
Regulation of Rac1 Activation in Choroidal Endothelial Cells: Insights into Mechanisms in Age-Related Macular Degeneration

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
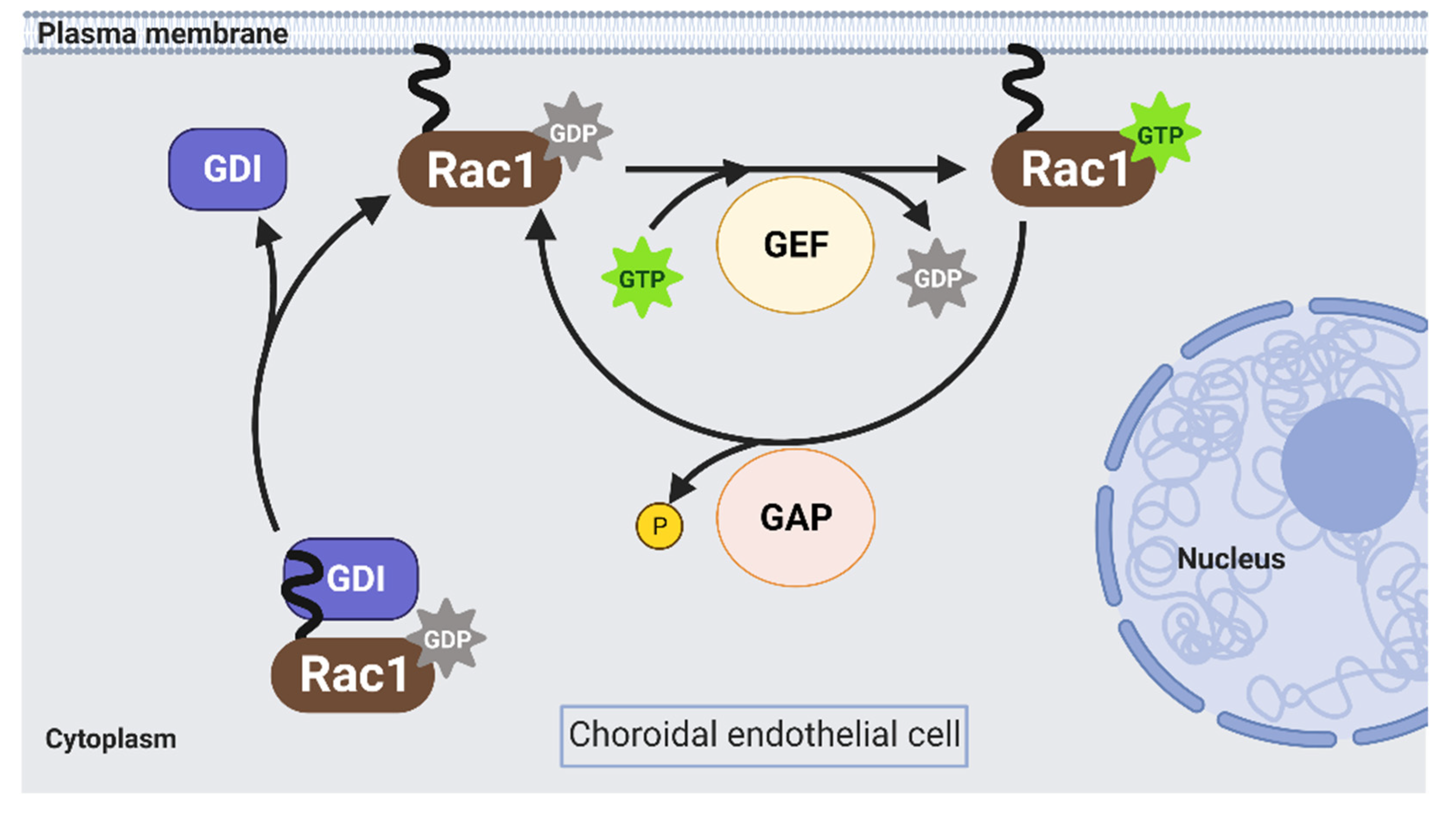
2. Activation of Rac1 GTPase in Endothelial Cells
2.1. Rho GEFs
2.2. Rho GAPs
2.3. Rho GDIs
2.4. Limitations of Regulating Rac1 Activation by Targeting Rho Gefs, Gaps and Gdis
3. Activated Rac1-Mediated Effectors in Endothelial Cells
3.1. PAKs
3.2. NADPH Oxidase (NOX)
3.3. IQGAPs
4. Active Rap1a Interferes with Rac1 Activation in Choroidal Endothelial Cells
4.1. Activation of Rap1 Reduces CNV
4.2. Active Rap1 Interacts with IQGAP1 to Interfere with Rac1 Activation in Choroidal Endothelial Cells
5. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wong, W.L.; Su, X.; Li, X.; Cheung, C.M.; Klein, R.; Cheng, C.Y.; Wong, T.Y. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta-analysis. Lancet. Glob. Health. 2014, 2, e106–e116. [Google Scholar] [CrossRef] [Green Version]
- Sassmannshausen, M.; Zhou, J.; Pfau, M.; Thiele, S.; Steinberg, J.; Fleckenstein, M.; Holz, F.G.; Schmitz-Valckenberg, S. Longitudinal Analysis of Retinal Thickness and Retinal Function in Eyes with Large Drusen Secondary to Intermediate Age-Related Macular Degeneration. Ophthalmology. Retin 2021, 5, 241–250. [Google Scholar] [CrossRef]
- Colijn, J.M.; Liefers, B.; Joachim, N.; Verzijden, T.; Meester-Smoor, M.A.; Biarnes, M.; Mones, J.; de Jong, P.; Vingerling, J.R.; Mitchell, P.; et al. Enlargement of Geographic Atrophy From First Diagnosis to End of Life. JAMA Ophthalmol. 2021, 139, 743–750. [Google Scholar] [CrossRef]
- Fleckenstein, M.; Mitchell, P.; Freund, K.B.; Sadda, S.; Holz, F.G.; Brittain, C.; Henry, E.C.; Ferrara, D. The Progression of Geographic Atrophy Secondary to Age-Related Macular Degeneration. Ophthalmology 2018, 125, 369–390. [Google Scholar] [CrossRef]
- Mittra, R.A.; Singerman, L.J. Recent advances in the management of age-related macular degeneration. Optom. Vis. Sci. 2002, 79, 218–224. [Google Scholar] [CrossRef] [Green Version]
- Gehrs, K.M.; Anderson, D.H.; Johnson, L.V.; Hageman, G.S. Age-related macular degeneration--emerging pathogenetic and therapeutic concepts. Ann. Med. 2006, 38, 450–471. [Google Scholar] [CrossRef]
- Wong, T.Y.; Chakravarthy, U.; Klein, R.; Mitchell, P.; Zlateva, G.; Buggage, R.; Fahrbach, K.; Probst, C.; Sledge, I. The natural history and prognosis of neovascular age-related macular degeneration: A systematic review of the literature and meta-analysis. Ophthalmology 2008, 115, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Pfau, M.; Möller, P.T.; Künzel, S.H.; von der Emde, L.; Lindner, M.; Thiele, S.; Dysli, C.; Nadal, J.; Schmid, M.; Schmitz-Valckenberg, S.; et al. Type 1 Choroidal Neovascularization Is Associated with Reduced Localized Progression of Atrophy in Age-Related Macular Degeneration. Ophthalmol. Retin. 2020, 4, 238–248. [Google Scholar] [CrossRef]
- Grossniklaus, H.E.; Gass, J.D. Clinicopathologic correlations of surgically excised type 1 and type 2 submacular choroidal neovascular membranes. Am. J. Ophthalmol. 1998, 126, 59–69. [Google Scholar] [CrossRef]
