar] [CrossRef]
- Jun, J.H.; Lee, S.-H.; Kwak, H.B.; Lee, Z.H.; Seo, S.-B.; Woo, K.M.; Ryoo, H.-M.; Kim, G.-S.; Baek, J.-H. N-Acetylcysteine Stimulates Osteoblastic Differentiation of Mouse Calvarial Cells. J. Cell. Biochem. 2008, 103, 1246–1255. [Google Scholar] [CrossRef]
- Kwak, H.B.; Lee, B.K.; Oh, J.; Yeon, J.-T.; Choi, S.-W.; Cho, H.J.; Lee, M.S.; Kim, J.-J.; Bae, J.-M.; Kim, S.H.; et al. Inhibition of Osteoclast Differentiation and Bone Resorption by Rotenone, through down-Regulation of RANKL-Induced c-Fos and NFATc1 Expression. Bone 2010, 46, 724–731. [Google Scholar] [CrossRef] [PubMed]
- Park, E.-J.; Pezzuto, J.M. The Pharmacology of Resveratrol in Animals and Humans. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2015, 1852, 1071–1113. [Google Scholar] [CrossRef] [PubMed]
- Cottart, C.-H.; Nivet-Antoine, V.; Beaudeux, J.-L. Review of Recent Data on the Metabolism, Biological Effects, and Toxicity of Resveratrol in Humans. Mol. Nutr. Food Res. 2014, 58, 7–21. [Google Scholar] [CrossRef]
- Tou, J.C. Resveratrol Supplementation Affects Bone Acquisition and Osteoporosis: Pre-Clinical Evidence toward Translational Diet Therapy. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2015, 1852, 1186–1194. [Google Scholar] [CrossRef] [PubMed]
- Boissy, P.; Andersen, T.L.; Abdallah, B.M.; Kassem, M.; Plesner, T.; Delaissé, J.-M. Resveratrol Inhibits Myeloma Cell Growth, Prevents Osteoclast Formation, and Promotes Osteoblast Differentiation. Cancer Res. 2005, 65, 9943–9952. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, K.; Ikeda, K.; Kawai, Y.; Yamori, Y. Resveratrol Stimulates the Proliferation and Differentiation of Osteoblastic MC3T3-E1 Cells. Biochem. Biophys. Res. Commun. 1998, 253, 859–863. [Google Scholar] [CrossRef]
- Dai, Z.; Li, Y.; Quarles, L.D.; Song, T.; Pan, W.; Zhou, H.; Xiao, Z. Resveratrol Enhances Proliferation and Osteoblastic Differentiation in Human Mesenchymal Stem Cells via ER-Dependent ERK1/2 Activation. Phytomedicine 2007, 14, 806–814. [Google Scholar] [CrossRef]
- Shakibaei, M.; Buhrmann, C.; Mobasheri, A. Resveratrol-Mediated SIRT-1 Interactions with P300 Modulate Receptor Activator of NF-ΚB Ligand (RANKL) Activation of NF-ΚB Signaling and Inhibit Osteoclastogenesis in Bone-Derived Cells. J. Biol. Chem. 2011, 286, 11492–11505. [Google Scholar] [CrossRef]
- Kupisiewicz, K.; Boissy, P.; Abdallah, B.M.; Hansen, F.D.; Erben, R.G.; Savouret, J.-F.; Søe, K.; Andersen, T.L.; Plesner, T.; Delaisse, J.-M. Potential of Resveratrol Analogues as Antagonists of Osteoclasts and Promoters of Osteoblasts. Calcif. Tissue Int. 2010, 87, 437–449. [Google Scholar] [CrossRef]
- Mobasheri, A.; Shakibaei, M. Osteogenic Effects of Resveratrol in Vitro: Potential for the Prevention and Treatment of Osteoporosis: Osteogenic Effects of Resveratrol. Ann. N. Y. Acad. Sci. 2013, 1290, 59–66. [Google Scholar] [CrossRef]
- Wang, X.; Chen, L.; Peng, W. Protective Effects of Resveratrol on Osteoporosis via Activation of the SIRT1-NF-κB Signaling Pathway in Rats. Exp. Ther. Med. 2017, 14, 5032–5038. [Google Scholar] [CrossRef]
- Liu, Z.P.; Li, W.X.; Yu, B.; Huang, J.; Sun, J.; Huo, J.S.; Liu, C.X. Effects of Trans -Resveratrol from Polygonum Cuspidatum on Bone Loss Using the Ovariectomized Rat Model. J. Med. Food 2005, 8, 14–19. [Google Scholar] [CrossRef]
- Zhao, H.; Li, X.; Li, N.; Liu, T.; Liu, J.; Li, Z.; Xiao, H.; Li, J. Long-Term Resveratrol Treatment Prevents Ovariectomy-Induced Osteopenia in Rats without Hyperplastic Effects on the Uterus. Br. J. Nutr. 2014, 111, 836–846. [Google Scholar] [CrossRef]
- Momken, I.; Stevens, L.; Bergouignan, A.; Desplanches, D.; Rudwill, F.; Chery, I.; Zahariev, A.; Zahn, S.; Stein, T.P.; Sebedio, J.L.; et al. Resveratrol Prevents the Wasting Disorders of Mechanical Unloading by Acting as a Physical Exercise Mimetic in the Rat. FASEB J. 2011, 25, 3646–3660. [Google Scholar] [CrossRef]