- Bressler, N.M.; Frost, L.A.; Bressler, S.B.; Murphy, R.P.; Fine, S.L. Natural course of poorly defined choroidal neovascularization associated with macular degeneration. Arch. Ophthalmol. 1988, 106, 1537–1542. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.S.; Freund, K.B.; de la Cruz, Z.; Yannuzzi, L.A.; Green, W.R. Clinicopathologic correlation of choroidal neovascularization demonstrated by indocyanine green angiography in a patient with retention of good vision for almost four years. Retin 1994, 14, 114–124. [Google Scholar] [CrossRef]
- Jalkh, A.E.; Nasrallah, F.P.; Marinelli, I.; Van de Velde, F. Inactive subretinal neovascularization in age-related macular degeneration. Ophthalmology 1990, 97, 1614–1618, Discuss 1618–1619. [Google Scholar] [CrossRef]
- Hartnett, M.E.; Elsner, A.E. Characteristics of exudative age-related macular degeneration determined in vivo with confocal and indirect infrared imaging. Ophthalmology 1996, 103, 58–71. [Google Scholar] [CrossRef]
- Stevens, T.S.; Bressler, N.M.; Maguire, M.G.; Bressler, S.B.; Fine, S.L.; Alexander, J.; Phillips, D.A.; Margherio, R.R.; Murphy, P.L.; Schachat, A.P. Occult choroidal neovascularization in age-related macular degeneration. A natural history study. Arch. Ophthalmol. 1997, 115, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Spaide, R.F.; Jaffe, G.J.; Sarraf, D.; Freund, K.B.; Sadda, S.R.; Staurenghi, G.; Waheed, N.K.; Chakravarthy, U.; Rosenfeld, P.J.; Holz, F.G.; et al. Consensus Nomenclature for Reporting Neovascular Age-Related Macular Degeneration Data: Consensus on Neovascular Age-Related Macular Degeneration Nomenclature Study Group. Ophthalmology 2020, 127, 616–636. [Google Scholar] [CrossRef]
- Seddon, J.M.; George, S.; Rosner, B. Cigarette smoking, fish consumption, omega-3 fatty acid intake, and associations with age-related macular degeneration: The US Twin Study of Age-Related Macular Degeneration. Arch. Ophthalmol. 2006, 124, 995–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seddon, J.M.; Silver, R.E.; Kwong, M.; Rosner, B. Risk Prediction for Progression of Macular Degeneration: 10 Common and Rare Genetic Variants, Demographic, Environmental, and Macular Covariates. Investig. Ophthalmol. Vis. Sci. 2015, 56, 2192–2202. [Google Scholar] [CrossRef]
- Sobrin, L.; Seddon, J.M. Nature and nurture- genes and environment- predict onset and progression of macular degeneration. Prog. Retin. Eye. Res. 2014, 40, 1–15. [Google Scholar] [CrossRef]
- Fritsche, L.G.; Igl, W.; Bailey, J.N.; Grassmann, F.; Sengupta, S.; Bragg-Gresham, J.L.; Burdon, K.P.; Hebbring, S.J.; Wen, C.; Gorski, M.; et al. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat. Genet. 2016, 48, 134–143. [Google Scholar] [CrossRef] [Green Version]
- Swaroop, A.; Branham, K.E.; Chen, W.; Abecasis, G. Genetic susceptibility to age-related macular degeneration: A paradigm for dissecting complex disease traits. Hum. Mol. Genet. 2007, 16, R174–R182. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Hartnett, M.E. Regulation of signaling events involved in the pathophysiology of neovascular AMD. Mol. Vis. 2016, 22, 189–202. [Google Scholar] [PubMed]
- Jaffe, G.J.; Ying, G.S.; Toth, C.A.; Daniel, E.; Grunwald, J.E.; Martin, D.F.; Maguire, M.G. Macular Morphology and Visual Acuity in Year Five of the Comparison of Age-related Macular Degeneration Treatments Trials. Ophthalmology 2019, 126, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Lamalice, L.; Le Boeuf, F.; Huot, J. Endothelial cell migration during angiogenesis. Circ. Res. 2007, 100, 782–794. [Google Scholar] [CrossRef]
- Geisen, P.; McColm, J.R.; King, B.M.; Hartnett, M.E. Characterization of barrier properties and inducible VEGF expression of several types of retinal pigment epithelium in medium-term culture. Curr. Eye. Res. 2006, 31, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Peterson, L.J.; Wittchen, E.S.; Geisen, P.; Burridge, K.; Hartnett, M.E. Heterotypic RPE-choroidal endothelial cell contact increases choroidal endothelial cell transmigration via PI 3-kinase and Rac1. Exp. Eye. Res. 2007, 84, 737–744. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Fotheringham, L.; Wittchen, E.S.; Hartnett, M.E. Rap1 GTPase Inhibits Tumor Necrosis Factor-alpha-Induced Choroidal Endothelial Migration via NADPH Oxidase- and NF-kappaB-Dependent Activation of Rac1. Am. J. Pathol. 2015, 185, 3316–3325. [Google Scholar] [CrossRef] [Green Version]
- Monaghan-Benson, E.; Hartmann, J.; Vendrov, A.E.; Budd, S.; Byfield, G.; Parker, A.; Ahmad, F.; Huang, W.; Runge, M.; Burridge, K.; et al. The role of vascular endothelial growth factor-induced activation of NADPH oxidase in choroidal endothelial cells and choroidal neovascularization. Am. J. Pathol. 2010, 177, 2091–2102. [Google Scholar] [CrossRef]
- Wang, H.; Geisen, P.; Wittchen, E.S.; King, B.; Burridge, K.; D’Amore, P.A.; Hartnett, M.E. The role of RPE cell-associated VEGF(1)(8)(9) in choroidal endothelial cell transmigration across the RPE. Investig. Ophthalmol. Vis. Sci. 2011, 52, 570–578. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Han, X.; Kunz, E.; Hartnett, M.E. Thy-1 Regulates VEGF-Mediated Choroidal Endothelial Cell Activation and Migration: Implications in Neovascular Age-Related Macular Degeneration. Investig. Ophthalmol. Vis. Sci. 2016, 57, 5525–5534. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Wittchen, E.S.; Jiang, Y.; Ambati, B.; Grossniklaus, H.E.; Hartnett, M.E. Upregulation of CCR3 by age-related stresses promotes choroidal endothelial cell migration via VEGF-dependent and -independent signaling. Investig. Ophthalmol. Vis. Sci. 2011, 52, 8271–8277. [Google Scholar] [CrossRef]
- Ramshekar, A.; Wang, H.; Kunz, E.; Pappas, C.; Hageman, G.S.; Chaqour, B.; Sacks, D.B.; Hartnett, M.E. Active Rap1-mediated inhibition of choroidal neovascularization requires interactions with IQGAP1 in choroidal endothelial cells. FASEB. J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2021, 35, e21642. [Google Scholar] [CrossRef]
- Wang, H.; Ramshekar, A.; Kunz, E.; Sacks, D.B.; Hartnett, M.E. IQGAP1 causes choroidal neovascularization by sustaining VEGFR2-mediated Rac1 activation. Angiogenesis 2020, 23, 685–698. [Google Scholar] [CrossRef]