- Durbin, S.M.; Jackson, J.R.; Ryan, M.J.; Gigliotti, J.C.; Alway, S.E.; Tou, J.C. Resveratrol Supplementation Preserves Long Bone Mass, Microstructure, and Strength in Hindlimb-Suspended Old Male Rats. J. Bone Miner. Metab. 2014, 32, 38–47. [Google Scholar] [CrossRef]
- Habold, C.; Momken, I.; Ouadi, A.; Bekaert, V.; Brasse, D. Effect of Prior Treatment with Resveratrol on Density and Structure of Rat Long Bones under Tail-Suspension. J. Bone Miner. Metab. 2011, 29, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Pearson, K.J.; Baur, J.A.; Lewis, K.N.; Peshkin, L.; Price, N.L.; Labinskyy, N.; Swindell, W.R.; Kamara, D.; Minor, R.K.; Perez, E.; et al. Resveratrol Delays Age-Related Deterioration and Mimics Transcriptional Aspects of Dietary Restriction without Extending Life Span. Cell Metab. 2008, 8, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Jiang, T.; Wang, Y.; Guo, L. The Role and Mechanism of SIRT1 in Resveratrol-Regulated Osteoblast Autophagy in Osteoporosis Rats. Sci. Rep. 2019, 9, 18424. [Google Scholar] [CrossRef]
- Tennen, R.I.; Michishita-Kioi, E.; Chua, K.F. Finding a Target for Resveratrol. Cell 2012, 148, 387–389. [Google Scholar] [CrossRef] [PubMed]
- Du, J.-K.; Cong, B.-H.; Yu, Q.; Wang, H.; Wang, L.; Wang, C.-N.; Tang, X.-L.; Lu, J.-Q.; Zhu, X.-Y.; Ni, X. Upregulation of MicroRNA-22 Contributes to Myocardial Ischemia-Reperfusion Injury by Interfering with the Mitochondrial Function. Free Radic. Biol. Med. 2016, 96, 406–417. [Google Scholar] [CrossRef]
- Lagouge, M.; Argmann, C.; Gerhart-Hines, Z.; Meziane, H.; Lerin, C.; Daussin, F.; Messadeq, N.; Milne, J.; Lambert, P.; Elliott, P.; et al. Resveratrol Improves Mitochondrial Function and Protects against Metabolic Disease by Activating SIRT1 and PGC-1α. Cell 2006, 127, 1109–1122. [Google Scholar] [CrossRef] [PubMed]
- Bellet, M.M.; Nakahata, Y.; Boudjelal, M.; Watts, E.; Mossakowska, D.E.; Edwards, K.A.; Cervantes, M.; Astarita, G.; Loh, C.; Ellis, J.L.; et al. Pharmacological Modulation of Circadian Rhythms by Synthetic Activators of the Deacetylase SIRT1. Proc. Natl. Acad. Sci. USA 2013, 110, 3333–3338. [Google Scholar] [CrossRef]
- Cheng, H.-L.; Mostoslavsky, R.; Saito, S.; Manis, J.P.; Gu, Y.; Patel, P.; Bronson, R.; Appella, E.; Alt, F.W.; Chua, K.F. Developmental Defects and P53 Hyperacetylation in Sir2 Homolog (SIRT1)-Deficient Mice. Proc. Natl. Acad. Sci. USA 2003, 100, 10794–10799. [Google Scholar] [CrossRef]
- Salminen, A.; Kauppinen, A.; Suuronen, T.; Kaarniranta, K. SIRT1 Longevity Factor Suppresses NF-ΚB -Driven Immune Responses: Regulation of Aging via NF-ΚB Acetylation? BioEssays 2008, 30, 939–942. [Google Scholar] [CrossRef]
- Zainabadi, K.; Liu, C.J.; Caldwell, A.L.M.; Guarente, L. SIRT1 Is a Positive Regulator of in Vivo Bone Mass and a Therapeutic Target for Osteoporosis. PLoS ONE 2017, 12, e0185236. [Google Scholar] [CrossRef] [PubMed]
- Shakibaei, M.; Shayan, P.; Busch, F.; Aldinger, C.; Buhrmann, C.; Lueders, C.; Mobasheri, A. Resveratrol Mediated Modulation of Sirt-1/Runx2 Promotes Osteogenic Differentiation of Mesenchymal Stem Cells: Potential Role of Runx2 Deacetylation. PLoS ONE 2012, 7, e35712. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Wang, Z.; Zhao, J.; Miao, W.; Ye, T.; Chen, A. Resveratrol Attenuates Lipopolysaccharides (LPS)-Induced Inhibition of Osteoblast Differentiation in MC3T3-E1 Cells. Med. Sci. Monit. 2018, 24, 2045–2052. [Google Scholar] [CrossRef]
- Zhao, M.; Ko, S.-Y.; Garrett, I.R.; Mundy, G.R.; Gutierrez, G.E.; Edwards, J.R. The Polyphenol Resveratrol Promotes Skeletal Growth in Mice through a Sirtuin 1-Bone Morphogenic Protein 2 Longevity Axis: Red Wine Products and Bone Health. Br. J. Pharmacol. 2018, 175, 4183–4192. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, L.-M.; Guo, C.; Han, J.-F. Resveratrol Promotes Osteoblastic Differentiation in a Rat Model of Postmenopausal Osteoporosis by Regulating Autophagy. Nutr. Metab. 2020, 17, 29. [Google Scholar] [CrossRef]