- Wang, H.; Ramshekar, A.; Kunz, E.; Hartnett, M.E. 7-ketocholesterol induces endothelial-mesenchymal transition and promotes fibrosis: Implications in neovascular age-related macular degeneration and treatment. Angiogenesis 2021, 24, 583–595. [Google Scholar] [CrossRef]
- Khan, K.A.; McMurray, J.L.; Mohammed, F.; Bicknell, R. C-type lectin domain group 14 proteins in vascular biology, cancer and inflammation. FEBS J. 2019, 286, 3299–3332. [Google Scholar] [CrossRef]
- Tosi, G.M.; Caldi, E.; Parolini, B.; Toti, P.; Neri, G.; Nardi, F.; Traversi, C.; Cevenini, G.; Marigliani, D.; Nuti, E.; et al. CD93 as a Potential Target in Neovascular Age-Related Macular Degeneration. J. Cell. Physiol. 2017, 232, 1767–1773. [Google Scholar] [CrossRef] [PubMed]
- Tosi, G.M.; Neri, G.; Barbera, S.; Mundo, L.; Parolini, B.; Lazzi, S.; Lugano, R.; Poletto, E.; Leoncini, L.; Pertile, G.; et al. The Binding of CD93 to Multimerin-2 Promotes Choroidal Neovascularization. Investig. Ophthalmol. Vis. Sci. 2020, 61, 30. [Google Scholar] [CrossRef] [PubMed]
- Barbera, S.; Lugano, R.; Pedalina, A.; Mongiat, M.; Santucci, A.; Tosi, G.M.; Dimberg, A.; Galvagni, F.; Orlandini, M. The C-type lectin CD93 controls endothelial cell migration via activation of the Rho family of small GTPases. Matrix. Biol. 2021, 99, 1–17. [Google Scholar] [CrossRef]
- van Buul, J.D.; Geerts, D.; Huveneers, S. Rho GAPs and GEFs: Controling switches in endothelial cell adhesion. Cell. Adh. Migr. 2014, 8, 108–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez-Garcia, R.; Iruela-Arispe, M.L.; Reyes-Cruz, G.; Vazquez-Prado, J. Endothelial RhoGEFs: A systematic analysis of their expression profiles in VEGF-stimulated and tumor endothelial cells. Vasc. Pharm. 2015, 74, 60–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrett, T.A.; Van Buul, J.D.; Burridge, K. VEGF-induced Rac1 activation in endothelial cells is regulated by the guanine nucleotide exchange factor Vav2. Exp. Cell. Res. 2007, 313, 3285–3297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Epting, D.; Wendik, B.; Bennewitz, K.; Dietz, C.T.; Driever, W.; Kroll, J. The Rac1 regulator ELMO1 controls vascular morphogenesis in zebrafish. Circ. Res. 2010, 107, 45–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanematsu, F.; Hirashima, M.; Laurin, M.; Takii, R.; Nishikimi, A.; Kitajima, K.; Ding, G.; Noda, M.; Murata, Y.; Tanaka, Y.; et al. DOCK180 is a Rac activator that regulates cardiovascular development by acting downstream of CXCR4. Circ. Res. 2010, 107, 1102–1105. [Google Scholar] [CrossRef]
- Wang, Y.; Yan, F.; Ye, Q.; Wu, X.; Jiang, F. PTP1B inhibitor promotes endothelial cell motility by activating the DOCK180/Rac1 pathway. Sci. Rep. 2016, 6, 24111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benson, C.E.; Southgate, L. The DOCK protein family in vascular development and disease. Angiogenesis 2021, 24, 417–433. [Google Scholar] [CrossRef] [PubMed]
- Aitsebaomo, J.; Wennerberg, K.; Der, C.J.; Zhang, C.; Kedar, V.; Moser, M.; Kingsley-Kallesen, M.L.; Zeng, G.Q.; Patterson, C. p68RacGAP is a novel GTPase-activating protein that interacts with vascular endothelial zinc finger-1 and modulates endothelial cell capillary formation. J. Biol. Chem. 2004, 279, 17963–17972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, Z.J.; Hahn, C.N.; Goodall, G.J.; Reck, N.M.; Leske, A.F.; Davy, A.; Kremmidiotis, G.; Vadas, M.A.; Gamble, J.R. A vascular cell-restricted RhoGAP, p73RhoGAP, is a key regulator of angiogenesis. Proc. Natl. Acad. Sci. USA 2004, 101, 12212–12217. [Google Scholar] [CrossRef] [Green Version]
- DerMardirossian, C.; Bokoch, G.M. GDIs: Central regulatory molecules in Rho GTPase activation. Trends. Cell. Biol. 2005, 15, 356–363. [Google Scholar] [CrossRef]
- Scheffzek, K.; Stephan, I.; Jensen, O.N.; Illenberger, D.; Gierschik, P. The Rac-RhoGDI complex and the structural basis for the regulation of Rho proteins by RhoGDI. Nat. Struct. Biol. 2000, 7, 122–126. [Google Scholar] [CrossRef]
- Majolee, J.; Podieh, F.; Hordijk, P.L.; Kovacevic, I. The interplay of Rac1 activity, ubiquitination and GDI binding and its consequences for endothelial cell spreading. PLoS ONE 2021, 16, e0254386. [Google Scholar] [CrossRef]
- Bid, H.K.; Roberts, R.D.; Manchanda, P.K.; Houghton, P.J. RAC1: An emerging therapeutic option for targeting cancer angiogenesis and metastasis. Mol. Cancer 2013, 12, 1925–1934. [Google Scholar] [CrossRef] [Green Version]
- Bergers, G.; Benjamin, L.E. Tumorigenesis and the angiogenic switch. Nat. Rev. Cancer 2003, 3, 401–410. [Google Scholar] [CrossRef]
- Akbar, H.; Cancelas, J.; Williams, D.A.; Zheng, J.; Zheng, Y. Rational design and applications of a Rac GTPase-specific small molecule inhibitor. Methods Enzym. 2006, 406, 554–565. [Google Scholar] [CrossRef]
- Grizot, S.; Faure, J.; Fieschi, F.; Vignais, P.V.; Dagher, M.C.; Pebay-Peyroula, E. Crystal structure of the Rac1-RhoGDI complex involved in nadph oxidase activation. Biochemistry 2001, 40, 10007–10013. [Google Scholar] [CrossRef] [PubMed]
- Manser, E.; Leung, T.; Salihuddin, H.; Zhao, Z.S.; Lim, L. A brain serine/threonine protein kinase activated by Cdc42 and Rac1. Nature 1994, 367, 40–46. [Google Scholar] [CrossRef]
- Lu, W.; Katz, S.; Gupta, R.; Mayer, B.J. Activation of Pak by membrane localization mediated by an SH3 domain from the adaptor protein Nck. Curr. Biol. 1997, 7, 85–94. [Google Scholar] [CrossRef] [Green Version]
- Kiosses, W.B.; Hood, J.; Yang, S.; Gerritsen, M.E.; Cheresh, D.A.; Alderson, N.; Schwartz, M.A. A dominant-negative p65 PAK peptide inhibits angiogenesis. Circ. Res. 2002, 90, 697–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stockton, R.A.; Schaefer, E.; Schwartz, M.A. p21-activated kinase regulates endothelial permeability through modulation of contractility. J. Biol. Chem. 2004, 279, 46621–46630. [Google Scholar] [CrossRef] [Green Version]
- Parrini, M.C.; Lei, M.; Harrison, S.C.; Mayer, B.J. Pak1 kinase homodimers are autoinhibited in trans and dissociated upon activation by Cdc42 and Rac1. Mol. Cell. 2002, 9, 73–83. [Google Scholar] [CrossRef]
- Lei, M.; Lu, W.; Meng, W.; Parrini, M.C.; Eck, M.J.; Mayer, B.J.; Harrison, S.C. Structure of PAK1 in an autoinhibited conformation reveals a multistage activation switch. Cell 2000, 102, 387–397. [Google Scholar] [CrossRef] [Green Version]