- Ornstrup, M.J.; Harsløf, T.; Kjær, T.N.; Langdahl, B.L.; Pedersen, S.B. Resveratrol Increases Bone Mineral Density and Bone Alkaline Phosphatase in Obese Men: A Randomized Placebo-Controlled Trial. J. Clin. Endocrinol. Metab. 2014, 99, 4720–4729. [Google Scholar] [CrossRef]
- Bo, S.; Gambino, R.; Ponzo, V.; Cioffi, I.; Goitre, I.; Evangelista, A.; Ciccone, G.; Cassader, M.; Procopio, M. Effects of Resveratrol on Bone Health in Type 2 Diabetic Patients. A Double-Blind Randomized-Controlled Trial. Nutr. Diabetes 2018, 8, 51. [Google Scholar] [CrossRef]
- Wong, R.H.; Thaung Zaw, J.J.; Xian, C.J.; Howe, P.R. Regular Supplementation With Resveratrol Improves Bone Mineral Density in Postmenopausal Women: A Randomized, Placebo-Controlled Trial. J. Bone Miner. Res. 2020, 35, 2121–2131. [Google Scholar] [CrossRef]
- Williams, L.D.; Burdock, G.A.; Edwards, J.A.; Beck, M.; Bausch, J. Safety Studies Conducted on High-Purity Trans-Resveratrol in Experimental Animals. Food Chem. Toxicol. 2009, 47, 2170–2182. [Google Scholar] [CrossRef]
- Groneberg, D.A.; Kindermann, B.; Althammer, M.; Klapper, M.; Vormann, J.; Littarru, G.P.; Döring, F. Coenzyme Q10 Affects Expression of Genes Involved in Cell Signalling, Metabolism and Transport in Human CaCo-2 Cells. Int. J. Biochem. Cell Biol. 2005, 37, 1208–1218. [Google Scholar] [CrossRef]
- Linnane, A.W.; Kios, M.; Vitetta, L. Coenzyme Q10—Its Role as a Prooxidant in the Formation of Superoxide Anion/Hydrogen Peroxide and the Regulation of the Metabolome. Mitochondrion 2007, 7, S51–S61. [Google Scholar] [CrossRef]
- Littarru, G.P.; Tiano, L. Bioenergetic and Antioxidant Properties of Coenzyme Q10: Recent Developments. Mol. Biotechnol. 2007, 37, 31–37. [Google Scholar] [CrossRef]
- Garrido-Maraver, J. Clinical Applications of Coenzyme Q10. Front. Biosci. 2014, 19, 619. [Google Scholar] [CrossRef]
- Acosta, M.J.; Vazquez Fonseca, L.; Desbats, M.A.; Cerqua, C.; Zordan, R.; Trevisson, E.; Salviati, L. Coenzyme Q Biosynthesis in Health and Disease. Biochim. Biophys. Acta BBA Bioenerg. 2016, 1857, 1079–1085. [Google Scholar] [CrossRef] [PubMed]
- Di Pierro, D.; Ciaccio, C.; Sbardella, D.; Tundo, G.R.; Bernardini, R.; Curatolo, P.; Galasso, C.; Pironi, V.; Coletta, M.; Marini, S. Effects of Oral Administration of Common Antioxidant Supplements on the Energy Metabolism of Red Blood Cells. Attenuation of Oxidative Stress-Induced Changes in Rett Syndrome Erythrocytes by CoQ10. Mol. Cell. Biochem. 2020, 463, 101–113. [Google Scholar] [CrossRef]
- Moon, H.-J.; Ko, W.-K.; Han, S.W.; Kim, D.-S.; Hwang, Y.-S.; Park, H.-K.; Kwon, I.K. Antioxidants, like Coenzyme Q10, Selenite, and Curcumin, Inhibited Osteoclast Differentiation by Suppressing Reactive Oxygen Species Generation. Biochem. Biophys. Res. Commun. 2012, 418, 247–253. [Google Scholar] [CrossRef]
- Moon, H.-J.; Ko, W.-K.; Jung, M.-S.; Kim, J.H.; Lee, W.-J.; Park, K.-S.; Heo, J.-K.; Bang, J.B.; Kwon, I.K. Coenzyme Q10 Regulates Osteoclast and Osteoblast Differentiation: Bone Effects of Coenzyme Q10. J. Food Sci. 2013, 78, H785–H791. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Cui, C.; Yu, M.; Li, X.; Wang, L.; Chen, X.; Lin, Y. Coenzyme Q10 Promotes Osteoblast Proliferation and Differentiation and Protects against Ovariectomy-Induced Osteoporosis. Mol. Med. Rep. 2017, 17, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Van Niel, G.; Raposo, G.; Candalh, C.; Boussac, M.; Hershberg, R.; Cerf–Bensussan, N.; Heyman, M. Intestinal Epithelial Cells Secrete Exosome–like Vesicles. Gastroenterology 2001, 121, 337–349. [Google Scholar] [CrossRef]
- Stahl, P.D.; Raposo, G. Extracellular Vesicles: Exosomes and Microvesicles, Integrators of Homeostasis. Physiology 2019, 34, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-Mediated Transfer of MRNAs and MicroRNAs Is a Novel Mechanism of Genetic Exchange between Cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef]
- Théry, C.; Ostrowski, M.; Segura, E. Membrane Vesicles as Conveyors of Immune Responses. Nat. Rev. Immunol. 2009, 9, 581–593. [Google Scholar] [CrossRef]
- Kowal, J.; Tkach, M.; Théry, C. Biogenesis and Secretion of Exosomes. Curr. Opin. Cell Biol. 2014, 29, 116–125. [Google Scholar] [CrossRef]
- Janas, T.; Janas, M.M.; Sapoń, K.; Janas, T. Mechanisms of RNA Loading into Exosomes. FEBS Lett. 2015, 589, 1391–1398. [Google Scholar] [CrossRef] [PubMed]
- Ge, M.; Wu, Y.; Ke, R.; Cai, T.; Yang, J.; Mu, X. Value of Osteoblast-Derived Exosomes in Bone Diseases. J. Craniofac. Surg. 2017, 28, 866–870. [Google Scholar] [CrossRef]
- Simons, M.; Raposo, G. Exosomes—Vesicular Carriers for Intercellular Communication. Curr. Opin. Cell Biol. 2009, 21, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; LeBleu, V.S. The Biology, Function and Biomedical Applications of Exosomes. Science 2020, 367, eaau6977. [Google Scholar] [CrossRef] [PubMed]
- Haraszti, R.A.; Didiot, M.-C.; Sapp, E.; Leszyk, J.; Shaffer, S.A.; Rockwell, H.E.; Gao, F.; Narain, N.R.; DiFiglia, M.; Kiebish, M.A.; et al. High-Resolution Proteomic and Lipidomic Analysis of Exosomes and Microvesicles from Different Cell Sources. J. Extracell. Vesicles 2016, 5, 32570. [Google Scholar] [CrossRef]
- Choudhry, H.; Harris, A.L. Advances in Hypoxia-Inducible Factor Biology. Cell Metab. 2018, 27, 281–298. [Google Scholar] [CrossRef]
- Yang, J.-X.; Xie, P.; Li, Y.-S.; Wen, T.; Yang, X.-C. Osteoclast-Derived MiR-23a-5p-Containing Exosomes Inhibit Osteogenic Differentiation by Regulating Runx2. Cell. Signal. 2020, 70, 109504. [Google Scholar] [CrossRef] [PubMed]
- Ambattu, L.A.; Ramesan, S.; Dekiwadia, C.; Hanssen, E.; Li, H.; Yeo, L.Y. High Frequency Acoustic Cell Stimulation Promotes Exosome Generation Regulated by a Calcium-Dependent Mechanism. Commun. Biol. 2020, 3, 553. [Google Scholar] [CrossRef]
- Zhao, L.; Jiang, S.; Hantash, B.M. Transforming Growth Factor Β1 Induces Osteogenic Differentiation of Murine Bone Marrow Stromal Cells. Tissue Eng. Part A 2010, 16, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Gao, W.; Papadimitriou, J.M.; Zhang, C.; Gao, J.; Zheng, M. Exosomes—the Enigmatic Regulators of Bone Homeostasis. Bone Res. 2018, 6, 36. [Google Scholar] [CrossRef]
- Cui, Y.; Luan, J.; Li, H.; Zhou, X.; Han, J. Exosomes Derived from Mineralizing Osteoblasts Promote ST2 Cell Osteogenic Differentiation by Alteration of MicroRNA Expression. FEBS Lett. 2016, 590, 185–192. [Google Scholar] [CrossRef]
- Zhang, J.; Tu, Q.; Bonewald, L.F.; He, X.; Stein, G.; Lian, J.; Chen, J. Effects of MiR-335-5p in Modulating Osteogenic Differentiation by Specifically Downregulating Wnt Antagonist DKK1. J. Bone Miner. Res. 2011, 26, 1953–1963. [Google Scholar] [CrossRef]
- You, L.; Gu, W.; Chen, L.; Pan, L.; Chen, J.; Peng, Y. MiR-378 Overexpression Attenuates High Glucose-Suppressed Osteogenic Differentiation through Targeting CASP3 and Activating PI3K/Akt Signaling Pathway. Int. J. Clin. Exp. Pathol. 2014, 7, 7249–7261. [Google Scholar]
- Wei, Y.; Tang, C.; Zhang, J.; Li, Z.; Zhang, X.; Miron, R.J.; Zhang, Y. Extracellular Vesicles Derived from the Mid-to-Late Stage of Osteoblast Differentiation Markedly Enhance Osteogenesis in Vitro and in Vivo. Biochem. Biophys. Res. Commun. 2019, 514, 252–258. [Google Scholar] [CrossRef]
- Deng, L.; Wang, Y.; Peng, Y.; Wu, Y.; Ding, Y.; Jiang, Y.; Shen, Z.; Fu, Q. Osteoblast-Derived Microvesicles: A Novel Mechanism for Communication between Osteoblasts and Osteoclasts. Bone 2015, 79, 37–42. [Google Scholar] [CrossRef]
- Solberg, L.B.; Stang, E.; Brorson, S.-H.; Andersson, G.; Reinholt, F.P. Tartrate-Resistant Acid Phosphatase (TRAP) Co-Localizes with Receptor Activator of NF-KB Ligand (RANKL) and Osteoprotegerin (OPG) in Lysosomal-Associated Membrane Protein 1 (LAMP1)-Positive Vesicles in Rat Osteoblasts and Osteocytes. Histochem. Cell Biol. 2015, 143, 195–207. [Google Scholar] [CrossRef]