- Panday, A.; Sahoo, M.K.; Osorio, D.; Batra, S. NADPH oxidases: An overview from structure to innate immunity-associated pathologies. Cell. Mol. Immunol. 2015, 12, 5–23. [Google Scholar] [CrossRef] [Green Version]
- Babior, B.M. The NADPH oxidase of endothelial cells. IUBMB Life 2000, 50, 267–269. [Google Scholar] [CrossRef]
- Li, J.M.; Shah, A.M. Endothelial cell superoxide generation: Regulation and relevance for cardiovascular pathophysiology. Am J Physiol Regul Integr Comp. Physiol. 2004, 287, R1014–R1030. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Hartnett, M.E. Roles of Nicotinamide Adenine Dinucleotide Phosphate (NADPH) Oxidase in Angiogenesis: Isoform-Specific Effects. Antioxidants 2017, 6, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, V.; Lecomte, J.; Hansen, S.; Blacher, S.; Gonzalez, M.L.; Struman, I.; Sounni, N.E.; Rozet, E.; de Tullio, P.; Foidart, J.M.; et al. Laser-induced choroidal neovascularization model to study age-related macular degeneration in mice. Nat. Protoc. 2013, 8, 2197–2211. [Google Scholar] [CrossRef]
- Tojo, T.; Ushio-Fukai, M.; Yamaoka-Tojo, M.; Ikeda, S.; Patrushev, N.; Alexander, R.W. Role of gp91phox (Nox2)-containing NAD(P)H oxidase in angiogenesis in response to hindlimb ischemia. Circ 2005, 111, 2347–2355. [Google Scholar] [CrossRef] [Green Version]
- Urao, N.; Inomata, H.; Razvi, M.; Kim, H.W.; Wary, K.; McKinney, R.; Fukai, T.; Ushio-Fukai, M. Role of nox2-based NADPH oxidase in bone marrow and progenitor cell function involved in neovascularization induced by hindlimb ischemia. Circ. Res. 2008, 103, 212–220. [Google Scholar] [CrossRef]
- Dong, F.; Li, L.; Chen, X.; Allen, T.; Liu, J. Glomerular endothelial cell IQGAP2 and filtration barrier function. Kidney. Int. 2016, 89, 1160–1161. [Google Scholar] [CrossRef]
- Hedman, A.C.; Smith, J.M.; Sacks, D.B. The biology of IQGAP proteins: Beyond the cytoskeleton. EMBO Rep. 2015, 16, 427–446. [Google Scholar] [CrossRef] [Green Version]
- Abel, A.M.; Schuldt, K.M.; Rajasekaran, K.; Hwang, D.; Riese, M.J.; Rao, S.; Thakar, M.S.; Malarkannan, S. IQGAP1: Insights into the function of a molecular puppeteer. Mol. Immunol. 2015, 65, 336–349. [Google Scholar] [CrossRef] [Green Version]
- Yamaoka-Tojo, M.; Ushio-Fukai, M.; Hilenski, L.; Dikalov, S.I.; Chen, Y.E.; Tojo, T.; Fukai, T.; Fujimoto, M.; Patrushev, N.A.; Wang, N.; et al. IQGAP1, a novel vascular endothelial growth factor receptor binding protein, is involved in reactive oxygen species--dependent endothelial migration and proliferation. Circ. Res. 2004, 95, 276–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeda, S.; Yamaoka-Tojo, M.; Hilenski, L.; Patrushev, N.A.; Anwar, G.M.; Quinn, M.T.; Ushio-Fukai, M. IQGAP1 regulates reactive oxygen species-dependent endothelial cell migration through interacting with Nox2. Arter. Thromb. Vasc. Biol. 2005, 25, 2295–2300. [Google Scholar] [CrossRef] [Green Version]
- Berdyshev, E.V.; Gorshkova, I.; Usatyuk, P.; Kalari, S.; Zhao, Y.; Pyne, N.J.; Pyne, S.; Sabbadini, R.A.; Garcia, J.G.; Natarajan, V. Intracellular S1P generation is essential for S1P-induced motility of human lung endothelial cells: Role of sphingosine kinase 1 and S1P lyase. PLoS ONE 2011, 6, e16571. [Google Scholar] [CrossRef] [Green Version]