- Xu, J.-F.; Yang, G.; Pan, X.-H.; Zhang, S.-J.; Zhao, C.; Qiu, B.-S.; Gu, H.-F.; Hong, J.-F.; Cao, L.; Chen, Y.; et al. Altered MicroRNA Expression Profile in Exosomes during Osteogenic Differentiation of Human Bone Marrow-Derived Mesenchymal Stem Cells. PLoS ONE 2014, 9, e114627. [Google Scholar] [CrossRef]
- Cappariello, A.; Loftus, A.; Muraca, M.; Maurizi, A.; Rucci, N.; Teti, A. Osteoblast-Derived Extracellular Vesicles Are Biological Tools for the Delivery of Active Molecules to Bone: Osteoblast-derived extracellular vesicles and bone. J. Bone Miner. Res. 2018, 33, 517–533. [Google Scholar] [CrossRef]
- Di Zazzo, E.; De Rosa, C.; Abbondanza, C.; Moncharmont, B. PRDM Proteins: Molecular Mechanisms in Signal Transduction and Transcriptional Regulation. Biology 2013, 2, 107–141. [Google Scholar] [CrossRef] [PubMed]
- Minamizaki, T.; Nakao, Y.; Irie, Y.; Ahmed, F.; Itoh, S.; Sarmin, N.; Yoshioka, H.; Nobukiyo, A.; Fujimoto, C.; Niida, S.; et al. The Matrix Vesicle Cargo MiR-125b Accumulates in the Bone Matrix, Inhibiting Bone Resorption in Mice. Commun. Biol. 2020, 3, 30. [Google Scholar] [CrossRef]
- Huynh, N.; VonMoss, L.; Smith, D.; Rahman, I.; Felemban, M.F.; Zuo, J.; Rody, W.J.; McHugh, K.P.; Holliday, L.S. Characterization of Regulatory Extracellular Vesicles from Osteoclasts. J. Dent. Res. 2016, 95, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Kaslan, M.; Lee, S.H.; Yao, J.; Gao, Z. Progress in Exosome Isolation Techniques. Theranostics 2017, 7, 789–804. [Google Scholar] [CrossRef] [PubMed]
- Zeringer, E.; Barta, T.; Li, M.; Vlassov, A.V. Strategies for Isolation of Exosomes. Cold Spring Harb. Protoc. 2015, 2015, pdb.top074476. [Google Scholar] [CrossRef] [PubMed]
- Baranyai, T.; Herczeg, K.; Onódi, Z.; Voszka, I.; Módos, K.; Marton, N.; Nagy, G.; Mäger, I.; Wood, M.J.; El Andaloussi, S.; et al. Isolation of Exosomes from Blood Plasma: Qualitative and Quantitative Comparison of Ultracentrifugation and Size Exclusion Chromatography Methods. PLoS ONE 2015, 10, e0145686. [Google Scholar] [CrossRef] [PubMed]
- Momen-Heravi, F.; Balaj, L.; Alian, S.; Mantel, P.-Y.; Halleck, A.E.; Trachtenberg, A.J.; Soria, C.E.; Oquin, S.; Bonebreak, C.M.; Saracoglu, E.; et al. Current Methods for the Isolation of Extracellular Vesicles. Biol. Chem. 2013, 394, 1253–1262. [Google Scholar] [CrossRef] [PubMed]
- Furuta, T.; Miyaki, S.; Ishitobi, H.; Ogura, T.; Kato, Y.; Kamei, N.; Miyado, K.; Higashi, Y.; Ochi, M. Mesenchymal Stem Cell-Derived Exosomes Promote Fracture Healing in a Mouse Model: MSC Exosomes Promote Fracture Healing. Stem Cells Transl. Med. 2016, 5, 1620–1630. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hao, Z.; Wang, P.; Xia, Y.; Wu, J.; Xia, D.; Fang, S.; Xu, S. Exosomes from Human Umbilical Cord Mesenchymal Stem Cells Enhance Fracture Healing through HIF-1α-mediated Promotion of Angiogenesis in a Rat Model of Stabilized Fracture. Cell Prolif. 2019, 52, e12570. [Google Scholar] [CrossRef]
- Chen, S.; Tang, Y.; Liu, Y.; Zhang, P.; Lv, L.; Zhang, X.; Jia, L.; Zhou, Y. Exosomes Derived from MiR-375-overexpressing Human Adipose Mesenchymal Stem Cells Promote Bone Regeneration. Cell Prolif. 2019, 52, e12669. [Google Scholar] [CrossRef]
- Hu, Y.; Xu, R.; Chen, C.-Y.; Rao, S.-S.; Xia, K.; Huang, J.; Yin, H.; Wang, Z.-X.; Cao, J.; Liu, Z.-Z.; et al. Extracellular Vesicles from Human Umbilical Cord Blood Ameliorate Bone Loss in Senile Osteoporotic Mice. Metabolism 2019, 95, 93–101. [Google Scholar] [CrossRef]
- Luo, Z.-W.; Li, F.-X.-Z.; Liu, Y.-W.; Rao, S.-S.; Yin, H.; Huang, J.; Chen, C.-Y.; Hu, Y.; Zhang, Y.; Tan, Y.-J.; et al. Aptamer-Functionalized Exosomes from Bone Marrow Stromal Cells Target Bone to Promote Bone Regeneration. Nanoscale 2019, 11, 20884–20892. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, X.; Li, H.; Chen, C.; Hu, B.; Niu, X.; Li, Q.; Zhao, B.; Xie, Z.; Wang, Y. Exosomes/Tricalcium Phosphate Combination Scaffolds Can Enhance Bone Regeneration by Activating the PI3K/Akt Signaling Pathway. Stem Cell Res. Ther. 2016, 7, 136. [Google Scholar] [CrossRef]
- Deng, L.; Peng, Y.; Jiang, Y.; Wu, Y.; Ding, Y.; Wang, Y.; Xu, D.; Fu, Q. Imipramine Protects against Bone Loss by Inhibition of Osteoblast-Derived Microvesicles. Int. J. Mol. Sci. 2017, 18, 1013. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, J.D.; Dufresne, E.R.; Schwartz, M.A. Mechanotransduction and Extracellular Matrix Homeostasis. Nat. Rev. Mol. Cell Biol. 2014, 15, 802–812. [Google Scholar] [CrossRef] [PubMed]
- Spyropoulou, A.; Karamesinis, K.; Basdra, E.K. Mechanotransduction Pathways in Bone Pathobiology. Biochim. Biophys. Acta 2015, 1852, 1700–1708. [Google Scholar] [CrossRef] [PubMed]










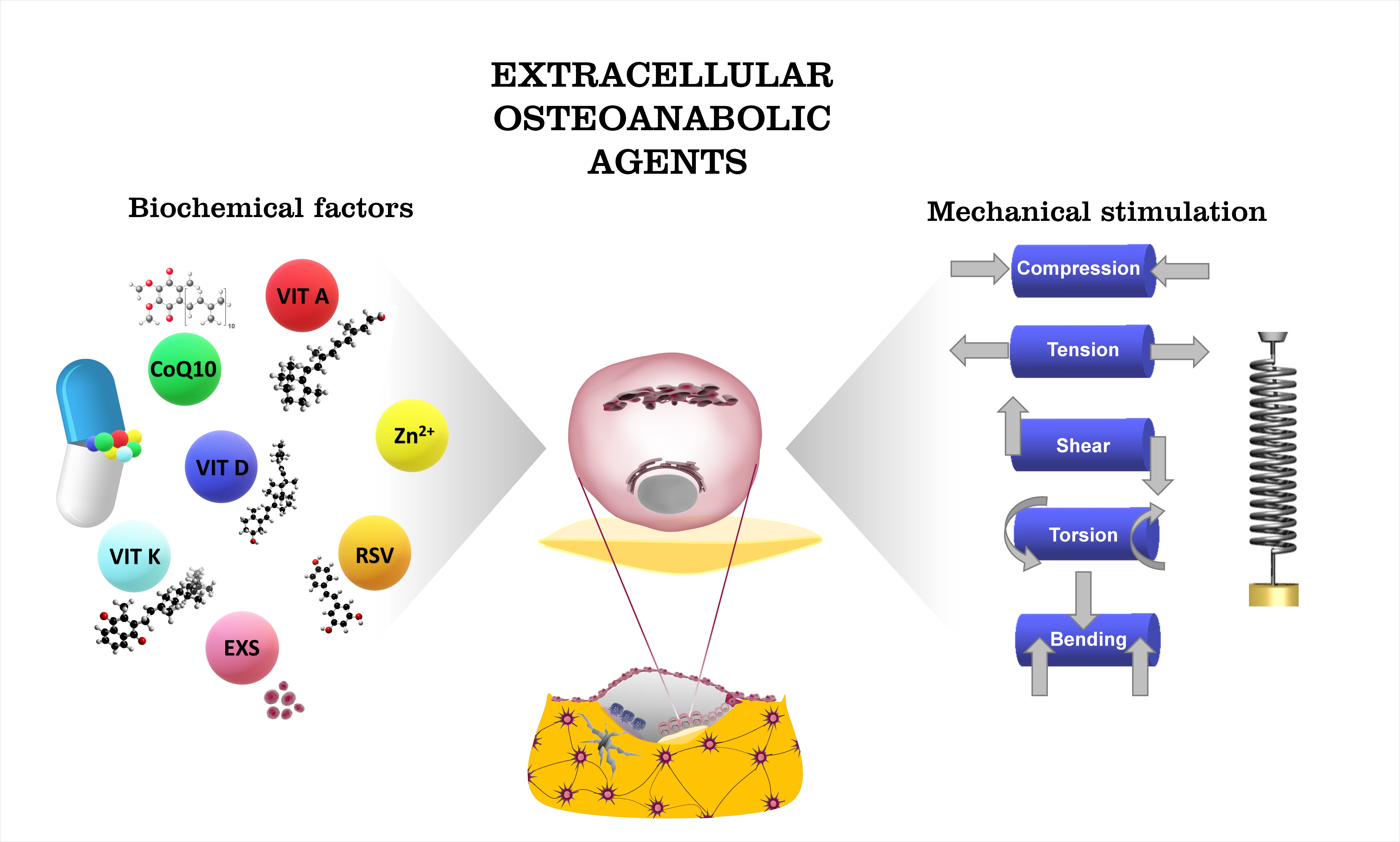{kind=link}
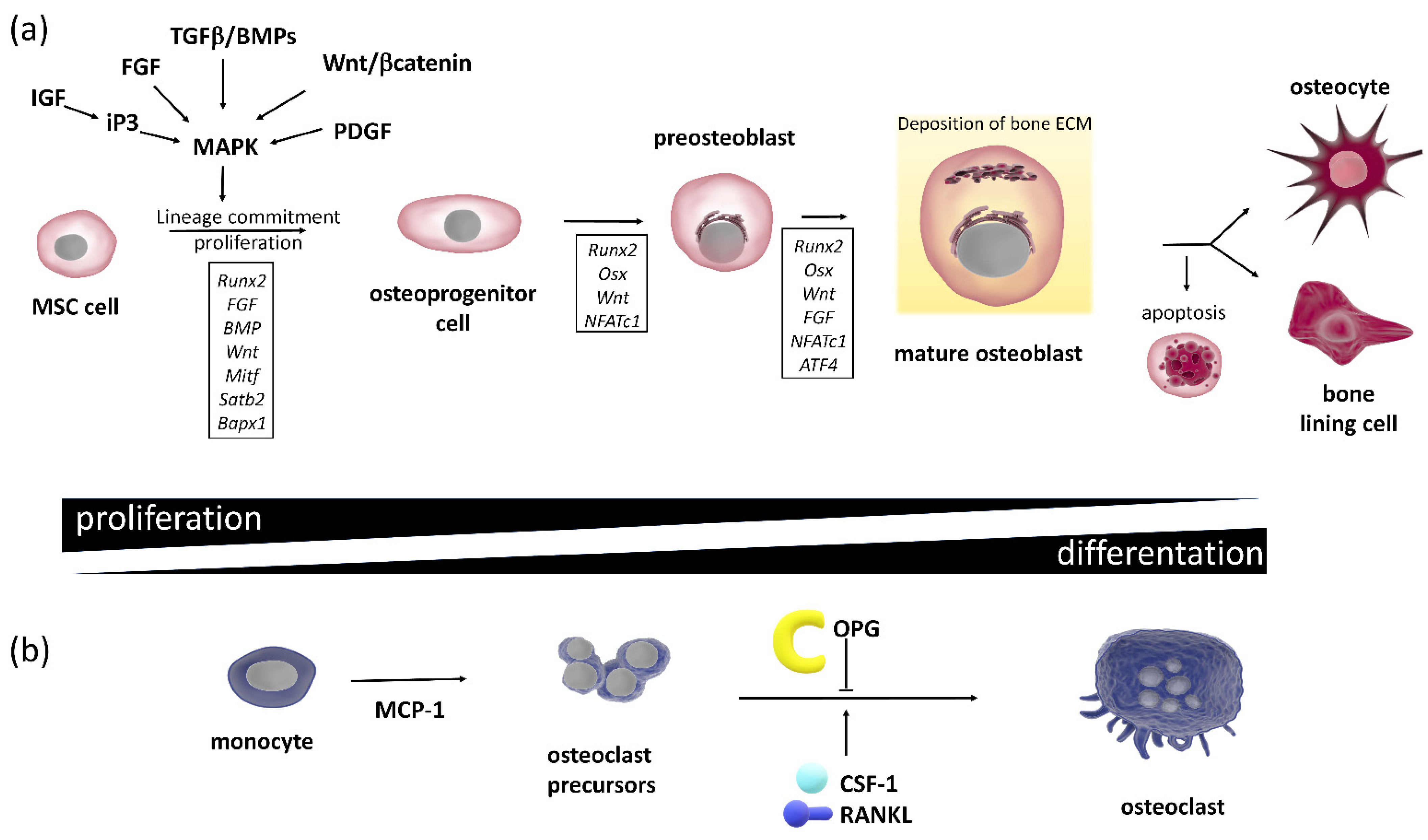{kind=link}
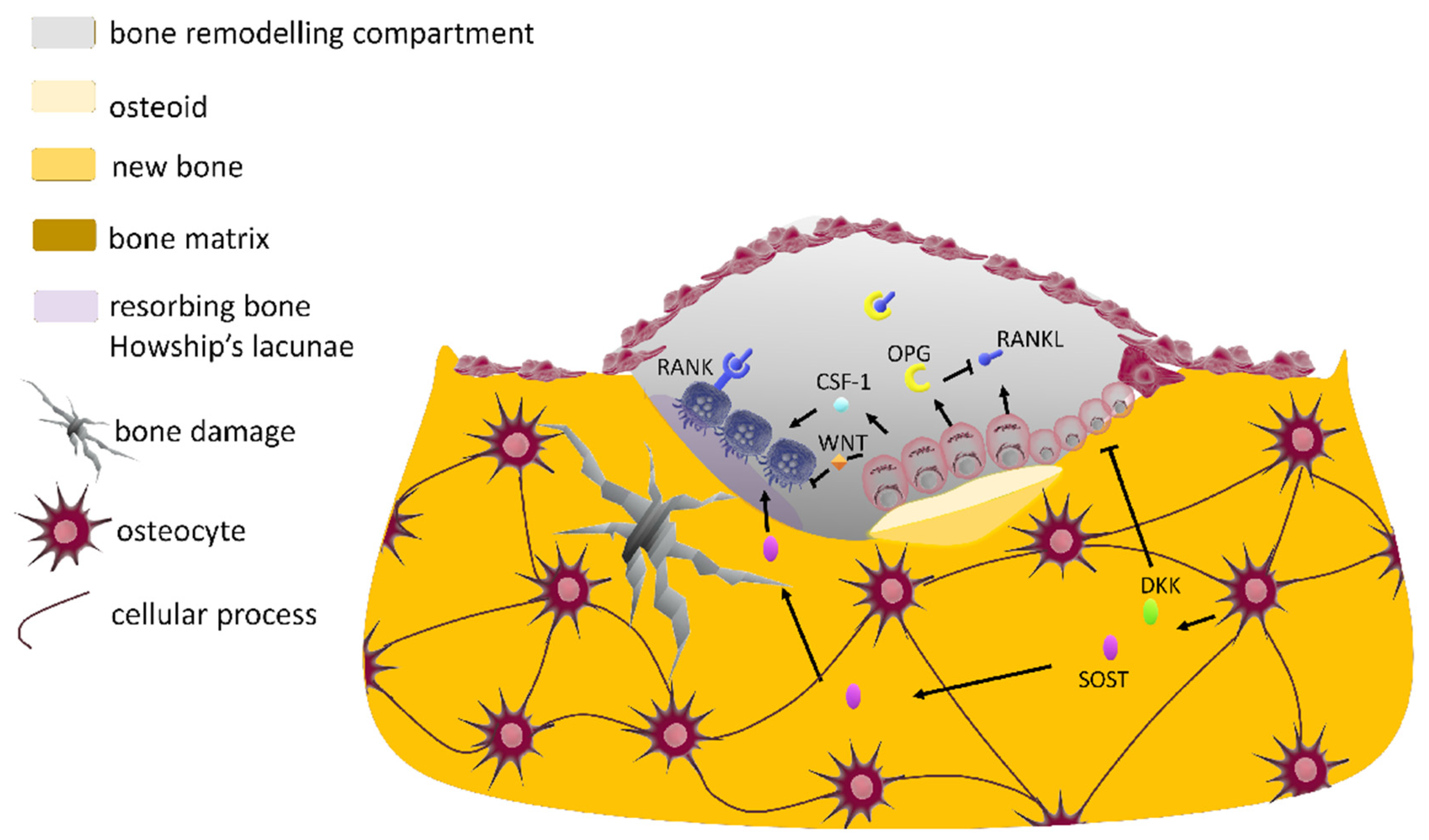{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
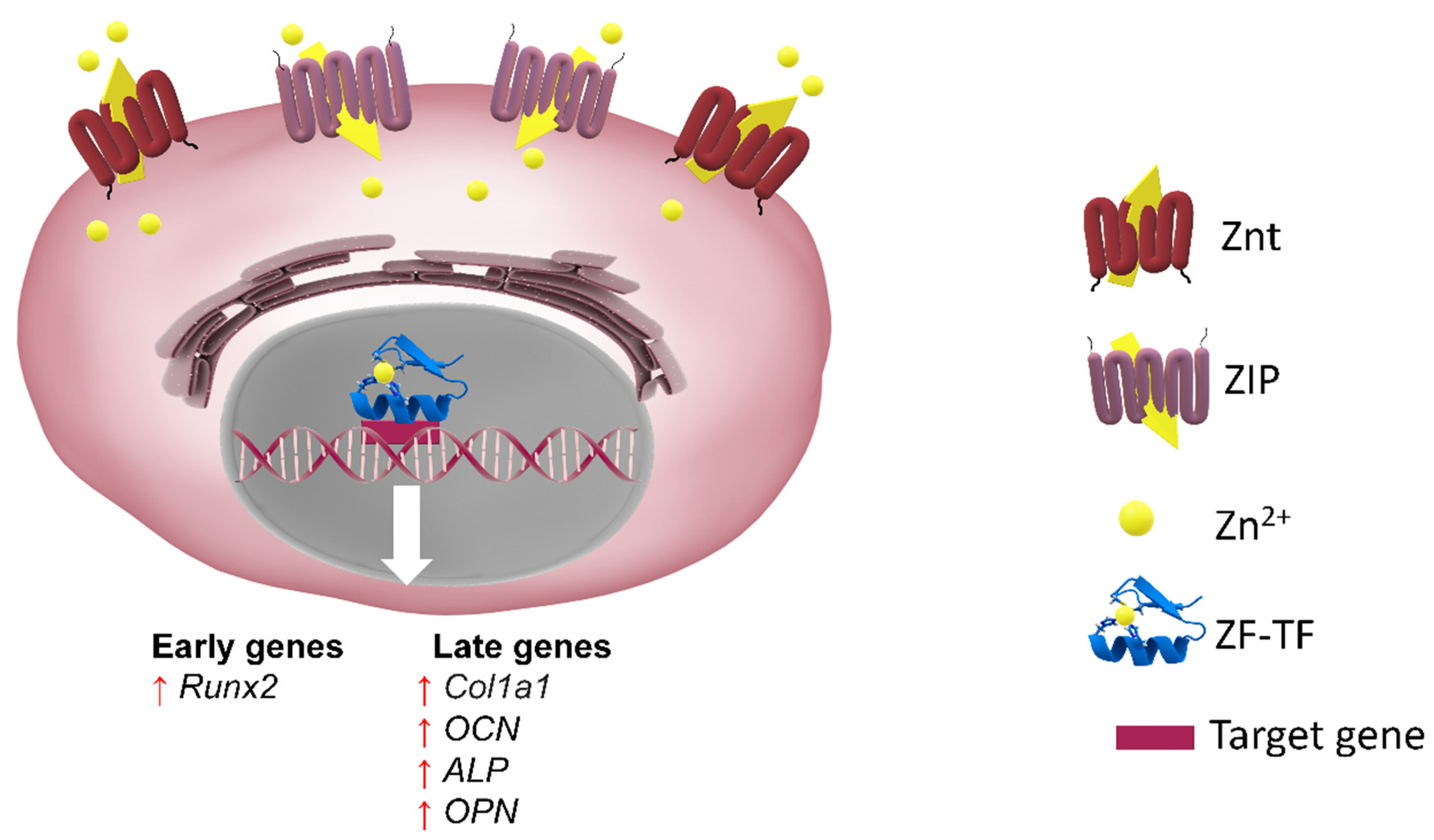{kind=link}
{kind=link}
{kind=link}
| Cell Type | Description | Major Functions | Key Signaling and Pathways |
|---|---|---|---|
| Osteoblasts | differentiate from MSCs but may also derive from bone lining cells [31]; may form a low columnar “epithelioid layer” at sites of bone deposition; are polarized cuboidal cells containing plenty of rough endoplasmic reticulum and large Golgi apparatus [32]; are responsible for bone calcification; once mature, cannot divide and have three possible fates: they can become a bone lining cell or an osteocyte or undergo apoptosis (Figure 1a) [33] | osteoid formation: secretion of type I collagen-rich bone matrix and regulation of matrix mineralization [34] | the RUNX2 transcription factor starts osteoblastogenesis [23]; OSX, a zinc finger transcription factor, regulates transition from osteoprogenitors to pre-osteoblasts; The canonical Wnt signaling pathway promotes OB differentiation, and it is antagonized by the secreted proteins SOST and members of the DKK family synthesized by osteocytes (Figure 1b) [24,35,36,37]; Hedgehog signaling, NOTCH, FGF and BMP [38] promote OB differentiation (Figure 1a) |
| Osteocytes | most abundant cells in bone, >90% of all adult bone cells [33]; derive from mature OBs that, once the osteoid (unmineralized matrix) is mineralized, terminally differentiate into osteocytes end up residing in small lacunae inside the calcified bone matrix; stellate cells with long dendritic processes that ramify in canaliculae; throughout the mineralized bone matrix, interconnection of osteocytes (Figure 1b) is mediated by GAP junctions, connecting osteocytes to bone lining cells and bone marrow cells, in a complex intercellular network [38] | mechanosensor cells that transduce bone loading signals to orchestrate the action of BMU [39,40]; are also involved in mineral homeostasis [41] | major source of RANKL required for osteoclastogenesis during bone remodeling [42,43]; secrete SOST and DKK-1, the negative regulators of Wnt signaling that limit osteoblastic bone formation (Figure 1a); secretion of SOST and DKK-1 is inhibited by mechanical loading, and thus an increased loading corresponds to a local apposition of bone mineralization (Figure 1b) [44] |
| Osteoclasts | multinucleated cells formed by fusion of precursors (derived from HSCs) that share precursors with macrophages; podosomes facilitate adhesion to the bone surface and formation of a sealing zone, providing an isolated acidic resorption bay within which OCs can dissolve calcium salts into soluble forms and digest the bone matrix [45] | bone minerals are dissolved though acidification, and bone matrix is broken down by secretion of lysosomal enzymes that proteolyze organic ECM [46] | differentiation is initiated by M-CSF factor and promoted by RANKL; upon the binding to its cognate receptor RANK on precursor cells [45], osteoclastogenesis is negatively regulated by osteoblast-derived decoy receptor OPG which binds RANKL to inhibit its binding to RANK (Figure 1b) [47] |