- Kuroda, S.; Fukata, M.; Kobayashi, K.; Nakafuku, M.; Nomura, N.; Iwamatsu, A.; Kaibuchi, K. Identification of IQGAP as a putative target for the small GTPases, Cdc42 and Rac1. J. Biol. Chem. 1996, 271, 23363–23367. [Google Scholar] [CrossRef] [Green Version]
- Hart, M.J.; Callow, M.G.; Souza, B.; Polakis, P. IQGAP1, a calmodulin-binding protein with a rasGAP-related domain, is a potential effector for cdc42Hs. EMBO J. 1996, 15, 2997–3005. [Google Scholar] [CrossRef]
- Kurella, V.B.; Richard, J.M.; Parke, C.L.; Lecour, L.F., Jr.; Bellamy, H.D.; Worthylake, D.K. Crystal structure of the GTPase-activating protein-related domain from IQGAP1. J. Biol. Chem. 2009, 284, 14857–14865. [Google Scholar] [CrossRef] [Green Version]
- Weissbach, L.; Settleman, J.; Kalady, M.F.; Snijders, A.J.; Murthy, A.E.; Yan, Y.X.; Bernards, A. Identification of a human rasGAP-related protein containing calmodulin-binding motifs. J. Biol. Chem. 1994, 269, 20517–20521. [Google Scholar] [CrossRef]
- Mataraza, J.M.; Briggs, M.W.; Li, Z.; Frank, R.; Sacks, D.B. Identification and characterization of the Cdc42-binding site of IQGAP1. Biochem. Biophys. Res. Commun. 2003, 305, 315–321. [Google Scholar] [CrossRef]
- Jameson, K.L.; Mazur, P.K.; Zehnder, A.M.; Zhang, J.; Zarnegar, B.; Sage, J.; Khavari, P.A. IQGAP1 scaffold-kinase interaction blockade selectively targets RAS-MAP kinase-driven tumors. Nat. Med. 2013, 19, 626–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mateer, S.C.; McDaniel, A.E.; Nicolas, V.; Habermacher, G.M.; Lin, M.J.; Cromer, D.A.; King, M.E.; Bloom, G.S. The mechanism for regulation of the F-actin binding activity of IQGAP1 by calcium/calmodulin. J. Biol. Chem. 2002, 277, 12324–12333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, C.D.; Erdemir, H.H.; Sacks, D.B. IQGAP1 and its binding proteins control diverse biological functions. Cell. Signal. 2012, 24, 826–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joyal, J.L.; Burks, D.J.; Pons, S.; Matter, W.F.; Vlahos, C.J.; White, M.F.; Sacks, D.B. Calmodulin activates phosphatidylinositol 3-kinase. J. Biol. Chem. 1997, 272, 28183–28186. [Google Scholar] [CrossRef] [Green Version]
- Ho, Y.D.; Joyal, J.L.; Li, Z.; Sacks, D.B. IQGAP1 integrates Ca2+/calmodulin and Cdc42 signaling. J. Biol. Chem. 1999, 274, 464–470. [Google Scholar] [CrossRef] [Green Version]
- Pannekoek, W.J.; Kooistra, M.R.; Zwartkruis, F.J.; Bos, J.L. Cell-cell junction formation: The role of Rap1 and Rap1 guanine nucleotide exchange factors. Biochim. Biophys. Acta. 2009, 1788, 790–796. [Google Scholar] [CrossRef] [Green Version]
- Bos, J.L.; de Rooij, J.; Reedquist, K.A. Rap1 signalling: Adhering to new models. Nature reviews. Mol. Cell. Biol. 2001, 2, 369–377. [Google Scholar] [CrossRef]
- Pannekoek, W.J.; Post, A.; Bos, J.L. Rap1 signaling in endothelial barrier control. Cell. Adh. Migr. 2014, 8, 100–107. [Google Scholar] [CrossRef] [Green Version]