| Osteoprogenitor cells | flat squamous cells located in the periosteum (external surface) and endosteum (internal surface)
| constant replenishment of these osteoblastic lineage cells | Ras-MAPK pathway regulates EPK signaling to form the skeletal structure, regulating differentiation of osteoprogenitor cells without changing proliferation [48]; signals transduced by TGFβ superfamily members control the formation of tissue differentiation; further, BMPs activate Smad 1 and 5 as extracellular signals through their effects on cell proliferation, differentiation and migration [49] |
| MSCs | once activated by active TGFβ, they migrate to bone-resorptive sites; can differentiate into osteoblastic lineage | all osteoblast progenitor cells present SOX9 transcription factor [38] | |
| Pre-osteoblasts | heterogeneous population of cells, including those transitioning from MSC cells to mature osteoblasts which express RUNX2 | are a key player in the osteogenic process | mechanistic target of rapamycin (mTOR) integrates both intracellular and extracellular signals to regulate cell growth and cell differentiation [50] |
| Bone lining cells | post-mitotic, long-lived flat osteoblast lineage cells lining the bone surface | can be a source of OBs in response to anabolic stimuli [31] | Wnt signaling [51] |
| Signal Mechanotransduction Mediators | Effects | References |
|---|---|---|
| Mitogen-activated protein kinase (MAPK) signaling pathway | Increases RUNX2, osterix, eNOS osteopontin, osteocalcin and CoX2 and MMP13 expression; RANKL downregulation; increases osteoblast commitment; ATP-dependent activation of calcium channels; integrin activation | [82,91,106,107] |
| PI3K/Akt signaling | Important mitogenic signaling which provokes rapid increase in intracellular calcium levels; activation of IP3, ATP and NO; release of PGE2 | [98,102,108,109,110,111] |
| G protein-mediated signaling | Activation of heterotrimeric GTPases via G protein coupling receptor rises intracellular calcium; cAMP and cGMP activation of rhoA GTPases | [96,112] |
| Wnt/beta-catenin pathway | Increases bone density; the amount of beta-catenin decreases, thus increasing its cytoplasmic concentration, possibly potentiating beta-catenin nuclear translocation; downregulation of sclerostin, thus increasing OB activity | [88,113,114,115,116] |
| Prostaglandins and prostacyclin (eicosanoid-derived phospholipids) | Their exogenous administration stimulates bone formation and increases the sensitivity of bone to external loads (PGE2); their release occurs concurrently with NO; PGE2 increases GAP junction communication and the formation of focal adhesions | [103,117,118] |
| Nitric oxide | Induces activity of NO synthase | [119,120] |
| Stromal cell-derived factor 1 (SDF-1) | Induces differentiation and recruitment of mesenchymal cells; influences cell adhesions and migration | [121,122] |
| Nucleotide signaling | Release of ATP into extracellular space; calcium mobilization; upregulation of RUNX2 | [123] |
| Estrogens | Activation of TGF1 receptor; COX2 gene is induced; ERα a downregulates sclerostin expression, whereas ERβ decreases the osteogenic response to loading | [124,125,126,127] |
| Parameter | Description | Symbol | Unit |
|---|---|---|---|
| Loading pressure | the mechanical stress is a measure of load per unit of area | P | Pa (N/m2) |
| Strain | the ratio of change in length to the original length, when a given body is subjected to some external force (expressed in percentage: change in length/the original length) | ε µε | % % × 10−6 |