- Wittchen, E.S.; Worthylake, R.A.; Kelly, P.; Casey, P.J.; Quilliam, L.A.; Burridge, K. Rap1 GTPase inhibits leukocyte transmigration by promoting endothelial barrier function. J. Biol. Chem. 2005, 280, 11675–11682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos, C.J.; Lin, C.; Liu, X.; Antonetti, D.A. The EPAC-Rap1 pathway prevents and reverses cytokine-induced retinal vascular permeability. J. Biol. Chem. 2018, 293, 717–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wittchen, E.S.; Nishimura, E.; McCloskey, M.; Wang, H.; Quilliam, L.A.; Chrzanowska-Wodnicka, M.; Hartnett, M.E. Rap1 GTPase Activation and Barrier Enhancement in RPE Inhibits Choroidal Neovascularization In Vivo. PLoS ONE 2013, 8, e73070. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zhang, R.; Wang, C.; Wang, X.; Xu, M.; Ma, J.; Shang, Q. Activation of the Small GTPase Rap1 Inhibits Choroidal Neovascularization by Regulating Cell Junctions and ROS Generation in Rats. Curr. Eye Res. 2018, 43, 934–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borland, G.; Smith, B.O.; Yarwood, S.J. EPAC proteins transduce diverse cellular actions of cAMP. Br. J. Pharm. 2009, 158, 70–86. [Google Scholar] [CrossRef] [Green Version]
- Carmona, G.; Gottig, S.; Orlandi, A.; Scheele, J.; Bauerle, T.; Jugold, M.; Kiessling, F.; Henschler, R.; Zeiher, A.M.; Dimmeler, S.; et al. Role of the small GTPase Rap1 for integrin activity regulation in endothelial cells and angiogenesis. Blood 2009, 113, 488–497. [Google Scholar] [CrossRef] [PubMed]
- Chrzanowska-Wodnicka, M.; Kraus, A.E.; Gale, D.; White, G.C., 2nd; Vansluys, J. Defective angiogenesis, endothelial migration, proliferation, and MAPK signaling in Rap1b-deficient mice. Blood 2008, 111, 2647–2656. [Google Scholar] [CrossRef] [Green Version]
- Chrzanowska-Wodnicka, M. Rap1 in endothelial biology. Curr. Opin. Hematol. 2017, 24, 248–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, H.W.; Li, Z.; Brown, M.D.; Sacks, D.B. IQGAP1 binds Rap1 and modulates its activity. J. Biol. Chem. 2007, 282, 20752–20762. [Google Scholar] [CrossRef] [Green Version]
- Malarkannan, S.; Awasthi, A.; Rajasekaran, K.; Kumar, P.; Schuldt, K.M.; Bartoszek, A.; Manoharan, N.; Goldner, N.K.; Umhoefer, C.M.; Thakar, M.S. IQGAP1: A regulator of intracellular spacetime relativity. J. Immunol. 2012, 188, 2057–2063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Sacks, D.B. Elucidation of the interaction of calmodulin with the IQ motifs of IQGAP1. J. Biol. Chem. 2003, 278, 4347–4352. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramshekar, A.; Wang, H.; Hartnett, M.E. Regulation of Rac1 Activation in Choroidal Endothelial Cells: Insights into Mechanisms in Age-Related Macular Degeneration. Cells 2021, 10, 2414. https://doi.org/10.3390/cells10092414
Ramshekar A, Wang H, Hartnett ME. Regulation of Rac1 Activation in Choroidal Endothelial Cells: Insights into Mechanisms in Age-Related Macular Degeneration. Cells. 2021; 10(9):2414. https://doi.org/10.3390/cells10092414
Chicago/Turabian StyleRamshekar, Aniket, Haibo Wang, and M. Elizabeth Hartnett. 2021. "Regulation of Rac1 Activation in Choroidal Endothelial Cells: Insights into Mechanisms in Age-Related Macular Degeneration" Cells 10, no. 9: 2414. https://doi.org/10.3390/cells10092414