| Frequency | number of applied cycles per second or per minute | n w | Hz (1/s) cycles/min |
| Strain rate | temporal change in strain magnitude within each strain cycle | µε/s | 1/s |
| Strain distribution | spatial change in strain magnitude across a given volume | Δµε/d | |
| Strain volume | expresses the total number of daily loading cycles | cpd | cycles/day |
| Trace Nutrients | Sources | Bone Effects |
|---|---|---|
| Boron | It is present mostly in soil and water, meaning the dietary sources are plant-based such as vegetables, fruits and nuts [303] | ↑ Mineralization [304] ↑ Regeneration of bone [305,306] |
| Copper | The best dietary sources are cereals, whole grain products, seeds, nuts and chocolate, as well as shellfish and animal offal [307] | ↑ Matrix stability and strength [308] ↑ Bone differentiation [309] ↑ Bone remodeling [310] |
| Iron | Foods containing the highest amounts of iron are red meat, especially offal, shellfish, pulses, fruits and especially nuts [307] | Maintains bone homeostasis [311] |
| Fluorine | It is present in soil and water; consequently, fruits and vegetables may contain traces of it [312] | ↑ Bone mass and density [313] ↑ Osteoblastogenesis [314,315] |
| Selenium | The main source of selenium is a proper diet, meaning the right selection of animal and plant products [316] | ↑ Protection against oxidative stress [317] ↑ Bone mass [318] |
| Chromium | Good sources are meat and whole grain cereals, some fruits and some vegetables [319] | ↓ Mineralization [320] ↑ Oxidative stress [321] |
| Cobalt | The main sources of Co in the diet are fish, green leafy vegetables and cereals [322] | ↓ Bone modeling [323] ↑ Oxidative stress [321] |
| Cadmium | The environment and smoking are the two main sources of Cd exposure in humans, specifically from contaminated food or drinking water [324] | ↑ Fracture risk [325] ↓ Bone formation [324] ↑ Bone resorption [324] |
| Cell of Origin | Exosomal Cargo | Target Cell | Biological Effect |
|---|---|---|---|
| BMSC | miR-196a, miR-27a, miR-206 | OB | ↑ osteogenesis |
| BMSC | MALAT1 | OB | ↑ osteogenesis |
| BMSC | miR-122-5p | OB | ↑ osteogenesis |
| BMSC | not specified | OB | ↑ osteogenesis |
| OB | miR-667-3p, miR-6769b-5p, miR-7044-5p, miR-7668-3p, miR-874-3p, OPG | BMSC | ↑ osteogenesis |
| OB | ECM proteins (tenascin C, fibronectin, collagen, TRIP1) | ECM; BMSC | ↑ osteogenesis |
| OB | RANKL, TRAP | BMM | ↑ OC genesis |
| OB | miR-125b | OC | ↓ OC genesis |
| OB | MMP2 | Endothelium | ↑ angiogenesis |
| OC | miR-214-3p | OB | ↓ osteogenesis |
| OC | miR-23a-5p | OB | ↓ osteogenesis |
| OC | RANK receptor | OC | ↑ osteogenesis |
| Osteocyte | SOST, RANKL, OPG | OB | ↑ osteogenesis |
| Osteocyte | miR-218 | OB | ↓ osteogenesis |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alloisio, G.; Ciaccio, C.; Fasciglione, G.F.; Tarantino, U.; Marini, S.; Coletta, M.; Gioia, M. Effects of Extracellular Osteoanabolic Agents on the Endogenous Response of Osteoblastic Cells. Cells 2021, 10, 2383. https://doi.org/10.3390/cells10092383
Alloisio G, Ciaccio C, Fasciglione GF, Tarantino U, Marini S, Coletta M, Gioia M. Effects of Extracellular Osteoanabolic Agents on the Endogenous Response of Osteoblastic Cells. Cells. 2021; 10(9):2383. https://doi.org/10.3390/cells10092383
Chicago/Turabian StyleAlloisio, Giulia, Chiara Ciaccio, Giovanni Francesco Fasciglione, Umberto Tarantino, Stefano Marini, Massimo Coletta, and Magda Gioia. 2021. "Effects of Extracellular Osteoanabolic Agents on the Endogenous Response of Osteoblastic Cells" Cells 10, no. 9: 2383. https://doi.org/10.3390/cells10092383
APA StyleAlloisio, G., Ciaccio, C., Fasciglione, G. F., Tarantino, U., Marini, S., Coletta, M., & Gioia, M. (2021). Effects of Extracellular Osteoanabolic Agents on the Endogenous Response of Osteoblastic Cells. Cells, 10(9), 2383. https://doi.org/10.3390/cells10092383